
High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases



1
Biomolecular Sciences Research Centre, Sheffield Hallam University, Sheffield S1 1WB, UK
2
College of Pharmacy, Nineveh University, Mosul 41002, Iraq
3
College of Pharmacy, University of Mosul, Mosul 41002, Iraq
4
School of Applied Sciences, Edinburgh Napier University, Edinburgh EH11 4BN, UK
5
School of Sciences, Research Institute in Healthcare Science, University of Wolverhampton, Wolverhampton WV1 1LY, UK
6
Oxford Parkinson’s Disease Centre, Department of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford OX1 3QX, UK
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Bioengineering 2021, 8(2), 30; https://doi.org/10.3390/bioengineering8020030
Submission received: 30 December 2020
/
Revised: 5 February 2021
/
Accepted: 18 February 2021
/
Published: 23 February 2021
Abstract
:Neurodegenerative diseases (NDDs) are incurable and debilitating conditions that result in progressive degeneration and/or death of nerve cells in the central nervous system (CNS). Identification of viable therapeutic targets and new treatments for CNS disorders and in particular, for NDDs is a major challenge in the field of drug discovery. These difficulties can be attributed to the diversity of cells involved, extreme complexity of the neural circuits, the limited capacity for tissue regeneration, and our incomplete understanding of the underlying pathological processes. Drug discovery is a complex and multidisciplinary process. The screening attrition rate in current drug discovery protocols mean that only one viable drug may arise from millions of screened compounds resulting in the need to improve discovery technologies and protocols to address the multiple causes of attrition. This has identified the need to screen larger libraries where the use of efficient high-throughput screening (HTS) becomes key in the discovery process. HTS can investigate hundreds of thousands of compounds per day. However, if fewer compounds could be screened without compromising the probability of success, the cost and time would be largely reduced. To that end, recent advances in computer-aided design, in silico libraries, and molecular docking software combined with the upscaling of cell-based platforms have evolved to improve screening efficiency with higher predictability and clinical applicability. We review, here, the increasing role of HTS in contemporary drug discovery processes, in particular for NDDs, and evaluate the criteria underlying its successful application. We also discuss the requirement of HTS for novel NDD therapies and examine the major current challenges in validating new drug targets and developing new treatments for NDDs.
1. Introduction
High-throughput screening (HTS) has increasingly been used for novel drug discovery in the field of pharmaceutics replacing the traditional “trial and error” approach to identify therapeutic targets and validate biological effects [1,2,3]. HTS involves assaying and screening a large number of biological effectors and modulators against designated and exclusive targets. Thus, HTS is generally favored when little is known of the target, which precludes structure-based drug design, but it can also be used in parallel with other strategies such as computational techniques and fragment-based drug design [4,5]. HTS comprises several steps including target recognition, compound management, reagent preparation, assay development, as well as the screening itself. Using HTS in conjunction with multi-well cell-based platforms allows for the identification of small molecule modulators of related biochemical and signal transduction pathways.
Current programs for central nervous system (CNS) drug development and discovery can be subdivided into four main areas: (1) Receptor and target engagement, (2) drug “hit” identification, (3) lead identification, and (4) drug lead optimization. Active compounds resulting from HTS screens—the so-called “hits”— are the prototypes from which drug “leads” are ultimately formed through additional combinatorial and medicinal chemistry. Following the screening of several hundred thousand small-molecules, a few hundred “hits” may be identified, leading potentially to viable drug compounds (Figure 1). Potential hits from the HTS must then be configured for delivery, metabolism, and pharmacokinetics to suggest initial lead compounds. However, these lead compounds generally require considerable medicinal chemistry optimization, for example, to block polar functional groups that may reduce high receptor affinity, in an effort to generate a medicine that has optimal drug metabolism and pharmacokinetic (DMPK) properties.
Neurodegenerative diseases (NDDs) are incurable and debilitating conditions that result in progressive degeneration and/or death of nerve cells in the CNS [6,7,8]. Developing drugs for CNS disorders; in particular NDDs, has become a risky business, where most candidates fail after years of costly clinical and non-clinical related activities. Thus, one of the critical first steps in the advancement of treatments of NDDs is the development of accurate assays for investigating neurodegeneration [9]. While the word “neurodegeneration” can be applied to a wide range of characteristics that cause the loss of neuronal activity, neuronal death is the most direct and prominent indicator of neurodegeneration.
The creation of successful assays includes the ability to identify the associated events that trigger and cause cell death. To this end, tests have been established to recognize biochemical events that contribute to neuronal death such as metabolic fluctuation, energy metabolism [10], and fragmentation of DNA [11]. Cytoprotective assays using dyes or fluorescent markers have been a crucial method in the past to classify therapeutics causing neuronal death [11,12,13]. Many of these tests have the benefit of being easily suited to HTS systems and are frequently used by pharmaceutical companies to investigate the neurotoxicity of drugs and their possible side effects. HTS in primary neurons combines the advantages of HTS with the biological importance of being able to capture critical cell events or homeostatic conditions that are present in disease states. Despite being difficult to transfect and requiring complicated culture protocols, HTS with primary neurons is still feasible, with increased biological and clinical relevance being worth the extra effort and expense [14]. For example, Sharma et al. (2013) developed a HTS method for primary neurons which is applicable for large-scale testing, ranging from compound libraries to whole-genome RNA interference (RNAi) [15].
Novel diagnostic technologies for temporal analysis of the neuronal region and consistency of the cell membrane have been developed, providing assays to track neurodegeneration over time [16]. Neurons may become defective in certain NDDs long before they die [17]. The detection of specific disease-related impairment, especially prior to associated cell death, is therefore an important step forward in the discovery of therapeutics. In the case of Amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD), for example, neurons in the nervous system’s most susceptible cell populations have been shown to become overactive years before noticeable clinical signs and neuropathology have been identified [18,19,20]. There are a number of abnormal characteristics and symptoms associated with NDDs, therefore, a major challenge remains to have a reliable screening phenotype when detecting complex disease-related signatures that can be distinct and predictive of disease and cell death.
In this review, we discuss the current challenges in validating new drug targets and developing new treatments for NDDs. Moreover, we review the increasing role of HTS in the drug discovery process focusing on existing platforms that mimic healthy and diseased states of the CNS. In addition, we identify the main strengths and limitations in their applications towards identifying new therapeutic targets and therapeutics for NDDs.
Druggable targets are scanned either virtually utilizing virtual compound structure libraries and/or by cell-based or biochemical testing of available peptide or chemical libraries via high-throughput screening (HTS). Abbreviations: Food and Drug Administration (FDA).
2. High-Throughput Screening (HTS)
2.1. Formats and Major Considerations for HTS Platforms
HTS involves in vitro, cell- or whole organism- based assays [21]. The most common readouts for biochemical assays in HTS are optical, including absorbance, fluorescence, luminescence, and scintillation. The efficiency of data production and cost per screen are the main determinants in the choice of the most suitable readout for a particular screen. However, the fluorescence-based techniques are considered as one of the primary detection methods [22]. This can mainly be attributed to the high sensitivity and diverse range of available fluorophores enabling multiplexed readouts which allow miniaturization, assay design stability, ease of handling, and the ability to simultaneously track several events in real time [23]. However, it is important to note that short wavelength excitation (particularly those under 400 nm) should be avoided during the development of functional assays in order to reduce interference from test compounds [24,25,26]. This direct screening approach has been applied to the selection of thrombin inhibitors, HIV-protease inhibitors, DNA gyrase inhibitors, etc. [27,28,29]. Quantitative kinetics of compound binding can be used to gain a higher level of understanding of binding mechanisms, as it is possible to investigate the effect of structural variations in a systematic way. Association and dissociation rates can vary independently for a specific lead series, resulting in the rapid evolution of sub nanomolar-affinity leads [30].
Data from screens can be archived and reviewed using information management systems [31] or more laboriously, in Excel spreadsheets. The data is evaluated in order to classify hits: Data points that surpass a certain specified threshold to determine a positive result. Importantly, the threshold limits can be quite subjective, but a value of three standard deviations from the mean signal of wells treated with DMSO, for example, is a fair and typical cut-off, since it offers a manageable false-positive statistical hit rate (0.15%) [32]. Alternatively, the maximum number of hits that can be processed may be increased by “cherry picking”, normally several hundred compounds can be simply picked for further evaluation. Additionally, the median rather than the mean for a single compound can be used to assess hits if the screening is done in triplicate together with the use of appropriate statistical methods [32]. This protects against the undue influence of significant outlier results, which are common in these techniques.
2.2. Main Types of HTS Assays
2.2.1. Cell-Based Assays
Using cell-based assays, whole pathways can be investigated generating numerous potential points of interest, as opposed to the analysis of particular predetermined steps as in biochemical assays. Moreover, cell-based assays may provide data that cannot be obtained from a biochemical assay, such as the existence of the pharmacological activity of the screened compound at a particular receptor or the intracellular target [33,34]. Consequently, cell-based platforms are especially promising as important tools in the study of cell growth and differentiation, in examining the influence of small molecules and cell growth conditions on cell function and physiology, and also in understanding signaling pathways in mammalian cells. They have also proven to be particularly valuable in studying complex conditions such as CNS injury and NDDs, as many factors can contribute to a specific cellular response [35].
HTS is frequently accomplished using scaled down cell-based methods. Cell-based tests enable chemical libraries to be tested for molecules that exhibit a diversity of biological activities. In the pharmaceutical industry, cellular microarrays utilizing 96- or 384-well microtiter plates with 2D cell monolayer cultures are commonly used [35]. Cellular microarrays consist of a solid framework wherein minute volumes of diverse biomolecules and cells can be presented, permitting the multiplexed examination of living cells and, subsequently, the assessment of cellular reactions [33,36]. Miscellaneous molecules such as antibodies, polymers and small molecules can be arrayed using automated spotting technology or soft lithography [37]. Cellular microarrays are also used in small molecule screening [38,39]. The screening of small molecules in mammalian cell lines, such as CHO cells, could be considered as an example of utilization of such a system [40,41]. There is flexibility in choosing the readout when using a cell-based assay focused on a signaling pathway. For example, if an antibody is available, every stage in which a protein is modified (e.g., phosphorylated), translocated [42] or changed in its abundance [43,44,45] can be possible readouts [46]. Multiple NDDs have been studied both with target-based and cell-based screens, including AD [47], PD [48], bipolar disease, autism and schizophrenia [49]. A key feature of cell-based screening is that multiple targets are screened at once, the readout being the outcome of a cellular pathway or network [50].
2.2.2. Biochemical Assays
Biochemical screening utilizes a purified target protein of interest and measures the binding of ligands or the inhibition of enzymatic activity in vitro [51]. These assays are generally conducted in a competition format, in which the compound under study displaces a known ligand or substrate. These assays are typically conducted in 384-well plates, which provide a good compromise between screening volumes (20–50 µL), throughput, and the cost of more sophisticated screening equipment. The readout is typically an optical method such as absorbance, fluorescence or luminescence [52]. Buratti et al. developed a method in which the activity of a specific RNA binding protein (RBP) (TDP-43) was measured, and due to the established activity of this protein, RBP was shown to be involved in the pathology of PD, AD, and other NDDs [53]. Additionally, Crowe et al. performed a novel study, screening almost 300,000 compounds to evaluate their effect on tau protein assembly. Formation of toxic tau oligomers in the brain is one of the main observed pathologic events of AD [54]. Using HTS assays based on complementary thioflavin T fluorescence and fluorescence polarization methods, the effects of inhibitors of tau oligomerization were determined. Specifically, that aminothienopyridazines (ATPZs) caused the inhibition of fibril assembly as well as fibrillization of tau. Additionally, the normal ability of tau to stabilize microtubules was not affected and ATPZs were shown to be promising drugs to treat AD [54,55]. Scaling down of bioanalytical activities, in order to decrease production expenses, as well as simplifying transport and saving space in the laboratory has led to a focus on laboratory-on-a-chip technology. Overall, scaling down improves the efficiency of required screening [56,57]. However, this could be complicated by extensive time implications, error-recovery rates, and complex experimental design often involving an error-prone robotic operation.
In summary, biochemical assays have the advantage that all hits found are against a known target by design. However, in those situations, the often costly and tedious determination of the molecular mechanisms of action would be needed, even though the target is known. Furthermore, due to the degree to which the predicted target was initially validated in the disease phase, the therapeutic potential of an in vitro hit can still be inconsistent. Even following the determination of such mechanistic details, it is difficult to predict the behavior of such compounds in a more complex cellular environment, due to variability in cellular permeability and metabolism, toxicity, selectivity, and the potential off-target activity of the compound [58]. However, cell-based assays have the benefit of detecting compounds that affect a phenotype in a complex cellular environment, but still suffer from a poor understanding of the target and mechanism of action. In addition, these experiments are usually more expensive and difficult to conform to miniature HTS assays [9]. Figure 2 summarizes the current classification of the main HTS assays.
2.3. Economics of HTS
HTS aims to decrease the costs of drug invention [59,60]. It is necessary to address the economics of HTS for NDDs drug discovery especially with the escalating yearly costs of mental and neurological pathologies (estimated to be around USD 1 trillion [61]) including drug sales figures (Figure 3). It is remarkable to note that 40% of these total costs were attributable to the lack of productivity of the affected population due to the presence of these diseases [62]. The financial burden of these pathologies is only likely to increase as they typically have long-term consequences combined with an increasingly aging population.
It is crucial to extensively enhance our knowledge and understanding of CNS diseases in order to be able to develop effective therapies. Interestingly, despite the number of individuals in the US who experience CNS disorders being more than double than people who suffer from cardiovascular diseases (CVDs), the global market for CNS therapeutics constitutes less than a third of the global drug market for CVDs [63]. Therefore, the CNS drug market would have to increase by over 5-fold just to correspond to the global market for CVDs.
The primary explanation for this under-development of the worldwide brain drug market is that the vast majority of CNS drugs do not cross the in vivo blood-brain barrier (BBB). The BBB is a unique and highly selective vascular interface that separates the peripheral blood circulation from the neural tissue in order to maintain an optimum homeostatic microenvironment for brain function and protection [64,65]. However, biology’s proverbial double-edged sword means that the protective nature of the BBB precludes almost all large-molecule neurotherapeutics and more than 98% of all small-molecules as viable drugs [66]. In one systematic medicinal research study, over 7000 drugs were evaluated in the comprehensive medicinal chemistry (CMC) database [67] and this suggested that the CNS was affected by just 5% of these medications. In another study, only one out of every eight medicines analyzed were active in the CNS and only 1% of the total number of drugs were clinically active in the CNS for diseases [68].
The procedure involved in developing a new drug is an elaborative effort which is often a costly and lengthy process. On average, the cost of developing a new medicine is around USD 1.3 billion (2018) [69]. However, the expenditure of the research and development (R&D) departments of the major pharmaceutical companies can be as high as USD 2.87 billion (2013) to discover and test a new drug [70]. Despite these huge investments in new treatments targeting NNDs and an expanding pipeline, there have been more failures and setbacks than overall treatment successes. The failure rate of clinical trials for new treatments targeting NDDs, for example AD, is exceptionally high and usually exceeds 99% [71]. For example, during the period 2010–2015, all the clinical trials of potential medicines for treating AD failed and were terminated after reaching phase three [72]. Recently, Biogen terminated both of the phase III, ENGAGE (NCT02477800), and the EMERGE (NCT02484547) clinical trials of Aducanumab [BIIB037], since it failed to demonstrate a superior activity compared to the placebo [73,74,75]. Consequently, Biogen lost more than 5 years and USD 2.5 billion on the failed experimental drug Aducanumab [BIIB037] [74]. It is clear that R&D expenditures over time have the most impact on the overall cost of drug development [76].
3. Drugs Discovery for NDDs
3.1. Challenges in the Discovery of CNS Drugs
CNS drugs face substantial developmental obstacles relative to non-CNS drugs, largely due to a limited understanding of the complex pathophysiology of many of the diseases they aim to treat, along with difficulties in identifying and assessing acceptable clinical endpoints. Approving a new drug for CNS diseases typically faces additional burdensome regulations. For instance, a study published by the Tufts Centre for the Study of Drug Development (Tufts CSDD) highlighted a real problem that hampers the discovery and subsequent development of CNS drugs. The study found that for the period 1995–2007, success rates for CNS drugs were less than half of non-CNS drug approval rates (6.2% vs. 13.3%, respectively). Additionally, between 2000 and 2017, the time for approval, after submission of a marketing application for CNS drugs, was 38% longer than for non-CNS drugs [78]. In 2017, Gribkoff and Kaczmarek analyzed the approval period and approval rates of clinical projects investigating 274 CNS and 1168 non-CNS drugs, in which 42 CNS and 345 non-CNS compounds finally got approved by the United States Food and Drug Administration (USFDA) [79]. Furthermore, a new Tufts CSDD report also revealed that the total time required for the development process was 20% higher for CNS drugs, and that the number of CNS drugs given the FDA priority review was considerably lower compared to the non-CNS drugs [80].
As a result, CNS drug research and development projects have been exposed to major layoffs and eliminations over the last decade. Although there has been recent revival of interest in CNS drug discovery, past shifts in the priorities of the pharmaceutical and biotech industries represent the well-documented reality of CNS-drug discovery projects. CNS drugs in general have higher failure rates than non-CNS drugs, both preclinically and clinically, and in certain cases, such as for the main NDD disorders, the clinical failure rate for disease-modifying medications has been 100% [37]. Compared to non-CNS drugs, the development periods for CNS drugs are slightly longer for those drugs which are approved, and post-development regulatory approval is also longer [37,60].
Although the last few decades have witnessed major developments in our understanding of basic neuroscience, such as neuropharmacology, most CNS pharmaceutical treatments are distinguished not by the treatment of the cause but rather of symptoms. For example, most pain drugs (used in the CNS disease treatment) minimize the discomfort, but do not permanently influence the cause of pain. This is often acceptable for acute pain, especially when the cause is self-limiting, but when the drug is withdrawn chronic and neuropathic pain often returns. The treatment of symptoms, even if followed by severe side effects, can be very effective in psychiatric conditions, but when the drug is stopped, the symptoms usually return without a decrease in severity [81]. The general degeneration (death) of neurons in AD or the more localized deaths of particular central cell populations in PD and ALS, contribute to increased impairment and eventual death in people. Currently, all of the approved treatments for these chronic NDDs are palliative and symptomatic therapies.
3.2. The Need for HTS in the Discovery of Drugs for NDDs
NDD, also known as “protein-misfolding disorders”, are a heterogeneous group of diseases characterized by extensive neuronal loss, cellular toxicity, and cell proteostatic impairment. Extensive neuropathological, biochemical, and molecular genetic studies indicate that the accumulation of proteins with altered physical and chemical characteristics is a fundamental phenomenon in many forms of NDDs in the human brain, as well as in peripheral organs [82,83].
The precise aetiology of the majority of NDDs is highly complicated and not fully understood. Studies performed in the last few decades have shown that abnormal protein folding and deposition is a common characteristic within the different types of NDDs (see Figure 4). For instance, the tau protein, FUS (fused in liposarcoma)/FET protein (FUS/FET), TAR DNA-binding protein 43 (TDP-43), and alpha-synuclein protein (α-syn) can accumulate intracellularly in the nervous system. Whereas, amyloid beta protein (Aβ) or prion protein (PrP) are examples of proteins that can accumulate extracellularly and also lead to NDDs [84,85]. AD is characterized by the presence of hyperphosphorylated and misfolded intraneuronal aggregates of tau protein, and by the extracellular deposition of amyloid plaques. Lewy body (LB)-associated diseases, which include PD and Lewy body dementia (LBD), display intraneuronal cytoplasmic inclusions. Whereas the sporadic, adult-onset degenerative motion disorder of unknown aetiology, known as multiple system atrophy (MSA) is characterized by a pathological aggregation of toxic forms of α-syn within oligodendrocytes and neurons. Deposition of neuronal tau is an important feature of AD, frontotemporal lobar degeneration (FTLD), primary age-related tauopathy (PART), neurofibrillary tangle (NFT)-dementia, and pick disease (PiD). Argyrophilic grain disease (AGD), progressive supra-nuclear palsy (PSP), and cortico-basal degeneration (CBD) all show both neuronal and glial tau aggregates, whereas globular glial tauopathies (GGT) are characterized by glial tau disorders [86,87]. Moreover, recent advances have demonstrated that glial cells (including astrocytes, oligodendrocytes, and microglia) are involved in mediating the pathophysiology of various CNS disorders including NDDs by activating neuroinflammation and disrupting the BBB function [88], thereby affecting brain water homeostasis [44] and impairing brain energy metabolism [89], all of which ultimately contribute to neuronal death and neurodegeneration (Figure 4).
The discovery and development of any new drug relies heavily on a detailed understanding of the underlying mechanisms of disease and a successful progression from the identification of candidates to the design of clinical trials [92]. However, our current knowledge and understanding of the precise aetiology of the majority of the NDDs is still incomplete. Even animal models recapitulate only limited aspects of each disease. The extent to which they can model human diseases involving complex and poorly defined factors is still limited and unclear due to differences in anatomy and physiology and hence, pathophysiological responses involved in the disease process [93,94,95]. This might partially explain the high failure of a large number of lead compounds during the in vivo part of clinical trials [96,97]. Lead compounds with promising safety and efficacy profiles can still fail during in vivo stages due to various physiochemical properties, for example, failing to cross the BBB due to poor lipophilicity, charge, hydrodynamic radius, and/or molecular weight. Therefore, it is not surprising that an extremely small number of drug candidates, which proceed from preclinical and animal studies to phase I clinical trials, have eventually made it to the bedside [98].
Only a small class of drugs actually cross the BBB which includes small molecules with high lipid solubility and a low molecular weight (Mw) of <400–500 Daltons (Da). However, There are a few brain diseases that reliably react to this category of small molecules, including depression, affective disorders, chronic pain, and epilepsy [67,99]. The classic experiment of intravenous injection of [14C]-histamine into mice has demonstrated the rate-limiting function of the BBB. Histamine is a small molecule of just 111 Da, but does not cross the BBB due to the presence of many hydrogen-bond-forming functional groups. In addition, BBB penetration is known to be inversely related to the number of hydrogen bonds (typically <8 hydrogen bonds to be able to cross BBB) [100]. Nevertheless, the vast majority of CNS drugs that evolved from HTS are either water soluble with a high degree of hydrogen bonding or have a Mw of more than 500 Da. Applying HTS to the discovery of CNS drugs has led to an increase in the molecular weight of drugs and a decrease in the lipid solubility of drugs. Without HTS, large molecular weight medicines would not have been developed for the CNS due to the BBB selectivity. In fact, potential large-molecular weight drugs that are found to be effective in the brain may still be neglected in favor of a quest for peptidomimetic small molecules [66]. Except for some endogenous ligands which are already small molecules, no small molecule peptidomimetics have been discovered to date that are able to transport through the BBB [66].
It is widely accepted that toxic misfolded proteins potentially underlie many NDDs. However, individual targets that regulate these proteins and their detrimental downstream effects are still not fully understood nor established. Phenotypic screening is an objective approach for identifying new targets and therapeutic molecules spanning a wide range of models from primitive organisms such as S. cerevisiae to more pathophysiologically relevant patient-derived cellular models.
The HTS of small molecules enables a rapid analysis of the effects of thousands or even millions of small molecules. Hence, it could be highly rewarding to exploit HTS in the field of drug discovery for NDDs. For example, AD is high on the list of leading causes of death in the United States and worldwide and therefore, there is a significant global market for disease-modifying medications for AD [101]. The number of people afflicted by AD is expected to increase dramatically as the “baby boom” generation ages and medical advances allow more people to live longer. For instance, in 2015, it was estimated that there is a new case of AD every 65 s in the US. However, by 2050, a new case of AD is predicted to occur every 33 s, or approximately 1,000,000 new cases every year [101,102]. Despite the presence of a number of FDA approved drugs to treat AD, such as galantamine, memantine, donepezil, tacrine, and rivastigmine, these drugs provide only symptomatic control. Lowering the AD prevalence rate and decelerating its progression will require new drugs that address the underlying pathophysiology of AD at a molecular level [93]. Therefore, drug discovery in the arena of NDDs will hugely benefit from efficient, rapid, and cost-effective approaches such as HTS to accelerate the discovery of potential new drugs for the increasing cases of NDD globally.
3.3. Modelling of NDDs for HTS
The ethical and financial concerns, time, and labor-intensive complexity of animal trials together with the obstacles associated with amending these models to the requirements of HTS have curtailed the wide use of such models for preliminary drug screening assays. Furthermore, the reported failure of many clinical trials addressing NDDs has raised doubts on the relevance of animal disease models to humans and implied the need for superior research tools [71,103,104,105,106]. Nonetheless, a number of in vivo models, such as Zebrafish, Drosophila melanogaster, and Caenorhabditis elegans, have been successfully used in modelling NDDs.
Zebrafish (Danio rerio) is a commonly used in vivo model for different NDDs [107,108]. Owing to their rapid development, small size, susceptibility to genetic manipulation, large numbers of offspring, and transparency during development, zebrafish are a useful model for advanced imaging [109,110,111]. Therefore, it has become an increasingly important in vivo model (over the traditional use of mammals) for HTS and effective in the determination of new compound safety and efficacy [112,113]. Zebrafish, however, have some inherent limitations when it comes to NDD modelling. For example, their CNS undergoes continuous growth and life-long development of new neurons, and displays remarkable potential for axon and neuron regeneration following focal lesion. This potential for continued development and repair may adversely affect their potential use as a model for certain aspects of neurodegeneration in humans [114,115,116].
Similarly, C. elegans offer an effective in vivo model for HTS [117,118,119]. The major advantages of C. elegans are their rapid growth rate and their cost-effectiveness as a model for the detection of compounds that have a protective effect(s) against the harmful impacts of the accumulated misfolded proteins in the neurons of patients suffering from NDDs such as ALS [120], Huntington’s disease (HD) [121], PD [122], and AD [123,124]. Another example is Drosophila melanogaster [125,126], which has similar advantages offered by zebrafish and C. elegans such as the rapid growth rate, amenability for genetic modifications, and successful application as a model of NDDs in HTS [127,128,129]. However, all of these models suffer from a common major drawback, the limited ability to effectively mimic the complex pathophysiological environment of NDDs in humans. For instance, all the above-mentioned models have a short lifespan which may lessen their relevancy in model lineage-related diseases in humans [130]. Moreover, C. elegans have a simplistic body plan, lack several defined tissues/organs such as blood, brain, and internal organs, and are evolutionarily distant from humans [131]. Moreover, Drosophila melanogaster do not have an adrenergic system [132]. Therefore, developing novel, and more physiologically relevant in vitro models, can bridge the gap between existing pre-clinical and human studies.
Human embryonic stem cells (ESCs) and subsequently human induced pluripotent stem cells (iPSCs) have emerged as powerful tools due their ability to be reprogrammed into several distinctive tissue-specific cell types making them an ideal model for connecting phenotype to genotype [133,134,135]. An increasing line of evidence suggests the successful implementation of iPSC-based models in HTS assays for discovering drug candidates for NDDs [136]. The introduction of CRISPR-Cas9 technology has revolutionized genome-editing through providing a more effective, cheaper, and faster technique than the earlier methods [137]. It has facilitated the generation of cell-based specific assays for various NDDs using genetically modified (knock-in and knockout) iPSCs [132,135,136]. Moreover, CRISPR-Cas9 has opened the door for developing humanized animal models of NDDs. Currently, humanized animal models for PD [138], AD [139], ALS [140], and HD [141] have been successfully generated. These models have become favored in contemporary studies to test promising drug candidates which have successfully passed the in vitro validation [142].
Advanced 3D culture models using hydrogels, extracellular matrix (ECM) scaffolds, spheroids, organoids or organ-on-a-chip models have been developed to emulate the physiological environment and functionality of human organs which can be lost in traditional 2D models [143,144,145]. 3D culture models of the brain, BBB, or brain spinal cord barrier (BSCB) have been recently validated for various NDDs [146]. These models promote multicellular tissue-like formation with distinctive cell–cell and cell–matrix interactions required for near physiological functionality [147]. Three-dimensional cell culture models allow for a more precise prediction of the effects of potential drugs by closely recapitulating essential facets of the brain environment, mimicking neuronal and glial cell interactions, and integrating the effect of physiological blood flow, unidirectionality and access to oxygen and nutrients [148]. One of the major exciting steps in HTS is the development and implementation of the organ-on-a-chip model. The organ-on-a-chip model is a miniaturized microfluidic perfusion tool that enables the in vitro culture of primary cells or tissues for an extended duration in a format which is applicable for high throughput research. These models not only preserve the cell-cell interactions and capture key structural and functional aspects of organs, but they also enable the use of very limited quantities of investigational drugs at a nano- to micro-liter scale [149]. The use of such microfluidic perfusion mechanisms in organ-on-a-chip models provides improved homeostatic activity through mimicking the flow of blood which not only provides nutrients, but also removes catabolic metabolites and waste products [150,151,152]. Therefore, brain- and microvessel-on-a-chip [65,153] and human organoids [154] have recently emerged as powerful tools for modelling the pathophysiology of various NDDs such as AD, PD, and HD since they enable a wide range of research applications, spanning the evaluation of disease progression, novel drug development, screening and non-invasive real-time monitoring of drug action [155,156,157].
4. Current Challenges and Future Perspectives
One of the major challenges facing the HTS in the discovery of promising “hits” for treating the NDDs is the limited capacity of the use of models to perfectly recapitulate the pathophysiological milieu of NDDs. The CNS is one of our body’s most heavily cellularized tissues, and the extracellular matrix, which occupies 20% of the CNS space, is an extremely significant additional layer of complexity. Therefore, current endeavors should indeed be targeted at developing new models and novel screening techniques that better recapitulate the in vivo physiologies [158]. The brain is the most sophisticated organ in the human body, summarized by theoretical physicist Michio Kaku who said “the human brain has 100 billion neurons, each neuron connected to 10,000 other neurons. Sitting on your shoulders is the most complicated object in the known universe” [159]. It is this complexity that means creating a model completely mimicking the physiological/pathophysiological conditions in the brain is not an easy nor straight forward task. However, recent breakthroughs in harnessing the advantages of the iPSCs in combination with the advancement in microfluidic systems and 3D culture models could lead to constructing brain-on-chip models that have a better ability in recapitulating pathophysiological condition(s) most closely related to those in NDDs patients [160]. For instance, currently, most in vitro models of NDDs are comprised largely from neuronal cells, therefore, their accuracy, and also complexity, would be increased through adding glial cells such as astrocytes, microglia, pericytes, and oligodendrocytes to better mimic the molecular and structural complexity [66,161,162,163]. Even after drug administration, the BBB can prevent the passage of over 98% of small molecule drugs and other therapies to the brain [66,164]. In addition, the BBB impairment is known to diminish its ability to prevent the peripheral immune cells from infiltrating the brain, so it is implicated in exacerbating the condition of patients with NDDs [66] such as AD and PD [165,166,167,168,169,170,171,172]. Therefore, future 3D models for NDDs should incorporate the BBB to more precisely model pathophysiological conditions and promote the targeted delivery of therapies, whilst lowering the potential for serious side-effects [151,173].
HTS is indispensable in the field of drug discovery for NDDs. However, the capability of HTS is not only restricted by the availability of human-relevant NDD models, but also limited by the quality and size of the library of the compounds screened in the HTS. Therefore, expanding the number of small molecules available for HTS increases the odds of discovering efficient disease-modifying drugs for the NDDs. Dynamic combinatorial chemistry (DCC) advancement, the introduction of cheminformatics to the pharmaceutical industry, along with the widespread use of artificial intelligence (AI) in the arena of drug discovery are expected to generate a huge number of compounds that may lead to more targeted drugs with activity in preliminary HTS assays [174,175,176].
Finally, in addition to the technical challenges and obstacles discussed previously, the elevated cost associated with the process of discovering a novel drug is one of the major factors that push the pharmaceutical industry away from the race to find novel drugs for NDDs. Interestingly, the recent advancements in exploiting AI and machine learning in pharmaceutics has shown enormous potential in making the process of novel drugs discovery cheaper and more effective [175]. Recently, a number of pharmaceutical companies have started to harness the power of AI through the development of algorithms in the quest for drug-structure prototypes within published research papers and curated databases. For example, Benevolent Bio Company (New York, NY, USA) is presently exploiting AI for discovering new ways to treat ALS. Researchers at Benevolent Bio identified 100 potential compounds for treating ALS, but the AI intervention meant that only five of these potential compounds were tested in patient derived neuronal cells. Researchers discovered that one of the five tested compounds had prominent activity in slowing the neurological symptoms of ALS in a mice model [177]. Therefore, with the aid of AI, the process of HTS assays for discovering a novel disease-modifying drug that targets NDDs is expected to be a less expensive and faster process in the very near future.
HTS informatics systems continue to embrace these new innovations and increasingly help extract HTS data more efficiently from the initial assays and subsequent platform analyses. In fact, researchers have also developed platforms to mimic multiorgan interactions that are not present in conventional tissue culture systems, using microfluidics with a co-culture system with the aim of studying the pharmacokinetics of drugs [178] or cell-based drug metabolism [179]. Although not yet applied to the CNS, “quasi-all-body” model systems may reveal novel key information in this field. Both fully automated robotic systems and dedicated workstations can be used in the automation of HTS units. There is an ongoing conversation around the most reliable and cost-effective automated HTS devices [180]. The selection between full robotic systems and assay specific workstations depends on multiple factors such as budget, shift patterns, and the available workforce.
In conclusion, NDDs are set to become a modern “silent epidemic” placing a major healthcare burden on countries with aging populations. Emerging advances in HTS combined with major developments in disease modelling and computational tools have become fundamental in tackling this unmet clinical demand and will help towards achieving more personalized treatments and effective precision medicine in the foreseeable future.
Author Contributions
H.A. and R.N.A.-Z. wrote the first draft of the manuscript together with M.M.S.; R.N.A.-Z. and M.M.S. composed the figures; M.M.S. and M.T.C. planned, edited, and supervised the writing and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Ilja Kusters, Harvard University, for the constructive discussions regarding the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A. Strategies for discovering drugs from previously unexplored natural products. Drug Discov. Today 2000, 5, 294–300. [Google Scholar] [CrossRef]
- Al-Ali, H. The evolution of drug discovery: From phenotypes to targets, and back. MedChemComm 2016, 7, 788–798. [Google Scholar] [CrossRef]
- Johnson, E.O.; Hung, D.T. A Point of Inflection and Reflection on Systems Chemical Biology. ACS Chem. Biol. 2019, 14, 2497–2511. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.G.; Pratt, M.R. Click Chemistry in Proteomic Investigations. Cell 2020, 180, 605–632. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Zalewska, A.; Gerreth, A.K. Salivary Redox Biomarkers in Selected Neurodegenerative Diseases. J. Clin. Med. 2020, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Rabanel, J.-M.; Perrotte, M.; Ramassamy, C. Nanotechnology at the Rescue of Neurodegenerative Diseases: Tools for Early Diagnostic. In Nanobiotechnology in Neurodegenerative Diseases; Springer International Publishing: New York, NY, USA, 2019; pp. 19–48. [Google Scholar]
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current Therapeutic Molecules and Targets in Neurodegenerative Diseases Based on in silico Drug Design. Curr. Neuropharmacol. 2018, 16, 649–663. [Google Scholar] [CrossRef]
- Varma, H.; Lo, D.C.; Stockwell, B.R. High Throughput Screening for Neurodegeneration and Complex Disease Phenotypes. Comb. Chem. High Throughput Screen. 2008, 11, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Seidenbecher, C.I. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef]
- Galluzzi, L.; Joza, N.; Tasdemir, E.; Maiuri, M.C.; Hengartner, M.O.; Abrams, J.M.; Tavernarakis, N.; Penninger, J.M.; Madeo, F.; Kroemer, G. No death without life: Vital functions of apoptotic effectors. Cell Death Differ. 2008, 15, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Daub, A.; Sharma, P.; Finkbeiner, S. High-content screening of primary neurons: Ready for prime time. Curr. Opin. Neurobiol. 2009, 19, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Ando, D.; Daub, A.; Kaye, J.A.; Finkbeiner, S. High-Throughput Screening in Primary Neurons. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 506, pp. 331–360. [Google Scholar]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Okano, H. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nat. Cell Biol. 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Shibuya, K.; Sato, Y.; Misawa, S.; Nasu, S.; Sekiguchi, Y.; Mitsuma, S.; Isose, S.; Fujimaki, Y.; Ohmori, S.; et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 734–738. [Google Scholar] [CrossRef] [Green Version]
- Kanai, K.; Kuwabara, S.; Misawa, S.; Tamura, N.; Ogawara, K.; Nakata, M.; Sawai, S.; Hattori, T.; Bostock, H. Altered axonal excitability properties in amyotrophic lateral sclerosis: Impaired potassium channel function related to disease stage. Brain 2006, 129, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Tang, I.C.; Wang, J.; Yang, S.T. Cell-based assays in high-throughput screening for drug discovery. Int. J. Biotechnol. Wellness Ind. 2012, 1, 31–51. [Google Scholar]
- Eggeling, C.; Brand, L.; Ullmann, D.; Jäger, S. Highly sensitive fluorescence detection technology currently available for HTS. Drug Discov. Today 2003, 8, 632–641. [Google Scholar] [CrossRef]
- An, W.F.; Tolliday, N.J. Introduction: Cell-Based Assays for High-Throughput Screening; Springer International Publishing: New York, NY, USA, 2009; pp. 1–12. [Google Scholar]
- Kaminski, T.; Geschwindner, S. Perspectives on optical biosensor utility in small-molecule screening. Expert Opin. Drug Discov. 2017, 12, 1083–1086. [Google Scholar] [CrossRef]
- Kaminski, T.; Gunnarsson, A.; Geschwindner, S. Harnessing the Versatility of Optical Biosensors for Target-Based Small-Molecule Drug Discovery. ACS Sens. 2016, 2, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Macarrón, R.; Hertzberg, R.P. Design and Implementation of High Throughput Screening Assays. Mol. Biotechnol. 2010, 47, 270–285. [Google Scholar] [CrossRef]
- El Harrad, L.; Bourais, I.; Mohammadi, H.; Amine, A. Recent Advances in Electrochemical Biosensors Based on Enzyme Inhibition for Clinical and Pharmaceutical Applications. Sensors 2018, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlarek, D.; Vorobii, M.; Ogieglo, W.; Knoll, W.; Rodriguez-Emmenegger, C.; Dostálek, J. Compact Grating-Coupled Biosensor for the Analysis of Thrombin. ACS Sens. 2019, 4, 2109–2116. [Google Scholar] [CrossRef]
- Pourbasheer, E.; Ganjali, M.R. Recent Advances in Biosensors Based Nanostructure for Pharmaceutical Analysis. Curr. Anal. Chem. 2019, 15, 152–158. [Google Scholar] [CrossRef]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [Green Version]
- Kelley, B.P.; Lunn, M.R.; Root, D.E.; Flaherty, S.P.; Martino, A.M.; Stockwell, B.R. A Flexible Data Analysis Tool for Chemical Genetic Screens. Chem. Biol. 2004, 11, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Varma, H.; Lo, D.C.; Stockwell, B.R. High-Throughput and High-Content Screening for Huntington’s Disease Therapeutics. In Neurobiology of Huntington’s Disease: Applications to Drug Discovery; Lo, D.C., Hughes, R.E., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 121–1461. [Google Scholar]
- Cader, Z.; Graf, M.; Burcin, M.; Mandenius, C.-F.; Ross, J.A. Cell-Based Assays Using Differentiated Human Induced Pluripotent Cells; Springer International Publishing: New York, NY, USA, 2019; Volume 1994, pp. 1–14. [Google Scholar]
- Mandenius, C.-F.; Ross, J.A. Cell-Based Assays Using IPSCs for Drug Development and Testing; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Lee, D.W.; Doh, I.; Nam, D.-H. Unified 2D and 3D cell-based high-throughput screening platform using a micropillar/microwell chip. Sens. Actuators B Chem. 2016, 228, 523–528. [Google Scholar] [CrossRef]
- Kelm, J.M.; Lal-Nag, M.; Sittampalam, G.S.; Ferrer, M. Translational in vitro research: Integrating 3D drug discovery and development processes into the drug development pipeline. Drug Discov. Today 2019, 24, 26–30. [Google Scholar] [CrossRef]
- Wildey, M.J.; Boyd, H.; Dale, I.L.; Dahl, G.; Nicolaus, F.; Bowen, W.; Lindmark, H. Chapter Five—High-Throughput Screening, in Annual Reports in Medicinal Chemistry; Goodnow, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 149–195. [Google Scholar]
- Fu, J.; Na, Z.; Peng, B.; Uttamchandani, M.; Yao, S.Q. Accelerated cellular on- and off-target screening of bioactive compounds using microarrays. Org. Biomol. Chem. 2015, 14, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Nierode, G.; Kwon, P.S.; Dordick, J.S.; Kwon, S.-J. Cell-Based Assay Design for High-Content Screening of Drug Candidates. J. Microbiol. Biotechnol. 2016, 26, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Rue, S.M.; Anderson, P.W.; Gaylord, M.R.; Miller, J.J.; Glaser, S.M.; Lesley, S.A. A High-Throughput System for Transient and Stable Protein Production in Mammalian Cells. In Methods in Molecular Biology; Springer International Publishing: New York, NY, USA, 2019; Volume 2025, pp. 93–142. [Google Scholar]
- Damavandi, N.; Raigani, M.; Joudaki, A.; Davami, F.; Zeinali, S. Rapid characterization of the CHO platform cell line and identification of pseudo attP sites for PhiC31 integrase. Protein Expr. Purif. 2017, 140, 60–64. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Taylor, L.H.J.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-induced Relocalization of the Aquaporin 4 Channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)-and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; Macdonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta BBA Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Wyler, M.R.; Smith, D.H.; Cayanis, E.; Többen, U.; Aulner, N.; Mayer, T. Cell-Based Assays to Probe the ERK MAP Kinase Pathway in Endothelial Cells. In Advanced Structural Safety Studies; Springer International Publishing: New York, NY, USA, 2009; Volume 486, pp. 29–41. [Google Scholar]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Current status on Alzheimer disease molecular genetics: From past, to present, to future. Hum. Mol. Genet. 2010, 19, R4–R11. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R.; Bandmann, O. Parkinson’s disease: Insights from pathways. Hum. Mol. Genet. 2010, 19, R21–R27. [Google Scholar] [CrossRef] [Green Version]
- Haggarty, S.J.; Silva, M.C.; Cross, A.; Brandon, N.J.; Perlis, R.H. Advancing drug discovery for neuropsychiatric disorders using patient-specific stem cell models. Mol. Cell. Neurosci. 2016, 73, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, F.; Kessler, T.; Weindl, D.; Shadrin, A.; Schmiester, L.; Hache, H.; Muradyan, A.; Schütte, M.; Lim, J.-H.; Heinig, M.; et al. Efficient Parameter Estimation Enables the Prediction of Drug Response Using a Mechanistic Pan-Cancer Pathway Model. Cell Syst. 2018, 7, 567–579.e6. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y. Ligand–receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D. Fluorescence-and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Romano, M.; Baralle, F.E. TDP-43 high throughput screening analyses in neurodegeneration: Advantages and pitfalls. Mol. Cell. Neurosci. 2013, 56, 465–474. [Google Scholar] [CrossRef]
- Ballatore, C.; Brunden, K.; Crowe, A.; Huryn, D.; Lee, V.; Trojanowski, J.; Smith, A.; Huang, R.; Huang, W.; Johnson, R.; et al. Aminothienopyridazine Inhibitors of Tau Assembly. Patent WO2011037985 A8, 31 March 2011. [Google Scholar]
- Brunden, K.R.; Ballatore, C.; Crowe, A.; Smith, A.B., III; Lee, V.M.Y.; Trojanowski, J.Q. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: A focus on tau assembly inhibitors. Exp. Neurol. 2010, 223, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mello, C.P.P.; Rumsey, J.; Slaughter, V.; Hickman, J.J. A human-on-a-chip approach to tackling rare diseases. Drug Discov. Today 2019, 24, 2139–2151. [Google Scholar] [CrossRef]
- Khan, N.I.; Song, E. Lab-on-a-Chip Systems for Aptamer-Based Biosensing. Micromachines 2020, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- MaCarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Pereira, A.D.; Williams, A.J. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, J.; Gustavsson, A.; Svensson, M.Y.; Wittchen, H.; Jonsson, B.H.; on behalf of the CDBE2010 Study Group; Council, T.E.B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2011, 19, 155–162. [Google Scholar] [CrossRef]
- Nutt, D.J. The full cost and burden of disorders of the brain in Europe exposed for the first time. Eur. Neuropsychopharmacol. 2011, 21, 715–717. [Google Scholar] [CrossRef]
- Pardridge, W.M. Why is the global CNS pharmaceutical market so under-penetrated? Drug Discov. Today 2002, 7, 5–7. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; De Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Biogen. 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02484547 (accessed on 23 February 2021).
- Bloomberg. Biogen to Spend $2.5 Billion Before Alzheimer’s Drug Results, in 2015. Available online: https://www.bloomberg.com/news/articles/2015-04-27/biogen-to-spend-2-5-billion-before-alzheimer-s-drug-results (accessed on 23 February 2021).
- Biogen. 221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02477800 (accessed on 23 February 2021).
- A DiMasi, J.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J. CNS Drugs Take Longer to Develop, Have Lower Success Rates, than Other Drugs. Tufts Center for the Study of Drug Development Website. 2014. Available online: http://csdd.tufts.edu/news/complete_story/pr_ir_nov_dec_ir (accessed on 23 February 2021).
- Gribkoff, V.K.; Kaczmarek, L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Take, C.D. 20% Longer to Develop and to Approve vs. Non-CNS Drugs. Tufts CSDD Impact Rep. 2018, 20, 5–8. [Google Scholar]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. P T A Peer-Rev. J. Formul. Manag. 2014, 39, 638–645. [Google Scholar]
- Cunningham, C.; Wilcockson, D.C.; Boche, D.; Perry, V.H. Comparison of Inflammatory and Acute-Phase Responses in the Brain and Peripheral Organs of the ME7 Model of Prion Disease. J. Virol. 2005, 79, 5174–5184. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Nirmalraj, P.N.; List, J.; Battacharya, S.; Howe, G.; Xu, L.; Thompson, D.; Mayer, M. Complete aggregation pathway of amyloid β (1-40) and (1-42) resolved on an atomically clean interface. Sci. Adv. 2020, 6, eaaz6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Heegaard, N.H.H. Analysis of Protein Aggregation in Neurodegenerative Disease. Anal. Chem. 2013, 85, 4215–4227. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Morrison, B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018, 309, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, T.N.; Athar, T.; Parveen, R.; Fatima, S. A review on protein misfolding, aggregation and strategies to prevent related ailments. Int. J. Biol. Macromol. 2017, 105, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, D.; de Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Neha; Sodhi, R.K.; Jaggi, A.S.; Singh, N. Animal models of dementia and cognitive dysfunction. Life Sci. 2014, 109, 73–86. [Google Scholar] [CrossRef]
- Bracken, M.B. Why animal studies are often poor predictors of human reactions to exposure. J. R. Soc. Med. 2009, 102, 120–122. [Google Scholar] [CrossRef] [Green Version]
- Greek, R.; Menache, A. Systematic Reviews of Animal Models: Methodology versus Epistemology. Int. J. Med Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ajay, G.W.B.; Murcko, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Oldendorf, W.H.; Cancilla, P.; Frank, H.J.L. Blood-Brain Barrier: Interface between Internal Medicine and the Brain. Ann. Intern. Med. 1986, 105, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s Disease Facts and Figures. In Alzheimer’s Dementia; Wiley: New Jersey, NY, USA, 2020; pp. 391–460. [Google Scholar]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. In Alzheimer’s Dementia; Wiley: New Jersey, NY, USA, 2018. [Google Scholar]
- Rolfes, L.; Pawlitzki, M.; Pfeuffer, S.; Huntemann, N.; Wiendl, H.; Ruck, T.; Meuth, S.G. Failed, Interrupted, or Inconclusive Trials on Immunomodulatory Treatment Strategies in Multiple Sclerosis: Update 2015–2020. BioDrugs 2020, 34, 587–610. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Kivipelto, M. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Olanow, C.W.; Kieburtz, K.; Schapira, A.H. Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, S101–S110. [Google Scholar] [CrossRef]
- Da Costa, M.M.J.; Allen, C.E.; Higginbottom, A.; Ramesh, T.; Shaw, P.J.; McDermott, C.J. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis. Model. Mech. 2013, 7, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGown, A.; McDearmid, J.R.; Panagiotaki, N.; Tong, H.; Al Mashhadi, S.; Redhead, N.; Lyon, A.N.; Beattie, C.E.; Shaw, P.J.; Ramesh, T.M. Early interneuron dysfunction in ALS: Insights from a mutant sod1 zebrafish model. Ann. Neurol. 2013, 73, 246–258. [Google Scholar] [CrossRef]
- Benedetti, L.; Ghilardi, A.; Rottoli, E.; De Maglie, M.; Prosperi, L.; Perego, C.; Francolini, M.I. NaP selective inhibition reverts precocious inter-and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci. Rep. 2016, 6, 1–20. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Zhang, P.-H.; Chen, Y.-J.; Wu, C.-L.; Tsai, H.-J. Conditional Overexpression of rtn4al in Muscle of Adult Zebrafish Displays Defects Similar to Human Amyotrophic Lateral Sclerosis. Mar. Biotechnol. 2018, 21, 52–64. [Google Scholar] [CrossRef]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Bugel, S.M.; Tanguay, R.L.; Planchart, A. Zebrafish: A Marvel of High-Throughput Biology for 21st Century Toxicology. Curr. Environ. Health Rep. 2014, 1, 341–352. [Google Scholar] [CrossRef] [Green Version]
- McGown, A.; Shaw, D.P.J.; Ramesh, T. ZNStress: A high-throughput drug screening protocol for identification of compounds modulating neuronal stress in the transgenic mutant sod1G93R zebrafish model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2016, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sager, J.J.; Bai, Q.; Burton, E.A. Transgenic zebrafish models of neurodegenerative diseases. Brain Struct. Funct. 2010, 214, 285–302. [Google Scholar] [CrossRef]
- De Abreu, M.S.; Friend, A.J.; Demin, K.A.; Amstislavskaya, T.G.; Bao, W.; Kalueff, A.V. Zebrafish models: Do we have valid paradigms for depression? J. Pharmacol. Toxicol. Methods 2018, 94, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, B. Sydney Brenner on the Genetics of Caenorhabditis elegans. Genetics 2016, 204, 1–2. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Carretero, M.; Solis, G.M.; Petrascheck, M.C. elegans as Model for Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Parker, J.A. Worming forward: Amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.P. Dissecting Age Associated Disease in C. elegans. 2013. Available online: https://digital.wpi.edu/concern/student_works/4f16c444r?locale=en (accessed on 23 February 2021).
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Parkinson’s Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [Green Version]
- Van Pelt, K.M.; Truttmann, M.C. Caenorhabditis elegans as a model system for studying aging-associated neurodegenerative diseases. Transl. Med. Aging 2020, 4, 60–72. [Google Scholar] [CrossRef]
- Solana-Manrique, C.; Moltó, M.D.; Calap-Quintana, P.; Sanz, F.J.; Llorens, J.V.; Paricio, N. Drosophila as a Model System for the Identification of Pharmacological Therapies in Neurodegenerative Diseases. In Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Springer International Publishing: New York, NY, USA, 2019; pp. 433–467. [Google Scholar]
- Sanz, F.J.; Solana-Manrique, C.; Muñoz-Soriano, V.; Calap-Quintana, P.; Moltó, M.D.; Paricio, N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic. Biol. Med. 2017, 108, 683–691. [Google Scholar] [CrossRef]
- Cho, Y.; Zhao, C.L.; Lu, H. Trends in high-throughput and functional neuroimaging inCaenorhabditis elegans. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1376. [Google Scholar] [CrossRef] [PubMed]
- Gosai, S.J.; Kwak, J.H.; Luke, C.J.; Long, O.S.; King, D.E.; Kovatch, K.J.; Johnston, P.A.; Shun, T.Y.; Lazo, J.S.; Perlmutter, D.H.; et al. Automated High-Content Live Animal Drug Screening Using C. elegans Expressing the Aggregation Prone Serpin α1-antitrypsin Z. PLoS ONE 2010, 5, e15460. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Hegarty, E.; Martin, C.; Gökçe, S.K.; Ghorashian, N.; Ben-Yakar, A. Large-scale microfluidics providing high-resolution and high-throughput screening of Caenorhabditis elegans poly-glutamine aggregation model. Nat. Commun. 2016, 7, 13023. [Google Scholar] [CrossRef]
- Johnson, T.E. Advantages and disadvantages of Caenorhabditis elegans for aging research. Exp. Gerontol. 2003, 38, 1329–1332. [Google Scholar] [CrossRef]
- Tissenbaum, H.A. Using C. elegans for aging research. Invertebr. Reprod. Develop. 2015, 59 (Suppl. S1), 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, C.D. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol. Ther. 2006, 112, 677–700. [Google Scholar] [CrossRef]
- Nierode, G.J.; Perea, B.C.; McFarland, S.K.; Pascoal, J.F.; Clark, D.S.; Schaffer, D.V.; Dordick, J.S. High-Throughput Toxicity and Phenotypic Screening of 3D Human Neural Progenitor Cell Cultures on a Microarray Chip Platform. Stem Cell Rep. 2016, 7, 970–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-D.; Li, H.-Q.; Cui, M.; Dong, Q.; Yu, J.-T. Pluripotent stem cells for neurodegenerative disease modeling: An expert view on their value to drug discovery. Expert Opin. Drug Discov. 2020, 15, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Nilsson, O.; Strecker, R.; Lindvall, O.; Astedt, B.; Björklund, A. Behavioural effects of human fetal dopamine neurons grafted in a rat model of Parkinson’s disease. Exp. Brain Res. 1986, 65, 235–240. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo-Arozena, A.; Kalmar, B.; Essa, S.; Ricketts, T.; Joyce, P.; Kent, R.; Rowe, C.; Parker, A.; Gray, A.; Hafezparast, M.; et al. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis. Model. Mech. 2011, 4, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Yang, W.; Yan, S.; Guo, X.; Li, X.-J. CRISPR/Cas9: A powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol. Neurodegener. 2015, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, K.; Kosic, M.; Lam, A.; Moradipour, A.; Zhao, Y.; Radisic, M. Biomaterials and Culture Systems for Development of Organoid and Organ-on-a-Chip Models. Ann. Biomed. Eng. 2020, 48, 2002–2027. [Google Scholar] [CrossRef]
- Lee, H.-K.; Sanchez, C.V.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; Van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.M.; Frega, M. Past, Present, and Future of Neuronal Models In Vitro. In Advances in Neurobiology; Springer International Publishing: New York, NY, USA, 2019; Volume 22, pp. 3–17. [Google Scholar]
- Gagliano, O.; Elvassore, N.; Luni, C. Microfluidic technology enhances the potential of human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 683–687. [Google Scholar] [CrossRef]
- Raasch, M.; Rennert, K.; Jahn, T.; Peters, S.; Henkel, T.; Huber, O.; Schulz, I.; Becker, H.; Lorkowski, S.; Funke, H.; et al. Microfluidically supported biochip design for culture of endothelial cell layers with improved perfusion conditions. Biofabrication 2015, 7, 015013. [Google Scholar] [CrossRef]
- Oddo, A.; Peng, B.; Tong, Z.; Wei, Y.; Tong, W.Y.; Thissen, H.; Voelcker, N.H. Advances in Microfluidic Blood–Brain Barrier (BBB) Models. Trends Biotechnol. 2019, 37, 1295–1314. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Bang, S.; Jeong, S.; Choi, N.; Kim, H.N. Brain-on-a-chip: A history of development and future perspective. Biomicrofluidics 2019, 13, 051301. [Google Scholar] [CrossRef]
- Chang, Y.; Kim, J.; Park, H.; Choi, H.; Kim, J. Modelling neurodegenerative diseases with 3D brain organoids. Biol. Rev. 2020, 95, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.H. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab. Chip 2015, 15, 141–150. [Google Scholar] [CrossRef]
- Cho, H.; Hashimoto, T.; Wong, E.; Hori, Y.; Wood, L.B.; Zhao, L.; Haigis, K.M.; Hyman, B.T.; Irimia, D. Microfluidic Chemotaxis Platform for Differentiating the Roles of Soluble and Bound Amyloid-β on Microglial Accumulation. Sci. Rep. 2013, 3, srep01823. [Google Scholar] [CrossRef] [Green Version]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting Corticostriatal Network on-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Kaku, M. The Future of the Mind: The Scientific Quest to Understand, enhance, and Empower the Mind; Anchor Books; Doubleday: New York, NY, USA, 2015. [Google Scholar]
- Haston, K.M.; Finkbeiner, S. Clinical Trials in a Dish: The Potential of Pluripotent Stem Cells to Develop Therapies for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 489–510. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Yuan, X.; Jones, Z.; Griffin, K.; Zhou, Y.; Ma, T.; Li, Y. Assembly of Human Stem Cell-Derived Cortical Spheroids and Vascular Spheroids to Model 3-D Brain-like Tissues. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46–116. [Google Scholar] [CrossRef]
- Ormel, P.R.; De Sá, R.V.; Van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; Van Dijk, R.E.; Scheefhals, N.; Van Berlekom, A.B.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; A Banks, W.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- McManus, R.M.; Heneka, M.T. T cells in Alzheimer’s disease: Space invaders. Lancet Neurol. 2020, 19, 285–287. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Sette, A. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Town, T.; Tan, J.; Flavell, R.A.; Mullan, M. T-Cells in Alzheimer’s Disease. NeuroMolecular Med. 2005, 7, 255–264. [Google Scholar] [CrossRef]
- Heneka, M.T. An immune-cell signature marks the brain in Alzheimer’s disease. Nat. Cell Biol. 2020, 577, 322–323. [Google Scholar] [CrossRef] [Green Version]
- Pietronigro, E.; Zenaro, E.; Della Bianca, V.; Dusi, S.; Terrabuio, E.; Iannoto, G.; Slanzi, A.; Ghasemi, S.; Nagarajan, R.; Piacentino, G.; et al. Blockade of α4 integrins reduces leukocyte–endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Mook-Jung, I. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gao, Y.; Zhu, G.; Wu, H.; Fang, Z.; Guo, K. Microfluidic synthesis of fatty acid esters: Integration of dynamic combinatorial chemistry and scale effect. Chem. Eng. J. 2020, 381, 122721. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nat. Cell Biol. 2018, 557, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic–pharmacodynamic (PK–PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef]
- Michael, S.; Auld, D.; Klumpp, C.; Jadhav, A.; Zheng, W.; Thorne, N.; Austin, C.P.; Inglese, J.; Simeonov, A. A Robotic Platform for Quantitative High-Throughput Screening. ASSAY Drug Dev. Technol. 2008, 6, 637–657. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Steps involved in the process of drug discovery.

Figure 2.
General classification of high-throughput screening (HTS) assays.

Figure 3.
Total expenditure on central nervous system (CNS)-related drugs in 2010. Adapted from Gustavsson et al., 2011 [77].
Figure 3.
Total expenditure on central nervous system (CNS)-related drugs in 2010. Adapted from Gustavsson et al., 2011 [77].


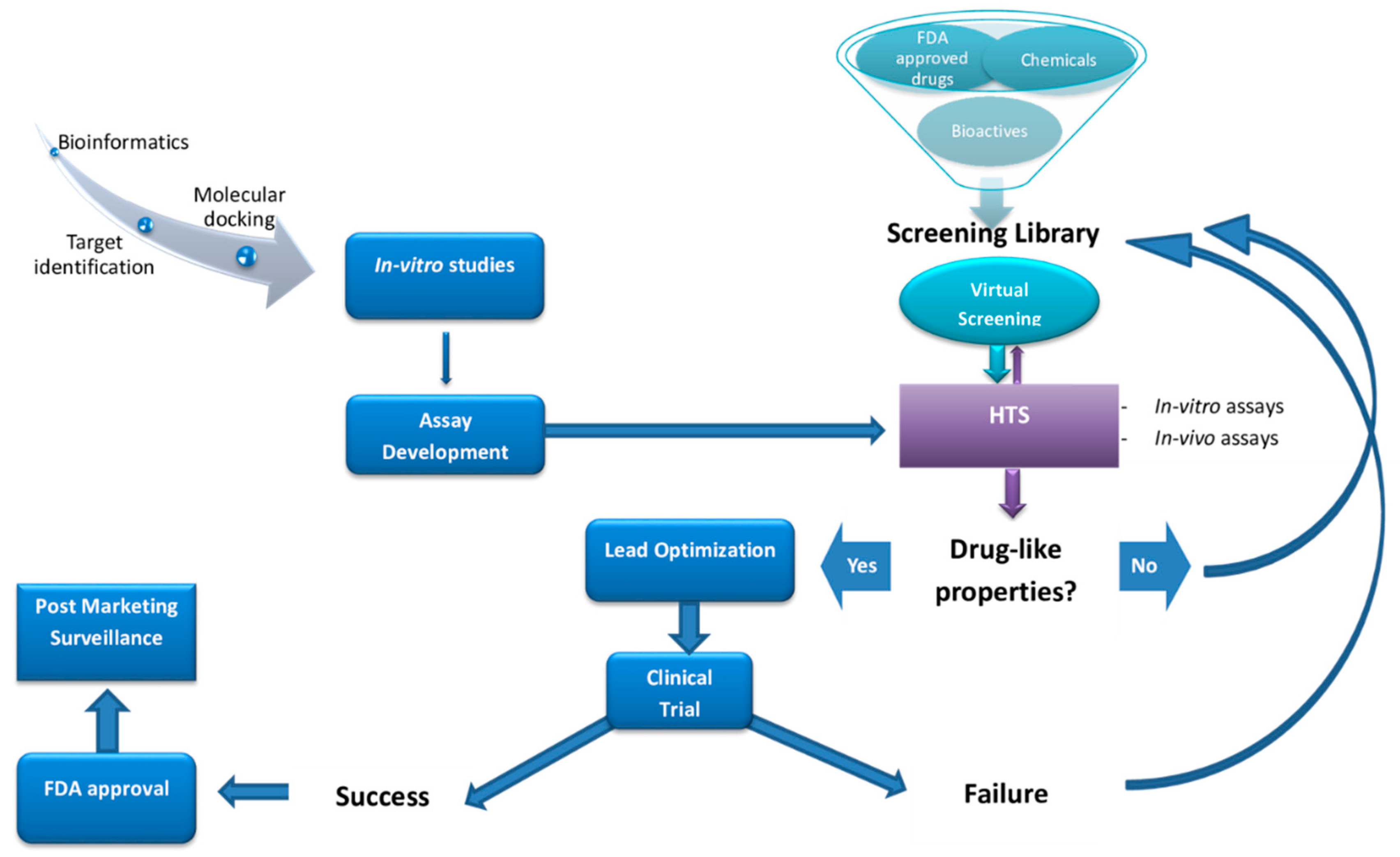{kind=link}
{kind=link}
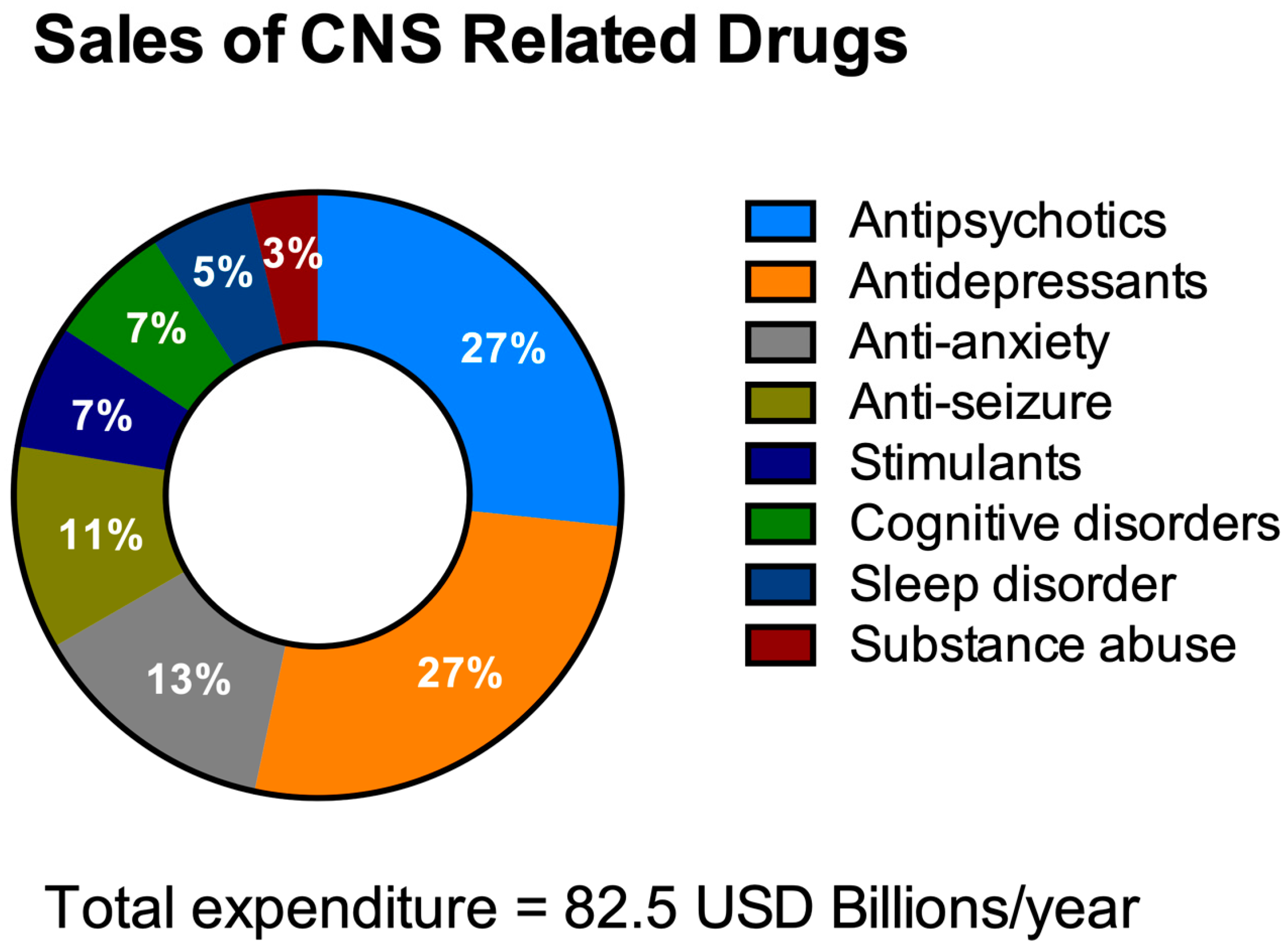{kind=link}
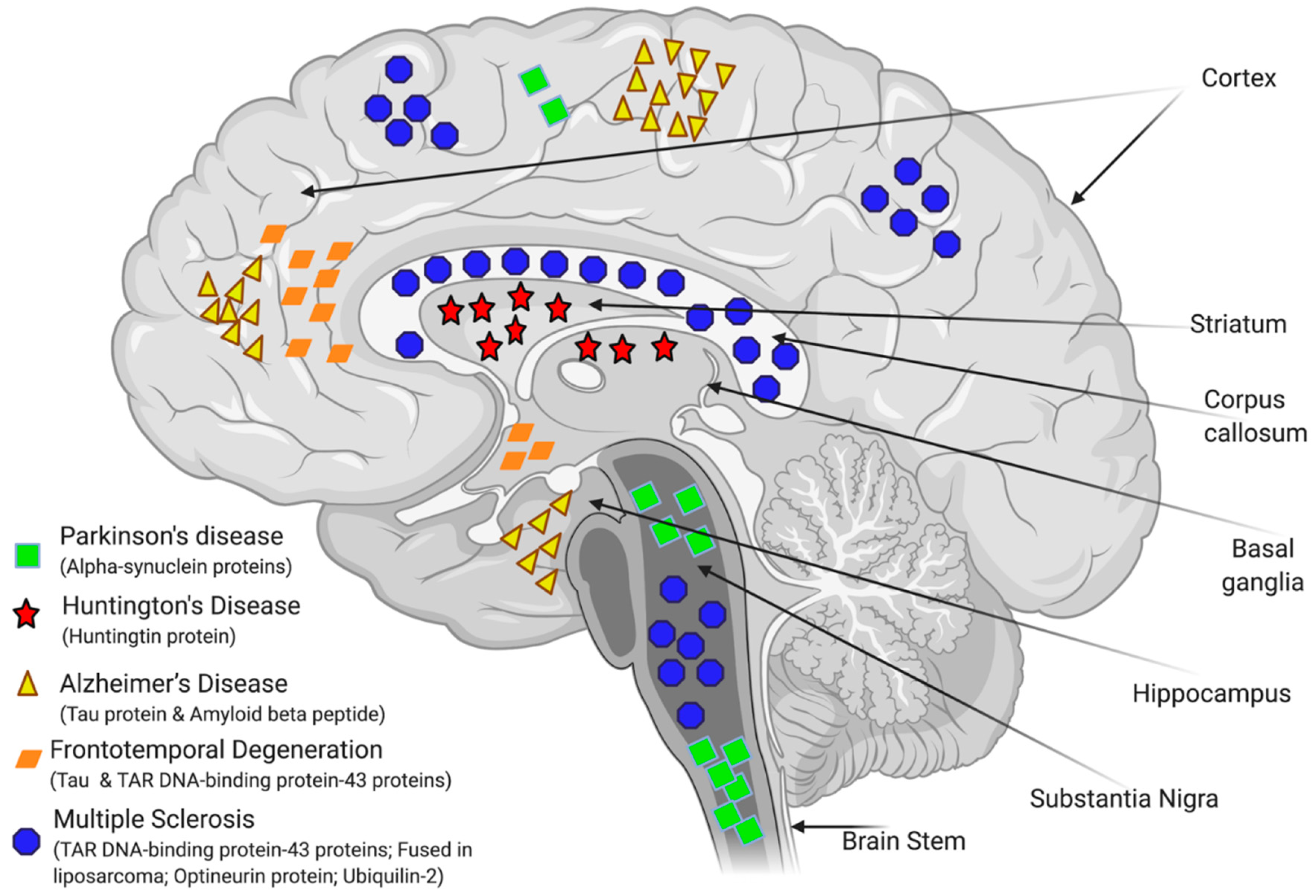{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. https://doi.org/10.3390/bioengineering8020030
AMA Style
Aldewachi H, Al-Zidan RN, Conner MT, Salman MM. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering. 2021; 8(2):30. https://doi.org/10.3390/bioengineering8020030
Chicago/Turabian StyleAldewachi, Hasan, Radhwan N. Al-Zidan, Matthew T. Conner, and Mootaz M. Salman. 2021. "High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases" Bioengineering 8, no. 2: 30. https://doi.org/10.3390/bioengineering8020030
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.