Chemoenzymatic Synthesis of Enantiomeric, Bicyclic δ-Halo-γ-lactones with a Cyclohexane Ring, Their Biological Activity and Interaction with Biological Membranes

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis
2.2. Chemicals
2.3. Synthesis of Lactones with Cyclohexane System
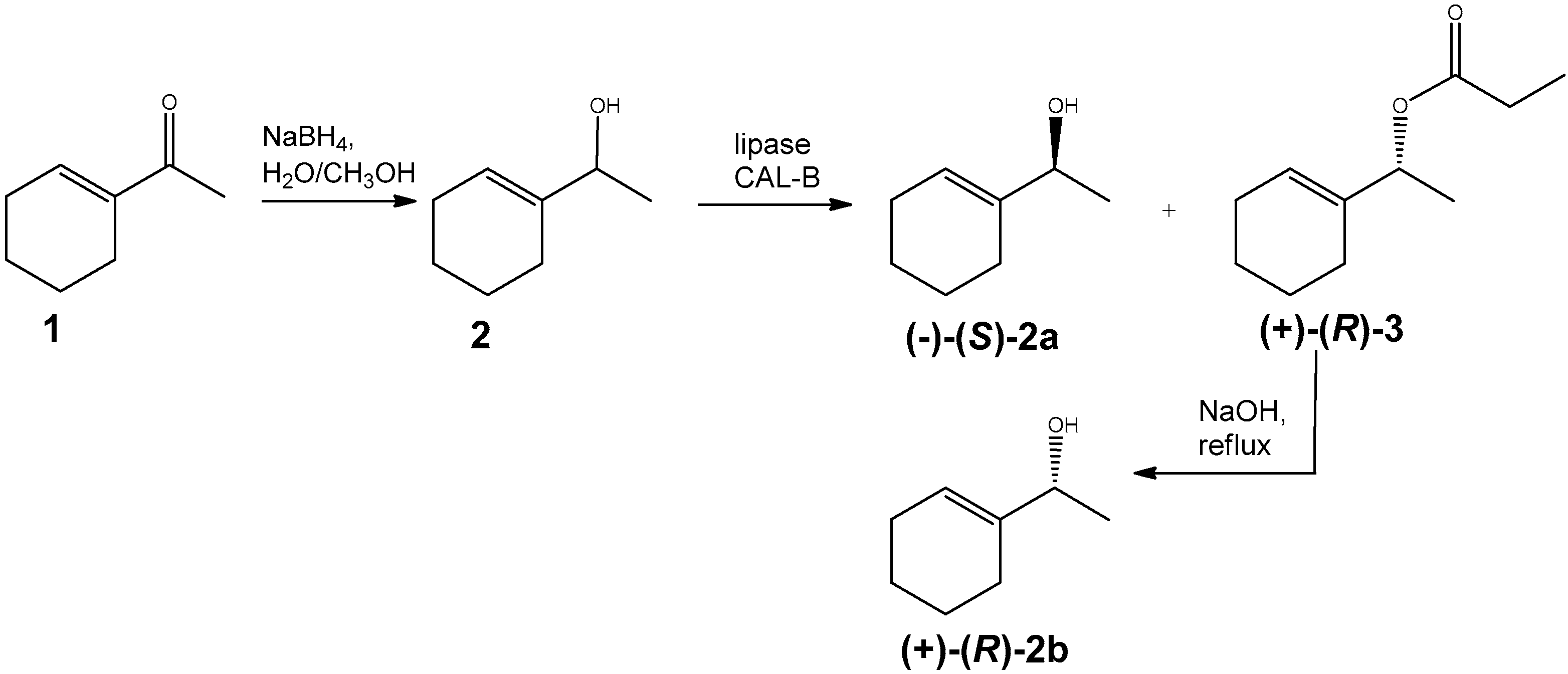
2.3.1. 1-(Cyclohex-1-en-1-yl) ethanol (2)
2.3.2. Kinetic Resolution of rac-2 by Enzymatic Transesterification
2.3.3. (R)-1-(cyclohex-1-en-1-yl)ethanol (2b)
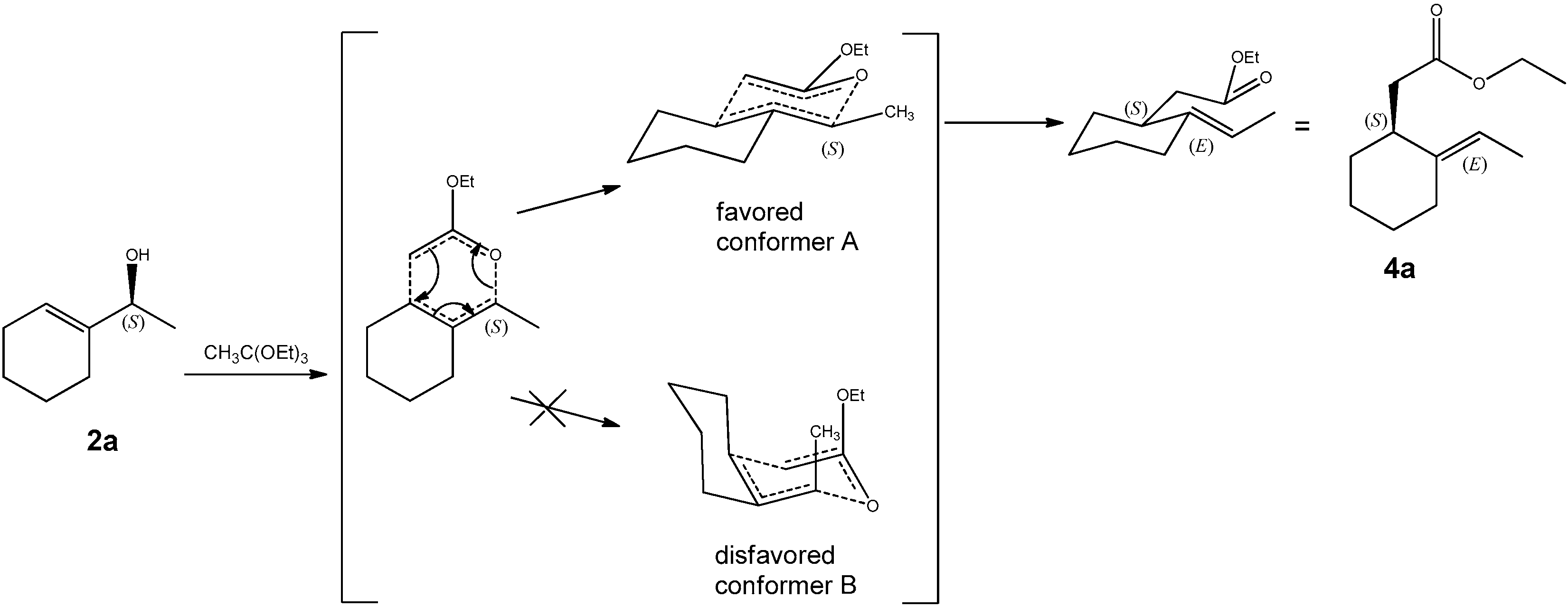
2.3.4. Ethyl 2-(2-ethylidenecyclohexyl) Acetates (4a and 4b)
General Procedure
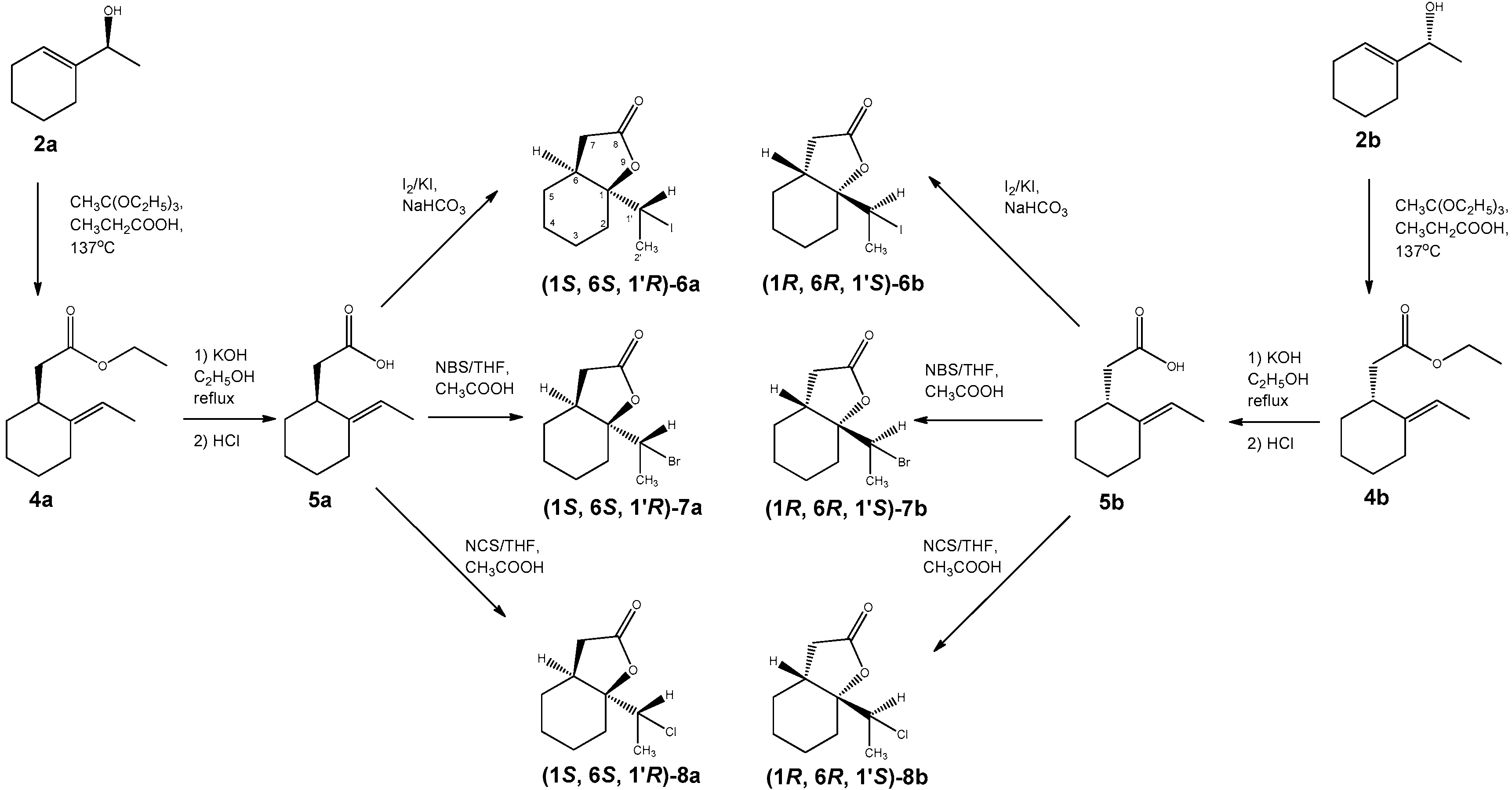
2.3.5. (E)-2-(2-Ethylidenecyclohexyl) Acetic Acid (5a and 5b)
General Procedure
2.3.6. 1-(1′-Iodoethyl)-9-Oxabicyclo[4.3.0]nonan-8-one (6a and 6b)
General Procedure
2.3.7. 1-(1′-Bromoethyl)-9-Oxabicyclo[4.3.0]nonan-8-one (7a and 7b)
General Procedure
2.3.8. 1-(1′-Chloroethyl)-9-Oxabicyclo[4.3.0]nonan-8-one (8a and 8b)
General Procedure
2.4. Antimicrobial Activity of Lactones
2.5. Antiproliferative Activity
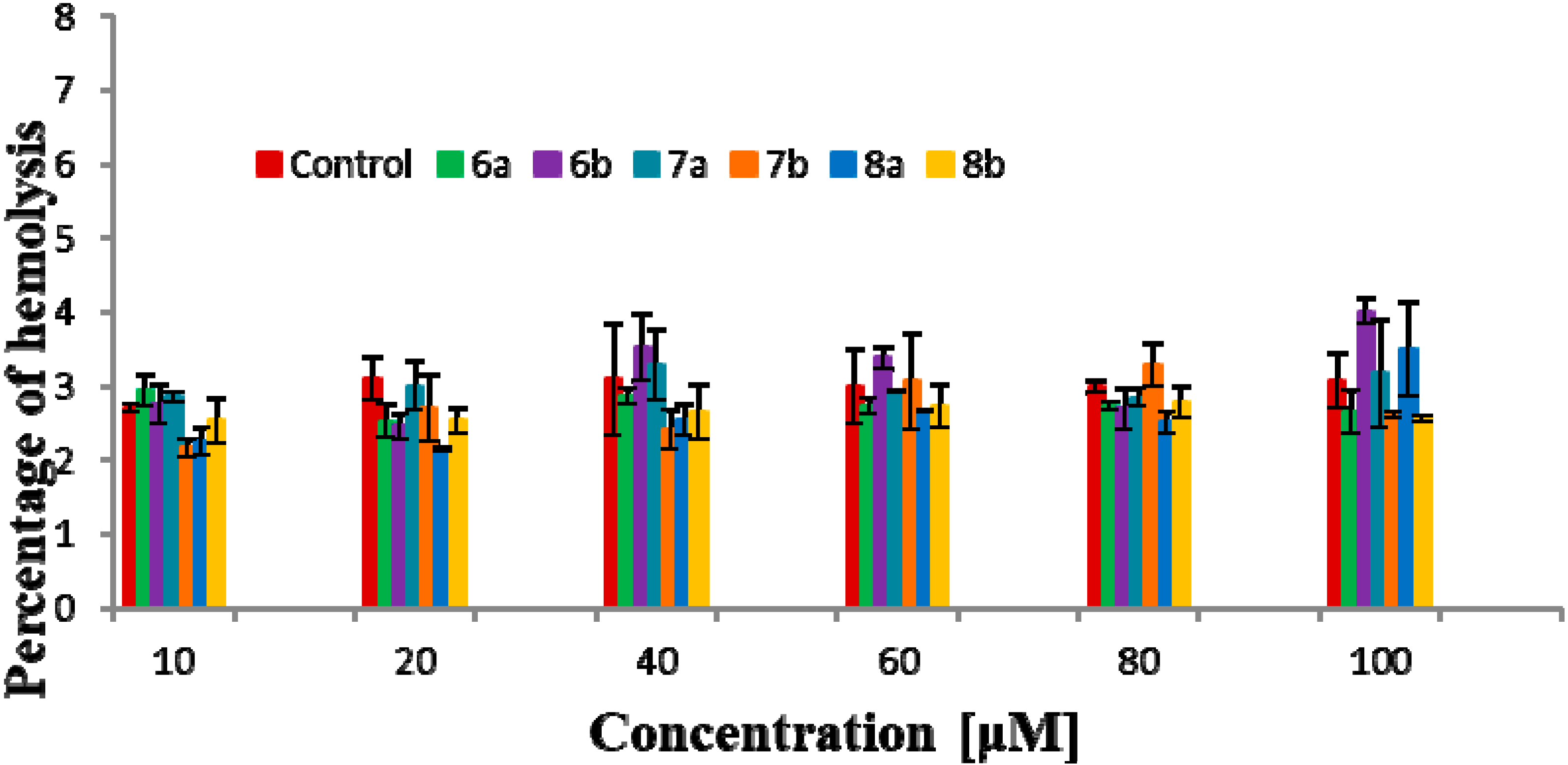
2.6. Hemolytic Activity
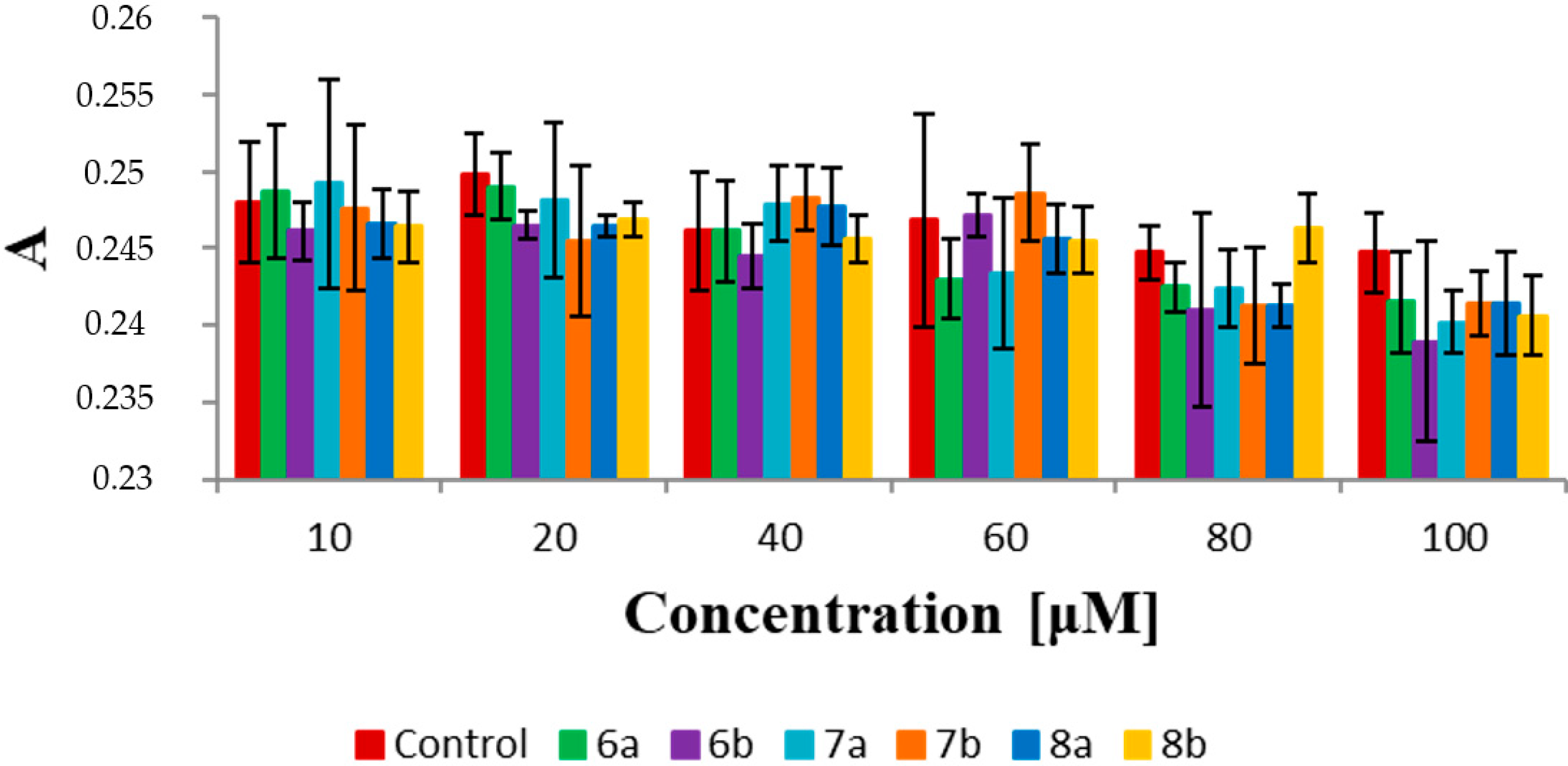
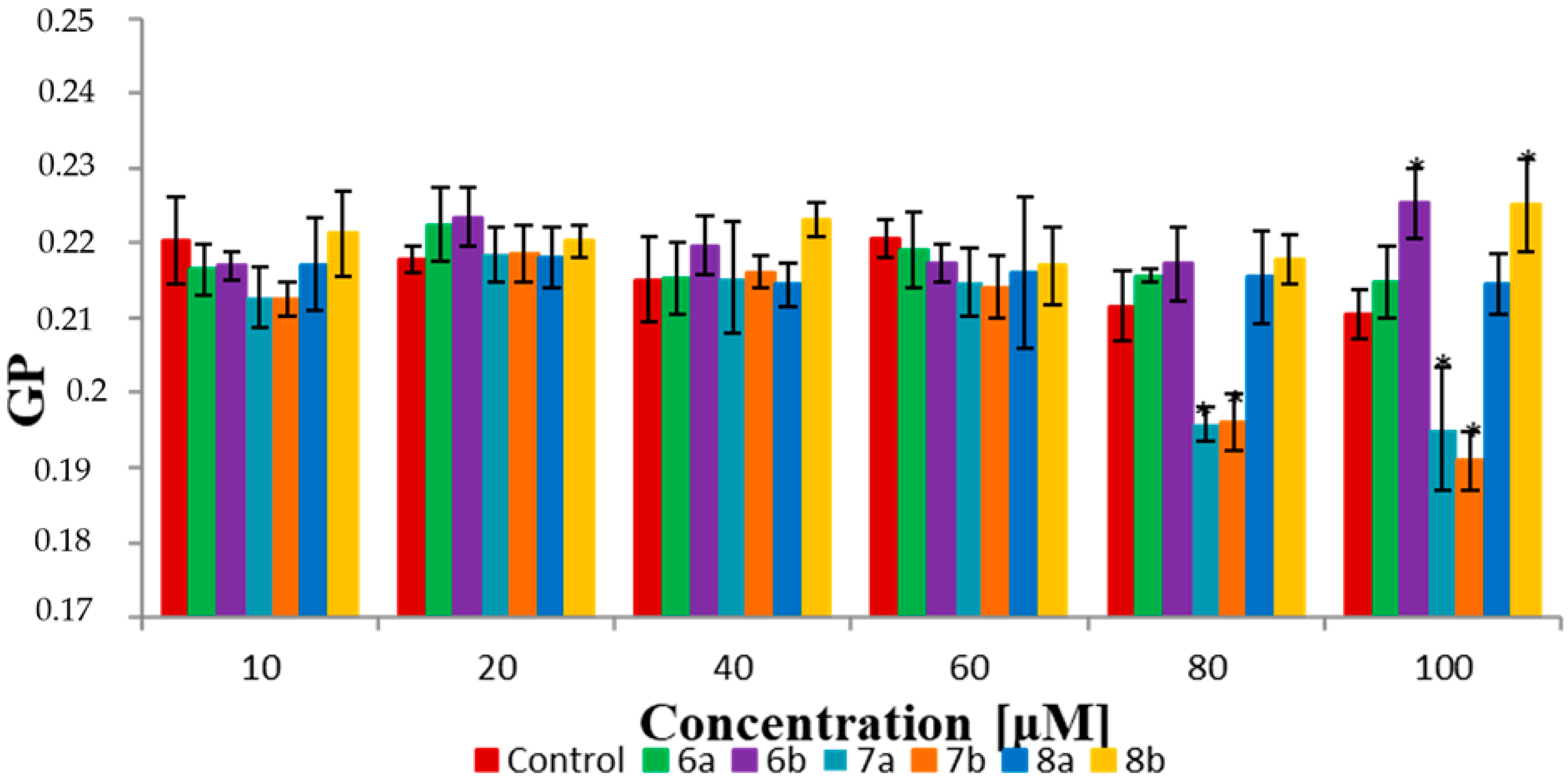
2.7. Fluorescence Spectroscopy
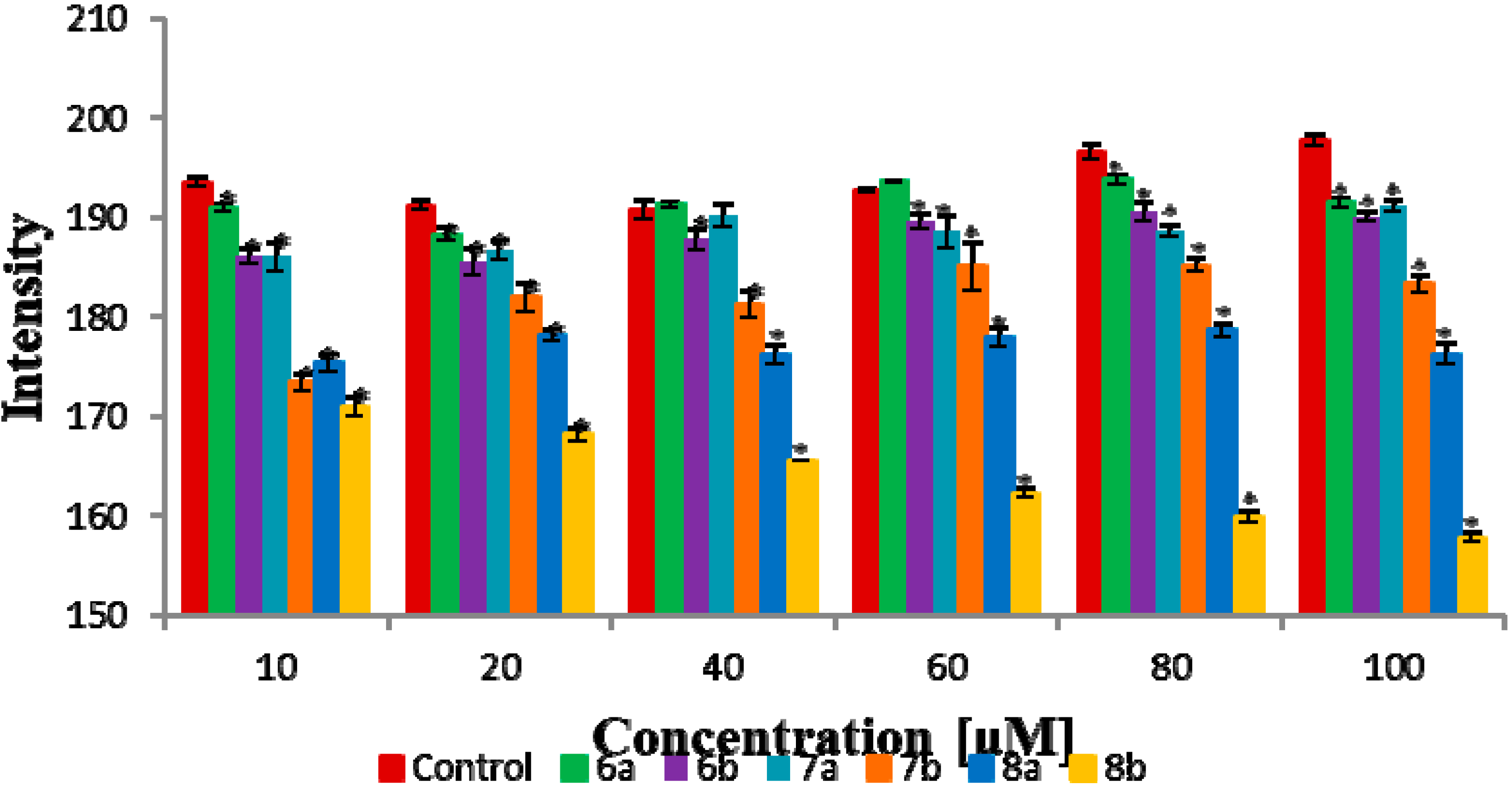
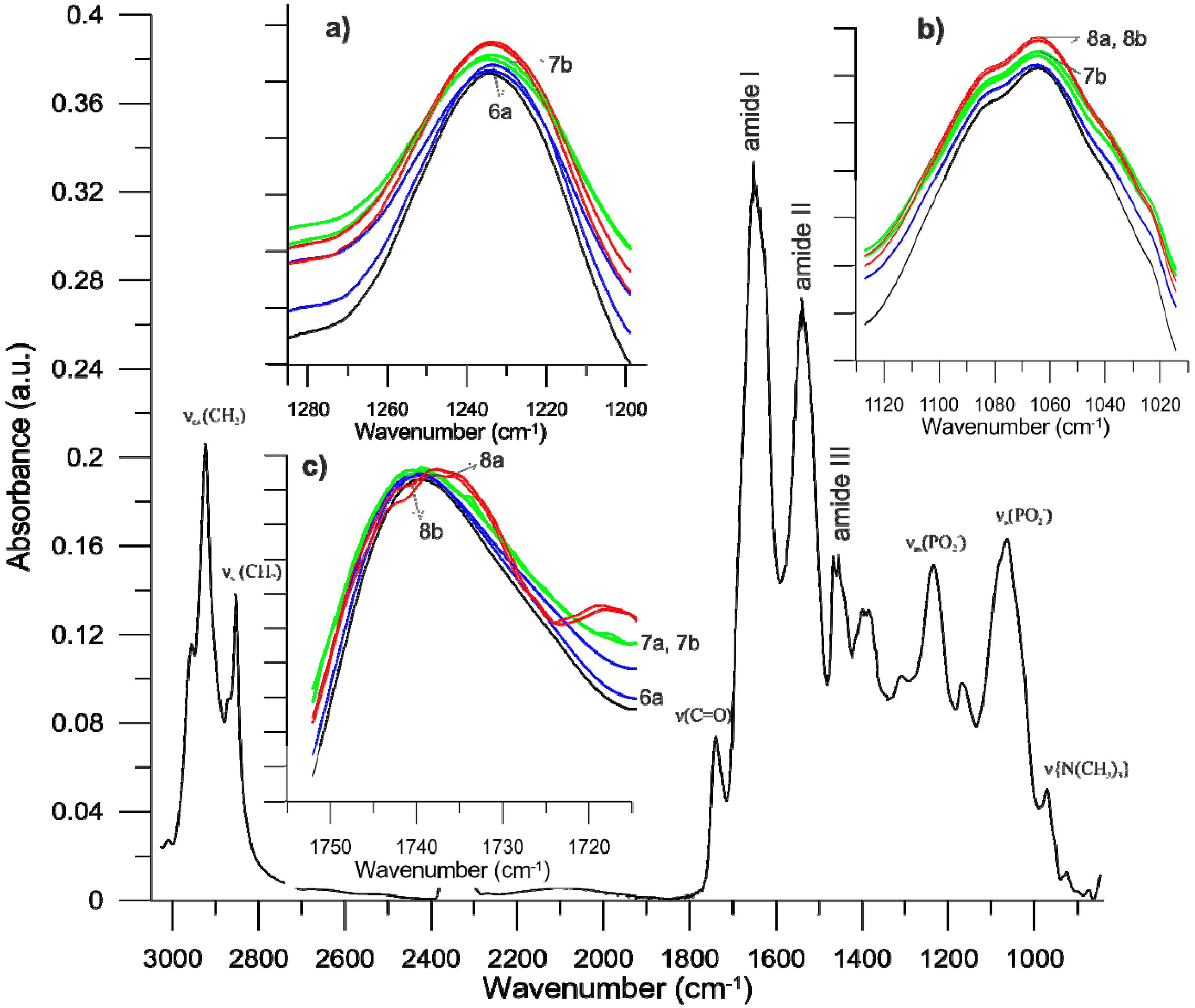
2.8. Fourier Transform Infrared Spectroscopy Method (FTIR)
3. Results and Discussion
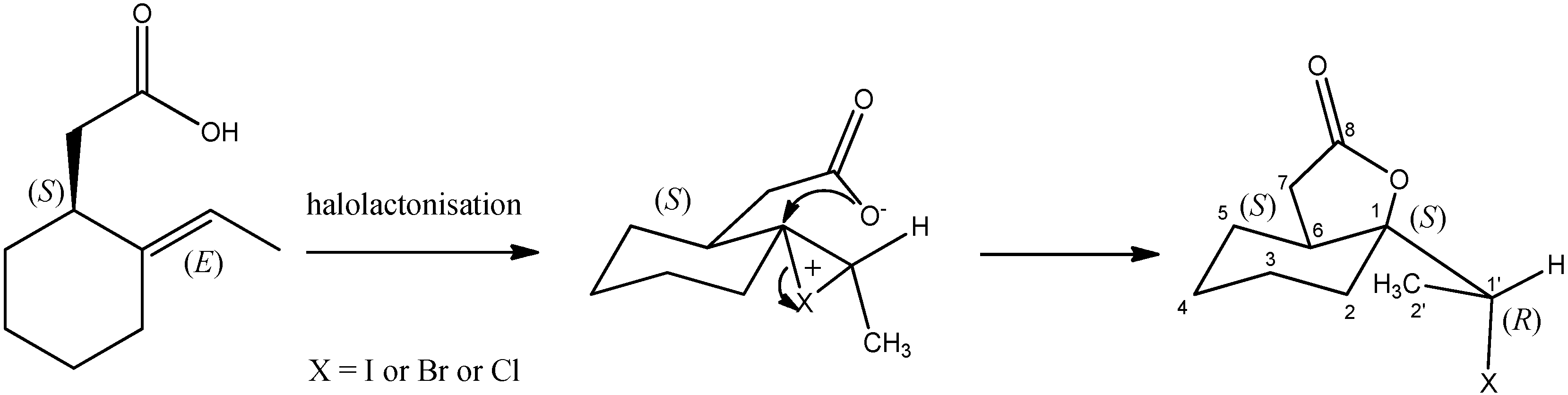
3.1. Synthesis of Halolactones
3.2. The Biological Activity
3.3. Biophysical Research
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gawdzik, B.; Kamizela, A.; Szyszkowska, A. Lactones with a fragrance properties. Chemik 2015, 69, 346–349. [Google Scholar]
- McGinty, D.; Letizia, C.S.; Api, A.M. Fragrance material review on ω-pentadecalactone. Food Chem. Toxicol. 2011, 49, S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Zope, D.D.; Patnekar, S.G.; Kanetkar, V.R. Novel synthesis of flavour quality γ-lactones. Flavour Fragr. J. 2006, 21, 395–399. [Google Scholar] [CrossRef]
- Nation, J.L. The Sex Pheromone Blend of Caribbean Fruit Fly Males: 1 Isolation Biological Activity, and Partial Chemical Characterization 2. Environ. Entomol. 1975, 4, 27–30. [Google Scholar] [CrossRef]
- Swedenborg, P.D.; Jones, R.L.; Zhou, H.Q.; Shin, I.; Liu, H.W. Biological activity of (3R,5S,6R)- and (3S,5R,6S)-3,5-dimethyl-6-(methylethyl)-3,4,5,6-tetrahydropyran-2-one, a pheromone of Macrocentrus grandii (Goidanich) (Hymenoptera: Braconidae). J. Chem. Ecol. 1994, 20, 3373–3380. [Google Scholar] [CrossRef] [PubMed]
- Grudniewska, A.; Kłobucki, M.; Dancewicz, K.; Szczepanik, M.; Gabryś, B.; Wawrzeńczyk, C. Synthesis and antifeedant activity of racemic and optically active hydroxy lactones with the p-Menthane system. PLoS ONE 2015, 10, e0131028. [Google Scholar] [CrossRef] [Green Version]
- Mazur, M.; Gładkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Białońska, A.; Wawrzeńczyk, C. Lactones 43. New biologically active lactones: β-cyclocitral derivatives. Pest Manag. Sci. 2014, 70, 286–294. [Google Scholar] [CrossRef]
- Mazur, M.; Skrobiszewski, A.; Gladkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Klejdysz, T.; Wawrzeńczyk, C. Lactones 46. Synthesis, antifeedant and antibacterial activity of γ-lactones with a p-methoxyphenyl substituent. Pest Manag. Sci. 2016, 72, 489–496. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.; Nadkarni, S.R.; Patel, M.V.; Bhat, R.G.; Desikan, K.R.; Ganguli, B.N.; Rupp, R.H.; Fehlhaber, H.W.; Kogler, H. Maclafungin, a new antifungal macrocyclic lactone from actinomycete sp. Y-8521050. Tetrahedron 1998, 54, 13621–13628. [Google Scholar] [CrossRef]
- Olejniczak, T.; Boraty, F.; Bia, A. Fungistatic Activity of Bicyclo [4.3.0]-γ-lactones. J. Agric. Food Chem. 2011; 59, 6071–6081. [Google Scholar] [CrossRef]
- Rabe, T.; Mullholland, D.; Van Staden, J. Isolation and identification of antibacterial compounds from Vernonia colorata leaves. J. Ethnopharmacol. 2002, 80, 91–94. [Google Scholar] [CrossRef]
- Habtamu, A.; Melaku, Y. Antibacterial and Antioxidant Compounds from the Flower Extracts of Vernonia amygdalina. Adv. Pharmacol. Sci. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, D. Sesquiterpene lactones: Structural diversity and their biological activities. Oppor. Chall. Scope Nat. Prod. Med. Chem. 2011, 661, 313–334. [Google Scholar]
- Alwaseem, H.; Frisch, B.J.; Fasan, R.; States, U.; States, U. Anticancer activity profiling of parthenolide analogs generated via P450-mediated chemoenzymatic synthesis. Bioorg. Med. Chem. 2019, 26, 1365–1373. [Google Scholar] [CrossRef]
- Rasul, A.; Parveen, S.; Ma, T. Costunolide: A novel anti-cancer sesquiterpene lactone. Bangladesh J. Pharmacol. 2012, 7, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Skrobiszewski, A.; Gładkowski, W.; Mazur, M.; Szczepanik, M.; Maciejewska, G.; Wawrzenczyk, C. Microbial hydrolysis of racemic β-aryl-γ-ethylidene-γ-lactones and antifeedant activity of the products against alphitobius diaperinus panzer. Molecules 2018, 23, 1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and anticancer activity of novel halolactones with β-aryl substituents from simple aromatic aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Pawlak, A.; Gładkowski, W.; Kutkowska, J.; Mazur, M.; Obmińska-Mrukowicz, B.; Rapak, A. Enantiomeric trans β-aryl-δ-iodo-γ-lactones derived from 2,5-dimethylbenzaldehyde induce apoptosis in canine lymphoma cell lines by downregulation of anti-apoptotic Bcl-2 family members Bcl-xL and Bcl-2. Bioorganic Med. Chem. Lett. 2018, 28, 1171–1177. [Google Scholar] [CrossRef]
- Gładkowski, W.; Włoch, A.; Pawlak, A.; Sysak, A.; Białońska, A.; Mazur, M.; Mituła, P.; Maciejewska, G.; Obmińska-Mrukowicz, B.; Kleszczyńska, H. Preparation of Enantiomeric β-(2′,5′-Dimethylphenyl)Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes. Molecules 2018, 23, 3035. [Google Scholar] [CrossRef] [Green Version]
- Mazur, M.; Gładkowski, W.; Pawlak, A.; Obminska-Mrukowicz, B.; Maciejewska, G.; Wawrzenczyk, C. Microbial asymmetric functionalization of β-cyclocitral-derived tetramethyl-substituted γ-lactone. Molecules 2019, 24, 666. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.A.; Michael, F.E.; Tedrow, J.S.; Campos, K.R. Application of chiral mixed phosphorus/sulfur ligands to enantioselective rhodium-catalyzed dehydroamino acid hydrogenation and ketone hydrosilylation processes. J. Am. Chem. Soc. 2003, 125, 3534–3543. [Google Scholar] [CrossRef]
- Dong, X.; Weickgenannt, A.; Oestreich, M. Broad-spectrum kinetic resolution of alcohols enabled by Cu-H-catalysed dehydrogenative coupling with hydrosilanes. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birman, V.B.; Jiang, H. Kinetic resolution of alcohols using a 1,2-dihydroimidazo[1,2-a]quinoline enantioselective acylation catalyst. Org. Lett. 2005, 7, 3445–3447. [Google Scholar] [CrossRef] [PubMed]
- Hazelden, I.R.; Carmona, R.C.; Langer, T.; Pringle, P.G.; Bower, J.F. Pyrrolidines and Piperidines by Ligand-Enabled Aza-Heck Cyclizations and Cascades of N-(Pentafluorobenzoyloxy)carbamates. Angew. Chemie-Int. Ed. 2018, 57, 5124–5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zulfiqar, F.; Malik, A. Facile approach to versatile chiral intermediates for fused cyclopentanoid natural products. Zeitschrift fur Naturforsch.-Sect. B J. Chem. Sci. 2001, 56, 1227–1234. [Google Scholar] [CrossRef]
- Liu, D.; Yu, X. Ireland-Claisen rearrangement of secondary allyl acetate revisited: Inevitable C-silylation circumvented by one-pot application of excessive LDA/TMSCl and TBAF. Tetrahedron Lett. 2012, 53, 2177–2180. [Google Scholar] [CrossRef]
- Nakaichi, M.; Taura, Y.; Kanki, M.; Mamba, K.; Momoi, Y.; Tsujimoto, H.; Nakama, H. Establishment and characterization of a new canine B-cell leukemia cell Line. J. Vet. Med. Sci. 1996, 58, 469–471. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, A.; Ziolo, E.; Kutkowska, J.; Blazejczyk, A.; Wietrzyk, J.; Krupa, A.; Hildebrand, W.; Dziegiel, P.; Dzimira, S.; Obminska-Mrukowicz, B.; et al. A novel canine B-cell leukaemia cell line. Establishment, characterisation and sensitivity to chemotherapeutics. Vet. Comp. Oncol. 2017, 15, 1218–1231. [Google Scholar] [CrossRef]
- Pruchnik, H.; Włoch, A.; Bonarska-Kujawa, D.; Kleszczyńska, H. An In Vitro Study of the Effect of Cytotoxic Triorganotin Dimethylaminophenylazobenzoate Complexes on Red Blood Cells. J. Membr. Biol. 2018, 251, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Dodge, J.T.; Mitchell, C.; Hanahan, D.J. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 1963, 100, 119–130. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Weissfloch, A.N.E.; Rappaport, A.T.; Cuccia, L.A. A Rule To Predict Which Enantiomer of a Secondary Alcohol Reacts Faster in Reactions Catalyzed by Cholesterol Esterase, Lipase from Pseudomonas cepacia, and Lipase from Candida rugosa. J. Org. Chem. 1991, 56, 2656–2665. [Google Scholar] [CrossRef]
- Gładkowski, W.; Gliszczyńska, A.; Siepka, M.; Czarnecka, M.; Maciejewska, G. Kinetic resolution of (E)-4-(2′,5′-dimethylphenyl)-but-3-en-2-ol and (E)-4-(benzo[d][1′,3′]dioxol-5′-yl)-but-3-en-2-ol through lipase-catalyzed transesterification. Tetrahedron Asymmetry 2015, 26, 702–709. [Google Scholar] [CrossRef]
- Ziegler, F.E. The Thermal, Aliphatic Claisen Rearrangement. Chem. Rev. 1988, 88, 1421–1452. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Białońska, A. Convenient chemoenzymatic route to optically active β-aryl-δ-iodo-γ-lactones and β-aryl-γ-iodo-δ-lactones with the defined configurations of stereogenic centers. Eur. J. Org. Chem. 2015, 2015, 605–615. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Gliszczyńska, A.; Czarnecka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Białońska, A. Chiral δ-iodo-γ-lactones derived from cuminaldehyde, 2,5-dimethylbenzaldehyde and piperonal: Chemoenzymatic synthesis and antiproliferative activity. Tetrahedron Asymmetry 2016, 27, 227–237. [Google Scholar] [CrossRef]
- Denmark, S.E.; Burk, M.T. Lewis base catalysis of bromo- and iodolactonization, and cycloetherification. Proc. Natl. Acad. Sci. USA 2010, 107, 20655–20660. [Google Scholar] [CrossRef] [Green Version]
- Ejchart, A. Substituent effects on 13C NMR. 2—Chemical shifts in the saturated framework of secondary aliphatic derivatives. Org. Magn. Reson. 1981, 15, 22–24. [Google Scholar] [CrossRef]
- Wińska, K.; Grabarczyk, M.; Maczka, W.; Zarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Szumny, A.; Anioł, M. Biotransformation of bicyclic halolactones with a methyl group in the cyclohexane ring into hydroxylactones and their biological activity. Molecules 2016, 21, 1453. [Google Scholar] [CrossRef] [Green Version]
- Mączka, W.; Wińska, K.; Grabarczyk, M.; Wińska, K.; Mączka, W.; Żarowska, B.; MacIejewska, G.; Dancewicz, K.; Gabryś, B.; Anioł, M.; et al. Synthesis, biotransformation and biological activity of halolactones obtained from β-ionone. Tetrahedron 2016, 72, 637–644. [Google Scholar]
- Wińska, K.; Anioł, M.; Żarowska, B.; Mączka, W.; Grabarczyk, M. The new halolactones and hydroxylactone with trimethylcyclohexene ring obtained through combined chemical and microbial processes. J. Mol. Catal. B Enzym. 2014, 102, 195–203. [Google Scholar]
- Mazur, M.; Gładkowski, W.; Srček, V.G.; Radošević, K.; Maciejewska, G.; Wawrzeńczyk, C. Regio- and enantioselective microbial hydroxylation and evaluation of cytotoxic activity of β-cyclocitral-derived halolactones. PLoS ONE 2017, 12, e0183429. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.; Faggio, C. The use of erythrocyte fragility to assess xenobiotic cytotoxicity. Cell Biochem. Funct. 2015, 33, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Krasnowska, E.K.; Bagatolli, L.; Gratton, E. Laurdan and Prodan as Polarity-Sensitive Fluorescent Membrane Probes. J. Fluoresc. 1998, 8, 365–373. [Google Scholar] [CrossRef]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.M.; Gratton, E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 1991, 60, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Alay, M.; Prat, J.; Haro, I.; Rojo, N.; Alsina, M.A.; Busquets, M.A. Spectroscopic analysis of the interaction of a peptide sequence of hepatitis G virus with bilayers. Talanta 2003, 60, 269–277. [Google Scholar] [CrossRef]
- Langner, M.; Hui, S.W. Merocyanine 540 as a fluorescence indicator for molecular packing stress at the onset of lamellar-hexagonal transition of phosphatidylethanolamine bilayers. Biochim. Biophys. Acta-Biomembr. 1999, 1415, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Manrique-Moreno, M.; Londoño-Londoño, J.; Jemioła-Rzemińska, M.; Strzałka, K.; Villena, F.; Avello, M.; Suwalsky, M. Structural effects of the Solanum steroids solasodine, diosgenin and solanine on human erythrocytes and molecular models of eukaryotic membranes. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelton, J.T.; McLean, L.R. Spectroscopic methods for analysis of protein secondary structure. Anal. Biochem. 2000, 277, 167–176. [Google Scholar] [CrossRef]
- Włoch, A.; Strugała, P.; Pruchnik, H.; Żyłka, R.; Oszmiański, J.; Kleszczyńska, H. Physical Effects of Buckwheat Extract on Biological Membrane In Vitro and Its Protective Properties. J. Membr. Biol. 2016, 249, 155–170. [Google Scholar] [CrossRef] [Green Version]
- Attar, M.; Kates, M.; Khalil, M.B.; Carrier, D.; Wong, P.T.T.; Tanphaichitr, N. A Fourier-transform infrared study of the interaction between germ-cell specific sulfogalactosylglycerolipid and dimyristoylglycerophosphocholine. Chem. Phys. Lipids 2000, 106, 101–114. [Google Scholar] [CrossRef]
- Lewis, R.N.; McElhaney, R.N.; Pohle, W.; Mantsch, H.H. Components of the carbonyl stretching band in the infrared spectra of hydrated 1,2-diacylglycerolipid bilayers: A reevaluation. Biophys. J. 1994, 67, 2367–2375. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.N.A.H.; Pohle, W.; McElhaney, R.N. The interfacial structure of phospholipid bilayers: Differential scanning calorimetry and Fourier transform infrared spectroscopic studies of 1,2-dipalmitoyl-sn-glycero-3-phosphorylcholine and its dialkyl and acyl-alkyl analogs. Biophys. J. 1996, 70, 2736–2746. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.N.A.H.; McElhaney, R.N. Calorimetric and spectroscopic studies of the thermotropic phase behavior of lipid bilayer model membranes composed of a homologous series of linear saturated phosphatidylserines. Biophys. J. 2000, 79, 2043–2055. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CH2-2 | CH2-3 | CH2-4 | CH2-5 | H-6 | CH2-7 | H-1′ | CH3-2′ | |
|---|---|---|---|---|---|---|---|---|
| C-1 | + | + | + | + | + | + | ||
| C-2 | + | + | + | |||||
| C-3 | + | + | ||||||
| C-4 | + | + | ||||||
| C-5 | + | + | + | |||||
| C-6 | + | + | + | + | ||||
| C-7 | + | |||||||
| C-8 | + | + | ||||||
| C-1′ | + | + | + | |||||
| C-2′ | + |
| Cell Line | Compound (IC50 µg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| 6a | 7a | 8a | 6b | 7b | 8b | Etoposide | |
| CLB70 | 19.59 ± 0.51 | 19.40 ± 0.35 | 23.93 ± 5.14 | 19.93 ± 0.07 | 28.57 ± 4.89 | 18.43 ± 1.46 | 14.31 ± 2.83 |
| GL-1 | 13.76 ± 1.08 | 15.55 ± 0.79 | 17.17 ± 3.62 | 14.53 ± 2.38 | 14.95 ± 1.93 | 11.40 ± 0.40 | 4.4 ± 1.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazur, M.; Włoch, A.; Bahri, F.; Pruchnik, H.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Gładkowski, W. Chemoenzymatic Synthesis of Enantiomeric, Bicyclic δ-Halo-γ-lactones with a Cyclohexane Ring, Their Biological Activity and Interaction with Biological Membranes. Biomolecules 2020, 10, 95. https://doi.org/10.3390/biom10010095
Mazur M, Włoch A, Bahri F, Pruchnik H, Pawlak A, Obmińska-Mrukowicz B, Maciejewska G, Gładkowski W. Chemoenzymatic Synthesis of Enantiomeric, Bicyclic δ-Halo-γ-lactones with a Cyclohexane Ring, Their Biological Activity and Interaction with Biological Membranes. Biomolecules. 2020; 10(1):95. https://doi.org/10.3390/biom10010095
Chicago/Turabian StyleMazur, Marcelina, Aleksandra Włoch, Fouad Bahri, Hanna Pruchnik, Aleksandra Pawlak, Bożena Obmińska-Mrukowicz, Gabriela Maciejewska, and Witold Gładkowski. 2020. "Chemoenzymatic Synthesis of Enantiomeric, Bicyclic δ-Halo-γ-lactones with a Cyclohexane Ring, Their Biological Activity and Interaction with Biological Membranes" Biomolecules 10, no. 1: 95. https://doi.org/10.3390/biom10010095