
Amyloids: The History of Toxicity and Functionality
by
 and
and
Elmira I. Yakupova
1,2,*, 
Liya G. Bobyleva
1, 
Sergey A. Shumeyko
1,
Ivan M. Vikhlyantsev
1 and
and
Alexander G. Bobylev
1 1
Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, 142290 Moscow, Russia
2
A. N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
*
Author to whom correspondence should be addressed.
Biology 2021, 10(5), 394; https://doi.org/10.3390/biology10050394
Submission received: 6 April 2021
/
Revised: 26 April 2021
/
Accepted: 29 April 2021
/
Published: 1 May 2021
(This article belongs to the Special Issue Protein Folding, Aggregation, and Cell Death)
Abstract
:Simple Summary
This review presents the beginning of the history of toxic properties of amyloids, especially on Aβ amyloids. We discuss anti-amyloid therapy and its problems and write about new views on amyloids that can play positive roles in different organisms including human.
Abstract
Proteins can perform their specific function due to their molecular structure. Partial or complete unfolding of the polypeptide chain may lead to the misfolding and aggregation of proteins in turn, resulting in the formation of different structures such as amyloid aggregates. Amyloids are rigid protein aggregates with the cross-β structure, resistant to most solvents and proteases. Because of their resistance to proteolysis, amyloid aggregates formed in the organism accumulate in tissues, promoting the development of various diseases called amyloidosis, for instance Alzheimer’s diseases (AD). According to the main hypothesis, it is considered that the cause of AD is the formation and accumulation of amyloid plaques of Aβ. That is why Aβ-amyloid is the most studied representative of amyloids. Therefore, in this review, special attention is paid to the history of Aβ-amyloid toxicity. We note the main problems with anti-amyloid therapy and write about new views on amyloids that can play positive roles in the different organisms including humans.
1. Introduction
The history of the study of amyloidosis dates back to the 17th century, when a woman was found to have a greatly enlarged spleen, which was crudely cut out with a knife [1]. In later works of the 19th century, the term “wax liver” was used for an organ in which large quantities of an unknown substance had collected [2]. This is the point from which researchers started studying amyloids deposited in different organs of the body, affecting the liver, spleen, kidneys and so forth [2,3]. Such diseases have subsequently been named “amyloidosis”.
Amyloids are defined as aggregates of misfolded peptides or proteins, with a quaternary cross-β structure, due to which they possess properties such as insolubility and resistance to proteolysis [4]. Because of these properties, accumulation/deposition of amyloids may occur in the body, thereby resulting in considerable changes in the metabolism of tissues and organs [4]. Amyloids are believed to lead to the development of amyloidoses. Amyloidoses are a group of incurable diseases. More than 30 different amyloidogenic proteins are currently associated with various diseases in humans [1,4]. Some of these are well known, for example AD. It is not only amyloids that are found in specific deposits in tissues with amyloidosis. Proteins such as glycosaminoglycans, apolipoprotein E and serum amyloid P components are also often present [4].
Due to the research carried out by Prusiner and his colleagues in the 1980s, the nature of infectious diseases such as Creutzfeldt–Jakob disease in humans, scrapie in sheep and bovine spongiform encephalopathy became better known [5,6]. It turns out that these diseases are caused by proteins called prions (or rather their altered form). Prions are a unique class of infectious agents. All of these diseases are related to the prion protein (PrP), which can change its conformation from a non-infected form (PrPSc) to an infected one [7]. This form is similar to an amyloid cross-β conformation. Therefore, we can say that another class of amyloid diseases (prion diseases) has emerged.
Since the beginning of the 21st century, our knowledge of amyloids has advanced, thanks to data on protein aggregates with an amyloid structure that do not cause dis-ease and may even have a specific role in the body. These are therefore called functional amyloids. Functional amyloids have been found in plants [8,9], prokaryotes [10,11,12] and eukaryotes [13,14,15,16,17,18,19], including mammals [16,17,18] and even humans [17,18]. All groups of organisms have adaptations that protect them from the toxic effects of amyloids. Here, we present a new perspective on amyloids that can play a positive role in different organisms.
2. Toxic Properties of Amyloids (the Beginning of Their History)
In the literature, amyloids are presented as being the main cause of development of diseases such as AD, amyloidosis of the liver and kidneys, etc. There is the main “cascade amyloid hypothesis” formulated by Hardy and Higgins (1992) [20]. According to this hypothesis, the cause of AD is formation and accumulation of amyloid plaques from fragments of the Aβ amyloid precursor protein (APP), which cause a cascade of molecular events, leading to loss of cell function and their death [20]. It was initially shown that Down syndrome (trisomy 21) patients have an extra copy of the amyloid precursor gene on chromosome 21. Their Aβ levels are also high, and, by age 40, they are likely to exhibit clinical symptoms of AD [21,22]. The second discovery was the familial type of AD, caused by mutations in the APP that lead to overproduction of Aβ [23,24,25,26,27]. Aβ is a 4.2-kDa short peptide of 40–42 amino acids, which is generated from intracellular cleavage of the APP by the sequential action of β-secretase and γ-secretase proteolytic enzymes [28].
In AD, two types of aggregates are found: extracellular Aβ deposits and intracellular tau protein. The toxicity of intracellular aggregates seems to be due to the sequestration of crucial proteins, together with amyloid, which leads to loss of cellular function and even cell death [29]. In AD, intracellular tau aggregates can, in theory, trap functional proteins and tau itself, which might induce microtubule destabilization [29]. Hardy’s hypothesis implies that, in AD, accumulation of Aβ-amyloids is the primary cause of neuronal death, and, for a post-mortem diagnosis, it is necessary to detect deposits of aggregated Aβ [20]. This is why Aβ-amyloid is the most studied representative of amyloids. Therefore, in this review, special attention is paid to Aβ-amyloid.
Aβ toxicity has been demonstrated on cultured nerve cells in vitro [29,30,31,32]. A study in 1989 showed that a peptide derived from the APP was toxic for hippocampal neurons in a culture [29]. Another study reported that a peptide ligand homologous to the first 28 residues of the Aβ-amyloid protein (Aβ1–28) increased the survival of cultured hippocampal neurons and appeared to have a neurotrophic effect [30]. In 1990, Whitson et al. detected that synthetic peptide β1–42 has a neurite-promoting effect, including extensive dendritic branching and axonal elongation [33]. In addition to the neurotrophic effect, a neurotoxic effect of Aβ has also been observed in other experiments [31]. For instance, in 1990, Yankner et al. showed that a portion of the amyloid beta protein, consisting of APP amino acids 25–35, contributed to trophic and, at the same time, toxic effects [31].
An article published in 1991 reported that the discovered in vitro effects could be caused by protein self-aggregation [32]. Data on the spontaneous assembly of a full-length β-amyloid peptide (β1–42) have already been reported in existing studies [33,34,35]. Based on this evidence, Pike et al. were perhaps the first researchers to assume that self-aggregation of β1–42 determines whether peptide trophic or toxic properties will be found [32]. To verify the hypothesis, Pike et al. demonstrated that β1–42 incubation influences the viability of hippocampal cultures in vitro. They found that pre-incubated β1–42 caused a neurotoxic effect, while recently solubilized β1–42 caused no toxicity but did promote neurite outgrowth [32]. Furthermore, toxicity of aggregates of the peptide that was incubated at 37 °C for 24 h increased the cell death effect [32,33]. Strong neurotoxicity in certain peptide sequences in Aβ1–42 (for example, β25–35), which exhibited rapid aggregation, was detected immediately upon solubilization [30]. Thus, it was concluded that Aβ toxicity is dependent on concentration, with low doses being protective and high doses being toxic [36]. A low dose of Aβ may also protect cells from the effects of a subsequent high dose [36].
The next stage in the history of Aβ toxicity included the hypothesis that negative effects on neurons are dependent on the ratio of Aβ1–42 to Aβ1–40 peptides. Aβ42:Aβ40 ratios of 3:7 (1 and 2 h) and 10:0 (1 and 2 h) were found to be associated with neurotoxicity in cultured mouse hippocampal neurons in vitro and to influence memory formation in mice in vivo. Aβ42:Aβ40 ratios of 1:9 and 0:10 did not show neurotoxicity at any stage of the aggregation process [37].
In many of the above studies, the toxic effect of Aβ was observed. However, what is the mechanism of βAP-mediated neurotoxicity? It is known that many aspects of calcium homeostasis change with aging (1987) [38] and that imbalances in calcium regulation lead to neural degeneration (1992) [39]. On this basis, in a 1993 study, it was suggested that one of the mechanisms behind the toxic effect of Aβ is disturbance of Ca2+ homeostasis [40]. Mattson et al. [40] noted that an experimental increase in [Ca2+] can induce antigenic and ultrastructural changes in neurons similar to those observed in neurofibrillary tangles in AD [32,41].
Continuing the research, Mattson concluded (in 1994) that Aβ which arises from alternative processing of beta APP aggregation destabilizes Ca2+ homeostasis [42]. Furthermore, vulnerable neurons possess high levels of glutamate receptors [42], and several growth factors can stabilize Ca2+ and protect neurons against excitotoxic injury and Aβ toxicity [42]. In the same year (1994), Weiss et al., studying the neurotoxicity of Aβ in primary murine cortical neurons, reported that Aβ (fragment 25–35) led to neurodegeneration that is concentration-dependent, and this effect decreased in the presence of Ca2+ channel blockers such as nimodipine (1–20 mM) and Co2+ (100 mM) [43]. In this context, neuronal Ca2+ homeostasis dysregulation is considered to be a common factor that underlies AD pathogenesis [44]. This effect is possible due to several mechanisms: direct interaction with membranes and further destabilization of the membrane structure [44,45,46,47,48]; formation of a cation-conducting pore [49,50,51,52,53,54,55]; and activation of cell surface receptors coupled with Ca2+ influx [56,57,58]. More recent studies also support the hypothesis of toxicity of Aβ amyloids, associated with changes in calcium homeostasis [59,60,61,62,63,64,65]. It is also assumed that neurotoxicity of Aβ might occur via generation of reactive free radicals [65,66,67,68,69,70,71,72]. In 1992, the cytotoxic effects of Aβ and an internal fragment encompassing residues 25–35 were shown using cultured cortical nerve cells. Vitamin E (known as an antioxidant and free radical scavenger) was found to inhibit Aβ-induced cell death [68]. At the same time (1992), five brain tissue sections from an AD case and five normal age-matched controls were studied with polyclonal antibodies against superoxide dismutase (CuZn- and Mn-forms) and catalase immunostaining [72]. As a result, a subgroup of neurofibrillary tangles (15–25%) and senile plaques (50%) showed immunoreactivity for both enzymes [72,73]. In 1994, Behl et al. suggested that Aβ causes increased levels of H2O2 [74,75].
In 1994, research provided evidence that beta-amyloid can inactivate oxidation-sensitive glutamine synthetase and creatine kinase enzymes [67]. A 1995 study investigated the release of nitric oxide (NO) from cultured rat microglia exposed to synthetic Aβ25–35 and Aβ1–40 (alone or in combination with cytokines IFN-α/β, IL-1β, TNF-α or TNF-β, including IFN-γ) [70]. Only IFN-γ among the cytokines being tested induced release of NO from the cells. β25–35 did not stimulate the release of nitric oxide by itself, but, in combination with IFN-γ, it caused NO release. In fact, β1–40 triggered NO production in microglia by itself, and this effect increased in conjunction with IFN-γ (100 U/mL) [70]. In 2020, it was shown that docosahexaenoic acid can suppress oligomeric Aβ-induced reactive oxygen species [71].
Further evidence indicating that generalized oxidative stress is important in pathogenesis effects was provided by a study of hippocampal cells. The study demonstrated that tyrosine nitration is increased in neuronal cytoplasm as well as in the nuclei of both neurons and glia in the regions of AD pathology. Thus, there is strong evidence that peroxynitrite is involved in oxidative damage of AD pathology [68,69].
In 1997, using primary cultures of cerebellar granule cells and astrocytes, it was shown that nanomolar concentrations of peptides such as Aβ-(1–40) and Aβ-(25–35) potently activate transcription factor NF-κB [76]. The authors of this work also conducted an immunohistochemical analysis of brain slices from patients with AD. In this case, activation of NF-κB was also observed in neurons and astroglia. Activated NF-κB was observed near the plaques [76]. NF-κB activation can lead to neuroprotection [37] via inhibition of amyloid β-mediated neuronal apoptosis [37].
Apoptosis is known to play a role in many neurologic disorders, including AD [77,78,79]. Dickson noted some problems in earlier experiments on apoptosis in AD: (1) the data were observed on cell cultures in vitro, which cannot necessarily be extrapolated to organisms in vivo; (2) in cell culture research, very high concentrations of Aβ are usually used, which do not exist in nature [80]; and (3) evidence for frank cellular apoptosis in AD is controversial [81].
Aβ is the main candidate for activation of apoptotic mechanisms in AD. It has recently been shown that insoluble Aβ complexes, including protofibrils and oligomers, play a role in this process [82]. Findings indicate that Aβ can activate caspases [83,84,85,86,87,88,89,90,91,92]. This mechanism can be triggered by the accumulation of Aβ at the site of its synthesis in endoplasmic reticulum or endosomes, which causes activation of apoptotic mechanisms through the unfolded protein response or endoplasmic reticulum stress. Apoptosis may also occur if Aβ activates alcohol dehydrogenase through mitochondrial stress [92].
The hypothesis of the toxic effects of APP protein fragments, having first undergone changes in favor of the toxic effects of Aβ aggregates (fibrils), has now been revised in favor of the highest toxicity of oligomers. It has already been demonstrated that Aβ oligomers exhibit neurotoxicity and cause neuronal damage [93,94,95,96,97,98,99,100,101,102,103,104,105,106,107]. Aβ protofibrils prepared in in vitro conditions have a smaller diameter than amyloid fibrils and high β-sheet content. It has been shown that a genetic risk factor for AD (APOJ; encoding clusterin) promotes formation of oligomeric structures with soluble properties [94]. Such oligomeric assemblies are involved in cell death both in vitro and in vivo [95,96]. A study in which soluble Aβ oligomeric species were extracted from the brain tissue of patients with AD showed that the presence of soluble oligomeric species correlates more closely with symptoms of the disease than with fibrils from amyloid plaques [97,98].
Mutations of the gene encoding APP are known to cause the development of AD [99]. It has been shown that two clinical mutations in the APP gene can increase the tendency of Aβ to oligomerize [100,101]. Benilova et al. concluded that, at the time of their study (2012), further evidence was needed to support the oligomeric hypothesis [93]. Indeed, there are a number of difficulties associated with research into oligomeric forms of the peptides. Firstly, the solution contains a mixture of water-soluble Aβ species [93]; and, secondly, as shown by Bitan et al., sodium dodecyl sulfate (SDS) can actually artificially induce oligomerization of Aβ [102]. It is known that the SDS-PAGE method used for obtained monomers, trimers and tetramers (as major bands) [102] is not a reliable method. The presence of SDS therefore makes it difficult to extrapolate in vivo effects from in vitro evidence [93]. It is still necessary to show whether similar modifications of Aβ monomers can occur in vivo. Some modifications of the peptide that can occur in vivo under the action of certain enzymes have already been discussed in the literature [104,105,106], but whether neurotoxicity in AD is associated with such modifications has not been fully elucidated.
The mechanisms behind the toxic effects of Aβ oligomers on cells may also lead to disruption of calcium homeostasis, and this has yet to be discussed in the literature [107]. The effects of oligomers of Aβ from both extracellular and intracellular Ca2+ sources have been shown [95,107]. The researchers also noted that Aβ42 and its oligomers caused increased membrane permeability in general [107]. This can cause unregulated flux of ions and molecules and, in turn, may be a common mechanism of oligomer toxicity [107]. However, we should note that high oligomer concentrations were used for the experiment to increase Ca2+ signals (100–1000 times greater than the levels of soluble Aβ measured in the brain of AD patients [107]).
Oligomers can show toxicity through membrane, intracellular and receptor-mediated mechanisms [78,108]. A study has been conducted on Aβ synaptotoxic effects, including disturbances in the functioning of N-methyl d-aspartate (NMDA) receptors that can affect calcium influx [79,108]. However, whether Aβ oligomers have a direct influence on NMDA receptors is a matter of controversy [97]. Alternative pathways suggest that Aβ induces synaptic failure due to apoptotic pathway activation [109] or upregulation of nicotinic acetylcholine receptor α7-nAcChR51 [109]. There is evidence of interactions with the insulin receptor [110]. Furthermore, the effects on the hypoxia-induced factor, clustering of angiotensin type 2 receptor and other mechanisms have also been described [111,112]. Some studies have also measured induction of apoptosis or other markers of cell death [108]. It is also interesting that astrocyte cell cultures have been found to be resistant to the action of Aβ [109]. Several reports in particular show that Aβ deposits in the brain do not necessarily correlate with AD symptoms [113,114]. Thus, we can conclude that there is still no consensus on the main mechanism of the toxic effects of amyloid fibrils or/and Aβ oligomers.
3. Anti-Amyloid Therapy and Its Problems
Amyloidogenesis, amyloid aggregates found in AD and the disease itself have gained increasing attention in the search for therapeutic drugs. Therapeutic agents/molecules have various properties, including the ability to do the following [111]:
- (1)
- Block β-sheet formation
- (2)
- Prevent fibrillogenesis
- (3)
- Dissolve Aβ aggregates into non-toxic species
- (4)
- Destabilize Aβ oligomers
- (5)
- Accelerate the conversion of Aβ oligomers to Aβ aggregates (modulators of Aβ aggregation)
However, despite the fact that amyloids are considered to be the main cause of AD development, therapeutic agents designed to reduce Aβ-peptide production or prevent Aβ-peptide aggregation have been found to be ineffective in phase III clinical trials [110,115]. At the same time, the number of developments in the search for drugs to treat AD has already exceeded 2300 [115]. However, AD remains an incurable disease.
Recent comparative studies of amyloid aggregates formed in vitro and isolated from tissues indicate morphological differences in amyloids. In particular, brain-derived amyloid fibrils of Aβ-peptides are right-twisted, while in vitro analogs are left-twisted [112]. This indicates that the study of amyloidogenesis in in vitro models has its own limitations, and the properties of “in vitro aggregates” may differ from their “in vivo analogs”. Moreover, the study of amyloidogenesis in vivo is a very complex task, requiring development of new research methods. All this raises the critical question of whether it is correct to extrapolate results obtained from in vitro studies relating to amyloids to in vivo systems. At the same time, the hypothesis about the main role of amyloids in development of AD is not confirmed and does not come without criticism [111,116,117].
The above data once again cast doubt on the fact that amyloids are harmful to the body and do not play any positive role in cells. In this regard, it is necessary to highlight interesting data showing that amyloids in brain cells accumulate at a certain time of the day in all healthy people (picomolar concentrations) [118,119]. This is believed to have some positive effect [111]. In favor of this assumption, we include reference to the results of experiments demonstrating that, when drugs capable of reducing the formation of amyloids are used, neurons die. In one study, neuronal death was prevented by adding amyloids [120]. It has been shown that all known developed drugs with an anti-amyloid effect cause death in people [121,122]. For example, active Aβ1–42 immunization (with AN-1792) resulted in 6% of patients developing meningoencephalitis [123]. At the same time, a reduction in senile plaques was observed [124], and none of these patients showed improved cognitive functioning [20]. Thus, should amyloids be treated as enemies or friends, and is it necessary to get rid of them?
To answer these questions, one of the strategies of anti-amyloid therapy, namely blocking the formation of β-sheets, should be discussed. This approach, in turn, depends on two membrane enzymes: β-secretase, also known as the β-site of the APP-cleaving enzyme (BACE1), and γ-secretase [23], consisting of five subunits, namely Pen-2, Presinilin 1 or Presinilin 2, Nicastrin and Aph1B. γ-secretase is involved in catalyzing the formation of several peptides, for instance, the Notch1 signal peptide, which plays a role in growth and proliferation of cellular processes [24]. It has been shown that subunit Presenilin 1 is important for the production of Aβ-peptide. One experiment found that knockout of the γ-secretase gene (in subunit Presenilin 1) caused the death of mice already at the embryonic stage [25]. Furthermore, inhibition of the entire γ-secretase gene by non-selective inhibitors led to various pathologies associated with Notch1 formation, including gastrointestinal tract disorders, impaired functioning of the immune system and skin pathologies [25,26]. In earlier studies, enzyme complex modulators instead of γ-secretase inhibitors were used to cause a shift in the production of Aβ-peptides from toxic Aβ fragments (1–42) to less toxic and shorter ones [20]. Inhibition of β-secretase with knockout of the gene in mice led to interruption of the accumulation of Aβ-peptides [21,27,28]. Mice lacking β-secretase secretion reproduced relatively healthy offspring with minor phenotypic abnormalities, such as hypomyelination [29,30] and behavior with schizophrenia-like symptoms [30]. Some experiments have focused on the search for selective inhibitors of BACE1. It has been shown that inhibitor OM99-2 polypeptide is capable of penetrating the blood–brain barrier, and, in 2008, clinical studies of this polypeptide began [31]. This inhibitor has a limitation, i.e., a “bulky” peptide structure for in vivo conditions [31]. A drug was subsequently created on the basis of OM99-2, which was able to stay in the bloodstream for a longer time, pass through the blood–brain barrier and reduce the level of Aβ-peptides (30–65%) in the plasma of transgenic mice modeling AD [32,39]. More than 10 drugs have been developed with similar characteristics. One of these has been found to be particularly successful (CTS-21166) and was tested on a group of volunteers suffering from Alzheimer’s disease. These drugs are promising, but none of them have gone through all the stages of clinical research. Most of the tested compounds were withdrawn from the initial stages of clinical studies for various reasons. Thus, are amyloid deposits the main cause of AD, and do their Aβ toxic properties really cause the death of nerve cells?
Understanding the exact mechanism of toxicity caused by Aβ oligomers or aggregates/fibrils (or both) is still a matter of discussion. In 2012, Benilova et al. presented an interesting point of view, asserting that proof of this “invisible toxin” is comparable to the (in)famous teapot of the British philosopher Bertrand Russell [28]. Results from clinical tests on anti-amyloid drugs also suggest that the approach adopted in AD treatment needs to be reviewed. To do this, the multifactorial nature of this disease needs to be taken into account. While researchers pay full attention to amyloid deposits, they ignore the mechanisms of development of cytoskeleton abnormalities, inflammation, oxidative stress, other metabolic abnormalities, etc. (Figure 1) [21,125,126].
We should not forget that the brain is extremely sensitive to fluctuations in blood glucose concentrations, and a lack of glucose for more than a few minutes can ultimately lead to cell death [127]. It is known that hypoglycemia [128], inadequate transportation of glucose across the blood–brain barrier [129], defective astroglial glutamate transport [130] and hyperammonemia [131] are associated with mental function. Comparison of the brains of patients who aged normally with the brains of AD patients has revealed certain metabolic differences, including glucose metabolism and cellular ATP, which decrease in the case of AD patients [132]. This supports the significant role of cerebral energy metabolism in AD pathology [119,133,134,135]. Thus, the cause of AD cannot only be due to amyloids but also other factors.
There is a hypothesis about the possible protective role of Aβ production in AD [136]. In the case of head trauma, neuroinflammation, ischemia-hypoxia/reperfusion, anesthesia or infectious agents, which are characteristically associated with neuronal damage [137,138], Aβ may exhibit trophic and neuroprotective [30], antioxidant [30,138] and antimicrobial properties [139]. Aβ can also be observed in tissue after metabolic impairment by non-specific stimuli [135].
Glucose and oxygen deregulation have been observed in patients with AD [126,140,141,142]. It has even been suggested that reduced mental capacity is caused by an imbalance of metabolic processes rather than the effects of Aβ deposits [135]. Red blood cells in the brains of AD patients have poorer oxygen transport efficiency than those in patients with normal brains [143]. A study conducted by Kosenko et al. on metabolites and enzymes in the main metabolic pathways [135] reported an increase in red blood cell glycolysis and ion fluxes in AD [137,144].
With AD, reduced brain glucose availability results in an increase in the cerebral ammonia concentration [145,146]. Moreover, ammonia is a powerful neurotoxin, and its accumulation in AD brains might be the reason for deterioration of memory and cognitive abilities.
It should also be noted that there is chronic inflammation in the brains of AD patients, and several components are involved. Activation of the brain’s resident macrophages (microglia) and specific cytokine signaling occurs in AD [147].
In 1992, immunohistochemical studies showed co-localization of the C1q component of the complement system, with amyloid deposits, in the brain of AD patients [148]. Researchers concluded that Aβ amyloids can directly activate the complement system. After this, in vitro studies started to research direct activation by amyloid fibrils in the absence of antibodies, according to the classical pathway [148,149,150,151,152,153,154,155]. Such activation was also observed for the alternative pathway [156,157,158]. It has been demonstrated that complement activation by Aβ fibrils in vitro results in the formation of C5a, as well as assembly of the C5b-9 proinflammatory membrane attack complex [152,155]. It has been shown that many complement system proteins, including C1q, C4, C3, C5, C6, C7, C8 and C5b-9, are also located near Aβ deposits and neurofibrillary tangles in the brains of AD patients [150,152,158,159,160,161,162]. This was followed by application of obtained data to new therapeutic approaches for reducing neuroinflammatory damage by influencing of the complement system response [161]. Recent in vitro studies do not confirm a direct antibody-independent complement system activated by Aβ fibrils [162]. However, the finding that complement system proteins are co-localized with amyloid deposit plaques needs to be explained further. It is worth noting that in addition to Aβ peptides, amyloid plaques contain other components, such as glycosaminoglycans, apopolyprotein E and amyloid serum component P [4]. Nevertheless, in this study [4], an association with the complement system was found only in the case of Aβ peptides. This raises the question of which factor it activates. Increased bacterial populations in the brain of patients with AD, compared with a normal brain, have been observed [163,164]. Therefore, Firmicutes, Actinobacteria and, in particular, P. acnes can directly play a role in neuroinflammation. In addition to co-localization of amyloids with the complement system, it has also been shown that amyloid aggregates have an antimicrobial function [164]. Thus, the complement system is more likely to be activated by microorganisms than by Aβ fibrils or other components of amyloid inclusions [162]. However, microbiota can influence the development of neurodegenerative diseases through amyloid and lipopolysaccharide formation in the gut, which trigger an increased inflammation response in the brain [165,166].
Thus, we can see that the primarily negative attention given to amyloids (especially Aβ) is changing now. There is no one uniform toxicity mechanism for Aβ and other amyloids. We still do not know why or how proteins/peptides aggregate in vivo and the reason for this process. In this review, we pay close attention to the history of Aβ toxicity in order to explain how researchers have studied amyloids and have come to a dead end, especially in AD treatment. Amyloids have still only been the subject of in vitro investigations. There are animal models of AD, but they are still far removed from human pathology. We highlight the main problems with anti-amyloid therapy above and next consider a new perspective on amyloids that can play a positive role in the functioning of different organisms, including humans.
4. Useful Properties of Amyloids. Functional Amyloids
Evidence that amyloids may have positive properties has been reported, and we now know that amyloids have antioxidant [137,138] and antibacterial effects [4,139,164]. Many amyloids are produced in the body, and it has been discovered that they play a vital role. Such amyloids are called functional amyloids. The first description of amyloidosis probably appeared in 1639 [1]. The first functional amyloid was discovered at the turn of the 20th century [10,167,168,169] (Figure 2). To date, more than 20 functional amyloids have been described.
Functional amyloids have various roles (Table 1), from formation of dense hydrophobic monolayers on the surface of spores and fruiting bodies of some fungi [166] to formation of the carcass of spider silk [15]; from protection of melanin toxic intermediates in melanosomes [16,17] to formation of long-term memory in animals [16,170]. They are involved in the fertilization process in mice [17] and regulation of the synthesis (or content) of hormones in humans [19]. Recent data indicate that amyloid aggregates can participate in immune responses because they are part of the extracellular neutrophil trap [171]. Functional amyloids are found in muscle tissue. It has been shown that, during regeneration of skeletal muscles in mice and humans, the cytoplasmic RNA-binding protein TDP-43 forms amyloid-like oligomers called myogranules [172]. Amyloids have also an RNA-modulating ability and play a role in transcription, translation, storage and degradation of RNA [173]. It has been shown that some motifs in prions of fungi and humans can be functionally related and be a model of amyloid signaling mechanisms from fungi to mammals [174,175]. Prokaryotic cells can also use such properties [8]. Just as people can store hormones in amyloids, plants can also store their protein using amyloid formation [8].
What is the reason for the non-toxicity properties of functional amyloids? Why can one be pathological but another functional? To date, the mechanisms behind the non-toxicity properties of functional amyloids are still unclear. However, Jackson and Hewitt suggested realistic ways to resolve this: (1) regulating the content of amyloidogenic peptides/proteins; (2) decreasing the time of the oligomers state during amyloidogenesis; (3) locating amyloids within membrane-bound organelles (e.g., melanosoma); and (4) regulating amyloid formation by other molecules and disassembling the fibrils under physiological conditions [229].
Might it be better to use the aggregation properties of amyloids? Scientists have now discovered how amyloid aggregates can inhibit VEGFR2-dependent tumor growth in a mouse tumor model [213], which was also observed using different human cell lines [226]. Oligomers can influence tumor growth too [230]. It has been shown that amyloids have antimicrobial properties [163,231], and these properties can be realized to β-sheet ion channel formation by amyloids [232].
5. Conclusions
The discovery of amyloids in organs and tissues in various diseases has tarnished the reputation of these formations for many years. Until now, amyloids have been considered to be harmful pathogenic aggregates, affecting cell homeostasis and, ultimately, resulting in cell death. There are numerous data on the toxicity of amyloids (Aβ amyloids in particular). Meanwhile, recent studies report that amyloids may have a beneficial role in cells and in organisms, and not just contribute to the development of amyloidosis, including AD. The results obtained in these experiments will enable many researchers to refocus their efforts on diligent study of the structural features and functional properties of amyloids. This will also yield important data to support selection of the most appropriate approaches for treatment of amyloidosis.
Author Contributions
E.I.Y. conceived and drafted the manuscript; S.A.S., L.G.B. and A.G.B. also wrote the part of the manuscript; and I.M.V. checked the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Russian Foundation for Basic Research (project №19-34-90054).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Aβ | β-amyloid protein |
| APP | amyloid precursor protein |
| AD | Alzheimer’s disease |
| CD | circular dichroism |
| FTIR | Fourier-transform infrared spectroscopy |
| NO | nitric oxide |
| PrP | prion protein |
| RC | random coil |
| RT | room temperature |
References
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G. Amyloids: From Pathogenesis to Function. Biochemistry 2015, 80, 1127–1144. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R. Amyloidosis: A Convoluted Story. Br. J. Haematol. 2001, 114, 529–538. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–89. [Google Scholar] [CrossRef] [Green Version]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.I.; Masters, C.L.; Merlini, G.; Sipe, J.D.A.; Saraiva, M.J. Primer of Amyloid Nomenclature. Amyloid 2007, 14, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Horwich, A.L.; Weissman, J.S. Deadly Conformations–Protein Misfolding in Prion Disease. Cell 1997, 89, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion Protein Biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Kushnirov, V.V.; Vishnevskaya, A.B.; Alexandrov, I.M.; Michael, D. Ter-Avanesyan Prion and Nonprion. Amyloids 2007, 1, 179–184. [Google Scholar]
- Antonets, K.S.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Kosolapova, A.O.; Sulatsky, M.I.; Andreeva, E.A.; Zykin, P.A.; Malovichko, Y.V.; Shtark, O.Y.; et al. Accumulation of Storage Proteins in Plant Seeds is Mediated by Amyloid Formation. PLoS Biol. 2020, 18, e3000564. [Google Scholar] [CrossRef]
- Surguchov, A.; Emamzadeh, F.N.; Surguchev, A.A. Amyloidosis and Longevity: A Lesson from Plants. Biology 2019, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Rçmling, U.; Bian, Z.; Hammar, M.; Sierralta, W.D.; Normark, S. Curli Fibers are Highly Conserved between Salmonella typhimurium and Escherichia coli with Respect to Open Structure and Regulation. J. Bacteriol. 1998, 180, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Claessen, D.; Rink, R.; de Jong, W.; Siebring, J.; de Vreughd, P.; Boersma, F.G.H.; Dijkhuizen, L.; Wçsten, H.A.B. A Novel Class of Secreted Hydrophobic Proteins is Involved in Aerial Hyphae Formation in Streptomyces coelicolor by Forming Amyloid-like Fibrils. Genes Dev. 2003, 17, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Otzen, D.; Nielsen, P.H. We Find Them Here, We Find Them There: Functional Bacterial Amyloid. Cell Mol Life Sci. 2008, 65, 910–927. [Google Scholar] [CrossRef]
- Wöesten, H.A.B.; de Vocht, M.L. Hydrophobins, the Fungal Coat Unraveled. Biochim. Biophys. Acta 2000, 1469, 79–86. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Galanis, A.S.; Cordopatis, P.; Hoenger, A.; Hamodrakas, S.J. Amyloid Fibril Formation Propensity is Inherent into the Hexapeptidetandemly Repeating Sequence of the Central Domain of Silk Moth Chorion Proteins of the A-family. J. Struct. Biol. 2006, 156, 480–488. [Google Scholar] [CrossRef]
- Slotta, U.; Hess, S.; Spiess, K.; Stromer, T.; Serpell, L.; Scheibel, T. Spider Silk and Amyloid Fibrils: A Structural Comparison. Macromol. Biosci. 2007, 7, 183–188. [Google Scholar] [CrossRef]
- Si, K.; Lindquist, S.; Kandel, E.R. A Neuronal Isoform of the Aplysia CPEB has Prion-like Properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Guyonnet, B.; Egge, N.; Cornwall, G.A. Functional Amyloids in the Mouse Sperm Acrosome. Mol. Cell Biol. 2014, 34, 2624–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional Amyloid Formation within Mammalian Tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional Amyloids as Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A. Alzheimer’s Disease and Down’s Syndrome. Histopathology 1988, 13, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Ud-Din, S.; Holmes, C. Genetics of Down’s Syndrome and Alzheimer’s Disease. Br. J. Psychiatry 2002, 181, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; et al. Early-onset Alzheimer’s Disease Caused by Mutations at Codon 717 of the f3-amyloid Precursor Protein Gene. Nature 1991, 353, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.-D.; Golde, T.; Younkin, S. Release of Excess Amyloid Beta-protein from a Mutant Amyloid Beta-protein Precursor. Science 1993, 259, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the Beta-amyloid Precursor Protein in Familial Alzheimer’s Disease Increases Beta-protein Production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef]
- Murrell, J.; Farlow, M.; Ghetti, B.; Benson, M.D. Amutation in the Amyloid Precursor Protein Associated with Hereditary Alzheimer’s Disease. Science 1991, 254, 97–99. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Vassar, R. BACE1: The Beta-secretase Enzyme in Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a Fragment of the Amyloid Precursor Associated with Alzheimer’s Disease. Science 1989, 245, 417–420. [Google Scholar] [CrossRef]
- Yankner, J.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and Neurotoxic Effects of Amyloid β Protein Reversal by Tachykinin Neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef]
- Mattson, M.P.; Tomaselli, K.J.; Rydel, R.E. Calcium De-stabilizing and Neurodegenerative Effects of Aggregated β-amyloid are Attenuated by Basic FGF. Brain Res. 1993, 621, 35–49. [Google Scholar] [CrossRef]
- Barrow, C.J.; Zagorski, M.G. Solution Structures of β Peptide and Its Constituent Fragments: Relation to Amyloid Deposition. Science 1991, 253, 179–182. [Google Scholar] [CrossRef]
- Whitson, J.S.; Glabe, C.G.; Shitani, E.; Abcar, A.; Cotman, C.W. β-Amyloid Protein Promotes Neuritic Branching in Hippocampal Cultures. Neurosci. Lett. 1990, 110, 319–324. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and Aggregation Properties of Synthetic Alzheimer’s A4/β Amyloid Peptide Analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [CrossRef]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Aggregation and Secondary Structure of Synthetic Amyloid flA4 Peptides of Alzheimer’s Disease. J. Mol. Biol. 1991, 218, 149–163. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Wellman, H.; Volk, B.; Kaltschmidt, C. Inhibition of NF-KB Potentates Amyloid β-mediatated Neuronal Apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 9409–9414. [Google Scholar] [CrossRef] [Green Version]
- Kuperstein, I.; Broersen, K.; Benilova, I.; Rozenski, J.; Jonckheere, W.; Debulpaep, M.; Vandersteen, A.; Segers-Nolten, I.; Van Der Werf, K.; Subramaniam, V.; et al. Neurotoxicity of Alzheimer’s Disease Aβ Peptides is Induced by Small Changes in the Aβ42 to Aβ40 Ratio. EMBO J. 2010, 29, 3408–3420. [Google Scholar] [CrossRef]
- Gibson, G.E.; Peterson, C. Calcium and the Aging Nervous System. Neurobiol. Aging 1987, 8, 329–343. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium as Sculptor and Destroyer of Neural Circuitry. Exp. Gerontol. 1992, 27, 29–49. [Google Scholar] [CrossRef]
- Mattson, M.P. Antigenic Changes Similiar to Those Seen in Neurofibrillary Tangles are Elicited by Glutamate and Calcium Influx in Cultured Hippocampal Neurons. Neuron 1990, 4, 105–117. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and Neuronal Injury in Alzheimer’s Disease. Contributions of Beta-amyloid Precursor Protein Mismetabolism, Free Radicals, and Metabolic Compromise. Ann. N. Y. Acad. Sci. 1994, 15, 50–76. [Google Scholar]
- Weiss, J.H.; Pike, C.J.; Cotman, C.W. Ca2+ Channel Blockers Attenuate Beta-amyloid Peptide Toxicity to Cortical Neurons in Culture. J. Neurochem. 1994, 62, 372–375. [Google Scholar] [CrossRef]
- Avdulov, N.A.; Chochina, S.V.; Igbavboa, U.; Warden, C.S.; Vassiliev, A.V.; Wood, W.G. Lipid Binding to Amyloid Beta-peptide Aggregates: Preferential Binding of Cholesterol as Compared with Phosphatidylcholine and Fatty Acids. J. Neurochem. 1997, 69, 1746–1752. [Google Scholar] [CrossRef]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [Green Version]
- Müller, W.E.; Koch, S.; Eckert, A.; Hartmann, H.; Scheuer, K. Beta-amyloid Peptide Decreases Membrane Fluidity. Brain Res. 1995, 674, 133–136. [Google Scholar] [CrossRef]
- Mason, R.P.; Trumbore, M.W.; Pettegrew, J.W. Molecular Membrane Interactions of a Phospholipid Metabolite. Implications for Alzheimer’s Disease Pathophysiology. Ann. N. Y. Acad. Sci. 1996, 777, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Green, J.D.; Kreplak, L.; Goldsbury, C.; Li Blatter, X.; Stolz, M.; Cooper, G.S.; Seelig, A.; Kistler, J.; Aebi, U. Atomic Force Microscopy Reveals Defects within Mica Supported Lipid Bilayers Induced by the Amyloidogenic Human Amylin Peptide. J. Mol. Biol. 2004, 342, 877–887. [Google Scholar] [CrossRef]
- Kawahara, M.; Kuroda, Y.; Arispe, N.; Rojas, E. Alzheimer’s Beta-amyloid, Human Islet Amylin, and Prion Protein Fragment Evoke Intracellular Free Calcium Elevations by a Common Mechanism in a Hypothalamic GnRH Neuronal Cell Line. J. Biol. Chem. 2000, 275, 14077–14083. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of Lipid Bilayers is a Common Conformation-dependent Activity of Soluble Amyloid Oligomers in Protein Misfolding Diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [Green Version]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Arispe, N. Architecture of the Alzheimer’s A Beta P Ion Channel Pore. J. Membr. Biol. 2004, 197, 33–48. [Google Scholar] [CrossRef]
- Lin, M.C.; Kagan, B.L. Electrophysiologic Properties of Channels Induced by Abeta25-35 in Planar Lipid Bilayers. Peptides 2002, 23, 1215–1228. [Google Scholar] [CrossRef]
- Kawahara, M.; Arispe, N.; Kuroda, Y.; Rojas, E. Alzheimer’s Disease Amyloid Beta-protein Forms Zn(2+)-sensitive, Cation-selective Channels Across Excised Membrane Patches from Hypothalamic Neurons. Biophys. J. 1997, 73, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Hirakura, Y.; Lin, M.C.; Kagan, B.L. Alzheimer Amyloid Abeta1-42 Channels: Effects of Solvent, pH, and Congo Red. J. Neurosci. Res. 1999, 57, 458–466. [Google Scholar] [CrossRef]
- Blanchard, B.J.; Chen, A.; Rozeboom, L.M.; Stafford, K.A.; Weigele, P.; Ingram, V.M. Efficient Reversal of Alzheimer’s Disease Fibril Formation and Elimination of Neurotoxicity by a Small Molecule. Proc. Natl. Acad. Sci. USA 2004, 101, 14326–14332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Furukawa, K.; Sopher, B.L.; Pham, D.G.; Xie, J.; Robinson, N.; Martin, G.M.; Mattson, M.P. Alzheimer’s PS-1 Mutation Perturbs Calcium Homeostasis and Sensitizes PC12 Cells to Death Induced by Amyloid Beta-peptide. Neuroreport 1996, 8, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Chan, S.L. Calcium Orchestrates Apoptosis. Nat. Cell. Biol. 2003, 5, 1041–1043. [Google Scholar] [CrossRef] [Green Version]
- Ferrarelli, L.K. New Connections: Amyloid-β, Calcium, and the Synapse. Sci. Signal. 2017, 10, eaao3024. [Google Scholar] [PubMed]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium Signaling in Alzheimer’s Disease & Therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and Beta-amyloid–Different Targets, Same Players: Calcium, Free Radicals and Mitochondria in the Mechanism of Neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martínez, J.; Botelho, H.M.; Göbl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The Neuronal S100B Protein is a Calcium-tuned Suppressor of Amyloid-β Aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, M.; Negishi-Kato, M.; Sadakane, Y. Calcium Dyshomeostasis and Neurotoxicity of Alzheimer’s Beta-amyloid Protein. Expert Rev. Neurother. 2009, 9, 681–693. [Google Scholar] [CrossRef]
- Sushma; Mondal, A.C. Role of GPCR Signaling and Calcium Dysregulation in Alzheimer’s Disease. Mol. Cell. Neurosci. 2019, 101, 103414. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.; Cole, G.M.; Schubert, D. Vitamin E Protects Nerve Cells from Amyloid Beta Protein Toxicity. Biochem. Biophys. Res. Commun. 1992, 186, 944–950. [Google Scholar] [CrossRef]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A Model for Beta-amyloid Aggregation and Neurotoxicity Based on Free Radical Generation by the Peptide: Relevance to Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread Peroxynitrite-mediated Damage in Alzheimer’s Disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative Damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef]
- Goodwin, J.L.; Uemura, E.; Cunnick, J.E. Microglial Release of Nitric Oxide by the Synergistic Action of Beta-amyloid and IFN-gamma. Brain Res. 1995, 692, 207–214. [Google Scholar] [CrossRef]
- Geng, X.; Yang, B.; Li, R.; Teng, T.; Ladu, M.J.; Sun, G.Y.; Greenlief, C.M.; Lee, J.C. Effects of Docosahexaenoic Acid and Its Peroxidation Product on Amyloid-β Peptide-Stimulated Microglia. Mol Neurobiol. 2020, 57, 1085–1098. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Omar, R.A.; Kim, K.S.; Robakis, N.K. Immunohistochemical Evidence of Oxidative (Corrected) Stress in Alzheimer’s Disease. Am. J. Pathol. 1992, 140, 621–628. [Google Scholar]
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s Disease an Inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen Peroxide Mediates Amyloid Beta Protein Toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription Factor NF-kappaB is Activated in Primary Neurons by Amyloid Beta Peptides and in Neurons Surrounding Early Plaques from Patients with Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- Pentreath, V.W. Responses of Cultured Astrocytes, C6 Glioma and 1321NI Astrocytoma Cells to Amyloid β-Peptide Fragments. Nonlinearity Biol. Toxicol. Med. 2004, 2, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Cotman, C.W.; Anderson, A.J. A Potential Role for Apoptosis in Neurodegeneration and Alzheimer’s Disease. Mol. Neurobiol. 1995, 10, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid Toxicity in Alzheimer’s Disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of Action of Amyloid-beta and Its Precursor Protein in Neuronal Cell Death. Metab. Brain Dis. 2020, 35, 11–30. [Google Scholar] [CrossRef]
- Dickson, D.W. Apoptotic Mechanisms in Alzheimer Neurofibrillary Degeneration: Cause or Effect? J. Clin. Investig. 2004, 114, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Bruck, W.; Bancher, C.; Jellinger, K.; Lassmann, H. Alzheimer Disease: DNA Fragmentation Indicates Increased Neuronal Vulnerability, but not Apoptosis. J. Neuropathol. Exp. Neurol. 1998, 57, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s Disease-affected Brain: Presence of Oligomeric A Beta Ligands (ADDLs) Suggests a Molecular Basis for Reversible Memory Loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical Evidence for Apoptosis in Alzheimer’s Disease. Neuroreport. 1994, 5, 2529–2533. [Google Scholar] [CrossRef] [PubMed]
- Smale, G.; Nichols, N.R.; Brady, D.R.; Finch, C.E.; Horton, W.E., Jr. Evidence for Apoptotic Cell Death in Alzheimer’s Disease. Exp. Neurol. 1995, 133, 225–230. [Google Scholar] [CrossRef]
- Anderson, A.J.; Su, J.H.; Cotman, C.W. DNA Damage and Apoptosis in Alzheimer’s Disease: Colocalization with c-Jun Immunoreactivity, Relationship to Brain Area, and Effect of Postmortem Delay. J. Neurosci. 1996, 16, 1710–1719. [Google Scholar] [CrossRef] [Green Version]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s Disease: An Understanding of the Physiology, Pathology and Therapeutic Avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Park, G.; Nhan, H.S.; Tyan, S.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase Activation and Caspase-Mediated Cleavage of APP Is Associated with Amyloid β-Protein-Induced Synapse Loss in Alzheimer’s Disease. Cell Reports 2020, 31, 107839. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; John, V.; Galvan, V. Importance of the Caspase Cleavage Site in Amyloid-β Protein Precursor. J. Alzheimers Dis. 2010, 22, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of Tau is an Early Event in Alzheimer Disease Tangle Pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C. Intracellular Mechanisms of Amyloid Accumulation and Pathogenesis in Alzheimer’s Disease. J. Mol. Neurosci. 2001, 17, 137–145. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Abeta to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar]
- Oda, T.; Wals, P.; Osterburg, H.H.; Johnson, S.A.; Pasinetti, G.M.; Morgan, T.E.; Rozovsky, I.; Stine, W.B.; Snyder, S.W.; Holzman, T.F.; et al. Clusterin (apoJ) Alters the Aggregation of Amyloid Beta-peptide (A beta 1-42) and Forms Slowly Sedimenting A Beta Complexes That Cause Oxidative Stress. Exp. Neurol. 1995, 136, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, Nonfibrillar Ligands Derived from Abeta1-42 are Potent Central Nervous System Neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally Secreted Oligomers of Amyloid Beta Protein Potently Inhibit Hippocampal Long-term Potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Abeta Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. Medical Research Council Cognitive Function and Ageing Study. The Presence of Sodium Dodecyl Sulphate-stable Abeta Dimers is Strongly Associated with Alzheimer-type Dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B. Proteases and Proteolysis in Alzheimer Disease: A Multifactorial View on the Disease Process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP Mutation (E693G) Causes Alzheimer’s Disease by Enhanced Abeta Protofibril Formation. Nat Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A New Amyloid Beta Variant Favoring Oligomerization in Alzheimer’s-type Dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic Protein Oligomers–What You See is not Always What You Get. Amyloid 2005, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Hepler, R.W.; Grimm, K.M.; Nahas, D.D.; Breese, R.; Dodson, E.C.; Acton, P.; Keller, P.M.; Yeager, M.; Wang, H.; Shughrue, P.; et al. Solution State Characterization of Amyloid Beta-derived Diffusible Ligands. Biochemistry 2006, 45, 15157–15167. [Google Scholar] [CrossRef]
- Hartley, D.M.; Zhao, C.; Speier, A.C.; Woodard, G.A.; Li, S.; Li, Z.; Walz, T. Transglutaminase Induces Protofibril-like Amyloid Beta-protein Assemblies That are Protease-resistant and Inhibit Long-term Potentiation. J. Biol. Chem. 2008, 283, 16790–16800. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.P.; Smith, D.G.; Curtain, C.C.; Boas, J.F.; Pilbrow, J.R.; Ciccotosto, G.D.; Lau, T.L.; Tew, D.J.; Perez, K.; Wade, J.D.; et al. Copper-mediated Amyloid-beta Toxicity is Associated with an Intermolecular Histidine Bridge. J. Biol. Chem. 2006, 281, 15145–15154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeazzi, L.; Ronchi, P.; Franceschi, C.; Giunta, S. In vitro Peroxidase Oxidation Induces Stable Dimers of Beta-amyloid (1-42) through Dityrosine Bridge Formation. Amyloid 1999, 6, 7–13. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksis, R.; Oleskovs, F.; Jaudzems, K.; Pahnke, J.; Biverstål, H. Structural Studies of Amyloid-β Peptides: Unlocking the Mechanism of Aggregation and the Associated Toxicity. Biochimie 2017, 140, 176–192. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; Strooper, B. The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug. Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Chakrabarti, M.; Govindaraju, T. Function and Toxicity of Amyloid Beta and Recent Therapeutic Interventions Targeting Amyloid Beta in Alzheimer’s Disease. Chem. Commun. 2015, 51, 13434–13450. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nature Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- ClinicalsTrails.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Alzheimer+Disease&term=&cntry=&state=&city=&dist= (accessed on 5 April 2021).
- Hardy, J.; Mayer, J. The Amyloid Cascade Hypothesis Has Misled the Pharmaceutical Industry. Biochem. Soc Trans. 2011, 39, 920–923. [Google Scholar]
- Kosenko, E.A.; Solomadin, I.N.; Tikhonova, L.A.; Reddy, V.P.; Aliev, G.; Kaminsky, Y.G. Pathogenesis of Alzheimer Disease: Role of Oxidative Stress, Amyloid-β Peptides, Systemic Ammonia and Erythrocyte Energy Metabolism. CNS Neurol. Disord. Drug. Targets. 2014, 13, 112–119. [Google Scholar] [CrossRef]
- Schmitt, F.A.; Davis, D.G.; Wekstein, D.R.; Smith, C.D.; Ashford, J.W.; Markesbery, W.R. “Preclinical” AD Revisited: Neuropathology of Cognitively Normal Older Adults. Neurology 2000, 55, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagust, W.J.; Mormino, E.C. Lifespan Brain Activity, β-amyloid, and Alzheimer’s Disease. Trends Cogn. Sci. 2011, 15, 520–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemming, M.L.; Selkoe, D.J. Amyloid Beta-protein is Degraded by Cellular Angiotensin-converting Enzyme (ACE) and Elevated by an ACE Inhibitor. J. Biol. Chem. 2005, 280, 37644–37650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term Effects of Abeta42 Immunisation in Alzheimer’s Disease: Follow-up of a Randomised, Placebo-controlled Phase I Trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Kurkinen, M. The Amyloid Hypothesis is too Good to be True. Alzheimers Dement. Cogn. Neurol. 2017, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute Meningoencephalitis in a Subset of Patients with AD after Abeta42 Immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.; Wilkinson, D.; Holmes, C.; Steart, P.; Markham, H.; Weller, R.O. Neuropathology of Human Alzheimer Disease after Immunization with Amyloid-beta Peptide: A Case Report. Nat. Med. 2003, 9, 448–452. [Google Scholar] [CrossRef]
- Tabira, T. Immunization Therapy for Alzheimer Disease: A Comprehensive Review of Active Immunization Strategies. Tohoku J. Exp. Med. 2010, 220, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blass, J.P. Immunologic Treatment of Alzheimer’s Disease. New Engl. J. Med. 1999, 341, 1694–1695. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Oelshlegel, F.J., Jr.; Moore, L.G.; Noble, N.A. In vivo Red Cell Glycolytic Control and DPG-ATP Levels. Ann. N. Y. Acad. Sci. 1974, 241, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Bruce, D.G.; Davis, W.A.; Casey, G.P.; Clarnette, R.M.; Brown, S.G.; Jacobs, I.G.; Almeida, O.P.; Davis, T.M. Severe Hypoglycaemia and Cognitive Impairment in Older Patients with Diabetes: The Fremantle Diabetes Study. Diabetologia 2009, 52, 1808–1815. [Google Scholar] [CrossRef] [Green Version]
- Klepper, J.; Voit, T. Facilitated Glucose Transporter Protein Type 1 (GLUT1) Deficiency Syndrome: Impaired Glucose Transport into Brain–A Review. Eur. J. Pediatr. 2002, 161, 295–304. [Google Scholar] [CrossRef]
- Ronnback, L.; Hansson, E. On the Potential Role of Glutamate Transport in Mental Fatigue. J. Neuroinflammation 2004, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Llansola, M.; Rodrigo, R.; Monfort, P.; Montoliu, C.; Kosenko, E.; Cauli, O.; Piedrafita, B.; Mlili, N.; Felipo, V. NMDA Receptors in Hyperammonemia and Hepatic Encephalopathy. Metab Brain Dis. 2007, 22, 321–335. [Google Scholar] [CrossRef]
- Hoyer, S. Oxidative Metabolism Deficiencies in Brains of Patients with Alzheimer’s Disease. Acta Neurol. Scand. Suppl. 1996, 165, 18–24. [Google Scholar] [CrossRef]
- Hoyer, S.; Oesterreich, K.; Wagner, O. Glucose Metabolism as the Site of the Primary Abnormality in Early-onset Dementia of Alzheimer Type? J. Neurol. 1988, 235, 143–148. [Google Scholar] [CrossRef]
- Aliev, G.; Palacios, H.H.; Walrafen, B.; Lipsitt, A.E.; Obrenovich, M.E.; Morales, L. Brain Mitochondria as a Primary Target in the Development of Treatment Strategies for Alzheimer Disease. Int. J. Biochem. Cell Biol. 2009, 41, 1989–2004. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Li, Y.; Palacios, H.H.; Obrenovich, M.E. Oxidative Stress Induced Mitochondrial DNA Deletion as a Hallmark for the Drug Development in the Context of the Cerebrovascular Diseases. Rec. Pat. Cardiovasc. Drug Discov. 2011, 6, 222–241. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Casadesus, G.; Zhu, X.; Takeda, A.; Perry, G.; Smith, M.A. Challenging the Amyloid Cascade Hypothesis: Senile Plaques and Amyloid-beta as Protective Adaptations to Alzheimer Disease. Ann. N. Y. Acad. Sci. 2004, 1019, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Zhu, X.; Castellani, R.J.; Nunomura, A.; Perry, G.; Smith, M.A. Amyloid-beta in Alzheimer Disease: The Null vs. the Alternate Hypotheses. J. Pharmacol. Exp. Ther. 2007, 321, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, C.S.; Robinson, S.R.; Smith, M.A. Amyloid-beta: Redox-metal Chelator and Antioxidant. J. Alzheimers Dis. 2002, 4, 203–214. [Google Scholar] [CrossRef]
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-beta and Tau Serve Antioxidant Functions in the Aging and Alzheimer Brain. Free Radic. Biol. Med. 2002, 33, 1194–1199. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-associated Amyloid Beta-protein is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- de la Torre, J.C. Impaired Cerebromicrovascular Perfusion. Summary of Evidence in Support of Its Causality in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2000, 924, 136–152. [Google Scholar] [CrossRef]
- de la Torre, J.C.; Aliev, G. Inhibition of Vascular Nitric Oxide after Rat Chronic Brain Hypoperfusion: Spatial Memory and Immunocytochemical Changes. J. Cereb. Blood Flow Metab. 2005, 25, 663–672. [Google Scholar] [CrossRef]
- Wen, Z.; Xie, J.; Guan, Z.; Sun, D.; Yao, W.; Chen, K.; Yan, Z.Y.; Mu, Q. A Study of Hemorheological Behaviour for Patients with Alzheimer’s Disease at the Early Stages. Clin. Hemorheol. Microcirc. 2000, 22, 261–266. [Google Scholar] [PubMed]
- Kaminsky, Y.; Poghosyan, A.; Tikhonova, L.; Palacios, H.H.; Kamal, M.A.; Kosenko, E.; Aliev, G. Glycolytic and Proteolytic Metabolism in Erythrocytes from Elderly and Demented Patients. Am. J. Neuroprotect. Neuroregener. 2012, 4, 73–77. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Reddy, V.P.; Ashraf, G.M.; Ahmad, A.; Benberin, V.V.; Kosenko, E.A.; Aliev, G. Age-related Defects in Erythrocyte 2,3-diphosphoglycerate Metabolism in Dementia. Aging Dis. 2013, 4, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Norberg, K.; Siesio, B.K. Oxidative Metabolism of the Cerebral Cortex of the Rat in Severe Insulin-induced Hypoglycaemia. J. Neurochem. 1976, 26, 345–352. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P. Complement Activation by Beta-amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Frederickson, R.C.; Brunden, K.R. Neuroglial-mediated Immunoinflammatory Responses in Alzheimer’s Disease: Complement Activation and Therapeutic Approaches. Neurobiol. Aging 1996, 17, 781–787. [Google Scholar] [CrossRef]
- Jiang, H.; Burdick, D.; Glabe, C.G.; Cotman, C.W.; Tenner, A.J. Beta-amyloid Activates Complement by Binding to a Specific Region of the Collagen-like Domain of the C1q A Chain. J. of Immunol. 1994, 152, 5050–5059. [Google Scholar]
- Afagh, A.; Cummings, B.J.; Cribbs, D.H.; Cotman, C.W.; Tenner, A.J. Localization and Cell Association of C1q in Alzheimer’s Disease Brain. Exp. Neurol. 1999, 6138, 22. [Google Scholar] [CrossRef]
- Webster, S.; Bradt, B.; Rogers, J.; Cooper, N.R. Aggregation State-dependent Activation of the Classical Complement Pathway by the Amyloid β Peptide (Aβ). J. Neurochem. 1997, 69, 388–398. [Google Scholar] [CrossRef]
- Webster, S.; Lue, L.F.; Brachova, L.; Tenner, A.; McGeer, P.L.; Walker, D.; Bradt, B.; Cooper, N.R.; Rogers, J. Molecular and Cellular Characterization of the Membrane Attack Complex, C5b-9, in Alzheimer’s Disease. Neurobiol. Aging 1997, 18, 415–421. [Google Scholar] [CrossRef]
- Veerhuis, R.; Van Breemen, M.J.; Hoozemans, J.M. Amyloid Beta Plaque-associated Proteins C1q and SAP Enhance the Abeta1-42 Peptide-induced Cytokine Secretion by Adult Human Microglia in vitro. Acta Neuropathol. 2003, 105, 135. [Google Scholar] [CrossRef] [PubMed]
- Tacnet-Delorme, P.; Chevallier, S.; Arlaud, G.J. β-Amyloid Fibrils Activate the C1 Complex of Complement Under Physiological Conditions: Evidence for a Binding Site for Aβ on the C1q Globular Regions. J. Immunol. 2001, 167, 6374. [Google Scholar] [CrossRef] [Green Version]
- Bradt, B.M.; Kolb, W.P.; Cooper, N.R. Complement-dependent Proinflammatory Properties of the Alzheimer’s Disease Beta-peptide. J. Exp. Med. 1998, 188, 431. [Google Scholar] [CrossRef] [PubMed]
- Strohmeyer, R.; Shen, Y.; Rogers, J. Detection of Complement Alternative Pathway mRNA and Proteins in the Alzheimer’s Disease Brain. Mol. Brain Res. 2000, 81, 7. [Google Scholar] [CrossRef]
- Watson, M.D.; Roher, A.E.; Kim, K.S.; Spiegel, K.; Emmerlin, M.R. Complement Interactions with Amyloid β1–42: A Nidus for Inflammation in AD Brains. Amyloid 1997, 4, 147–156. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Stam, F.C. Immunoglobulins and Complement Factors in Senile Plaques. An Immunoperoxidase Study. Acta Neuropathol. 1982, 57, 239. [Google Scholar] [CrossRef]
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Activation of the Classical Complement Pathway in Brain Tissue of Alzheimer Patients. Neurosci. Lett. 1989, 107, 341. [Google Scholar] [CrossRef]
- Shen, Y.; Li, R.; McGeer, E.G.; McGeer, P.L. Neuronal Expression of mRNAs for Complement Proteins of the Classical Pathway in Alzheimer Brain. Brain Res. 1997, 769, 391. [Google Scholar] [CrossRef]
- Landlinger, C.; Oberleitner, L.; Gruber, P.; Noiges, B.; Yatsyk, K.; Santic, R.; Mandler, M.; Staffler, G. Active Immunization against Complement Factor C5a: A New Therapeutic Approach for Alzheimer’s Disease. J. Neuroinflammation 2015, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Complement System Activation by Amyloid Aggregates of Aβ(1-40) and Aβ(1-42) Peptides: Facts and Hypotheses. Biophysics 2020, 65, 18–21. [Google Scholar] [CrossRef]
- Emery, D.C.; Shoemark, D.K.; Batstone, T.E.; Waterfall, C.M.; Coghill, J.A.; Cerajewska, T.L.; Davies, M.; West, N.X.; Allen, S.J. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer’s Post-Mortem Brain. Front. Aging Neurosci. 2017, 9, 195. [Google Scholar] [CrossRef]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [Green Version]
- Daskalov, A.; Saupe, S.J. The Expanding Scope of Amyloid Signalling. Prion 2021, 15, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, L.; Tükel, Ç. Bacterial Amyloids: The Link between Bacterial Infections and Autoimmunity. Trends Microbiol. 2019, 27, 954–963. [Google Scholar] [CrossRef]
- Olsen, A.; Jonsson, A.; Normark, S. Fibronectin Binding Mediated by a Novel Class of Surface Organelles on Escherichia coli. Nature 1976, 338, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Mena, A.; Cámara-Almirón, J.; de Vicente, A.; Romero, D. Multifunctional Amyloids in the Biology of Gram-Positive Bacteria. Microorganisms 2020, 8, 2020. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.; Kwan, A.H.; Sunde, M. Functional Amyloid: Widespread in Nature, Diverse in Purpose. Essays Biochem. 2014, 56, 207–219. [Google Scholar] [PubMed]
- Balistreri, A.; Goetzler, E.; Chapman, M. Functional Amyloids Are the Rule Rather Than the Exception in Cellular Biology. Microorganisms 2020, 8, 1951. [Google Scholar] [CrossRef]
- Pulze, L.; Bassani, B.; Gini, E.; D’Antona, P.; Grimaldi, A.; Luini, A.; Marino, F.; Noonan, D.M.; Tettamanti, G.; Valvassori, R.; et al. NET Amyloidogenic Backbone in Human Activated Neutrophils. Clin. Exp. Immunol. 2016, 183, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA Form Amyloid-like Myo-granules in Regenerating Muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Antonets, K.S.; Bondarev, S.A.; Inge-Vechtomov, S.G.; Derkatch, I.L. Prions, Amyloids, and RNA: Pieces of a Puzzle. Prion 2016, 10, 182–206. [Google Scholar] [CrossRef] [Green Version]
- Bardin, T.; Daskalov, A.; Barrouilhet, S.; Granger-Farbos, A.; Salin, B.; Blancard, C.; Kauffmann, B.; Saupe, S.J.; Cousto, U.V. Partial Prion Cross-Seeding between Fungal and Mammalian Amyloid Signaling Motifs. mBio 2021, 12, e02782-20. [Google Scholar] [CrossRef] [PubMed]
- Kosolapova, A.O.; Antonets, K.S.; Belousov, M.V.; Nizhnikov, A.A. Biological Functions of Prokaryotic Amyloids in Interspecies Interactions: Facts and Assumptions. Int. J. Mol. Sci. 2020, 21, 7240. [Google Scholar] [CrossRef]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, J.P.; Matthews, J.M.; Winefield, R.D.; Mackay, L.G.; Haverkamp, R.G.; Templeton, M.D. The Hydrophobin EAS is Largely Unstructured in Solution and Functions by Forming Amyloid-like Structures. Structure 2001, 9, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Kenney, J.M.; Knight, D.; Wise, M.J.; Vollrath, F. Amyloidogenic Nature of Spider Silk. Eur. J. Biochem. 2002, 269, 4159–4163. [Google Scholar] [CrossRef]
- Berson, J.F.; Theos, A.C.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Proprotein Convertase Cleavage Liberates a Fibrillogenic Fragment of a Resident Glycoprotein to Initiate Melanosome Biogenesis. J. Cell Biol. 2003, 161, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Chien, P.; Weissman, J.S.; DePace, A.H. Emerging Principles of Conformation-based Prion Inheritance. Annu. Rev. Biochem. 2004, 73, 617–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaglestone, S.S.; Cox, B.S.; Tuite, M.F. Translation Termination Efficiency Can be Regulated in Saccharomyces cerevisiae by Environmental Stress through a Prion-mediated Mechanism. EMBO J. 1999, 18, 1974–1981. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B.; et al. Functional Profiling of the Saccharomyces cerevisiae Genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Nakayashiki, T.; Kurtzman, C.P.; Edskes, H.K.; Wickner, R.B. Yeast Prions [URE3] and [PSI+] are Diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 10575–10580. [Google Scholar] [CrossRef] [Green Version]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The Protein Product of the Het-s Heterokaryon Incompatibility Gene of the Fungus Podospora anserina Behaves as a Prion Analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [Green Version]
- Saupe, S.J. Molecular Genetics of Heterokaryon Incompatibility in Filamentous Ascomycetes. Microbiol. Mol. Biol. Rev. 2000, 64, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dueholm, M.S.; Petersen, S.V.; Sonderkaer, M.; Larsen, P.; Christiansen, G.; Hein, K.L.; Enghild, J.J.; Nielsen, J.L.; Nielsen, K.L.; Nielsen, P.H.; et al. Functional Amyloid in Pseudomonas. Mol. Microbiol. 2010, 77, 1009–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otzen, D. Functional Amyloid: Turning Swords into Plowshares. Prion 2010, 4, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Shewmaker, F.; McGlinchey, R.P.; Wickner, R.B. Structural Insights into Functional and Pathological Amyloid. J. Biol. Chem. 2011, 286, 16533–16540. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, K.; Boles, B.R. Microbial Amyloids: Functions and Interactions within the Host. Curr. Opin. Microbiol. 2013, 16, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adda, C.G.; Murphy, V.J.; Sunde, M.; Waddington, L.J.; Schloegel, J.; Talbo, G.H.; Vingas, K.; Kienzle, V.; Masciantonio, R.; Howlett, G.J.; et al. Plasmodium Falciparum Merozoite Surface Protein 2 is Unstructured and Forms Amyloid-like Fibrils. Mol. Biochem. Parasitol. 2009, 166, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, K.; Lecomte, S.; Estevez, Y.; Coulary-Salin, B.; Bentaleb, A.; Cullin, C.; Deffieux, A.; Peruch, F. Rubber Elongation Factor (REF), a Major Allergen Component in Hevea brasiliensis latex Has Amyloid Properties. PLoS ONE 2012, 7, e48065. [Google Scholar] [CrossRef] [Green Version]
- Gossler-Schofberger, R.; Hesser, G.; Reif, M.M.; Friedmann, J.; Duscher, B.; Toca-Herrera, J.L.; Oostenbrink, C.; Jilek, A. A Stereochemical Switch in the aDrs Model System, a Candidate for a Functional Amyloid. Arch. Biochem. Biophys. 2012, 522, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhart, M.M.; Chapman, M.R. Curli Biogenesis and Function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherny, I.; Rockah, L.; Levy-Nissenbaum, O.; Gophna, U.; Ron, E.Z.; Gazit, E. The Formation of Escherichia coli Curli Amyloid Fibrils is Mediated by Prion-like Peptide Repeats. J. Mol. Biol. 2005, 352, 245–252. [Google Scholar] [CrossRef]
- Shewmaker, F.; McGlinchey, R.P.; Thurber, K.R.; McPhie, P.; Dyda, F.; Tycko, R.; Wickner, R.B. The Functional Curli Amyloid is not Based on In-register Parallel Beta-sheet Structure. J. Biol. Chem. 2009, 284, 25065–25076. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, E.B.; Claessen, D.; Haas, M.; Hurgobin, B.; Gras, S.L. The Assembly of Individual Chaplin Peptides from Streptomyces coelicolor into Functional Amyloid Fibrils. PLoS ONE 2011, 6, e18839. [Google Scholar] [CrossRef] [Green Version]
- Kosolapova, A.O.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Sulatsky, M.I.; Antonets, K.S.; Volkov, K.V.; Lykholay, A.N.; Shtark, O.Y.; Vasileva, E.N.; et al. Two Novel Amyloid Proteins, RopA and RopB, from the Root Nodule Bacterium Rhizobium leguminosarum. Biomolecules 2019, 9, 694. [Google Scholar] [CrossRef] [Green Version]
- Bieler, S.; Estrada, L.; Lagos, R.; Baeza, M.; Castilla, J.; Soto, C. Amyloid Formation Modulates the Biological Activity of a Bacterial Protein. J. Biol. Chem. 2005, 280, 26880–26885. [Google Scholar] [CrossRef] [Green Version]
- Shahnawaz, M.; Park, K.W.; Mukherjee, A.; Diaz-Espinoza, R.; Soto, C. Prion-like Characteristics of the Bacterial Protein Microcin E492. Sci. Rep. 2017, 7, 45720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz, R.; Mercado, G.; Martín-Benito, J.; Giraldo, R.; Monasterio, O.; Lagos, R.; Valpuesta, J.M. Structural Characterization of Microcin E492 Amyloid Formation: Identification of the Precursors. J. Struct. Biol. 2012, 178, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Kim, J.G.; Jeon, E.; Yoo, C.H.; Moon, J.S.; Rhee, S.; Hwang, I. Amyloidogenesis of Type III-dependent Harpins from Plant Pathogenic Bacteria. J. Biol. Chem. 2007, 282, 13601–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Reis, S.; Coulary-Salin, B.; Forge, V.; Lascu, I.; Bégueret, J.; Saupe, S.J. The HET-s Prion Protein of the Filamentous Fungus Podospora Anserina Aggregates in vitro into Amyloid-like Fibrils. J. Biol. Chem. 2002, 277, 5703–5706. [Google Scholar] [CrossRef] [Green Version]
- Balguerie, A.; Dos Reis, S.; Ritter, C.; Chaignepain, S.; Coulary-Salin, B.; Forge, V.; Bathany, K.; Lascu, I.; Schmitter, J.M.; Riek, R.; et al. Domain Organization and Structure-function Relationship of the HET-s Prion Protein of Podospora Anserina. EMBO J. 2003, 22, 2071–2081. [Google Scholar] [CrossRef]
- Ritter, C.; Maddelein, M.L.; Siemer, A.B.; Lührs, T.; Ernst, M.; Meier, B.H.; Saupe, S.J.; Riek, R. Correlation of Structural Elements and Infectivity of the HET-s Prion. Nature 2005, 435, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Wille, H.; Stöhr, J.; Baxa, U.; Prusiner, S.B.; Stubbs, G. Degradation of Fungal Prion HET-s(218-289) Induces Formation of a Generic Amyloid Fold. Biophys. J. 2012, 102, 2339–2344. [Google Scholar] [CrossRef] [Green Version]
- Baxa, U.; Cheng, N.; Winkler, D.C.; Chiu, T.K.; Davies, D.R.; Sharma, D.; Inouye, H.; Kirschner, D.A.; Wickner, R.B.; Steven, A.C. Filaments of the Ure2p Prion Protein Have a Cross-β Core Structure. J. Struct. Biol. 2005, 150, 170–179. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Fernández, M.; Batlle, C.; Navarro, S.; De Groot, N.S.; Serpell, L.; Ventura, S. Characterization of Amyloid Cores in Prion Domains. Sci. Rep. 2016, 6, 34274. [Google Scholar] [CrossRef] [PubMed]
- King, C.Y.; Tittmann, P.; Gross, H.; Gebert, R.; Aebi, M.; Wüthrich, K. Prion-inducing Domain 2–114 of Yeast Sup35 Protein Transforms in vitro into Amyloid-like Filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 6618–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic Regulation of Translation Reveals Hidden Genetic Variation to Produce Complex Traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef]
- Du, Z.; Park, K.W.; Yu, H.; Fan, Q.; Li, L. Newly Identified Prion Linked to the Chromatin-remodeling Factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Butko, P.; Buford, J.P.; Goodwin, J.S.; Stroud, P.A.; McCormick, C.L.; Cannon, G.C. Spectroscopic Evidence for Amyloid-like Interfacial Self-assembly of Hydrophobin Sc3. Biochem. Biophys. Res. Commun. 2001, 280, 212–215. [Google Scholar] [CrossRef] [PubMed]
- de Vocht, M.L.; Reviakine, I.; Wösten, H.A.; Brisson, A.; Wessels, J.G.; Robillard, G.T. Structural and Functional Role of the Disulfide Bridges in the Hydrophobin SC3. J. Biol. Chem. 2000, 275, 28428–28432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, A.H.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural Basis for Rodlet Assembly in Fungal Hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iconomidou, V.A.; Vriend, G.; Hamodrakas, S.J. Amyloids Protect the Silkmoth Oocyte and Embryo. FEBS Lett. 2000, 479, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Podrabsky, J.E.; Carpenter, J.F.; Hand, S.C. Survival of Water Stress in Annual Fish Embryos: Dehydration Avoidance and Egg Envelope Amyloid Fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R123–R131. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Vriend, G.; Hoenger, A.; Hamodrakas, S.J. Amyloid-like Fibrils from an 18-residue Peptide Analogue of a Part of the Central Domain of the B-family of Silkmoth Chorion Proteins. FEBS Lett. 2001, 499, 268–273. [Google Scholar] [CrossRef] [Green Version]
- Hamodrakas, S.J.; Jones, C.W.; Kafatos, F.C. Secondary Structure Predictions for Silkmoth Chorion Proteins. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1982, 700, 42–51. [Google Scholar] [CrossRef]
- Dicko, C.; Knight, D.; Kenney, J.M.; Vollrath, F. Structural Conformation of Spidroin in Solution: A Synchrotron Radiation Circular Dichroism Study. Biomacromolecules 2004, 5, 758–767. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal Domains Elicit Formation of Functional Pmel17 Amyloid Fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [Green Version]
- Si, K.; Giustetto, M.; Etkin, A.; Hsu, R.; Janisiewicz, A.M.; Miniaci, M.C.; Kim, J.H.; Zhu, H.; Kandel, E.R. A Neuronal Isoform of CPEB Regulates Local Protein Synthesis and Stabilizes Synapse-specific Long-term Facilitation in Aplysia. Cell 2003, 115, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM Structure of a Neuronal Functional Amyloid Implicated in Memory Persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Ulamec, S.M.; Radford, S.E. Spot the Difference: Function vs. Toxicity in Amyloid Fibrils. Trends Biochem. Sci. 2020, 45, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, S.; Bajakian, T.; Soria, M.; Falk, A.S.; Service, R.J.; Langen, R.; Siemer, A.B. Identification and Structural Characterization of the N-terminal Amyloid Core of Orb2 isoform A. Sci. Rep. 2016, 6, 38265. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, R.; Ramakers, M.; De Smet, F.; Claes, F.; Khodaparast, L.; Khodaparast, L.; Couceiro, J.R.; Langenberg, T.; Siemons, M.; Nyström, S.; et al. De novo Design of a Biologically Active Amyloid. Science 2016, 354, aah4949. [Google Scholar] [CrossRef] [PubMed]
- Latza, V.; Guerette, P.; Ding, D.; Amini, S.; Kumar, A.; Schmidt, I.; Keating, S.; Oxman, N.; Weaver, J.C.; Masic, A. Multi-scale Thermal Stability of a Hard Thermoplastic Protein-based Material. Nat. Commun. 2015, 6, 8313. [Google Scholar] [CrossRef]
- Maji, S.K.; Schubert, D.; Rivier, C.; Lee, S.; Rivier, J.E.; Riek, R. Amyloid as a Depot for the Formulation of Long-acting Drugs. PLoS Biol. 2008, 6, e17. [Google Scholar] [CrossRef]
- Cui, M.; Qi, Q.; Gurry, T.; Zhao, T.; An, B.; Pu, J.; Gui, X.; Cheng, A.A.; Zhang, S.; Xun, D.; et al. Modular Genetic Design of Multi-domain Functional Amyloids: Insights into Self-assembly and Functional Properties. Chem. Sci. 2019, 10, 4004–4014. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.P.; Hewitt, E.W. Why are Functional Amyloids Non-Toxic in Humans? Biomolecules 2017, 7, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavliukeviciene, B.; Zentelyte, A.; Jankunec, M.; Valiuliene, G.; Talaikis, M.; Navakauskiene, R.; Niaura, G.; Valincius, G. Amyloid β Oligomers Inhibit Growth of Human Cancer Cells. PLoS ONE 2019, 14, e0221563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, B.L.; Jang, H.; Capone, R.; Teran Arce, F.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial Properties of Amyloid Peptides. Mol Pharm. 2012, 9, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The mechanisms of AD.

Figure 2.
Timeline of history of functional amyloid discoveries. The name, host and function are presented.
Figure 2.
Timeline of history of functional amyloid discoveries. The name, host and function are presented.


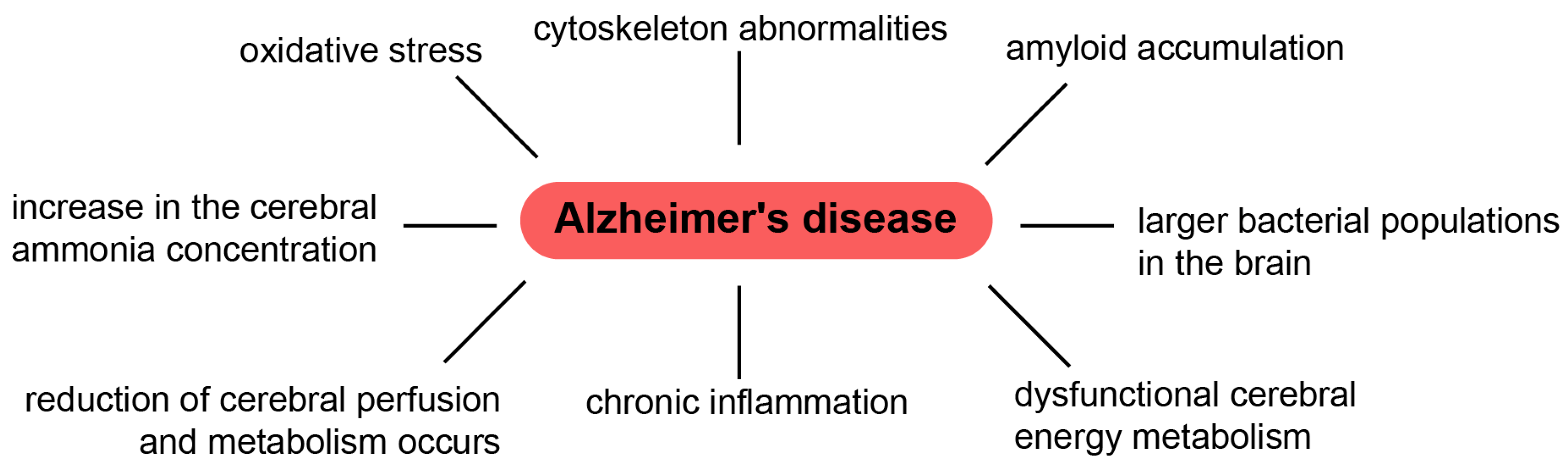{kind=link}
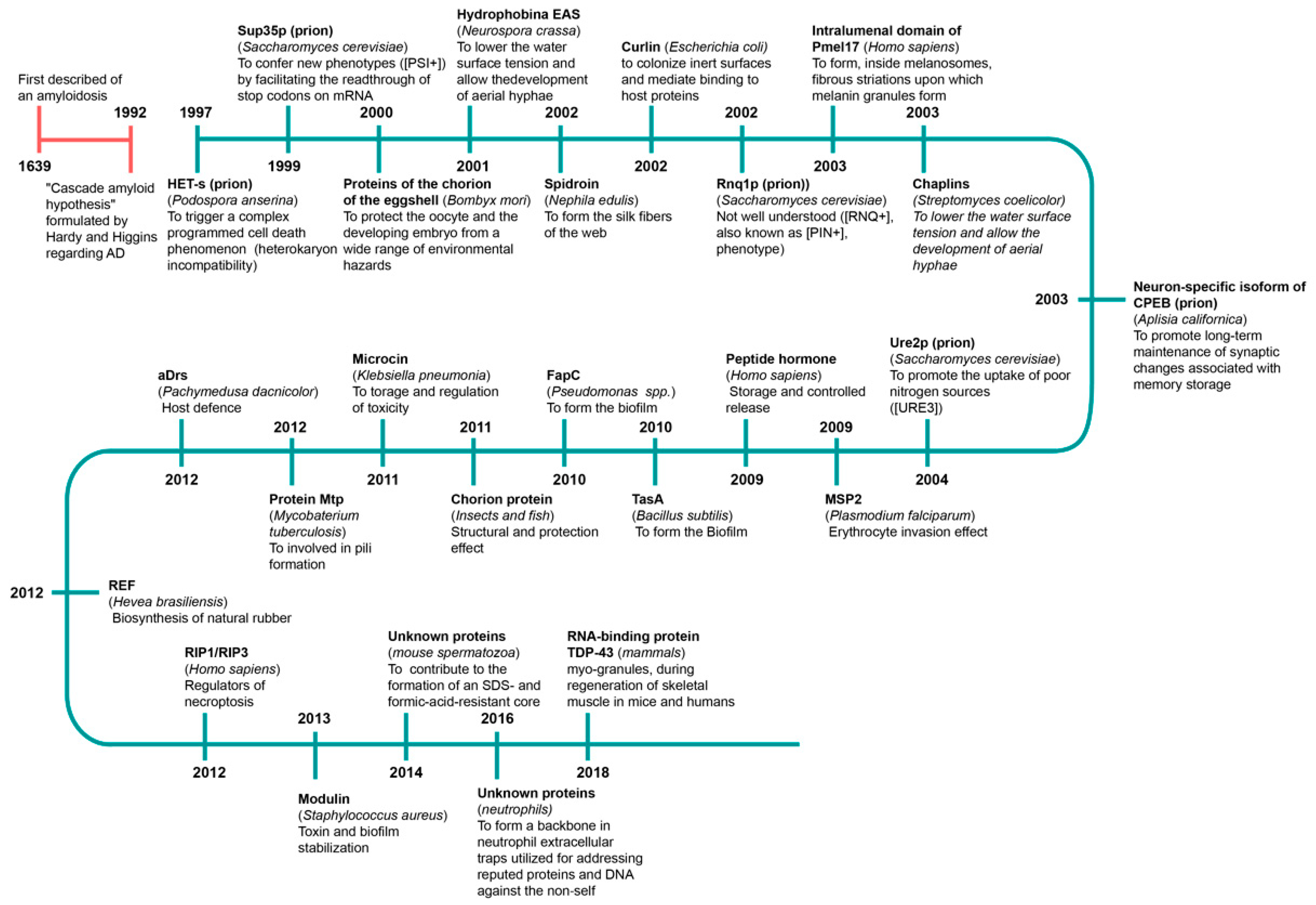{kind=link}
Table 1.
The prevalence of functional amyloids and features of their research [176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228].
| Species or Organisms | Protein or Peptide | Function | Mol. Weight | Structure | Evidence of Cross β-Structure Presence | Secondary Structure Changes | Congo Red and ThT Binding | Condition of in vitro Amyloid Fibril Forming | References |
|---|---|---|---|---|---|---|---|---|---|
| Bacteria | |||||||||
| Escherichia coli, Salmonella spp. | Curli | Biofilm formation, host invasion. | CsgA (main damain of curlin) ~17.5 kDa. | Previously β-structure. | X-ray diffraction for CsgA. | CD method: CsgA fibrils are as follows: 16 ± 2% α-helix, 40 ± 2% β-sheet, 13 ± 2% β-turn and 31 ± 2% remainder. | CR, ThT | CsgA fibrils were prepared by dialyzing purified protein into 25 mM Tris, pH 7.5, 100 mM NaCl and 0.5 mM EDTA and incubating at room temperature (RT) for several days. | [176,193,194,195] |
| Streptomyces coelicolor | Chaplins | Modulation of water surface tension (i.e., development of aerial structures). | ChpD-H up to 6 kDa ChpA-C ~17–20 kDa. | ChpD and ChpF comprise β-sheet; ChpE is random coil (RC); ChpG and ChpH have mixed secondary structure comprising elements of both β-sheet and RC. | X-ray diffraction. | CD method: the protein mixture adopted a conformation rich in β-sheet. | ThT | Synthetic chaplin peptides were dissolved at a final concentration of 0.5 mg/mL in water and the pH adjusted by titration of NaOH/HCl. | [11,196] |
| Rhizobium leguminosarum | RopA and RopB | Possibility role in the control of plant-microbial symbiosis. | RopA 38.97 kDa RopB 22 kDa. | Previously β- structure. | none | CD method: Before aggregation: RopA more than 40% β-structure, RopB more than 30% β-structure After aggregation: 42% and 38% β-structure for RopA and RopB aggregates respectively. | CR, ThT | Proteins were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and incubated for seven days. Afterward, HFIP was evaporated under a stream of nitrogen, and the samples were stirred for an additional seven days. | [197] |
| Klebsiella pneumoniae | Microcin E492 (Mcc) | Bacteriocin, membrane pore-forming peptide, amyloid form is inactive. | ~7.8 kDa. | RC conformation in aqueous buffer and α-helix in methanol. | X-Ray diffraction. | CD method: Aggregated Mcc is rich in β-sheet structures. | CR, ThT | Purified Mcc a (400 μg/mL) were incubated in aggregation buffer (50 mM PIPES-NaOH, pH 6.5, 0.5 M NaCl) for 48 h at 37 °C with vigorous shaking. | [198,199,200] |
| Xanthomonas species | Harpins (HpaG) | Secreted by plant pathogenic bacteria, destabilize plant membranes, induce cell death. | 15.6 kDa. | Previously α-helix. | non | CD method: After 3 days, the CD spectrum of HpaG changed to a minimum at 220 nm, which is indicative of transition to a β-sheet. | CR | Harpin samples were incubated without agitation in 20 mm Tris-HCl (pH 8.0) containing 10 mm NaCl to mimic the salt concentration in the intercellular space of plant tissues at 27 °C for 14 days. | [201] |
| Fungi | |||||||||
| Podospora anserine | HET-s | Regulation of heterokaryon formation. | ~32 kDa. | Estimated content of 34% α-helical, 16% β-sheet and 50% RC structure. | X-ray diffraction of HET-s (218–289). | CD method: 17% α-helix, 32% β-sheet and 50% random coil. FTIR: In the infrared spectrum of the soluble form the amide I′ band reached a maximum at 1650 cm−1. In the spectrum of the aggregated form this maximum was shifted at 1643 cm−1 and a shoulder around 1625 cm−1 was observed. | CR, ThT of HET-s (218–289). | The HET-s (218–289) peptide was soluble at pH 2.5 in 150 mM acetic acid, but, under non-denaturing conditions at pH 8.0, in a time course of a few hours, the peptide spontaneously formed aggregates. | [202,203,204,205] |
| Saccharomyces cerevisiae | URE2p | Regulation of nitrogen catabolism. | ~38 kDa. | β-strands, α-helix and RC. | Electron diffraction, X-ray diffraction and X-ray diffraction (PFD domain). | CD method of PFD domain: Switching from an initially disordered, RC structure, to a β-sheet enriched conformation. FTIR of PFD domain: A band at ∼1625 cm−1 dominates the spectrum (the presence of intermolecular β-sheet structure). | CR, ThT | Filaments were made by incubation of protein solutions (usually at about 1 mg/mL) on a shaker for 16 h at 4 °C. | [206,207] |
| Sup35p (Prion-inducing domain 2–114 and PFD domain) | Regulation of stop-codon read-through. | ~75 kDa. | A freshly prepared solution exhibits a far UV CD spectrum that indicates little α-helix or β-sheet content. | X-ray diffraction (PFD domain). | CD method (PFD domain): Switching from an initially disordered, RC structure, to a β-sheet enriched conformation. FTIR: A band at ∼1625 cm−1 dominates the spectrum. | CR | Filaments of Sup35pN (Prion-inducing domain 2–114) were prepared in 0.1% (vol/vol) TFA/40% (vol/vol) acetonitrile using reverse-phase HPLC fractions containing isocratically eluted Sup35pN. Preparation of a 100 μM solution of Sup35pN yielded filaments after 1 week of incubation at 4 °C. Spontaneous filament formation exists in 50 mM sodium phosphate buffer (pH 2.0) with 40% acetonitrile. | [207,208,209] | |
| Swi1p | Chromatin remodeling factor, prion form inactive. | ~140 kDa. | none | X-ray diffraction (PFD domain). | CD method (PFD domain): Switching from an initially disordered, RC structure, to a β-sheet enriched conformation. FTIR: A band at ∼1625 cm−1 dominates the spectrum. | none | none | [209,210] | |
| Mot3 | Transcriptional regulator of cell wall remodeling genes, prion form is inactive. | ~55 kDa. | none | X-ray diffraction (PFD domain). | CD method (PFD domain) Switching from an initially disordered, random coil structure, to a β-sheet enriched conformation. FTIR (PFD domain) A band at ∼1625 cm−1 dominates the spectrum. | CR, ThT. | none | [207,211] | |
| Most fungi | Hydrophobins | Fungal coat formation, modulation of adhesion and surface tension. | 7–9 kDa. | Previously RC and small core of antiparallel β-sheet. | X-ray. | CD method: β sheet is the predominant element of secondary structure in polymerized hydrophobin rodlets. | CR, ThT | For Hydrophobin SC3 Schizophyllum commune: Upon binding to a hydrophobic solid surface, the protein is arrested in an intermediate α-helical state, whereas, upon self-assembly at the air–water interface, rodlets are formed in a β-sheet conformation. | [176,212,213,214] |
| Animal | |||||||||
| Insects and fish | Chorion proteins (central domain of silkmoth chorion proteins of the A and B -family) | Structural and protective functions in the eggshell. | 34 and 24 kDa. | In both families of proteins β-sheet structure predominates. | X-ray diffraction | FTIR method ATR FT-IR supports the presence of uniform β-sheets in the structure of cA_m1 peptide fibrils; β-sheet structure also suggested by X-ray diffraction ATR FT-IR data: 64% antiparallel β-sheet and 30% β-turns in the central domain of silkmoth chorion proteins. | CR | cA peptide (central domain of the A class of silkmoth chorion proteins) was dissolved in a 50 mM sodium acetate buffer (pH 5) at a concentration of 9 mg/mL to produce amyloid-like fibrils after 3–4 weeks incubation. | [14,215,216,217,218] |
| Nephila clavipes Nephila edulis Araneus diadematus | Spidroins and Araneus diadematus fibroin | Structural (i.e., spider silk). | ~320 kDa (spidroin). | β-sheet or β-turn and RC. | X-Ray diffraction. | CD method: increasing of β-sheet structures. | CR, ThT | Lyophilized protein was dissolved in 6 M guanidinium thiocyanate at a concentration of 10 mg/mL−1 and dialyzed against 10 × 10−3 M potassium phosphate for several days at RT. For acceleration of fibril formation, 10 vol.-% methanol was added. | [15,177,219] |
| All mammalians including Homo sapiens | Non-glycosylated, 442-residue lumenal fragment of Pmel17 (rMα) | Pmel17 amyloid templates and accelerates the covalent polymerization of reactive small molecules into melanin. | 110 kDa (28-kDa transmembrane fragment (Mβ) and an 80-kDa lumenal fragment (Mα)). | β-strands, α-helix and RC | X-ray diffraction | CD and FTIR: Mα aggregates are approximately 11% α-helix, 32% β-sheet, 23% β-turn and 33% disordered, based on curve fitting with a basis set of 43 soluble proteins. | CR, ThT | rMα fibers were generated by diluting (from concentrated 8 M GdmCl, 50 mM KH2PO4/K2HPO 4 [pH 7.4], 100 mM KCl stock) rMα into 125 mM CH3COOH/ CH3COOK buffer (pH 5.0) at a final concentration of 10 μM and allowing it to stand at RT for 24 h. | [18,220] |
| Drosophila melanogaster | CPEEB (Orb2) | Memory consolidation Cytoplasmic polyadenylation element-binding protein regulates mRNA translation. | ~62 kDa | The protofilament core adopts a simple hairpin-like fold, composed of two β-strands, b1 (residues 176 to 186) and b2 (residues 197 to 206). | CryoEM | Only 31 residues (176–206) of the 704-residue protein form the amyloid core. N650 residues are dynamically disordered. | ThT | Recombinant Orb2A and Orb2A88 samples were exchanged into 10 mM HEPES, pH 7.6, 100 mM KCl, 1 M Urea and 1 mM DTT using dialysis and a PD-10 desalting column, respectively. Samples were then incubated on a shaker at RT for up to 2 weeks. | [221,222,223,224] |
| Plants | |||||||||
| Pisum sativum L. | Vicilin (Cupin-1.1 ((19–166 aa) and Cupin-1.2 (229–394 aa)) | Amyloid formation in charge of the accumulation of storage proteins in plant seeds. | ~50 kDa. | β-barrel domains. | X-Ray diffraction. | CD method Before aggregation Cupin-1.1 and Cupin-1.2 (4–12% β-content), Vicilin (39% β-content) After aggregation Cupin-1.1,Cupin-1.2 and Vicilin (40–42% β-content). | CR, ThT. | 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) solvent for the proteins dissolution with its subsequent removal from the sample and incubation of dissolved proteins in the distilled water at 37 °C for 7 days for Vicilin, Cupin-1.1, Cupin-1.2 and 5 mM phosphate buffered saline (PBS) [pH 7.4]) for one day at 25 °C) for Cupin-1.2 | [8] |
| Synthetic amyloid aggregates | |||||||||
| Synthesized peptides (Homo sapiens and Mouse) | Vascin (Peptide based on an amyloidogenic sequence in the vascular endothelial growth factor receptor (VEGFR2) | Inhibited VEGFR2-dependent tumor growth. | 2272.15 Da. | Secondary structures are absent. | X-Ray diffraction. | FTIR method b-sheet structure change in b-structured conformation. | ThT. | 300 mM vascin in 1% (w/v) NH4CO3 after 24 h incubation at room temperature. | [225] |
| Dosidicus gigas (D. gigas) | Sucker ring teeth (SRT) from squid. SRT are assembled entirely from a protein family | Molecular design of biomimetic protein- and peptide-based thermoplastic structural biopolymers with potential biomedical and 3D printing applications. | none | Previously β-structure. | X-Ray diffraction. | FTIR method: the β-sheet-specific infrared band was centered at 1.235 cm−1 from RT up to 150 °C, at which point it shifted gradually to 1.220 cm−1, which was still within the β-sheet- region. | none | none | [226] |
| Synthesized peptides | Gonadotropin-releasing hormone analog (GnRH) | Use of amyloids in the formulation of long-acting drugs. Sorting, storage, and release of diverse hormones. | 1183.27 Da. | Secondary structures are absent. | none | none | CR, ThT. | GnRH analogs were dissolved in a glass tube in 1 mL of 5% D-mannitol and 0.01% sodium azide at a concentration of 1 mg/mL. The GnRH analogs were then incubated at RT without stirring. | [227] |
| Escherichia coli and Bacillus circus and Mytilus galloprovincialis | CsgA (as amyloidogenic cores) + chitin-binding domains (CBDs) + mussel foot proteins (Mfp3/Mfp5) two-domain and three-domain constructions with constant presence of CsgA | Development of multifunctional molecular materials with individual structure and characteristics based on amyloid. | CsgA ~17.5 kDa Mfp3 5–7.5 kDa Mfp5 9.5 CBD 6 kDa. | β-strands and RC. | X-Ray diffraction. | The two-domain proteins contained 60% of β-sheet/β-turn structures and 40% of RC, owing to the introduction of RC Mfps. Compared with their two-domain counterparts, the three-domain fibrils possess more β-sheet structures. | CR, ThT | Proteins were either dialyzed against PBS solutions (pH = 5.0 or 2.5) for 2 days or were incubated at 4 °C under acidic conditions for 3 days to promote the formation of amyloid fibers, followed by redissolving in hexafluoro-2-propanol (HFIP) solvent. | [228] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yakupova, E.I.; Bobyleva, L.G.; Shumeyko, S.A.; Vikhlyantsev, I.M.; Bobylev, A.G. Amyloids: The History of Toxicity and Functionality. Biology 2021, 10, 394. https://doi.org/10.3390/biology10050394
AMA Style
Yakupova EI, Bobyleva LG, Shumeyko SA, Vikhlyantsev IM, Bobylev AG. Amyloids: The History of Toxicity and Functionality. Biology. 2021; 10(5):394. https://doi.org/10.3390/biology10050394
Chicago/Turabian StyleYakupova, Elmira I., Liya G. Bobyleva, Sergey A. Shumeyko, Ivan M. Vikhlyantsev, and Alexander G. Bobylev. 2021. "Amyloids: The History of Toxicity and Functionality" Biology 10, no. 5: 394. https://doi.org/10.3390/biology10050394
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.