
FAK Inhibition Attenuates Corneal Fibroblast Differentiation In Vitro

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. 3D Construct Assembly
2.3. Human Fibrosis and Wound Healing RT2 Profiler PCR Array
2.4. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.5. Western Blots
2.6. Immunofluorescence Staining
2.7. Statistical Analysis
3. Results
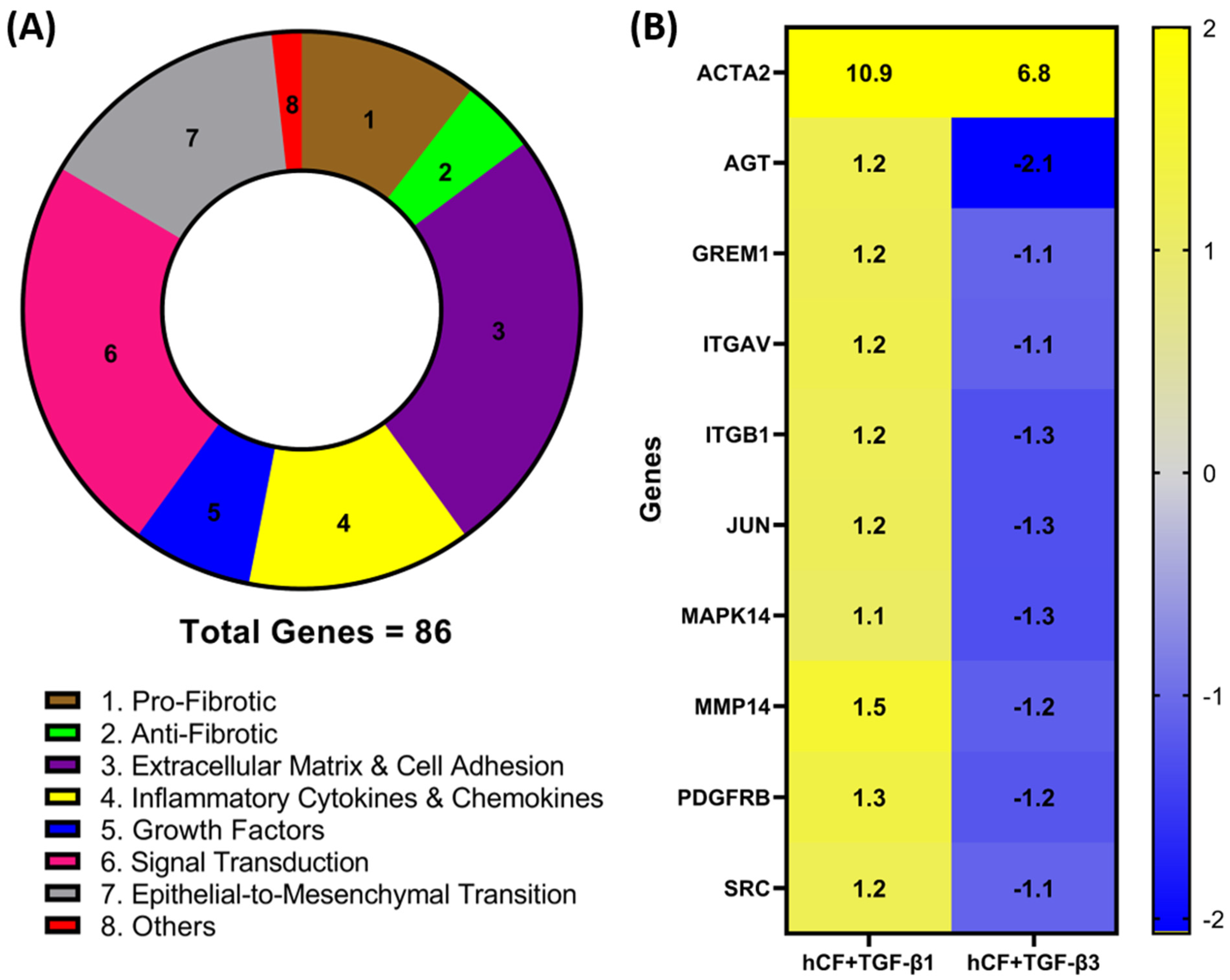
3.1. Identification of Differentially Expressed Genes in hCF Constructs following TGF-β1 or TGF-β3 Treatment
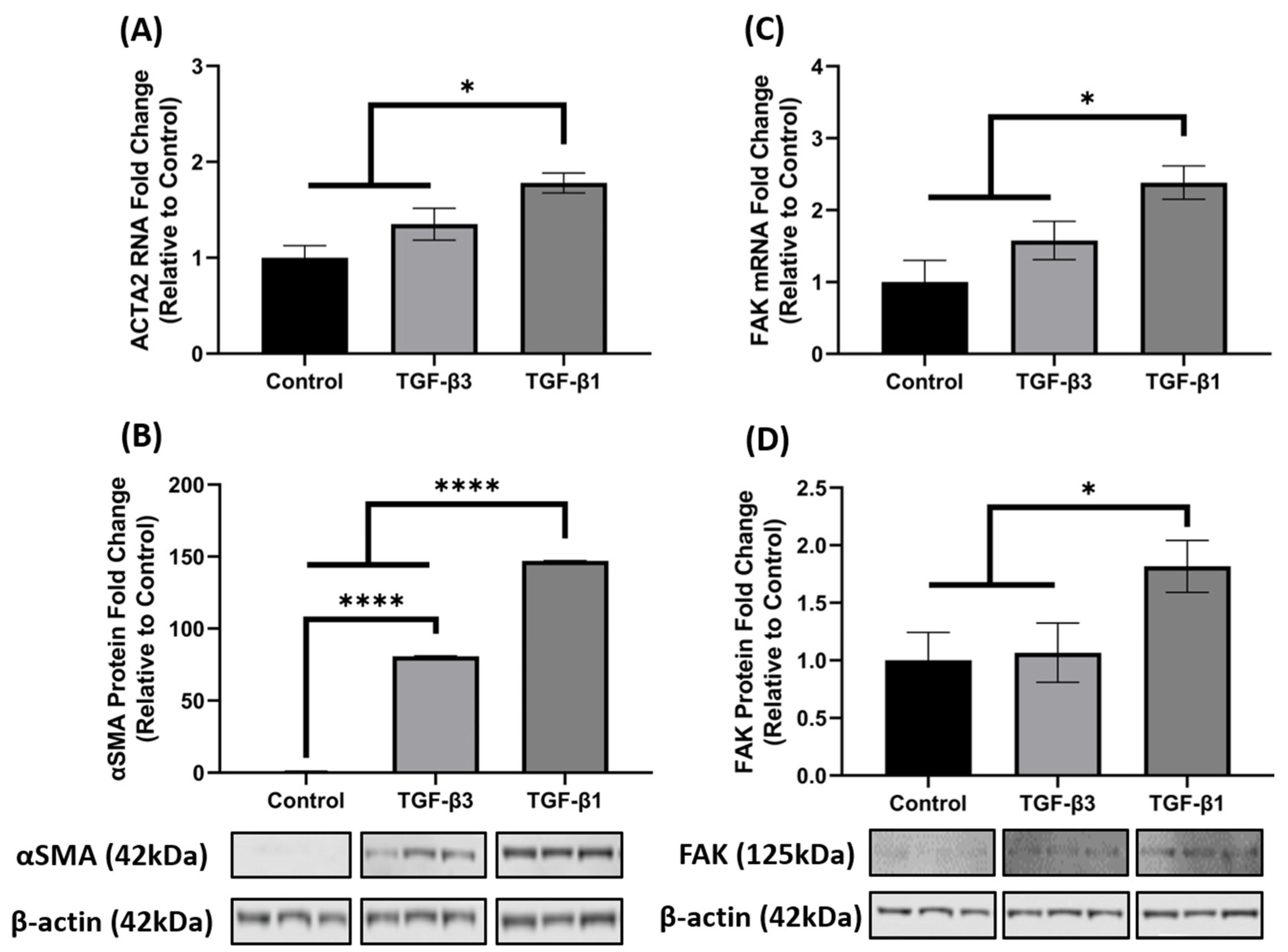
3.2. TGF-β1 Enhances FAK mRNA and Protein Expression
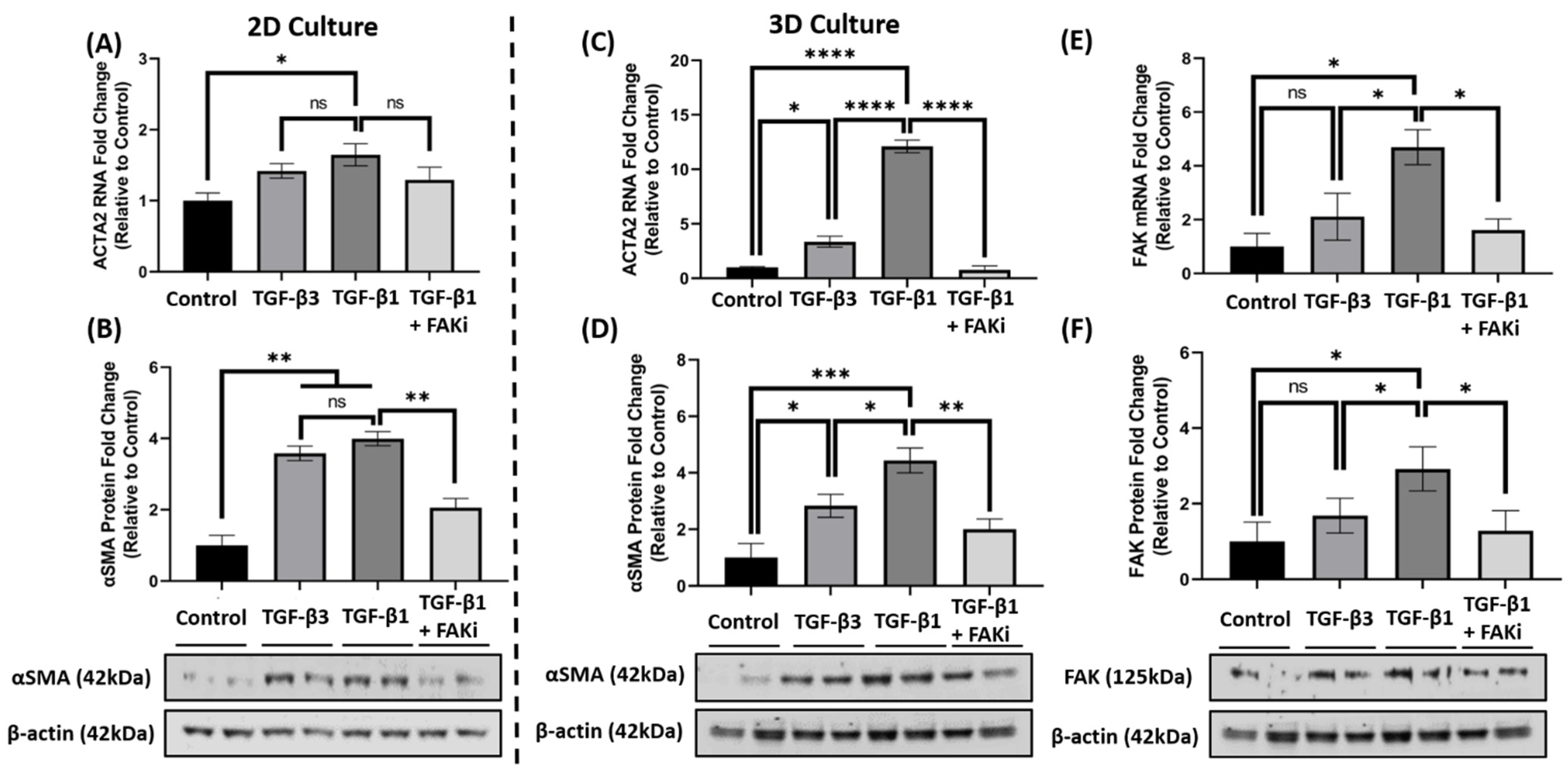
3.3. FAK Inhibition Attenuates TGF-β1-Mediated αSMA Expression in 3D Constructs
3.4. FAK Inhibition Attenuates αSMA Expression at the mRNA and Protein Level in 3D Constructs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | hCF-TGF-β1 | STDEV | hCF-TGF-β3 | STDEV |
|---|---|---|---|---|
| ACTA2 | 10.8745 | 0.000127 | 6.8149 | 0.000623 |
| AGT | 1.2251 | 0.994969 | −2.0676 | 0.179804 |
| AKT1 | 1.2624 | 0.157868 | 1.0415 | 0.764383 |
| AKT2 | 1.0494 | 0.593021 | 1.1086 | 0.415904 |
| AKT3 | 1.5187 | 0.018033 | 1.0224 | 0.909048 |
| BCL2 | 1.1247 | 0.815784 | −1.2408 | 0.702404 |
| BMP7 | −1.2372 | 0.659927 | −1.0507 | 0.913471 |
| CAV1 | −1.1099 | 0.579853 | −1.4155 | 0.277854 |
| CEBPB | 1.3161 | 0.459956 | 1.1477 | 0.810001 |
| COL1A2 | 1.4502 | 0.052828 | 1.006 | 0.997354 |
| COL3A1 | 1.2194 | 0.250259 | 1.0201 | 0.945166 |
| CTGF | 2.5601 | 0.003845 | 1.5678 | 0.17133 |
| DCN | −1.5022 | 0.071293 | −1.5779 | 0.055471 |
| EDN1 | −1.0573 | 0.727419 | −1.0055 | 0.993908 |
| EGF | −3.4833 | 0.013729 | −3.9394 | 0.022065 |
| ENG | −1.4049 | 0.026476 | −1.8463 | 0.007247 |
| FASLG | N/A | N/A | N/A | N/A |
| FN1 | 3.1085 | 0.00139 | 2.1416 | 0.007745 |
| GREM1 | 1.2054 | 0.09758 | −1.0953 | 0.463088 |
| HGF | −1.1759 | 0.20697 | −1.0802 | 0.652778 |
| IL10 | −1.5953 | 0.04713 | −2.5163 | 0.025841 |
| IL13 | N/A | N/A | N/A | N/A |
| IL13RA2 | −7.6059 | 0.07401 | −7.9519 | 0.072558 |
| IL2 | −1.2545 | 0.188189 | −1.0079 | 0.874455 |
| INHBE | 1.8655 | 0.280818 | 1.3153 | 0.51604 |
| ITGA1 | 1.9135 | 0.017726 | 1.7638 | 0.174106 |
| ITGA2 | −1.9059 | 0.000659 | −2.1654 | 0.000417 |
| ITGA3 | 1.7245 | 0.031304 | 1.4228 | 0.114872 |
| ITGAV | 1.2451 | 0.005626 | −1.1313 | 0.23991 |
| ITGB1 | 1.1644 | 0.45432 | −1.2876 | 0.283416 |
| ITGB3 | 3.5379 | 0.013586 | 2.9053 | 0.140037 |
| ITGB5 | 2.6504 | 0.001845 | 1.7679 | 0.028366 |
| ITGB6 | 36.8318 | 0.012598 | 15.4767 | 0.087959 |
| ITGB8 | −2.0474 | 0.034861 | −2.9717 | 0.021873 |
| JUN | 1.159 | 0.500013 | −1.2876 | 0.269455 |
| LOX | 3.1157 | 0.00742 | 2.2481 | 0.055475 |
| LTBP1 | −1.4016 | 0.008138 | −2.3641 | 0.00093 |
| MAPK14 | 1.0739 | 0.663648 | −1.3055 | 0.140065 |
| MMP1 | 1.6429 | 0.083457 | 1.1799 | 0.452964 |
| MMP13 | 1.1351 | 0.999602 | 1.1009 | 0.955945 |
| MMP14 | 1.5013 | 0.107373 | −1.1551 | 0.723604 |
| MMP2 | 1.3975 | 0.17182 | 1.0512 | 0.843387 |
| MMP3 | −1.6477 | 0.188291 | −1.5742 | 0.302279 |
| MMP8 | 3.1959 | 0.015414 | 1.6881 | 0.332422 |
| MMP9 | −2.4974 | 0.184368 | −2.5692 | 0.172014 |
| MYC | −1.0476 | 0.765856 | −1.1604 | 0.43132 |
| NFKB1 | −1.1228 | 0.277577 | −1.5312 | 0.017509 |
| PDGFA | 3.3859 | 0.007316 | 2.5764 | 0.054476 |
| PDGFB | N/A | N/A | N/A | N/A |
| PDGFRA | −1.5769 | 0.046534 | −1.9788 | 0.017984 |
| PDGFRB | 1.3437 | 0.077322 | −1.2351 | 0.510587 |
| PIK3CA | 1.0043 | 0.99742 | −1.0314 | 0.881799 |
| PLAT | −2.0427 | 0.236592 | −2.3696 | 0.203052 |
| PLAU | 3.9711 | 0.000363 | 2.2795 | 0.00642 |
| PLG | N/A | N/A | N/A | N/A |
| SERPINA1 | 2.7249 | 0.143343 | 3.3373 | 0.026145 |
| SERPINE1 | 3.1592 | 0.017261 | 1.5933 | 0.280042 |
| SERPINH1 | 1.2595 | 0.236419 | 1.3336 | 0.042944 |
| SMAD2 | −1.0026 | 0.949866 | −1.479 | 0.010164 |
| SMAD3 | −1.0404 | 0.639796 | −1.6794 | 0.225547 |
| SMAD4 | −1.1074 | 0.146352 | −1.549 | 0.021287 |
| SMAD6 | −1.3017 | 0.146467 | −1.5525 | 0.080067 |
| SMAD7 | −1.3048 | 0.412951 | −2.2731 | 0.077446 |
| SNAI1 | −1.1979 | 0.35866 | −1.4654 | 0.24799 |
| SP1 | 1.1752 | 0.49412 | −1.4927 | 0.095233 |
| SRC | 1.1943 | 0.03643 | −1.108 | 0.153392 |
| STAT1 | −1.625 | 0.099674 | −2.3751 | 0.037132 |
| STAT3 | −1.2868 | 0.021921 | −1.4553 | 0.006344 |
| STAT6 | −1.5232 | 0.016405 | −1.8764 | 0.005402 |
| TGFB1 | 2.1085 | 0.000675 | 1.5109 | 0.174072 |
| TGFB2 | −1.9369 | 0.023158 | −2.7094 | 0.009191 |
| TGFB3 | −1.1202 | 0.386433 | −1.4319 | 0.078127 |
| TGFBR1 | 2.3234 | 0.000288 | 1.3398 | 0.037444 |
| TGFBR2 | −1.1438 | 0.350833 | −1.5597 | 0.026326 |
| TGIF1 | −1.1385 | 0.448445 | −1.1821 | 0.410261 |
| THBS1 | 2.8275 | 0.002341 | 1.4162 | 0.093332 |
| THBS2 | 2.4109 | 0.000965 | 1.539 | 0.161535 |
| TIMP1 | 1.2683 | 0.210136 | 1.0708 | 0.765096 |
| TIMP2 | 1.5471 | 0.016557 | 1.1424 | 0.471441 |
| TIMP3 | 4.1879 | 0.003398 | 3.3451 | 0.006523 |
| TIMP4 | −1.9595 | 0.078666 | −2.2211 | 0.052237 |
| TNF | N/A | N/A | N/A | N/A |
| VEGFA | 2.5959 | 0.059826 | 2.6611 | 0.252738 |
| ZFYVE9 | −1.0921 | 0.180398 | −1.5418 | 0.058137 |
References
- Khalil, N.; O’Connor, R.N.; Flanders, K.C.; Unruh, H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: An immunohistochemical study. Am. J. Respir. Cell Mol. Biol. 1996, 14, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Jester, J.V.; Huang, J.; Petroll, W.M.; Cavanagh, H.D. TGFβ Induced Myofibroblast Differentiation of Rabbit Keratocytes Requires Synergistic TGFβ, PDGF and Integrin Signaling. Exp. Eye Res. 2002, 75, 645–657. [Google Scholar] [CrossRef]
- Yu, L.; Border, W.A.; Huang, Y.; Noble, N.A. TGF-β isoforms in renal fibrogenesis. Kidney Int. 2003, 64, 844–856. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Hutcheon, A.E.; Zieske, J.D. Molecular insights on the effect of TGF-β1/-β3 in human corneal fibroblasts. Exp. Eye Res. 2016, 146, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M. Beyond mitomycin: TGF-β and wound healing. Prog. Retin. Eye Res. 2002, 21, 75–89. [Google Scholar] [CrossRef]
- Sriram, S.; Gibson, D.; Robinson, P.; Tuli, S.; Lewin, A.; Schultz, G. Reduction of corneal scarring in rabbits by targeting the TGFB1 pathway with a triple siRNA combination. Adv. Biosci. Biotechnol. 2013, 4, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.J.; Pi, L.; Sriram, S.; Mao, C.; Petersen, B.E.; Scott, E.W.; Leask, A.; Schultz, G.S. Conditional Knockout of CTGF Affects Corneal Wound Healing. Investig. Opthalmology Vis. Sci. 2014, 55, 2062–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, T.B.; Yeung, V.; Hutcheon, A.E.K.; Guo, X.; Zieske, J.D.; Ciolino, J.B. Extracellular Vesicles in the Cornea: Insights from Other Tissues. Anal. Cell. Pathol. 2021, 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, R.; Nonaka, K.; Morifuji, M.; Shum, L.; Ohishi, M. TGF-β3 Decreases Type I Collagen and Scarring after Labioplasty. J. Dent. Res. 2003, 82, 558–564. [Google Scholar] [CrossRef]
- Carrington, L.M.; Albon, J.; Anderson, I.; Kamma, C.; Boulton, M. Differential Regulation of Key Stages in Early Corneal Wound Healing by TGF-β Isoforms and Their Inhibitors. Investig. Opthalmol. Vis. Sci. 2006, 47, 1886–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Occleston, N.L.; Laverty, H.G.; O’Kane, S.; Ferguson, M.W.J. Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGFβ3): From laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 2008, 19, 1047–1063. [Google Scholar] [CrossRef]
- So, K.; McGrouther, D.A.; Bush, J.A.; Durani, P.; Taylor, L.; Skotny, G.; Mason, T.; Metcalfe, A.; O’kane, S.; Ferguson, M.W.J. Avotermin for Scar Improvement following Scar Revision Surgery: A Randomized, Double-Blind, Within-Patient, Placebo-Controlled, Phase II Clinical Trial. Plast. Reconstr. Surg. 2011, 128, 163–172. [Google Scholar] [CrossRef]
- Ohno, S.; Hirano, S.; Kanemaru, S.-I.; Kitani, Y.; Kojima, T.; Ishikawa, S.; Mizuta, M.; Tateya, I.; Nakamura, T.; Ito, J. Transforming growth factor β3 for the prevention of vocal fold scarring. Laryngoscope 2012, 122, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Karamichos, D.; Hutcheon, A.; Zieske, J. Reversal of fibrosis by TGF-β3 in a 3D in vitro model. Exp. Eye Res. 2014, 124, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, S.; Tran, J.A.; Guo, X.Q.; Lei, H.; Kazlauskas, A.; Zieske, J.D. Identifying the role of PDGFRα in the fibrotic pathway of TGF-β3. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2355. [Google Scholar]
- Li, M.; Qiu, L.; Hu, W.; Deng, X.; Xu, H.; Cao, Y.; Xiao, Z.; Peng, L.; Johnson, S.; Alexey, L.; et al. Genetically-modified bone mesenchymal stem cells with TGF-β 3 improve wound healing and reduce scar tissue formation in a rabbit model. Exp. Cell Res. 2018, 367, 24–29. [Google Scholar] [CrossRef]
- Feldman, D.S.; McCauley, J.F. Mesenchymal Stem Cells and Transforming Growth Factor-β3 (TGF-β3) to Enhance the Regenerative Ability of an Albumin Scaffold in Full Thickness Wound Healing. J. Funct. Biomater. 2018, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Yeung, V.; Webber, J.P.; Dunlop, E.A.; Morgan, H.; Hutton, J.; Gurney, M.; Jones, E.; Falcon-Perez, J.; Tabi, Z.; Errington, R.; et al. Rab35-dependent extracellular nanovesicles are required for induction of tumour supporting stroma. Nanoscale 2018, 10, 8547–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmouliere, A.; Darby, I.A.; Laverdet, B.; Bonté, F. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkett, E.A. Role of growth factors in lung repair and diseases. Curr. Opin. Pediatr. 1995, 7, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.U.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nat. Cell Biol. 1992, 360, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Yeung, V.; Clayton, A. Extracellular vesicles as modulators of the cancer microenvironment. Semin. Cell Dev. Biol. 2015, 40, 27–34. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Z.; Wu, X.; Zhang, W.; Zhou, G.; Liu, W. Inhibitory effect of TGF-βpeptide antagonist on the fibrotic phenotype of human hypertrophic scar fibroblasts. Pharm. Biol. 2015, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shephard, A.P.; Yeung, V.; Clayton, A.; Webber, J.P. Prostate cancer exosomes as modulators of the tumor microenvironment. J. Cancer Metastasis Treat. 2017, 3, 288. [Google Scholar] [CrossRef]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, V.; Willis, G.R.; Taglauer, E.; Mitsialis, S.A.; Kourembanas, S. Paving the Road for Mesenchymal Stem Cell-Derived Exosome Therapy in Bronchopulmonary Dysplasia and Pulmonary Hypertension. In Stem Cell-Based Therapy for Lung Disease; Springer: Cham, Switzerland, 2019; pp. 131–152. [Google Scholar] [CrossRef]
- Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Yeung, V.; Liu, X.; Ericsson, M.; Andrews, N.A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J. Extracell. Vesicles 2020, 9, 1790874. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Willis, G.R.; Yeung, V.; Reis, M.; Liu, X.; Mitsialis, S.A.; Kourembanas, S. Therapeutic Effects of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles in Oxygen-Induced Multi-Organ Disease: A Developmental Perspective. Front. Cell Dev. Biol. 2021, 9, 647025. [Google Scholar] [CrossRef]
- Karamichos, D.; Guo, X.Q.; Hutcheon, A.E.K.; Zieske, J.D. Human Corneal Fibrosis: An In Vitro Model. Investig. Opthalmol. Vis. Sci. 2010, 51, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Karamichos, D.; Hutcheon, A.E.K.; Zieske, J.D. Transforming growth factor-β3 regulates assembly of a non-fibrotic matrix in a 3D corneal model. J. Tissue Eng. Regen. Med. 2011, 5, e228–e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, A.; Tovey, J.C.K.; Sharma, A.; Gupta, R.; Mohan, R.R. Role of Transforming Growth Factor Beta in Corneal Function, Biology and Pathology. Curr. Mol. Med. 2010, 10, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast Differentiation by Transforming Growth Factor-β1 Is Dependent on Cell Adhesion and Integrin Signaling via Focal Adhesion Kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.C.; Watt, F.M. Regulation of development and differentiation by the extracellular matrix. Development 1993, 117, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta 2013, 1832, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-Y.; Zhao, X.-K.; Yu, L.; Qi, F.; Zhai, B.; Gao, C.-Q.; Ding, Q. Interaction of Src and Alpha-V Integrin Regulates Fibroblast Migration and Modulates Lung Fibrosis in A Preclinical Model of Lung Fibrosis. Sci. Rep. 2017, 7, srep46357. [Google Scholar] [CrossRef] [Green Version]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGFβ-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef]
- Thakur, R.; Trivedi, R.; Rastogi, N.; Singh, M.; Mishra, D.P. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci. Rep. 2015, 5, 10194. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, X.; Wu, X.; Hui, B.; Han, S.; Gao, J.; Li, Y.; Shi, J.; Zhu, H.; Zhao, B.; et al. Simultaneous deactivation of FAK and Src improves the pathology of hypertrophic scar. Sci. Rep. 2016, 6, 26023. [Google Scholar] [CrossRef] [Green Version]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. The TGF-β1/p53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules 2019, 9, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Hutcheon, A.E.K.; Melotti, S.A.; Zieske, J.D.; Trinkaus-Randall, V.; Ruberti, J.W. Morphologic characterization of organized extracellular matrix deposition by ascorbic acid-stimulated human corneal fibroblasts. Investig. Opthalmol. Vis. Sci. 2007, 48, 4050–4060. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zieske, J.D.; Hutcheon, A.E.; Guo, X.; Chung, E.H.; Joyce, N.C. TGF-beta receptor types I and II are differentially expressed during corneal epithelial wound repair. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1465–1471. [Google Scholar]
- Saika, S. TGFβ pathobiology in the eye. Lab. Investig. 2005, 86, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Whitby, D.J.; Ferguson, M.W. Immunohistochemical localization of growth factors in fetal wound healing. Dev. Biol. 1991, 147, 207–215. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Duncan, J.; Bond, J.; Bush, J.; Durani, P.; So, K.; Taylor, L.; Chantrey, J.; Mason, T.; James, G.; et al. Prophylactic administration of avotermin for improvement of skin scarring: Three double-blind, placebo-controlled, phase I/II studies. Lancet 2009, 373, 1264–1274. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Z.; Sun, J. Human bone marrow mesenchymal stem cell-derived exosomes stimulate cutaneous wound healing mediates through TGF-β/Smad signaling pathway. Stem Cell Res. Ther. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Schrementi, M.E.; Ferreira, A.M.; Zender, C.; DiPietro, L.A. Site-specific production of TGF-β in oral mucosal and cutaneous wounds. Wound Repair Regen. 2008, 16, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Kishimoto, Y.; Hasan, A.; Welham, N.V. TGF-β3 modulates the inflammatory environment and reduces scar formation following vocal fold mucosal injury in rats. Dis. Model. Mech. 2014, 7, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Kuwano, K.; Maeyama, T.; Yoshimi, M.; Hamada, N.; Fukumoto, J.; Egashira, K.; Hiasa, K.; Takayama, K.; Nakanishi, Y. Gene transfer of soluble transforming growth factor type II receptor by in vivo electroporation attenuates lung injury and fibrosis. J. Clin. Pathol. 2006, 60, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, J.; Berg, P.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ding, Z.; Wu, Z.; Xu, Y.; Yao, H.; Lin, K. Targeting the TGF-β signaling pathway for fibrosis therapy: A patent review (2015–2020). Expert Opin. Ther. Patents 2021, 31, 723–743. [Google Scholar] [CrossRef]
- Sriram, S.; Tran, J.A.; Guo, X.; Hutcheon, A.E.K.; Lei, H.; Kazlauskas, A.; Zieske, J.D. PDGFRα Is a Key Regulator of T1 and T3’s Differential Effect on SMA Expression in Human Corneal Fibroblasts. Investig. Opthalmol. Vis. Sci. 2017, 58, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.W.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef]
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ask, K.; Bonniaud, P.; Maass, K.; Eickelberg, O.; Margetts, P.J.; Warburton, D.; Groffen, J.; Gauldie, J.; Kolb, M. Progressive pulmonary fibrosis is mediated by TGF-β isoform 1 but not TGF-β3. Int. J. Biochem. Cell Biol. 2008, 40, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, B.; Hagood, J.S.; Zhuo, Y.; Nguyen, H.T.; MacEwen, M.; Morris, G.F.; Lasky, J.A. Thy-1 Attenuates TNF-α-Activated Gene Expression in Mouse Embryonic Fibroblasts via Src Family Kinase. PLoS ONE 2010, 5, e11662. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cai, G.-Q.; Hu, M.; Yang, Y.; Zheng, A.; Tang, Q.; Gladson, C.L.; Hayasaka, H.; Wu, H.; You, Z.; et al. FAK-Related Nonkinase Is a Multifunctional Negative Regulator of Pulmonary Fibrosis. Am. J. Pathol. 2013, 182, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Siesser, P.M.; Hanks, S.K. The Signaling and Biological Implications of FAK Overexpression in Cancer. Clin. Cancer Res. 2006, 12, 3233–3237. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K.; Aono, Y.; Azuma, M.; Kishi, J.; Takezaki, A.; Kishi, M.; Makino, H.; Okazaki, H.; Uehara, H.; Izumi, K.; et al. Antifibrotic Effects of Focal Adhesion Kinase Inhibitor in Bleomycin-Induced Pulmonary Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 536–543. [Google Scholar] [CrossRef]
- Zhao, X.-K.; Cheng, Y.; Cheng, M.L.; Yu, L.; Mu, M.; Li, H.; Liu, Y.; Zhang, B.; Yao, Y.; Guo, H.; et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci. Rep. 2016, 6, 19276. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, Z.; Xue, Z.; Wang, L.; Fu, M.; Lu, Y.; Bai, L.; Zhang, D.; Fan, Z. Focal adhesion kinase (FAK) siRNA inhibits human hypertrophic scar by suppressing integrin α, TGF-β and α-SMA. Cell Biol. Int. 2014, 38, 803–808. [Google Scholar] [CrossRef]
- Fan, G.-P.; Wang, W.; Zhao, H.; Cai, L.; Zhang, P.-D.; Yang, Z.-H.; Zhang, J.; Wang, X. Pharmacological Inhibition of Focal Adhesion Kinase Attenuates Cardiac Fibrosis in Mice Cardiac Fibroblast and Post-Myocardial-Infarction Models. Cell. Physiol. Biochem. 2015, 37, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, K.; Miyagawa-Tomita, S.; Matsumoto, H.; Koyama, H.; Nakanishi, T.; Hirose, H. Effects of transforming growth factor-?3 and matrix metalloproteinase-3 on the pathogenesis of chronic mitral valvular disease in dogs. Am. J. Vet. Res. 2011, 72, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.E.; Boraas, L.C.; Sowder, M.; Bechtel, M.K.; Orwin, E.J. Three-Dimensional Cell Culture Environment Promotes Partial Recovery of the Native Corneal Keratocyte Phenotype from a Subcultured Population. Tissue Eng. Part A 2013, 19, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- McKay, T.B.; Hutcheon, A.E.; Guo, X.; Zieske, J.D.; Karamichos, D. Modeling the cornea in 3-dimensions: Current and future perspectives. Exp. Eye Res. 2020, 197, 108127. [Google Scholar] [CrossRef]
- Petroll, W.M.; Miron-Mendoza, M. Mechanical interactions and crosstalk between corneal keratocytes and the extracellular matrix. Exp. Eye Res. 2015, 133, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Cen, L.; Liu, W.; Cui, L.; Zhang, W.; Cao, Y. Collagen Tissue Engineering: Development of Novel Biomaterials and Applications. Pediatr. Res. 2008, 63, 492–496. [Google Scholar] [CrossRef]
- Chen, S.; Mienaltowski, M.J.; Birk, D.E. Regulation of corneal stroma extracellular matrix assembly. Exp. Eye Res. 2015, 133, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Meek, K.M.; Knupp, C. Corneal structure and transparency. Prog. Retin. Eye Res. 2015, 49, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Puperi, D.S.; Grande-Allen, K.J.; West, J.L. Ascorbic acid promotes extracellular matrix deposition while preserving valve interstitial cell quiescence within 3D hydrogel scaffolds. J. Tissue Eng. Regen. Med. 2015, 11, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Jhanji, V.; Billig, I.; Yam, G.H.-F. Cell-Free Biological Approach for Corneal Stromal Wound Healing. Front. Pharmacol. 2021, 12, 1339. [Google Scholar] [CrossRef]
- Ren, R.; Hutcheon, A.; Guo, X.; Saeidi, N.; Melotti, S.; Ruberti, J.; Zieske, J.; Trinkaus-Randall, V. Human primary corneal fibroblasts synthesize and deposit proteoglycans in long-term 3-D cultures. Dev. Dyn. 2008, 237, 2705–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamichos, D.; Zareian, R.; Guo, X.; Hutcheon, A.E.; Ruberti, J.W.; Zieske, J.D. Novel in Vitro Model for Keratoconus Disease. J. Funct. Biomater. 2012, 3, 760–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, T.B.; Hjortdal, J.; Sejersen, H.; Karamichos, D. Differential Effects of Hormones on Cellular Metabolism in Keratoconus In Vitro. Sci. Rep. 2017, 7, srep42896. [Google Scholar] [CrossRef]
- McKay, T.B.; Priyadarsini, S.; Rowsey, T.; Karamichos, D. Arginine Supplementation Promotes Extracellular Matrix and Metabolic Changes in Keratoconus. Cells 2021, 10, 2076. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.E.; Rowsey, T.G.; Priyadarsini, S.; Mandal, N.A.; Karamichos, D. Unravelling the interplay of sphingolipids and TGF-β signaling in the human corneal stroma. PLoS ONE 2017, 12, e0182390. [Google Scholar] [CrossRef] [Green Version]
- Priyadarsini, S.; Sarker-Nag, A.; Rowsey, T.G.; Ma, J.-X.; Karamichos, D. Establishment of a 3D In Vitro Model to Accelerate the Development of Human Therapies against Corneal Diabetes. PLoS ONE 2016, 11, e0168845. [Google Scholar] [CrossRef] [PubMed]
- Whelchel, A.E.; McKay, T.B.; Priyadarsini, S.; Rowsey, T.; Karamichos, D. Association between Diabetes and Keratoconus: A Retrospective Analysis. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Whelchel, A.E.; Nicholas, S.E.; Ma, J.-X.; Karamichos, D. Nerve influence on the metabolism of type I and type II diabetic corneal stroma: An in vitro study. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marlo, T.L.; Giuliano, E.A.; Sharma, A.; Mohan, R.R. Development of a novelex vivoequine corneal model. Vet. Ophthalmol. 2016, 20, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Lagares, D.; Busnadiego, O.; García-Fernández, R.A.; Kapoor, M.; Liu, S.; Carter, D.E.; Abraham, D.; Shi-Wen, X.; Carreira, P.; Fontaine, B.A.; et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2011, 64, 1653–1664. [Google Scholar] [CrossRef]
- Du, G.; Wang, J.; Zhang, T.; Ding, Q.; Jia, X.; Zhao, X.; Dong, J.; Yang, X.; Lu, S.; Zhang, C.; et al. Targeting Src family kinase member Fyn by Saracatinib attenuated liver fibrosis in vitro and in vivo. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.E.; Xiang, B.; Zent, R.; Quaranta, V.; Pozzi, A.; Arteaga, C.L. Transforming Growth Factor β Induces Clustering of HER2 and Integrins by Activating Src-Focal Adhesion Kinase and Receptor Association to the Cytoskeleton. Cancer Res. 2009, 69, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Gladson, C.L.; Wu, H.; Hayasaka, H.; Olman, M.A. Focal Adhesion Kinase (FAK)-related Non-kinase Inhibits Myofibroblast Differentiation through Differential MAPK Activation in a FAK-dependent Manner. J. Biol. Chem. 2008, 283, 26839–26849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Che, P.; Han, X.; Cai, G.-Q.; Liu, G.; Antony, V.; Luckhardt, T.; Siegal, G.P.; Zhou, Y.; Liu, R.-M.; et al. Therapeutic Targeting of Src Kinase in Myofibroblast Differentiation and Pulmonary Fibrosis. J. Pharmacol. Exp. Ther. 2014, 351, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.-K.; Yu, L.; Cheng, M.-L.; Che, P.; Lu, Y.-Y.; Zhang, Q.; Mu, M.; Li, H.; Zhu, L.-L.; Zhu, J.-J.; et al. Focal Adhesion Kinase Regulates Hepatic Stellate Cell Activation and Liver Fibrosis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lim, W.; Nam, J.-S.; Sheen, Y.Y. TIMP-1 mediates TGF-β-dependent crosstalk between hepatic stellate and cancer cells via FAK signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Subramanian, I.; Luckhardt, T.R.; Che, P.; Waghray, M.; Zhao, X.-K.; Bone, N.; Kurundkar, A.R.; Hecker, L.; Hu, M.; et al. Focal adhesion kinase signaling determines the fate of lung epithelial cells in response to TGF-β. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L926–L935. [Google Scholar] [CrossRef] [Green Version]
- Rudmann, D.G. On-target and Off-target-based Toxicologic Effects. Toxicol. Pathol. 2012, 41, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, T.; Honma, M.; Kariya, Y.; Ghosh, S.; Kitano, H.; Kurachi, Y.; Fujita, K.-I.; Sasaki, Y.; Homma, Y.; Abernethy, D.R.; et al. Elucidation of the molecular mechanisms underlying adverse reactions associated with a kinase inhibitor using systems toxicology. Npj Syst. Biol. Appl. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Furuno, M.; Arakawa, T.; Takizawa, S.; De Hoon, M.; Suzuki, H.; Arner, E. A framework for identification of on- and off-target transcriptional responses to drug treatment. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Thordarson, P.; Marquis, A.; Crossley, M.J. The synthesis and studies towards the self-replication of bis(capped porphyrins). Org. Biomol. Chem. 2003, 1, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A Small Molecule Inhibitor, 1,2,4,5-Benzenetetraamine Tetrahydrochloride, Targeting the Y397 Site of Focal Adhesion Kinase Decreases Tumor Growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef] [Green Version]
- Hochwald, S.N.; Nyberg, C.; Zheng, M.; Zheng, D.; Wood, C.; Massoll, N.A.; Magis, A.; Ostrov, D.; Cance, W.G.; Golubovskaya, V.M. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle 2009, 8, 2435–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beierle, E.A.; Ma, X.; Stewart, J.; Nyberg, C.; Trujillo, A.; Cance, W.G.; Golubovskaya, V.M. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle 2010, 9, 1005–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeung, V.; Sriram, S.; Tran, J.A.; Guo, X.; Hutcheon, A.E.K.; Zieske, J.D.; Karamichos, D.; Ciolino, J.B. FAK Inhibition Attenuates Corneal Fibroblast Differentiation In Vitro. Biomolecules 2021, 11, 1682. https://doi.org/10.3390/biom11111682
Yeung V, Sriram S, Tran JA, Guo X, Hutcheon AEK, Zieske JD, Karamichos D, Ciolino JB. FAK Inhibition Attenuates Corneal Fibroblast Differentiation In Vitro. Biomolecules. 2021; 11(11):1682. https://doi.org/10.3390/biom11111682
Chicago/Turabian StyleYeung, Vincent, Sriniwas Sriram, Jennifer A. Tran, Xiaoqing Guo, Audrey E. K. Hutcheon, James D. Zieske, Dimitrios Karamichos, and Joseph B. Ciolino. 2021. "FAK Inhibition Attenuates Corneal Fibroblast Differentiation In Vitro" Biomolecules 11, no. 11: 1682. https://doi.org/10.3390/biom11111682