
The Mitochondrial Carnitine Acyl-carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Functional Role of CAC
3. Structure-Function Relationships
3.1. Substrate Binding Site and Translocation Events
3.2. The Molecular Basis of the Antiport Mode
4. CAC as a Redox Sensor
4.1. Regulation of CAC by H2O2
4.2. Regulation of CAC by Glutathione
4.3. Modulation of CAC by NO
4.4. Modulation of CAC by H2S
5. Other Regulatory Mechanisms
6. Interaction of CAC with Xenobiotics
6.1. Polyphenols
6.2. Dantrolene
6.3. β-Lactam Antibiotics
6.4. Proton Pump Inhibitors
6.5. Mildronate
6.6. Ingenols
6.7. Heavy Metals
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monne, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [Green Version]
- Agrimi, G.; Russo, A.; Scarcia, P.; Palmieri, F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem. J. 2012, 443, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e415. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.A.; Palmieri, F.; Bennett, M.J. Disorders of the mitochondrial carnitine shuttle. Online Metab. Mol. Bases Inherit. Dis. 2013. [Google Scholar] [CrossRef]
- Bhuiyan, J.; Pritchard, P.H.; Pande, S.V.; Seccombe, D.W. Effects of high-fat diet and fasting on levels of acyl-coenzyme A binding protein in liver, kidney, and heart of rat. Metabolism 1995, 44, 1185–1189. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Asp. Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine palmitoyltransferase 1C: From cognition to cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [Green Version]
- De Pinto, V. Renaissance of VDAC: New Insights on a Protein Family at the Interface between Mitochondria and Cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mitochondrial membrane. Mol. Cell Biochem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Muller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin Nutr 2010, 92, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Scalise, M.; Giangregorio, N.; Tonazzi, A.; Barile, M.; Indiveri, C. The Link Between the Mitochondrial Fatty Acid Oxidation Derangement and Kidney Injury. Front. Physiol. 2020, 11, 794. [Google Scholar] [CrossRef]
- Talley, J.T.; Mohiuddin, S.S. Biochemistry, Fatty Acid Oxidation; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Nałecz, K.A.; Miecz, D.; Berezowski, V.; Cecchelli, R. Carnitine: Transport and physiological functions in the brain. Mol. Aspects Med. 2004, 25, 551–567. [Google Scholar] [CrossRef]
- Tonazzi, A.; Mantovani, C.; Colella, M.; Terenghi, G.; Indiveri, C. Localization of mitochondrial carnitine/acylcarnitine translocase in sensory neurons from rat dorsal root Ganglia. Neurochem. Res. 2013, 38, 2535–2541. [Google Scholar] [CrossRef]
- Kaminska, J.; Nalecz, K.A.; Azzi, A.; Nalecz, M.J. Purification of carnitine carrier from rat brain mitochondria. Biochem. Mol. Biol. Int. 1993, 29, 999–1007. [Google Scholar]
- Huizing, M.; Iacobazzi, V.; Ijlst, L.; Savelkoul, P.; Ruitenbeek, W.; van den Heuvel, L.; Indiveri, C.; Smeitink, J.; Trijbels, F.; Wanders, R.; et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am. J. Hum. Genet. 1997, 61, 1239–1245. [Google Scholar] [CrossRef] [Green Version]
- IJlst, L.; Ruiter, J.P.; Oostheim, W.; Niezen-Koning, K.E.; Palmieri, F.; Wanders, R.J. Identification of a missense mutation in a patient with lethal carnitine acyl-carnitine carrier deficiency. Adv. Exp. Med. Biol. 1999, 466, 347–351. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Pasquali, M.; Singh, R.; Matern, D.; Rinaldo, P.; Amat di San Filippo, C.; Palmieri, F.; Longo, N. Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am. J. Med. Genet. A 2004, 126A, 150–155. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Huertas, M.D.; et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.Y.; Iacobazzi, V.; Wang, Z.; Harvie, H.; Chalmers, R.A.; Saudubray, J.M.; Palmieri, F.; Ganguly, A.; Stanley, C.A. Aberrant mRNA splicing associated with coding region mutations in children with carnitine-acylcarnitine translocase deficiency. Mol. Genet. Metab. 2001, 74, 248–255. [Google Scholar] [CrossRef]
- Huizing, M.; Wendel, U.; Ruitenbeek, W.; Iacobazzi, V.; Lglst, L.; Veenhuizen, P.; Savelkoul, P.; van den Heuvel, L.P.; Smeitink, J.A.; Wanders, R.J.; et al. Carnitine-acylcarnitine carrier deficiency: Identification of the molecular defect in a patient. J. Inherit. Metab. Dis. 1998, 21, 262–267. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Mazza, T.; Pochini, L.; Galluccio, M.; Giangregorio, N.; Tonazzi, A.; Indiveri, C. Carnitine Traffic in Cells. Link With Cancer. Front. Cell Dev. Biol. 2020, 8, 583850. [Google Scholar] [CrossRef]
- Magoulas, P.L.; El-Hattab, A.W. Systemic primary carnitine deficiency: An overview of clinical manifestations, diagnosis, and management. Orphanet. J. Rare Dis. 2012, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, M.S.; Rabaglia, M.E.; Bhatnagar, S.; Shang, J.; Ilkayeva, O.; Mynatt, R.; Zhou, Y.P.; Schadt, E.E.; Thornberry, N.A.; Muoio, D.M.; et al. Downregulation of carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion. Diabetes 2014, 63, 3805–3814. [Google Scholar] [CrossRef] [Green Version]
- Peluso, G.; Petillo, O.; Margarucci, S.; Mingrone, G.; Greco, A.V.; Indiveri, C.; Palmieri, F.; Melone, M.A.; Reda, E.; Calvani, M. Decreased mitochondrial carnitine translocase in skeletal muscles impairs utilization of fatty acids in insulin-resistant patients. Front. Biosci. J. Virtual Libr. 2002, 7, a109–a116. [Google Scholar] [CrossRef]
- Monne, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Characterization of the unidirectional transport of carnitine catalyzed by the reconstituted carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta 1991, 1069, 110–116. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. The reconstituted carnitine carrier from rat liver mitochondria: Evidence for a transport mechanism different from that of the other mitochondrial translocators. Biochim. Biophys. Acta 1994, 1189, 65–73. [Google Scholar] [CrossRef]
- Palmieri, L.; Lasorsa, F.M.; Iacobazzi, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae. FEBS Lett. 1999, 462, 472–476. [Google Scholar] [CrossRef] [Green Version]
- De Lucas, J.R.; Indiveri, C.; Tonazzi, A.; Perez, P.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Functional characterization of residues within the carnitine/acylcarnitine translocase RX(2)PANAAXF distinct motif. Mol. Membr. Biol. 2008, 25, 152–163. [Google Scholar] [CrossRef]
- Ramon De Lucas, J.; Martinez, O.; Perez, P.; Isabel Lopez, M.; Valenciano, S.; Laborda, F. The Aspergillus nidulans carnitine carrier encoded by the acuH gene is exclusively located in the mitochondria. FEMS Microbiol. Lett. 2001, 201, 193–198. [Google Scholar] [CrossRef]
- Perez, P.; Martinez, O.; Romero, B.; Olivas, I.; Pedregosa, A.M.; Palmieri, F.; Laborda, F.; Ramon De Lucas, J. Functional analysis of mutations in the human carnitine/acylcarnitine translocase in Aspergillus nidulans. Fungal Genet. Biol. 2003, 39, 211–220. [Google Scholar] [CrossRef]
- Palmieri, F.; Agrimi, G.; Blanco, E.; Castegna, A.; Di Noia, M.A.; Iacobazzi, V.; Lasorsa, F.M.; Marobbio, C.M.; Palmieri, L.; Scarcia, P.; et al. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim. Biophys. Acta 2006, 1757, 1249–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacques, F.; Rippa, S.; Perrin, Y. Physiology of L-carnitine in plants in light of the knowledge in animals and microorganisms. Plant. Sci. 2018, 274, 432–440. [Google Scholar] [CrossRef]
- Porcelli, V.; Vozza, A.; Calcagnile, V.; Gorgoglione, R.; Arrigoni, R.; Fontanesi, F.; Marobbio, C.M.T.; Castegna, A.; Palmieri, F.; Palmieri, L. Molecular identification and functional characterization of a novel glutamate transporter in yeast and plant mitochondria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J.A.; Ranea-Robles, P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165720. [Google Scholar] [CrossRef]
- Juraszek, B.; Nałęcz, K.A. SLC22A5 (OCTN2) Carnitine Transporter-Indispensable for Cell Metabolism, a Jekyll and Hyde of Human Cancer. Molecules 2019, 25, 14. [Google Scholar] [CrossRef] [Green Version]
- Pande, S.V. A mitochondrial carnitine acylcarnitine translocase system. Proc. Natl. Acad. Sci. USA 1975, 72, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, I.B.; Yue, K.T. Long-Chain Carnitine Acyltransferase and the Role of Acylcarnitine Derivatives in the Catalytic Increase of Fatty Acid Oxidation Induced by Carnitine. J. Lipid Res. 1963, 4, 279–288. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Tubbs, P.K. The mechanism of fatty acid uptake by heart mitochondria: An acylcarnitine-carnitine exchange. FEBS Lett. 1975, 54, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta 1990, 1020, 81–86. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Prezioso, G.; Palmieri, F. Kinetic characterization of the reconstituted carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta 1991, 1065, 231–238. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Dierks, T.; Krämer, R.; Palmieri, F. The mitochondrial carnitine carrier: Characterization of SH-groups relevant for its transport function. Biochim. Biophys. Acta 1992, 1140, 53–58. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Giangregorio, N.; Palmieri, F. Probing the active site of the reconstituted carnitine carrier from rat liver mitochondria with sulfhydryl reagents. A cysteine residue is localized in or near the substrate binding site. Eur. J. Biochem. 1995, 228, 271–278. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. The mitochondrial carnitine carrier protein: cDNA cloning, primary structure and comparison with other mitochondrial transport proteins. Biochem. J. 1997, 321 Pt 3, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. Bacterial overexpression, purification, and reconstitution of the carnitine/acylcarnitine carrier from rat liver mitochondria. Biochem. Biophys. Res. Commun. 1998, 249, 589–594. [Google Scholar] [CrossRef]
- Fiermonte, G.; Walker, J.E.; Palmieri, F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem. J. 1993, 294 Pt 1, 293–299. [Google Scholar] [CrossRef]
- Palmieri, F.; Monne, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Giangregorio, N.; Tonazzi, A.; Indiveri, C. Proteoliposomes as tool for assaying membrane transporter functions and interactions with xenobiotics. Pharmaceutics 2013, 5, 472–497. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Galluccio, M.; Pochini, L.; Console, L.; Barile, M.; Giangregorio, N.; Tonazzi, A.; Indiveri, C. Studying Interactions of Drugs with Cell Membrane Nutrient Transporters: New Frontiers of Proteoliposome Nanotechnology. Curr. Pharm. Des. 2017, 23, 3871–3883. [Google Scholar] [CrossRef]
- Parker, C.G.; Kuttruff, C.A.; Galmozzi, A.; Jørgensen, L.; Yeh, C.H.; Hermanson, D.J.; Wang, Y.; Artola, M.; McKerrall, S.J.; Josyln, C.M.; et al. Chemical Proteomics Identifies SLC25A20 as a Functional Target of the Ingenol Class of Actinic Keratosis Drugs. ACS Cent. Sci. 2017, 3, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Tonazzi, A.; Eberini, I.; Indiveri, C. Molecular mechanism of inhibition of the mitochondrial carnitine/acylcarnitine transporter by omeprazole revealed by proteoliposome assay, mutagenesis and bioinformatics. PLoS ONE 2013, 8, e82286. [Google Scholar] [CrossRef] [Green Version]
- Pochini, L.; Galluccio, M.; Scumaci, D.; Giangregorio, N.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Interaction of beta-lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem. Biol. Interact. 2008, 173, 187–194. [Google Scholar] [CrossRef]
- Bolognino, I.; Giangregorio, N.; Pisani, L.; de Candia, M.; Purgatorio, R.; Tonazzi, A.; Altomare, C.D.; Cellamare, S.; Catto, M. A Prospective Repurposing of Dantrolene as a Multitarget Agent for Alzheimer’s Disease. Molecules 2019, 24, 4298. [Google Scholar] [CrossRef] [Green Version]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Scalise, M.; La Russa, D.; Notaristefano, C.; Brunelli, E.; Barca, D.; Indiveri, C. Mitochondrial carnitine/acylcarnitine transporter, a novel target of mercury toxicity. Chem. Res. Toxicol. 2015, 28, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C. Post-translational modification by acetylation regulates the mitochondrial carnitine/acylcarnitine transport protein. Mol. Cell Biochem. 2017, 426, 65–73. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Lorusso, I.; De Palma, A.; Indiveri, C. The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim. Biophys. Acta 2016, 1860, 20–27. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; De Palma, A.; Indiveri, C. Nitric oxide inhibits the mitochondrial carnitine/acylcarnitine carrier through reversible S-nitrosylation of cysteine 136. Biochim. Biophys. Acta 2017, 1858, 475–482. [Google Scholar] [CrossRef]
- Tonazzi, A.; Console, L.; Indiveri, C. Inhibition of mitochondrial carnitine/acylcarnitine transporter by H(2)O(2): Molecular mechanism and possible implication in pathophysiology. Chem. Biol. Interact. 2013. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Prejanò, M.; Marino, T.; Russo, N.; Indiveri, C. Effect of Copper on the Mitochondrial Carnitine/Acylcarnitine Carrier Via Interaction with Cys136 and Cys155. Possible Implications in Pathophysiology. Molecules 2020, 25, 820. [Google Scholar] [CrossRef] [Green Version]
- Murthy, M.S.; Pande, S.V. Mechanism of carnitine acylcarnitine translocase-catalyzed import of acylcarnitines into mitochondria. J. Biol. Chem. 1984, 259, 9082–9089. [Google Scholar] [CrossRef]
- Tonazzi, A.; Console, L.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis of a hydrophobic binding site of the mitochondrial carnitine/acylcarnitine carrier involved in the interaction with acyl groups. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Bartelds, B.; Takens, J.; Smid, G.B.; Zammit, V.A.; Prip-Buus, C.; Kuipers, J.R.; van der Leij, F.R. Myocardial carnitine palmitoyltransferase I expression and long-chain fatty acid oxidation in fetal and newborn lambs. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2243–H2248. [Google Scholar] [CrossRef]
- Eaton, S. Control of mitochondrial beta-oxidation flux. Prog. Lipid Res. 2002, 41, 197–239. [Google Scholar] [CrossRef]
- Violante, S.; Ijlst, L.; Te Brinke, H.; Tavares de Almeida, I.; Wanders, R.J.; Ventura, F.V.; Houten, S.M. Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines. FASEB J. 2013, 27, 2039–2044. [Google Scholar] [CrossRef]
- Arduini, A.; Zammit, V. Acetate transport into mitochondria does not require a carnitine shuttle mechanism. Magn. Reson. Med. 2017, 77, 11. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.; Arduini, A. The carnitine acyltransferases and their role in modulating acyl-CoA pools. Arch. Biochem. Biophys. 1993, 302, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Kjalarsdottir, L.; Thompson, J.W.; Dubois, L.G.; Stevens, R.D.; Ilkayeva, O.R.; Brosnan, M.J.; Rolph, T.P.; Grimsrud, P.A.; Muoio, D.M. The Acetyl Group Buffering Action of Carnitine Acetyltransferase Offsets Macronutrient-Induced Lysine Acetylation of Mitochondrial Proteins. Cell Rep. 2016, 14, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bisaccia, F.; De Palma, A.; Dierks, T.; Krämer, R.; Palmieri, F. Reaction mechanism of the reconstituted tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1993, 1142, 139–145. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP, ATP shuttle of the mitochondrion. Trends Biochem. Sci. 1979, 4, 249–252. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, S.V.; Parvin, R. Carnitine-acylcarnitine translocase catalyzes an equilibrating unidirectional transport as well. J. Biol. Chem. 1980, 255, 2994–3001. [Google Scholar] [CrossRef]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Galluccio, M.; Scalise, M.; Console, L.; Indiveri, C. OCTN: A Small Transporter Subfamily with Great Relevance to Human Pathophysiology, Drug Discovery, and Diagnostics. SLAS Discov. 2019, 24, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Noland, R.C.; Koves, T.R.; Seiler, S.E.; Lum, H.; Lust, R.M.; Ilkayeva, O.; Stevens, R.D.; Hegardt, F.G.; Muoio, D.M. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 2009, 284, 22840–22852. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 2015, 116, 107–112. [Google Scholar] [CrossRef]
- Pande, S.V.; Parvin, R. Characterization of carnitine acylcarnitine translocase system of heart mitochondria. J. Biol. Chem. 1976, 251, 6683–6691. [Google Scholar] [CrossRef]
- Tonazzi, A.; Indiveri, C. Chemical modification of the mitochondrial ornithine/citrulline carrier by SH reagents: Effects on the transport activity and transition from carrier to pore-like function. Biochim. Biophys. Acta Biomembr. 2003, 1611, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Krämer, R. Mitochondrial carrier proteins can reversibly change their transport mode: The cases of the aspartate/glutamate and the phosphate carrier. Exp. Physiol. 1998, 83, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stappen, R.; Kramer, R. Functional properties of the reconstituted phosphate carrier from bovine heart mitochondria: Evidence for asymmetric orientation and characterization of three different transport modes. Biochim. Biophys. Acta 1993, 1149, 40–48. [Google Scholar] [CrossRef]
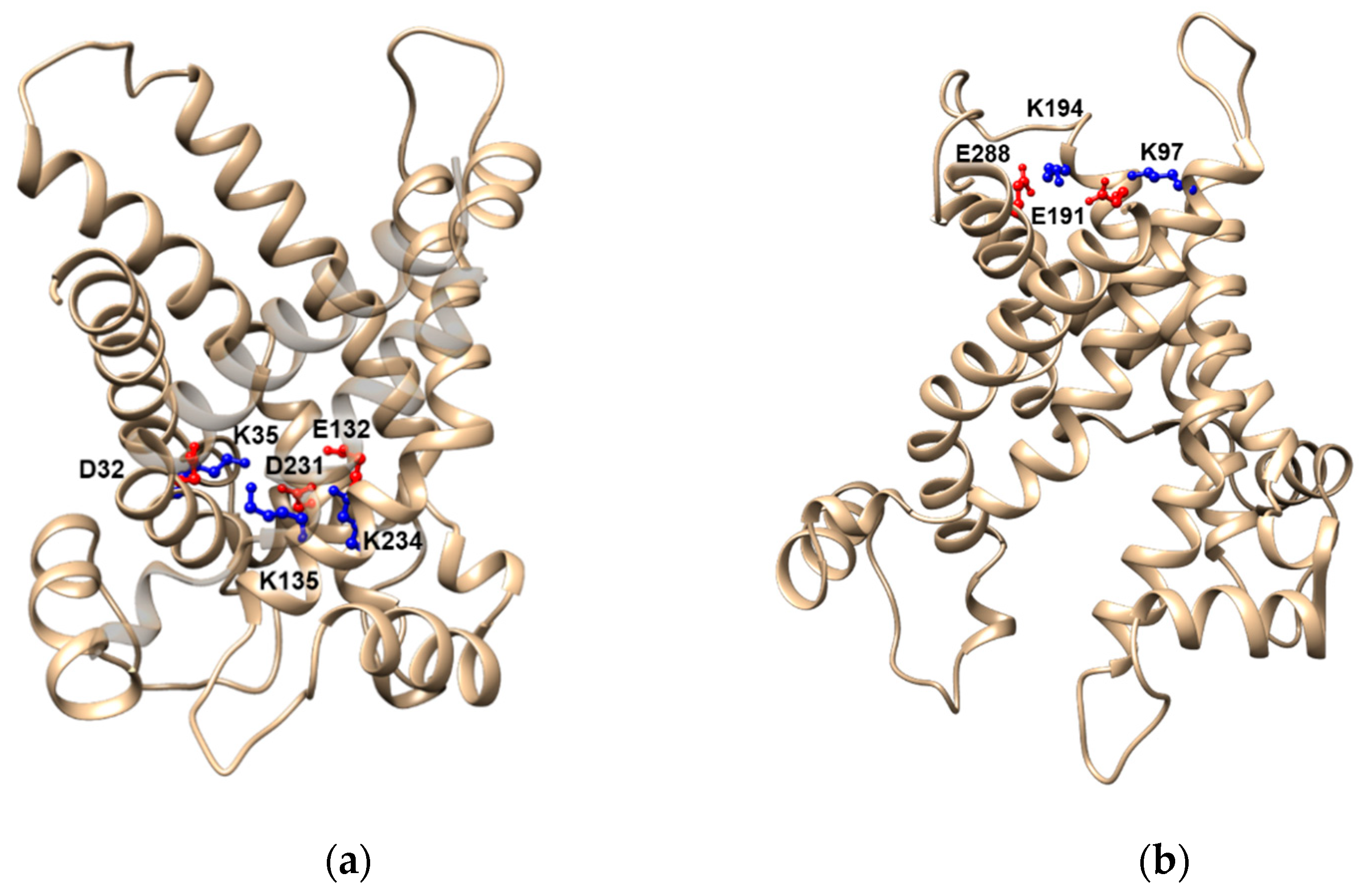
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C.; Palmieri, F. Site-directed mutagenesis of charged amino acids of the human mitochondrial carnitine/acylcarnitine carrier: Insight into the molecular mechanism of transport. Biochim. Biophys. Acta Biomembr. 2010, 1797, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Indiveri, C.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Site-directed mutagenesis and chemical modification of the six native cysteine residues of the rat mitochondrial carnitine carrier: Implications for the role of cysteine-136. Biochemistry 2002, 41, 8649–8656. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Indiveri, C.; Palmieri, F. Conformation-dependent accessibility of Cys-136 and Cys-155 of the mitochondrial rat carnitine/acylcarnitine carrier to membrane-impermeable SH reagents. Biochim. Biophys. Acta Biomembr. 2007, 1767, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Dolce, V.; Palmieri, F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 1998, 273, 22782–22787. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.; Palmieri, F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [Green Version]
- Di Noia, M.A.; Todisco, S.; Cirigliano, A.; Rinaldi, T.; Agrimi, G.; Iacobazzi, V.; Palmieri, F. The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J. Biol. Chem. 2014, 289, 33137–33148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jussupow, A.; Di Luca, A.; Kaila, V.R.I. How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 2019, 5, eaav1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, M. Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 2009, 1788, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miniero, D.V.; Cappello, A.R.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Functional and structural role of amino acid residues in the matrix alpha-helices, termini and cytosolic loops of the bovine mitochondrial oxoglutarate carrier. Biochim. Biophys. Acta 2011, 1807, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Cappello, A.R.; Curcio, R.; Valeria Miniero, D.; Stipani, I.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Functional and structural role of amino acid residues in the even-numbered transmembrane alpha-helices of the bovine mitochondrial oxoglutarate carrier. J. Mol. Biol. 2006, 363, 51–62. [Google Scholar] [CrossRef]
- Cappello, A.R.; Miniero, D.V.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Functional and structural role of amino acid residues in the odd-numbered transmembrane alpha-helices of the bovine mitochondrial oxoglutarate carrier. J. Mol. Biol. 2007, 369, 400–412. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Kunji, E.R. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunji, E.R.; Robinson, A.J. The conserved substrate binding site of mitochondrial carriers. Biochim. Biophys. Acta 2006, 1757, 1237–1248. [Google Scholar] [CrossRef] [Green Version]
- Chinen, Y.; Yanagi, K.; Nakamura, S.; Nakayama, N.; Kamiya, M.; Nakayashiro, M.; Kaname, T.; Naritomi, K.; Nakanishi, K. A novel homozygous missense SLC25A20 mutation in three CACT-deficient patients: Clinical and autopsy data. Human Genome Var. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Site-directed mutagenesis of the His residues of the rat mitochondrial carnitine/acylcarnitine carrier: Implications for the role of His-29 in the transport pathway. Biochim. Biophys. Acta Biomembr. 2009, 1787, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Kunji, E.R. Structural changes in the transport cycle of the mitochondrial ADP/ATP carrier. Curr. Opin. Struct. Biol. 2019, 57, 135–144. [Google Scholar] [CrossRef]
- Robinson, A.J.; Overy, C.; Kunji, E.R. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. USA 2008, 105, 17766–17771. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Pierri, C.L. Structure and function of mitochondrial carriers-role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, A.; Pierri, C.L.; Palmieri, F.; Klingenberg, M. The switching mechanism of the mitochondrial ADP/ATP carrier explored by free-energy landscapes. Biochim. Biophys. Acta 2016, 1857, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Giangregorio, N.; Console, L.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Identification of amino acid residues underlying the antiport mechanism of the mitochondrial carnitine/acylcarnitine carrier by site-directed mutagenesis and chemical labeling. Biochemistry 2014, 53, 6924–6933. [Google Scholar] [CrossRef] [PubMed]
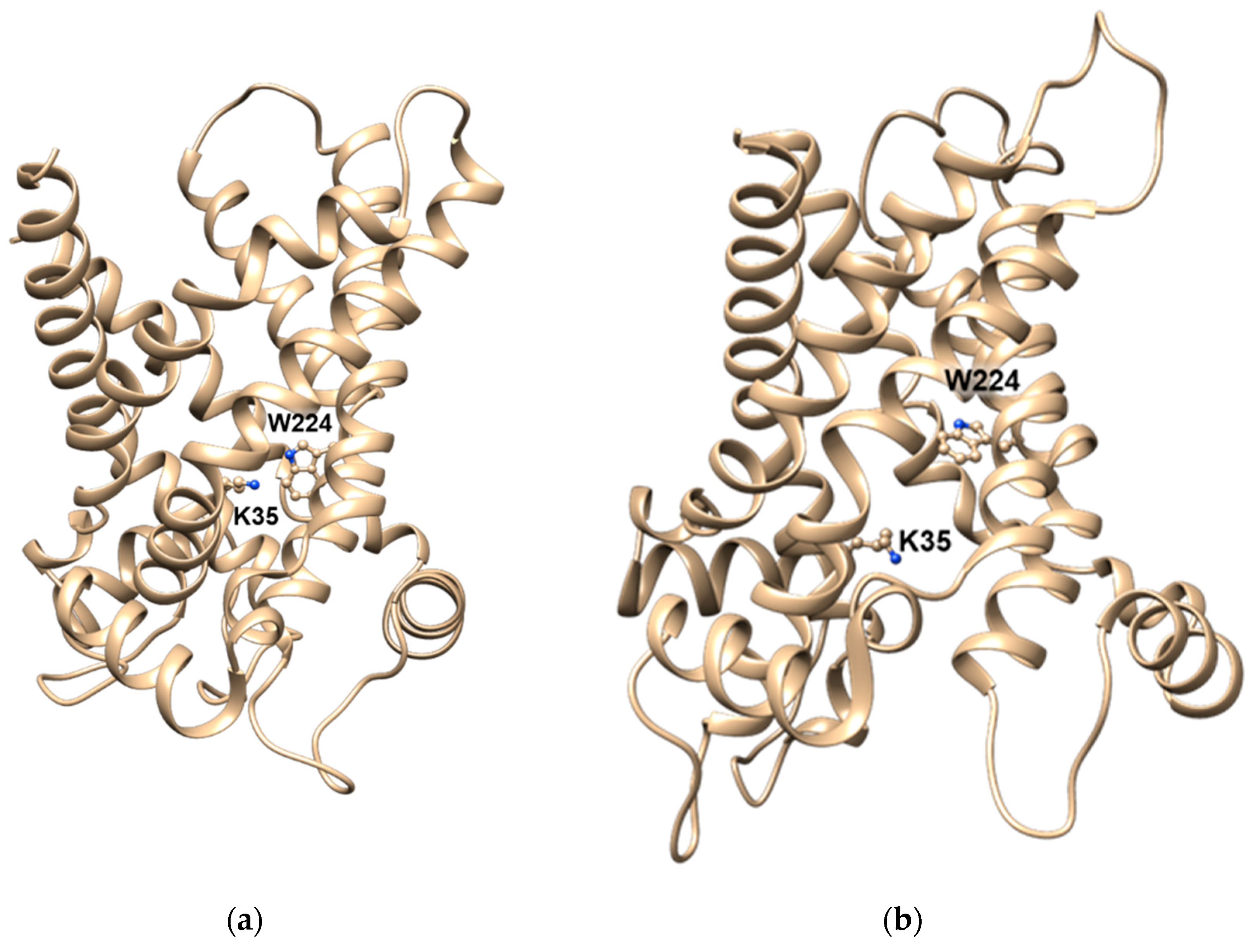
- Giangregorio, N.; Tonazzi, A.; Console, L.; Pistillo, M.; Scalera, V.; Indiveri, C. Tryptophan 224 of the rat mitochondrial carnitine/acylcarnitine carrier is crucial for the antiport mechanism. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J. Biol. Chem. 2012, 287, 7925–7934. [Google Scholar] [CrossRef] [Green Version]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis and chemical modification of three vicinal cysteine residues in rat mitochondrial carnitine/acylcarnitine transporter. J. Biol. Chem. 2005, 280, 19607–19612. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Schroder, E.; Eaton, P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: Issues and considerations. Curr. Opin. Pharmacol. 2008, 8, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Galluccio, M.; Accardi, R.; Cornet, I.; Tommasino, M.; Indiveri, C. Human OCTN2 (SLC22A5) is down-regulated in virus- and nonvirus-mediated cancer. Cell Biochem. Funct. 2012, 30, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Giangregorio, N.; Palmieri, F.; Indiveri, C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta 2013, 1830, 5299–5304. [Google Scholar] [CrossRef] [PubMed]
- Pai, H.V.; Starke, D.W.; Lesnefsky, E.J.; Hoppel, C.L.; Mieyal, J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 2007, 9, 2027–2033. [Google Scholar] [CrossRef]
- Stroher, E.; Millar, A.H. The biological roles of glutaredoxins. Biochem. J. 2012, 446, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [Green Version]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yu, H.; Chen, J.; Sun, J.; Guo, L.; Huang, P.; Zhong, Y. Hydrogen Sulfide and Endoplasmic Reticulum Stress: A Potential Therapeutic Target for Central Nervous System Degeneration Diseases. Front. Pharmacol. 2020, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef]
- Olas, B. Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 2015, 439, 212–218. [Google Scholar] [CrossRef]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Giuffre, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, E.M.; Spera, I.; Menga, A.; Infantino, V.; Porcelli, V.; Iacobazzi, V.; Pierri, C.L.; Hooper, D.C.; Palmieri, F.; Castegna, A. Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim. Biophys. Acta 2015, 1847, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Naglieri, M.A.; Stanley, C.A.; Wanders, R.J.; Palmieri, F. The structure and organization of the human carnitine/acylcarnitine translocase (CACT1) gene. Biochem. Biophys. Res. Commun. 1998, 252, 770–774. [Google Scholar] [CrossRef]
- Viggiano, L.; Iacobazzi, V.; Marzella, R.; Cassano, C.; Rocchi, M.; Palmieri, F. Assignment of the carnitine/acylcarnitine translocase gene (CACT) to human chromosome band 3p21.31 by in situ hybridization. Cytogenet. Cell Genet. 1997, 79, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Ruitenbeek, W.; van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.; Palmieri, F.; Trijbels, J.M. Human mitochondrial transmembrane metabolite carriers: Tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284. [Google Scholar] [CrossRef]
- Tachibana, K.; Takeuchi, K.; Inada, H.; Yamasaki, D.; Ishimoto, K.; Tanaka, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Doi, T. Regulation of the human SLC25A20 expression by peroxisome proliferator-activated receptor alpha in human hepatoblastoma cells. Biochem. Biophys. Res. Commun. 2009, 389, 501–505. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Palmieri, F. Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation. Biology 2013, 2, 284–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobazzi, V.; Convertini, P.; Infantino, V.; Scarcia, P.; Todisco, S.; Palmieri, F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem. Biophys. Res. Commun. 2009, 388, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Giangregorio, N.; Cellamare, S.; Bolognino, I.; Palasciano, M.; Indiveri, C.; Incampo, G.; Campana, S.; Tonazzi, A. Human mitochondrial carnitine acylcarnitine carrier: Molecular target of dietary bioactive polyphenols from sweet cherry (Prunus avium L.). Chem. Biol. Interact. 2019, 307, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Tune, B.M.; Hsu, C.Y. Toxicity of cephaloridine to carnitine transport and fatty acid metabolism in rabbit renal cortical mitochondria: Structure-activity relationships. J. Pharmacol. Exp. Ther. 1994, 270, 873–880. [Google Scholar]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Indiveri, C. Mitochondrial carnitine/acylcarnitine translocase: Insights in structure/ function relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015, 15, 396–405. [Google Scholar] [CrossRef]
- Oppedisano, F.; Fanello, D.; Calvani, M.; Indiveri, C. Interaction of mildronate with the mitochondrial carnitine/acylcarnitine transport protein. J. Biochem. Mol. Toxicol. 2008, 22, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Godinho-Santos, A.; Goncalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, W.; Gwynn, R.C.; Jeffery, N.; Kass, D.; Thorpe, L.E.; Garg, R.K.; Palmer, C.D.; Parsons, P.J. A biomonitoring study of lead, cadmium, and mercury in the blood of New York city adults. Environ. Health Perspect. 2007, 115, 1435–1441. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonazzi, A.; Giangregorio, N.; Console, L.; Palmieri, F.; Indiveri, C. The Mitochondrial Carnitine Acyl-carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs. Biomolecules 2021, 11, 521. https://doi.org/10.3390/biom11040521
Tonazzi A, Giangregorio N, Console L, Palmieri F, Indiveri C. The Mitochondrial Carnitine Acyl-carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs. Biomolecules. 2021; 11(4):521. https://doi.org/10.3390/biom11040521
Chicago/Turabian StyleTonazzi, Annamaria, Nicola Giangregorio, Lara Console, Ferdinando Palmieri, and Cesare Indiveri. 2021. "The Mitochondrial Carnitine Acyl-carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs" Biomolecules 11, no. 4: 521. https://doi.org/10.3390/biom11040521