
Challenges and Strategies for a Thorough Characterization of Antibody Acidic Charge Variants
 , , ,
, , ,
Abstract
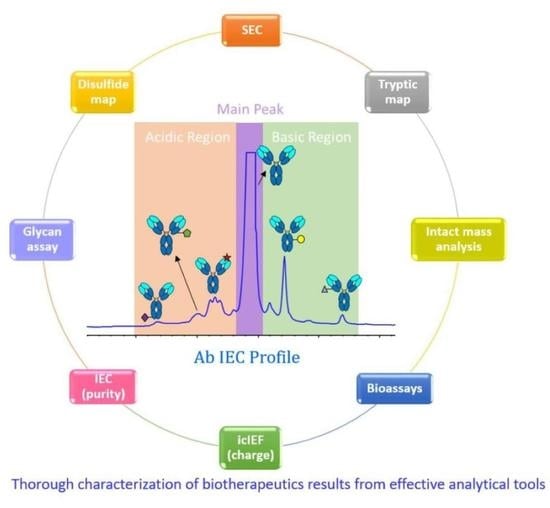
:
1. Introduction
2. Materials and Methods
2.1. Material
2.2. IEC Method
2.3. Fraction Collection by IEC
2.4. SEC Method
2.5. icIEF Method
2.6. DSC Analysis
2.7. HDX-MS Analysis
2.8. Generating Antibody Fragments Using Enzyme IdeS and IdeE
2.9. Desialylation
2.10. LC/MS Analysis for Intact Protein and mAb1 Fragments
2.11. Chip Native CE/MS Analysis for Intact Protein
2.12. Tryptic Peptide Map Analysis
2.12.1. Peptide Map by LC/MS
2.12.2. mD LC/MS Analysis
3. Results
3.1. Purity and Quality Check of Collected Acidic and Main Fractions
3.2. Charge Variant Determination by icIEF Method
3.3. HOS Studies by DSC and HDX-MS
3.3.1. DSC Analysis
3.3.2. HDX-MS
3.4. LC/MS Analysis of Additional Potential Acidic Charge Variants
3.4.1. Sialylated Glycan
3.4.2. Lysine Succinylation
3.4.3. Pyroglutamate
3.5. Glycation Quantification by Intact Mass Analysis
4. Discussion
4.1. Closing the Gaps of mAb1 Acidic Variant Quantification
4.2. Recommend Strategies for Characterization of Protein Charge Variants
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorai, H.; Ganguly, S. Mammalian cell-produced therapeutic proteins: Heterogeneity derived from protein degradation. Curr. Opin. Biotechnol. 2014, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Protein heterogeneity and the immunogenicity of biotherapeutics. GaBI J. 2018, 7, 63–69. [Google Scholar] [CrossRef]
- Shi, R.L.; Xiao, G.; Dillon, T.M.; Ricci, M.S.; Bondarenko, P.V. Characterization of therapeutic proteins by cation exchange chromatography-mass spectrometry and top-down analysis. mAbs 2020, 12, 1739825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, D.; Peng, Y.; Berkowitz, S.A.; Engen, J.R. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol. Cell Proteom. 2010, 9, 1716. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Walsh, A.; Ehrick, R.; Xu, W.; May, K.; Liu, H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. mAbs 2012, 4, 578–585. [Google Scholar] [CrossRef]
- Vlasak, J.; Ionescu, R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr. Pharm. Biotechnol. 2008, 9, 468–481. [Google Scholar] [CrossRef]
- Talebi, M.; Nordborg, A.; Gaspar, A.; Lacher, N.A.; Wang, Q.; He, X.Z.; Haddad, P.R.; Hilder, E.F. Charge heterogeneity profiling of monoclonal antibodies using low ionic strength ion-exchange chromatography and well-controlled pH gradients on monolithic columns. J. Chromatogr. A. 2013, 1317, 148–154. [Google Scholar] [CrossRef]
- Vlasak, J.; Bussat, M.C.; Wang, S.; Wagner-Rousset, E.; Schaefer, M.; Klinguer-Hamour, C.; Kirchmeier, M.; Corvaïa, N.; Ionescu, R.; Beck, A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal. Biochem. 2009, 392, 145–154. [Google Scholar] [CrossRef]
- Neill, A.; Nowak, C.; Patel, R.; Ponniah, G.; Gonzalez, N.; Miano, D.; Liu, H. Characterization of recombinant monoclonal antibody charge variants using OFFGEL fractionation, weak anion exchange chromatography, and mass spectrometry. Anal. Chem. 2015, 87, 6204–6211. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, D.; Malani, H.; Rathore, A.S. LC–MS based case-by-case analysis of the impact of acidic and basic charge variants of bevacizumab on stability and biological activity. Sci. Rep. 2021, 11, 2487–2498. [Google Scholar] [CrossRef]
- Ambrogellya, A.; Gozob, S.; Katiyarc, A.; Dellatored, S.; Kunee, Y.; Bhatf, R.; Sung, J.; Lih, N.; Wangi, D.; Nowakj, C.; et al. Analytical comparability study of recombinant monoclonal antibody therapeutic. mAbs 2018, 10, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. mAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.D.; Enk, J.Z.; Flynn, G.C. Human antibody Fc deamidation in vivo. Biologicals 2009, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Dada, O.O.; Jaya, N.; Valliere-Douglass, J.; Salas-Solano, O. Characterization of acidic and basic variants of IgG1 therapeutic monoclonal antibodies based on non-denaturing IEF fractionation. Electrophoresis 2015, 36, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; May, K. Disulfide bond structures of IgG molecules: Structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012, 4, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Glover, Z.K.; Sreedhara, A. Assessing the Utility of Circular Dichroism and FTIR Spectroscopy in Monoclonal-Antibody Comparability Studies. J. Pharm. Sci. 2015, 104, 4459–4466. [Google Scholar] [CrossRef]
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. MAbs 2016, 8, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein. Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.R.; Zhang, M.M.; Gross, M.L. Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev. 2020, 120, 4355–4454. [Google Scholar] [CrossRef]
- Engen, J.R.; Botzanowski, T.; Peterle, D.; Georgescauld, F.; Wales, T.E. Developments in Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2021, 93, 567–582. [Google Scholar] [CrossRef]
- Liu, Y.D.; Smith, D.L. Probing High Order Structure of Proteins by Fast-Atom Bombardment Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1994, 5, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.L.; Zhang, Z.; Liu, Y.D. Amide hydrogen exchange and mass spectrometry: A probe of high-order structure in proteins. Pure Appl. Chem. 1994, 66, 89–94. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Huang, K.Y.; Hsu, J.B.-K.; Lee, T.Y. Characterization and Identifcation of Lysine Succinylation Sites based on Deep Learning Method. Sci. Rep. 2019, 9, 16175–16186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A.; Schauer, R. Sialic Acids. In Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; ISBN 978-1-621821-32-8. [Google Scholar]
- Shah, P.; Yang, S.; Sun, S.; Aiyetan, P.; Yarema, K.J.; Zhang, H. Mass Spectrometric Analysis of Sialylated Glycans with Use of Solid-Phase Labeling of Sialic Acids. Anal. Chem. 2013, 85, 3606–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, G.C.; Chen, X.; Liu, Y.D.; Shah, B.; Zhang, Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol. Immunol. 2010, 47, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Boons, G.-J.; Demchenko, A.V. Recent Advances in O-Sialylation. Chem. Rev. 2000, 100, 4539–4566. [Google Scholar] [CrossRef]
- Bunn, H.F.; Higgins, P.J. Reaction of monosaccharides with proteins: Possible evolutionary significance. Science 1981, 213, 222–224. [Google Scholar] [CrossRef]
- Saleem, R.A.; Affholter, B.R.; Deng, S.; Campbell, P.C.; Matthies, K.; Eakin, C.M.; Wallace, A. A chemical and computational approach to comprehensive glycation characterization on antibodies. MAbs 2015, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Arroll, T.; Chu, L.; Flynn, G.C. Rates and impact of human antibody glycation in vivo. Glycobiology 2012, 22, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Alcala, E.; Petkovska, E.; Matthews, D.; Canova-Davis, E.; Taticek, R.; Ma, S. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: Where it is, how it got there, and how it affects charge-based behaviour. Anal. Biochem. 2008, 373, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priego Capote, F.; Sanchez, J.C. Strategies for proteomic analysis of non-enzymatically glycated proteins. Mass Spectr. Rev. 2009, 28, 135–146. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, T.; Jiang, L.; Hewitt, D.; Huang, Y.; Kao, Y.H.; Katta, V. Rapid identification of low level glycation sites in recombinant antibodies by isotopic labeling with 13C6-reducing sugars. Anal. Chem. 2012, 84, 2313–2320. [Google Scholar] [CrossRef]
- Valliere-Douglass, J.F.; Kodama, P.; Mujacic, M.; Brady, L.J.; Wang, W.; Wallace, A.; Yan, B.; Reddy, P.; Treuheit, M.J.; Balland, A. Asparagine-linked Oligosaccharides Present on a Non-consensus Amino Acid Sequence in the CH1Domain of Human Antibodies. J. Biol. Chem. 2009, 284, 32493–32506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Willison, L.N.; Tripathi, P.; Sathe, S.K.; Roux, K.H.; Emmett, M.R.; Blakney, G.T.; Zhang, H.M.; Marshall, A.G. Epitope Mapping of a 95 kDa Antigen in Complex with Antibody by Solution-Phase Amide Backbone Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2011, 83, 7129–7136. [Google Scholar] [CrossRef] [Green Version]
- Camperi, J.; Goyon, A.; Guillarme, D.; Zhang, K.; Stella, C. Multi-dimensional LC-MS: The next generation characterization of antibody-based therapeutics by unified online bottom-up, middle-up and intact approaches. Analyst 2021, 146, 747–769. [Google Scholar] [CrossRef]
- Durowoju, I.B.; Bhandal, K.S.; Hu, J.; Carpick, B.; Kirkitadze, M. Differential Scanning Calorimetry—A Method for Assessing the Thermal Stability and Conformation of Protein Antigen. J. Vis. Exp. 2017, 4, 55262–55281. [Google Scholar]
- Ozohanics, O.; Ambrus, A. Hydrogen-Deuterium Exchange Mass pectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10, 286. [Google Scholar] [CrossRef]
- Zhang, H.M.; Li, C.; Lei, M.; Lundin, V.; Lee, H.Y.; Ninonuevo, M.; Lin, K.; Han, G.; Sandoval, W.; Lei, D.; et al. Structural and Functional Characterization of a Hole–Hole Homodimer Variant in a “Knob-Into-Hole” Bispecific Antibody. Anal. Chem. 2017, 89, 13494–13501. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gao, X.; Lundin, V.; Shi, C.; Adem, Y.; Lin, K.; Jiang, G.; Kao, Y.H.; Yang, F.; Michels, D.; et al. Probing the Impact of the Knob-into-Hole Mutations on the Structure and Function of a Therapeutic Antibody. Anal. Chem. 2020, 92, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Goetze, A.M.; Bass, R.B.; Flynn, G.C. N-terminal Glutamate to Pyroglutamate Conversion in vivo. JBC 2011, 286, 11211–11217. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Steen, S.; Hambly, D.; Valliere-Douglass, J.; Bos, T.V.; Smallwood, S.; Yates, Z.; Arroll, T.; Han, Y.; Gadgil, H.; et al. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J. Pharm. Sci. 2009, 98, 3509–3521. [Google Scholar] [CrossRef] [PubMed]
- Lyubarskaya, Y.; Houde, D.; Woodard, J.; Murphy, D.; Mhatre, R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Anal. Biochem. 2006, 348, 24–39. [Google Scholar] [CrossRef]
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D.; et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs 2010, 2, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Weitzhandler, M.; Farnan, D.; Rohrer, J.S.; Avdalovic, N. Protein variant separations using cation exchange chromatography on grafted, polymeric stationary phases. Proteomics 2001, 1, 179–185. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attributes | Percentage | Assays |
|---|---|---|
| Deamidation | 17.2 | |
| Oxidation | 21.6 | |
| Hydroxylation | 0.6 | Peptide Mapping |
| Fab Glycan | 0.6 | |
| Lys Succinylation | ND * | |
| Pyroglutamate | 3.6 | |
| LMWS | 10.0 | SEC |
| Glycation | 37.4 | Intact mass analysis |
| AGE | ND * | |
| O-glycan | ND * | |
| HOS | ND * | DSC, HDX-MS |
| Total identified species | 91.0 |
| Modifications Observed in Acidic IEC Species | Assay |
|---|---|
| Deamidation | Tryptic map |
| N-terminal cyclization (Q to pE) | Tryptic map |
| Modification by maleuric acid | Tryptic map |
| Lysine succinylation | Tryptic map |
| Hydroxylation | Tryptic map |
| Sialylation | Glycan assay |
| High mannose | Glycan assay |
| Glycation | Intact mass analysis |
| Glucuronidation | Intact mass analysis |
| Fragments (LMWS) | SEC |
| Cysteinylation | Non-reducing Lys-C map |
| Glutathionylation | Non-reducing Lys-C map |
| Disulfide isoforms | Non-reducing Lys-C map |
| Trisulfide | Non-reducing Lys-C map |
| Reduced disulfide bonds (free-thiol) | RP-HPLC with NcME-tagging |
| Oxidation | Tryptic map |
| N-terminal cyclization (E to pE) | Tryptic map |
| Modifications Observed in IEC Basic Species | Assay |
|---|---|
| C-terminal un-processing Lys | IEC with/without cpb |
| C-terminal Pro amidation | Tryptic map |
| N-terminal signal peptide | Tryptic map |
| Isomerization | Tryptic map |
| Aggregates (HMWS) | SEC |
| Glycosylation | Glycan assay |
| Succinimide | Tryptic map |
| Oxidation | Tryptic map |
| Fragments (LMWS) | SEC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.D.; Cadang, L.; Bol, K.; Pan, X.; Tschudi, K.; Jazayri, M.; Camperi, J.; Michels, D.; Stults, J.; Harris, R.J.; et al. Challenges and Strategies for a Thorough Characterization of Antibody Acidic Charge Variants. Bioengineering 2022, 9, 641. https://doi.org/10.3390/bioengineering9110641
Liu YD, Cadang L, Bol K, Pan X, Tschudi K, Jazayri M, Camperi J, Michels D, Stults J, Harris RJ, et al. Challenges and Strategies for a Thorough Characterization of Antibody Acidic Charge Variants. Bioengineering. 2022; 9(11):641. https://doi.org/10.3390/bioengineering9110641
Chicago/Turabian StyleLiu, Y. Diana, Lance Cadang, Karenna Bol, Xiao Pan, Katherine Tschudi, Mansour Jazayri, Julien Camperi, David Michels, John Stults, Reed J. Harris, and et al. 2022. "Challenges and Strategies for a Thorough Characterization of Antibody Acidic Charge Variants" Bioengineering 9, no. 11: 641. https://doi.org/10.3390/bioengineering9110641