Deciphering Master Gene Regulators and Associated Networks of Human Mesenchymal Stromal Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Multiple Cell Sample Series with Transcriptomic Data Collected and Unified in a Compendium Set
2.2. Regulatory Networks Based on Mutual Information
2.3. Differential Expression Between Six Types of Human Cells Related to Bone-Marrow MSCs
2.4. Detection of Master Regulators in the Regulatory Networks
2.5. Gene Set Functional Enrichment Analysis
2.6. DNA Methylation Analysis
3. Results and Discussion
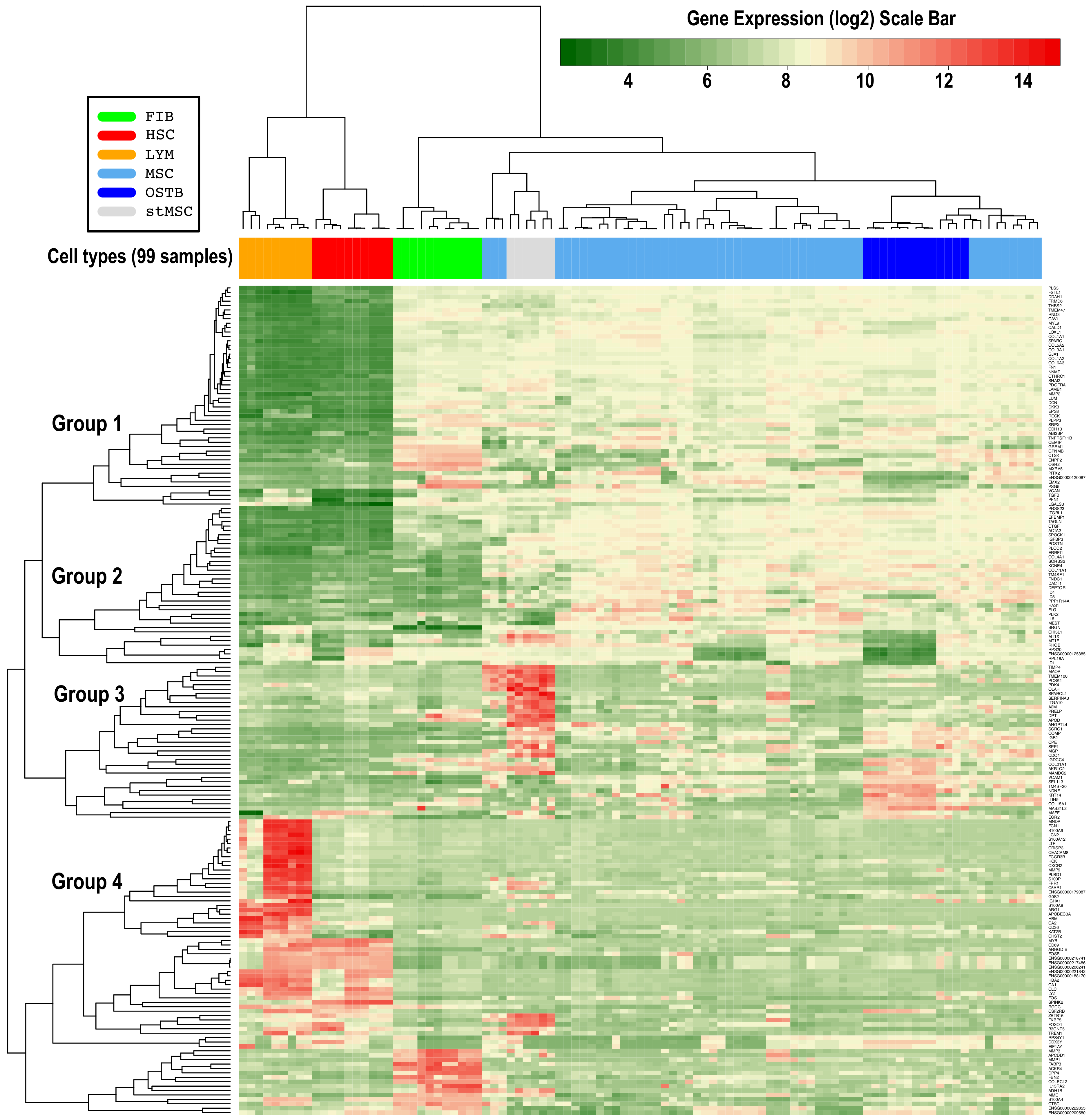
3.1. Differential Gene Expression Profiling of MSCs versus Related Cell Types
3.2. Identification of Master Regulators of MSCs and of Other Related Cell Types
3.3. Differential Expression Analysis of the MSCs Master Regulators
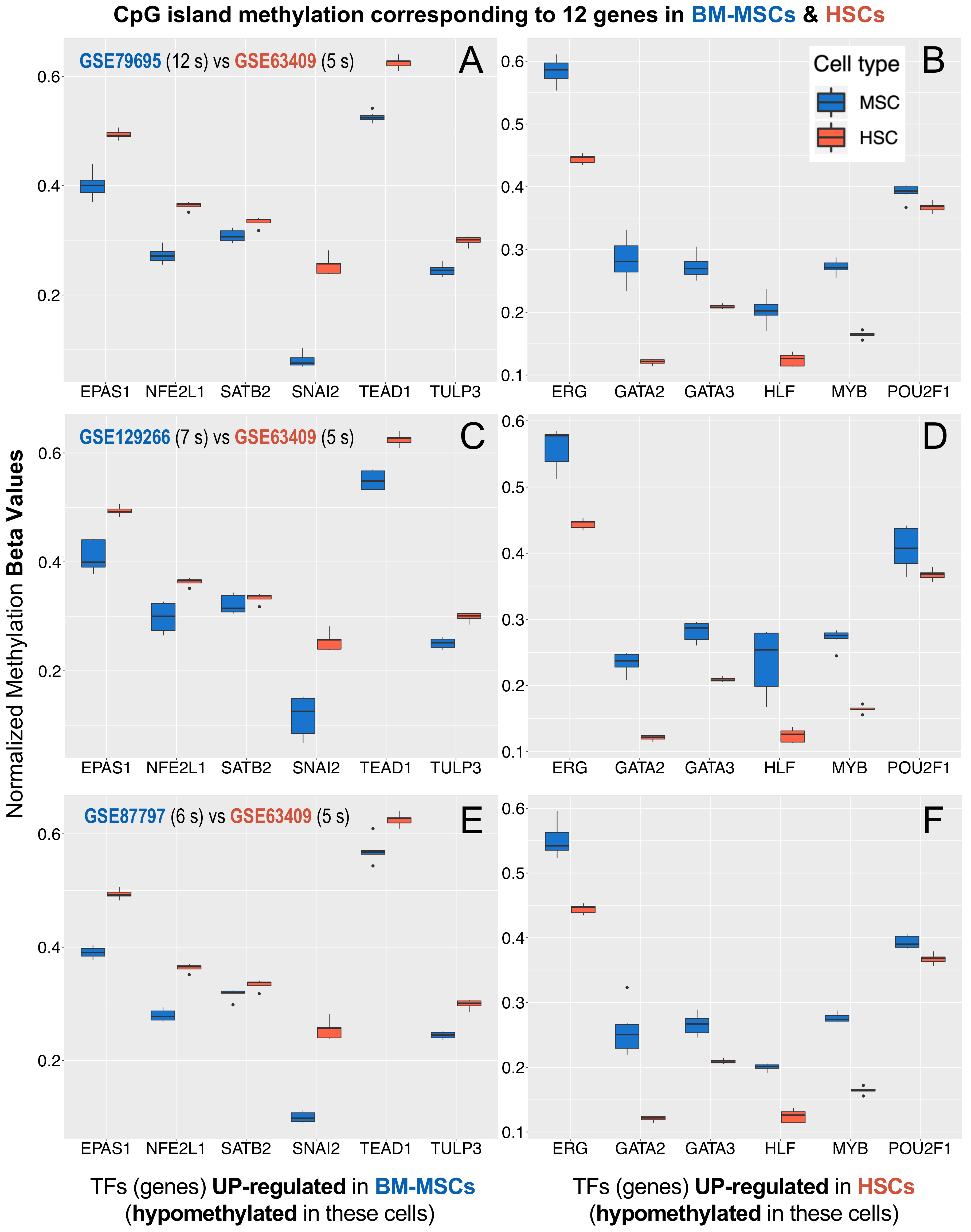
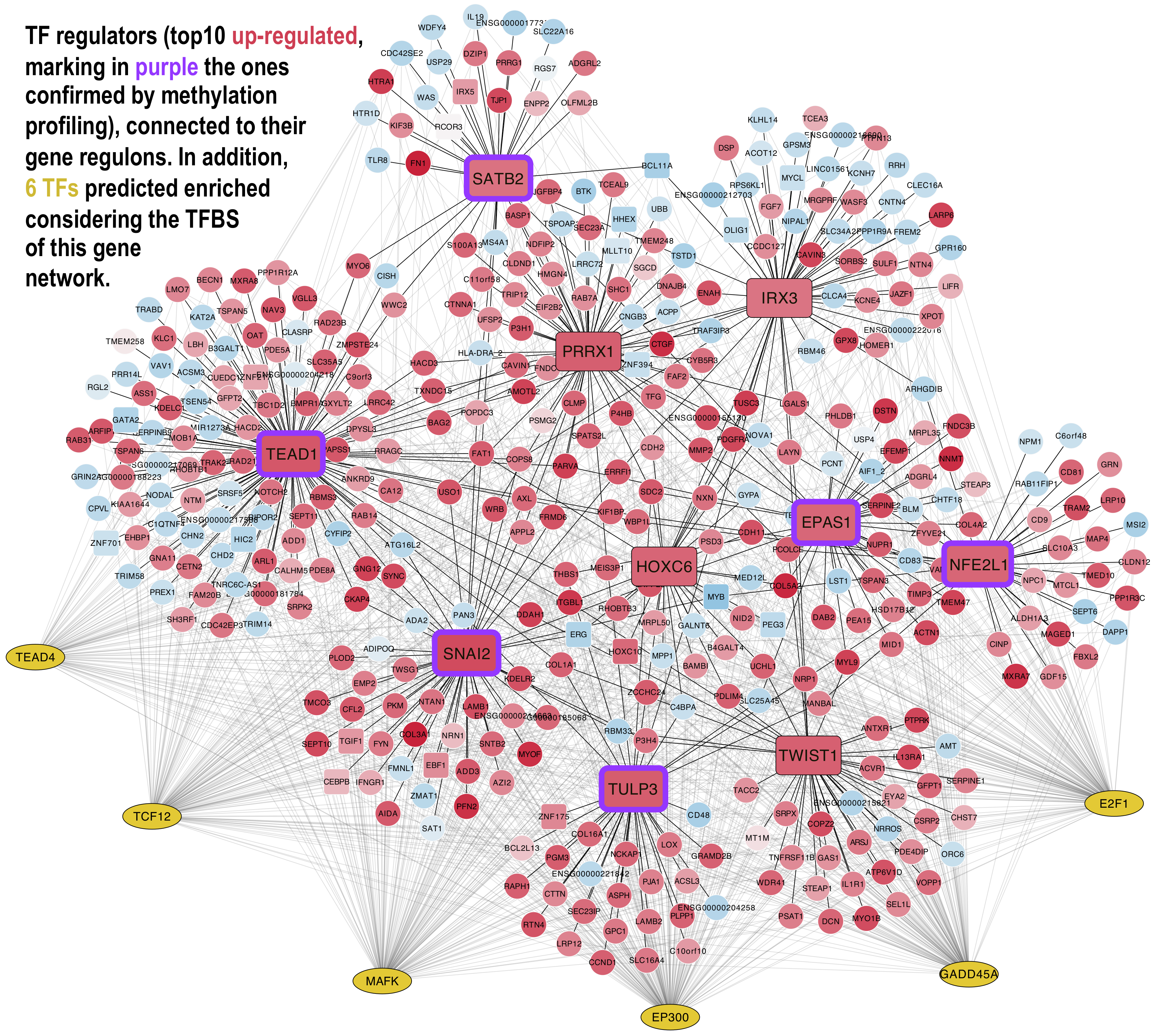
3.4. Methylation Profiles of the MSCs Master Regulators
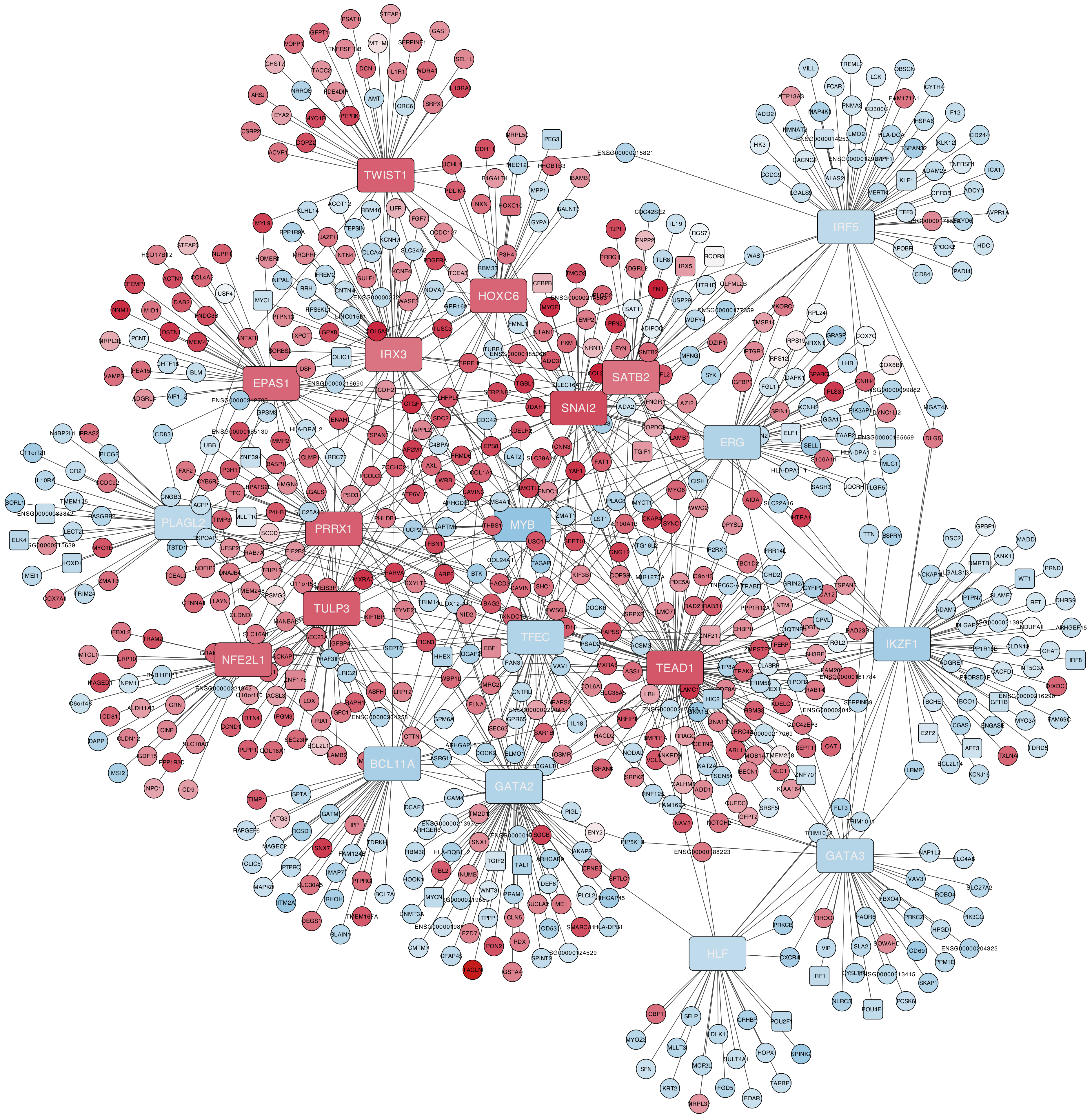
3.5. Construction of Bipartite Networks Including Regulators and Regulons
3.6. Functional Enrichment Analysis of the Master Regulators and Their Regulons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yu, V.W.C.; Scadden, D.T. Hematopoietic Stem Cell and Its Bone Marrow Niche. Current Top. Dev. Biol. 2016, 118, 21–44. [Google Scholar] [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Keating, A. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A.; Nemcek, S.; Bloom, D.D.; Goodrich, A.D.; Kim, J.; Mosher, D.F.; Hematti, P. Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematol. 2016, 136, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roson-Burgo, B.; Sanchez-Guijo, F.; Del Cañizo, C.; De Las Rivas, J. Insights into the human mesenchymal stromal/stem cell identity through integrative transcriptomic profiling. BMC Genom. 2016, 17, 944. [Google Scholar] [CrossRef] [Green Version]
- Waterman, R.S.; Henkle, S.L.; Betancourt, A.M. Mesenchymal Stem Cell 1 (MSC1)-Based Therapy Attenuates Tumor Growth Whereas MSC2-Treatment Promotes Tumor Growth and Metastasis. PLoS ONE 2012, 7, e45590. [Google Scholar] [CrossRef] [Green Version]
- Najar, M.; Krayem, M.; Meuleman, N.; Bron, D.; Lagneaux, L. Mesenchymal stromal cells and toll-like receptor priming: A critical review. Immune Netw. 2017, 17, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Network-based inference of protein activity helps functionalize the genetic landscape of cancer. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Risueño, A.; Fontanillo, C.; Dinger, M.E.; De Las Rivas, J. GATExplorer: Genomic and transcriptomic explorer; mapping expression probes to gene loci, transcripts, exons and ncRNAs. BMC Bioinform. 2010, 11, 221. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. Minet: A r/bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinform. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Sales, G.; Romualdi, C. Parmigene-a parallel R package for mutual information estimation and gene network reconstruction. Bioinformatics 2011, 27, 1876–1877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Liu, T.; Liu, C.J.; Song, S.; Zhang, X.; Liu, W.; Guo, A.Y. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 7, e47. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Nucleic Acids Res. 2003, 13, 2498–2504. [Google Scholar]
- Janky, R.; Verfaillie, A.; Imrichová, H.; van de Sande, B.; Standaert, L.; Christiaens, V.; Aerts, S. iRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Fontanillo, C.; Nogales-Cadenas, R.; Pascual-Montano, A.; De Las Rivas, J. Functional analysis beyond enrichment: Non-redundant reciprocal linkage of genes and biological terms. PLoS ONE 2011, 6, e24289. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatic 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatic 2017, 33, 558–560. [Google Scholar] [CrossRef]
- Cheng, W.-Y.; Kandel, J.; Yamashiro, D.; Canoll, P.; Anastassiou, D. Slug-based epithelial-mesenchymal transition gene signature is associated with prolonged time to recurrence in glioblastoma. Nat. Preced. 2011. [Google Scholar] [CrossRef]
- Herpin, A.; Lelong, C.; Favrel, P. Transforming growth factor-β-related proteins: An ancestral and widespread superfamily of cytokines in metazoans. Dev. Comp. Immunol. 2004, 28, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.J.; Zhao, K.Q.; Wang, Z.Q.; Feng, Y.; Xing, S.C. Mesenchymal stem cells display different gene expression profiles compared to hyaline and elastic chondrocytes. Int. J. Clin. Exp. Med. 2011, 4, 81–90. [Google Scholar] [PubMed]
- Xu, C.; Sun, L.; Jiang, C.; Zhou, H.; Gu, L.; Liu, Y.; Xu, Q. SPP1, analyzed by bioinformatics methods, promotes the metastasis in colorectal cancer by activating EMT pathway. Biomed. Pharmacother. 2017, 91, 1167–1177. [Google Scholar] [CrossRef]
- Ward, A.; Hudson, J.W. p53-dependent and cell specific epigenetic regulation of the polo-like kinases under oxidative stress. PLoS ONE 2014, 9, e87918. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Canalis, E.; Parker, K.; Zanotti, S. Gremlin1 is required for skeletal development and postnatal skeletal homeostasis. J. Cel. Physiol. 2012, 227, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Zaman, G.; Sunters, A.; Galea, G.L.; Javaheri, B.; Saxon, L.K.; Moustafa, A.; Lanyon, L.E. Loading-related regulation of transcription factor EGR2/Krox-20 in bone cells is ERK1/2 protein-mediated and prostaglandin, Wnt signaling pathway-, and insulin-like growth factor-i axis-dependent. J. Biol. Chem. 2012, 289, 25509. [Google Scholar] [CrossRef] [Green Version]
- González, A.J.; Setty, M.; Leslie, C.S. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat. Genet. 2015, 47, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Bujanover, N.; Goldstein, O.; Greenshpan, Y.; Turgeman, H.; Klainberger, A.; Scharff, Y.; Gazit, R. Identification of immune-activated hematopoietic stem cells. Leukemia 2018, 32, 2016–2020. [Google Scholar] [CrossRef]
- Ganesan, R.; Mallets, E.; Gomez-Cambronero, J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 2016, 10, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, P.I.; Brendeford, E.M.; Gilfillan, S.; Gavrilov, A.A.; Leedsak, M.; Razin, S.V.; Gabrielsen, O.S. Identification of c-Myb Target Genes in K562 Cells Reveals a Role for c-Myb as a Master Regulator. Genes Cancer. 2011, 2, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froidure, A.; Marchal-Duval, E.; Ghanem, M.; Gerish, L.; Jaillet, M.; Crestani, B.; Mailleux, A. Mesenchyme associated transcription factor PRRX1: A key regulator of IPF fibroblast. Eur. Respir. J. 2016, 48, OA506. [Google Scholar]
- Tijchon, E.; Havinga, J.; Van Leeuwen, F.N.; Scheijen, B. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia. 2013, 27, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, K.; Qian, C.N.; Leach, R. DNA methylation is associated with transcription of Snail and Slug genes. Biochem. Biophys. Res. Commun. 2013, 430, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gomez, A.; Li, T.; Rodríguez-Ubreva, J.; Ciudad, L.; Català-Moll, F.; Martín-Sánchez, M.; San-Segundo, L.; Morales, X.; Ortiz de Solórzano, C.; Oyarzabal, J.; et al. Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. bioRxiv 2019, 767897. [Google Scholar] [CrossRef] [Green Version]
- Aranda, P.; Agirre, X.; Ballestar, E.; Andreu, E.J.; Román-Gómez, J.; Prieto, I.; Martín-Subero, J.I.; Cigudosa, J.C.; Siebert, R.; Esteller, M.; et al. Epigenetic signatures associated with different levels of differentiation potential in human stem cells. PLoS ONE 2009, 4, e7809. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Yang, P.; Wu, Y.; Meng, S.; Sui, L.; Zhang, L.; Yu, L.; Tang, Y.; Jiang, H.; Xuan, D.; et al. Epigenetically modified bone marrow stromal cells in silk scaffolds promote craniofacial bone repair and wound healing. Tissue Eng. Part A 2015, 21, 2156–2165. [Google Scholar] [CrossRef]
- Krampera, M.; Marconi, S.; Pasini, A.; Galiè, M.; Rigotti, G.; Mosna, F.; Bonetti, B. Induction of neural-like differentiation in human mesenchymal stem cells derived from bone marrow, fat, spleen and thymus. Bone 2007, 40, 382–390. [Google Scholar] [CrossRef]
- Calloni, G.W.; Glavieux-Pardanaud, C.; Le Douarin, N.M.; Dupin, E. Sonic Hedgehog promotes the development of multipotent neural crest progenitors endowed with both mesenchymal and neural potentials. Proc. Natl. Acad. Sci. USA 2007, 104, 19879–19884. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, K.J.; Dovat, S. Ikaros and tumor suppression in acute Lymphoblastic leukemia. Crit. Rev. Oncog. 2011, 16, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Zhang, F.; Tao, H.; Ma, X.; Su, G.; Xie, X.; Yin, B. BCL11A expression in acute phase chronic myeloid leukemia. Leuk. Res. 2016, 47, 88–92. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enriched Functional Term | N Genes (in the Function) | N Genes (in the Query) | N in Function/N in Query (%) | Regulation (UP/DOWN) | p-value (adj. Benjamini) |
|---|---|---|---|---|---|
| generation of neurons | 26 | 283 | 7.34 | UP | 0.015168 |
| neurogenesis | 30 | 283 | 8.47 | UP | 0.001735 |
| nervous system development | 42 | 283 | 11.86 | UP | 0.007296 |
| cell-substrate adhesion | 10 | 283 | 2.82 | UP | 0.014732 |
| extracellular matrix | 22 | 323 | 6.21 | UP | 0.001305 |
| cytoskeleton | 51 | 323 | 14.41 | UP | 0.003200 |
| face morphogenesis | 4 | 283 | 1.13 | UP | 0.047319 |
| embryonic development | 29 | 283 | 8.19 | UP | 0.001836 |
| organ morphogenesis | 31 | 283 | 8.76 | UP | 0.000405 |
| organ development | 57 | 283 | 16.10 | UP | 0.016957 |
| cell differentiation | 61 | 283 | 17.23 | UP | 0.000488 |
| T cell activation | 17 | 293 | 5.00 | DOWN | 0.000004 |
| lymphocyte activation | 21 | 293 | 6.18 | DOWN | 0.000005 |
| hemopoiesis | 22 | 293 | 6.47 | DOWN | 0.000010 |
| hemopoietic or lymphoid organ development | 22 | 293 | 6.47 | DOWN | 0.000043 |
| immune system development | 22 | 293 | 6.47 | DOWN | 0.000074 |
| regulation of immune system process | 23 | 293 | 6.76 | DOWN | 0.002043 |
| calponin-like actin-binding | 10 | 323 | 2.94 | DOWN | 0.002577 |
| actin filament-based process | 19 | 293 | 5.59 | DOWN | 0.000509 |
| cytoskeleton organization | 26 | 293 | 7.65 | DOWN | 0.000664 |
| GTPase regulator activity | 24 | 304 | 7.06 | DOWN | 0.001932 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Luis, E.; Joaquín-García, A.; Campos-Laborie, F.J.; Sánchez-Guijo, F.; De las Rivas, J. Deciphering Master Gene Regulators and Associated Networks of Human Mesenchymal Stromal Cells. Biomolecules 2020, 10, 557. https://doi.org/10.3390/biom10040557
Sánchez-Luis E, Joaquín-García A, Campos-Laborie FJ, Sánchez-Guijo F, De las Rivas J. Deciphering Master Gene Regulators and Associated Networks of Human Mesenchymal Stromal Cells. Biomolecules. 2020; 10(4):557. https://doi.org/10.3390/biom10040557
Chicago/Turabian StyleSánchez-Luis, Elena, Andrea Joaquín-García, Francisco J. Campos-Laborie, Fermín Sánchez-Guijo, and Javier De las Rivas. 2020. "Deciphering Master Gene Regulators and Associated Networks of Human Mesenchymal Stromal Cells" Biomolecules 10, no. 4: 557. https://doi.org/10.3390/biom10040557