
C-Ring Oxidized Estrone Acetate Derivatives: Assessment of Antiproliferative Activities and Docking Studies

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction

2. Materials and Methods
2.1. The Chemical Synthesis and Structural Characterization
2.1.1. Synthesis of 17-Oxoestra-1,3,5(10)-trien-3-yl Acetate (Estrone Acetate, 2)
2.1.2. Synthesis of 9α-Hydroxy-17-oxoestra-1,3,5(10)-trien-3-yl Acetate (4)
2.1.3. Synthesis of 9α-Hydroxy-11β-nitrooxy-17-oxoestra-1,3,5(10)-trien-3-yl Acetate (5)
2.1.4. Synthesis of 17-Oxoestra-1,3,5(10),9(11)-trien-3-yl Acetate (6)
2.2. Bioactivity Assays
2.2.1. Cell Culture
2.2.2. Stock Solutions
2.2.3. Antiproliferative Assay
2.2.4. E-Screening Assay
2.2.5. Cell Viability Evaluation
2.2.6. Cell Cycle Distribution Study
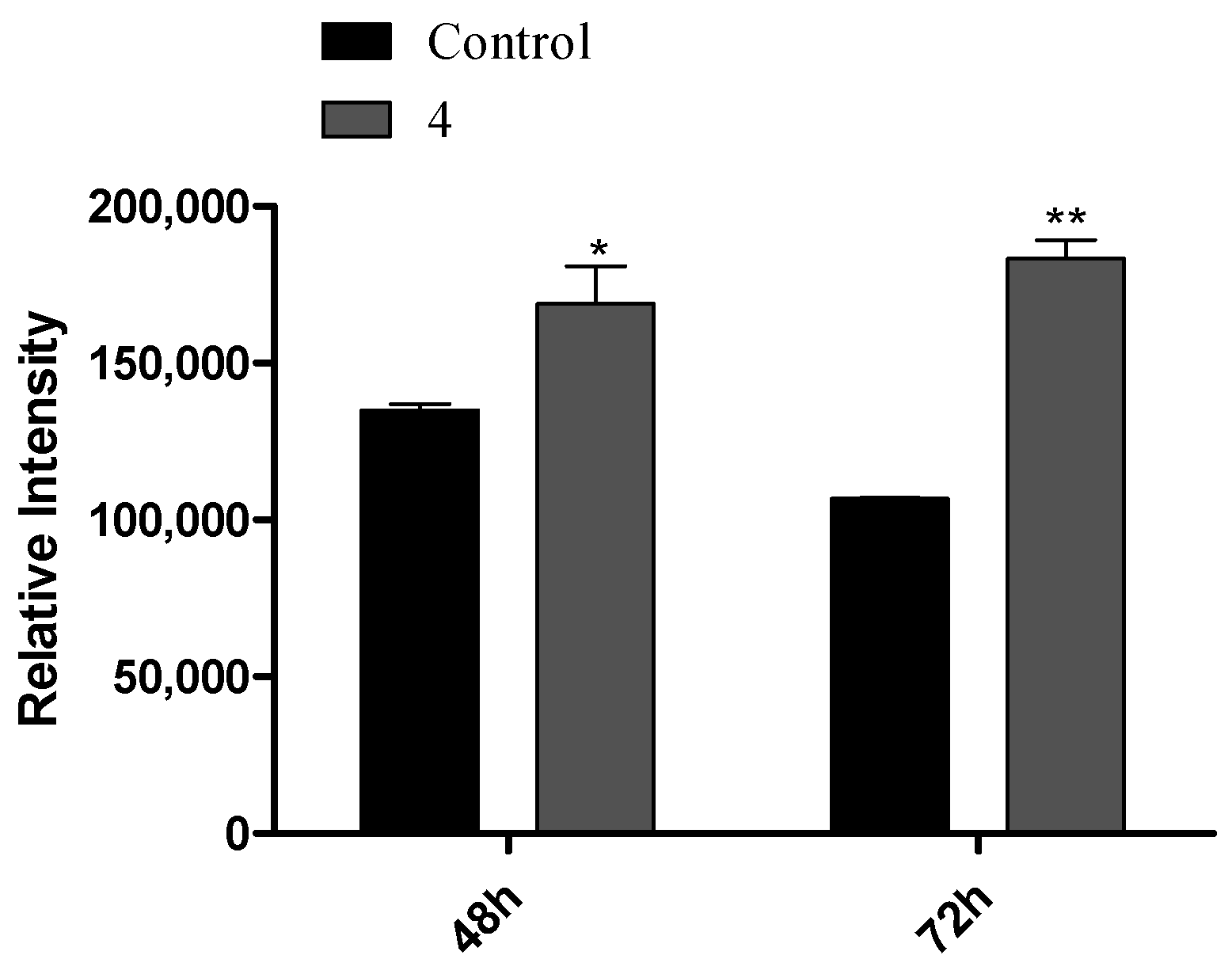
2.2.7. Cell Proliferation Analysis by the Carboxyfluorescein Succinimidyl Ester Assay
2.3. Molecular Docking
2.3.1. Preparation of Proteins for Molecular Docking
2.3.2. Preparation of Ligands
2.3.3. Grid Parameters
2.3.4. Docking Simulations
2.3.5. Validation of the Molecular Docking Performance
2.4. Statistical Analysis
3. Results
3.1. Chemistry
3.2. Cell Proliferation
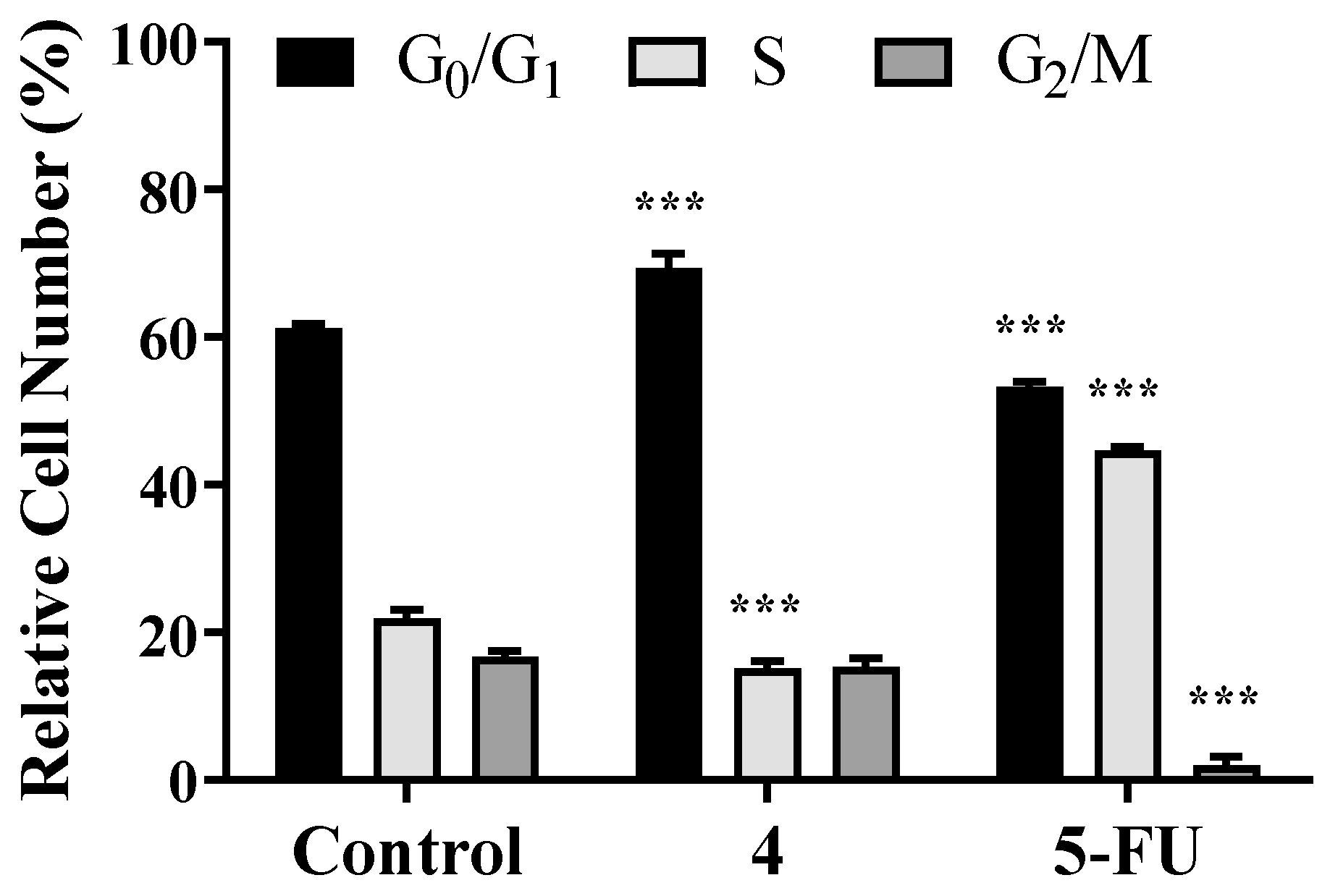
3.3. Flow Cytometry Experiments
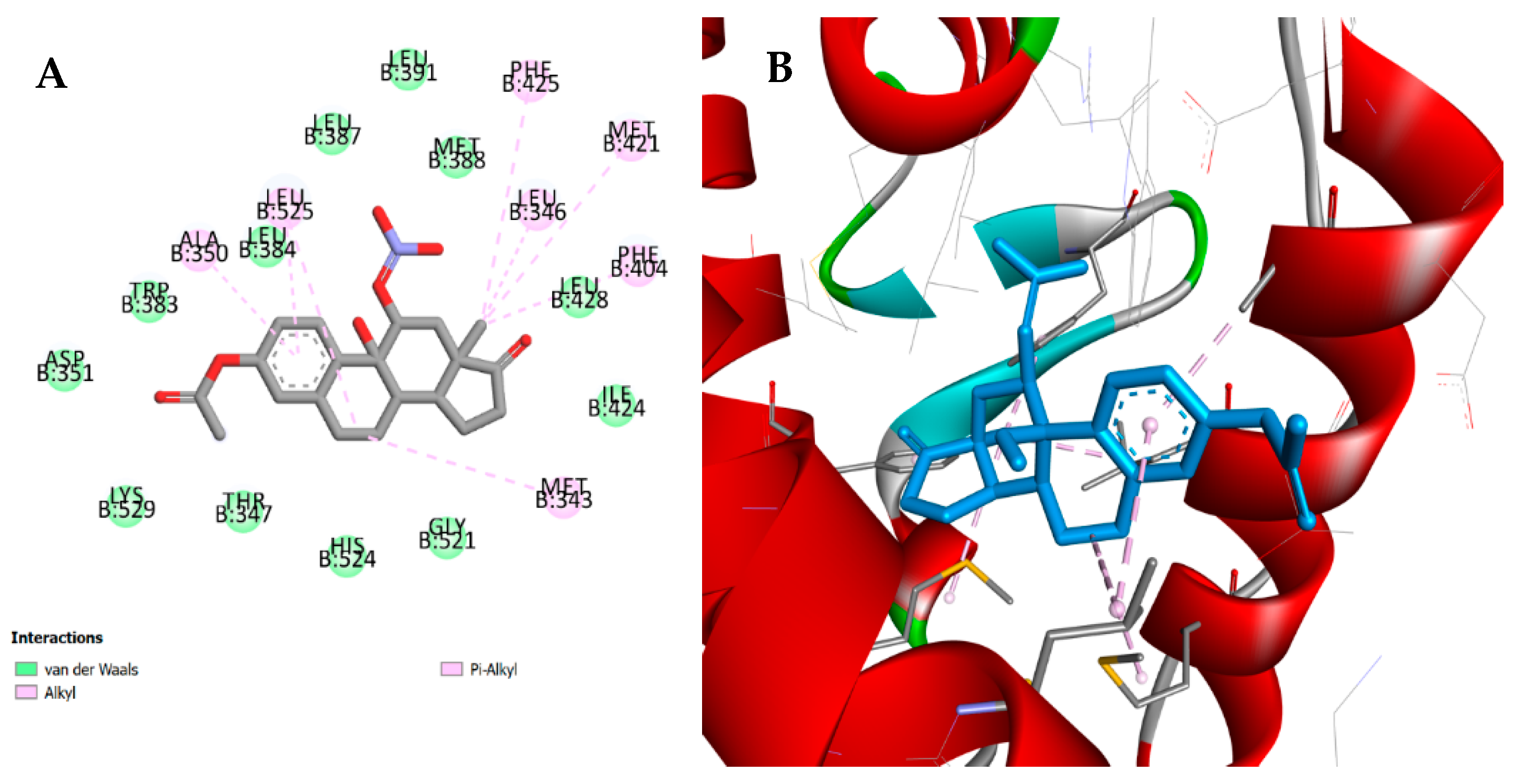
3.4. Molecular Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soronen, P.; Laiti, M.; Törn, S.; Härkönen, P.; Patrikainen, L.; Li, Y.; Pulkka, A.; Kurkela, R.; Herrala, A.; Kaija, H.; et al. Sex steroid hormone metabolism and prostate cancer. Steroid Biochem. Mol. Biol. 2004, 92, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Capper, C.P.; Rae, J.M.; Auchus, R.J. The metabolism, analysis, and targeting of steroid hormones in breast and prostate cancer. Horm. Cancer 2016, 7, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, G.; Sutton, E.L. Oral contraception. Med. Clin. N. Am. 2015, 99, 479–503. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.; Wang, Y.; Chen, M.; Zhang, J.; You, Q. Design, synthesis and biological evaluation of novel 2-methoxyestradiol analogs as dual selective estrogen receptor modulators (SERMs) and antiangiogenic agents. Eur. J. Med. Chem. 2017, 139, 390–400. [Google Scholar] [CrossRef]
- Lee, C.I.; Goodwin, A.; Wilcken, N. Fulvestrant for hormone-sensitive metastatic breast cancer (review). Cochrane Database Syst. Rev. 2017, 3, CD011093. [Google Scholar]
- Salvador, J.A.R.; Carvalho, J.F.S.; Neves, M.A.C.; Silvestre, S.M.; Leitão, A.J.; Silva, M.M.C.; Sá e Melo, M.L. Anticancer steroids: Linking natural and semi-synthetic compounds. Nat. Prod. Rep. 2013, 30, 324–374. [Google Scholar] [CrossRef]
- de Almeida Chuffa, L.G.; Lupi-Júnior, L.A.; Costa, A.B.; de Arruda Amorim, J.P.; Seiva, F.R.F. The role of sex hormones and steroid receptors on female reproductive cancers. Steroids 2017, 118, 93–108. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, E.; Fiaschi, R.; Carlson, S.K.E.; Katzenellenbogen, J.A. 11β-Substituted Estradiol Derivatives. 2. Potential Carbon-11- and Iodine-Labeled Probes for the Estrogen Receptor. J. Med. Chem. 1995, 38, 2774–2779. [Google Scholar] [CrossRef] [PubMed]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids 1997, 62, 268–303. [Google Scholar] [CrossRef]
- Claussner, A.; Nédélec, L.; Nique, F.; Philibert, D.; Teutsch, G.; Velde, P. Van 11β-Amidoalkyl Estradiols, A New Series of Pure Antiestrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 609–614. [Google Scholar] [CrossRef]
- Poirier, D.; Mérand, Y.; Labrie, C.; Labrie, F. D-ring alkylamide derivatives of estradiol: Effect on ER-binding affinity and antiestrogenic activity. Bioorg. Med. Chem. Lett. 1996, 6, 2537–2542. [Google Scholar] [CrossRef]
- Lobaccaro, C.; Pons, J.F.; Duchesne, M.J.; Auzou, G.; Pons, M.; Nique, F.; Teutsch, G.; Borgna, J.L. Steroidal affinity labels of the estrogen receptor. 3. Estradiol 11β-n-alkyl derivatives bearing a terminal electrophilic group: Antiestrogenic and cytotoxic properties. J. Med. Chem. 1997, 40, 2217–2227. [Google Scholar] [CrossRef]
- Aliau, S.; Delettre, G.; Mattras, H.; El Garrouj, D.; Nique, F.; Teutsch, G.; Borgna, J.-L. Steroidal affinity labels of the estrogen receptor α. 4. Electrophilic 11β-Aryl derivatives of estradiol. J. Med. Chem. 2000, 43, 613–628. [Google Scholar] [CrossRef]
- Hanson, R.N.; Hua, E.; Adam Hendricks, J.; Labaree, D.; Hochberg, R.B. Synthesis and evaluation of 11β-(4-Substituted phenyl) estradiol analogs: Transition from estrogen receptor agonists to antagonists. Bioorg. Med. Chem. 2012, 20, 3768–3780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-X.; Labaree, D.C.; Hochberg, R.B. Nonpolar and short side chain groups at C-11β of estradiol result in antiestrogens. J. Med. Chem. 2005, 48, 1428–1447. [Google Scholar] [CrossRef]
- Dwivedy, I.; Gupta, A.; Grover, A.; Srivastava, V.; Singh, M.M.; Ray, S. Synthesis and in vivo evaluation of 11-substituted estradiol derivatives as anti-implantation agents. Bioorg. Med. Chem. Lett. 2008, 18, 4102–4105. [Google Scholar] [CrossRef]
- Wang, P.; McInnes, C.; Zhu, B.T. Structural Characterization of the Binding Interactions of Various Endogenous Estrogen Metabolites with Human Estrogen Receptor α and β Subtypes: A Molecular Modeling Study. PLoS ONE 2013, 8, e74615. [Google Scholar] [CrossRef] [Green Version]
- Alsayari, A.; Kopel, L.; Ahmed, M.S.; Pay, A.; Carlson, T.; Halaweish, F.T. Design, synthesis, and biological evaluation of steroidal analogs as estrogenic/anti-estrogenic agents. Steroids 2017, 118, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.H.; Crowe, D.F.; Avery, M.A.; Chong, W.K.M.; Tanabe, M. 11β-Nitrate Estrane Analogues: Potent Estrogens. J. Med. Chem. 1989, 32, 2306–2310. [Google Scholar] [CrossRef] [PubMed]
- Rzheznikov, V.M.; Golubovskaya, L.E.; Minailova, O.N.; Osetrova, I.P.; Smirnova, Z.S. Steroidal Nitrates: Synthesis and Antitumor Activity of of 9α,11β-Dihydroxyestra-1,3,5(10)-triene 11-nitrates. Pharm. Chem. J. 2003, 37, 10–12. [Google Scholar] [CrossRef]
- Canário, C.; Matias, M.; Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. Δ9,11-Estrone derivatives as potential antiproliferative agents: Synthesis, in vitro biological evaluation and docking studies. C. R. Chim. 2020, 23, 201–217. [Google Scholar] [CrossRef]
- Brito, V.; Santos, A.O.; Almeida, P.; Silvestre, S. Novel 4-azaandrostenes as prostate cancer cell growth inhibitors: Synthesis, antiproliferative effects, and molecular docking studies. C. R. Chim. 2019, 22, 73–83. [Google Scholar] [CrossRef]
- Canário, C.; Silvestre, S.; Falcão, A.; Alves, G. Steroidal Oximes: Useful Compounds with Antitumor Activities. Curr. Med. Chem. 2018, 25, 660–686. [Google Scholar] [CrossRef] [PubMed]
- Canário, C.; Matias, M.; Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. New Estrone Oxime Derivatives: Synthesis, Cytotoxic Evaluation and Docking Studies. Molecules 2021, 26, 2687. [Google Scholar] [CrossRef]
- Simeón, J.L.L.; Morales, J.E.T.; Navarro, F.A.V.; Manchado, F.C.; Montoto, L.G.P. Actividad catalítica del acetato de vanadilo en la acetilación de alcoholes secundarios. Rev. CENIC Cienc. Químicas 2004, 35, 141–145. [Google Scholar]
- D’Accolti, L.; Fusco, C.; Lampignano, G.; Capitelli, F.; Curci, R. Oxidation of natural targets by dioxiranes. Part 6: On the direct regio- and site-selective oxyfunctionalization of estrone and of 5α-androstane steroid derivatives. Tetrahedron Lett. 2008, 49, 5614–5617. [Google Scholar] [CrossRef]
- Quinkert, G.; Weber, W.-D.; Schwartz, U. Process for Synthesizing Estrone or Estrone Derivatives. U.S. Patent 4,357,278, 2 November 1982. [Google Scholar]
- Bovicelli, P.; Lupattelli, P.; Mincione, E.; Prencipe, T.; Curci, R. Oxidation of Natural Targets by Dioxiranes. Oxyfunctionalization of Steroids. J. Org. Chem. 1992, 57, 2182–2184. [Google Scholar] [CrossRef]
- Stéphan, E.; Zen, R.; Authier, L.; Jaouen, G. Improved synthesis of a protected 11-oxoestrone. Steroids 1995, 60, 809–811. [Google Scholar] [CrossRef]
- Ayan, D.; Maltais, R.; Roy, J.; Poirier, D. A new nonestrogenic steroidal inhibitor of 17β-hydroxysteroid dehydrogenase type I blocks the estrogen-dependent breast cancer tumor growth induced by estrone. Mol. Cancer Ther. 2012, 11, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Guzman, F.G.; Higashiyama, T.; Pangborn, W.; Osawa, Y.; Ghosh, D. Structure of human estrone sulfatase suggests functional roles of membrane association. J. Biol. Chem. 2003, 278, 22989–22997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aka, J.A.; Mazumdar, M.; Chen, C.-Q.; Poirier, D.; Lin, S.-X. 17β-Hydroxysteroid Dehydrogenase Type 1 Stimulates Breast Cancer By Dihydrotestosterone Inactivation in Addition To Estradiol Production. Mol. Endocrinol. 2010, 24, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Carugo, O. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Murugan, R.; Scriven, E.F.V. Applications of dialkylaminopyridine (DMAP) catalysts in organic synthesis. Aldrichima Acta 2003, 36, 21–27. [Google Scholar] [CrossRef]
- Schwarz, S.; Schumacher, M.; Nanninga, A.; Weber, G.; Thieme, I.; Undeutsch, B.; Elger, W. 17β-Hydroxy-11α-(3’-sulfanylpropyl)oxy-estra-1,3,5(10)-trien-3-yl sulfamate—A novel hapten structure: Toward the development of a specific enzyme immunoassay (EIA) for estra-1,3,5(10)-triene-3-yl sulfamates. Steroids 1999, 64, 460–471. [Google Scholar] [CrossRef]
- Sykes, P.J.; Rutherford, F.J.; Laing, S.B.; Phillipps, G.H.; Turnbull, J.P. Oxidation of ring a-aromatic steroids to 9,11β-diol 11-nitrates with ceric ammonium nitrate. Tetrahedron Lett. 1971, 12, 3393–3396. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Silvestre, S.M. Bismuth-catalyzed allylic oxidation using t-butyl hydroperoxide. Tetrahedron Lett. 2005, 46, 2581–2584. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.D.; Baran, J.S. Synthesis and Conformational Stabilities of 11-Oxo-9α- and 9β-Estradiol 3-Benzyl Ether. Tetrahedron 1976, 32, 2067–2069. [Google Scholar] [CrossRef]
- Gao, H. Approaches to partial syntheses of 11-oxo steroids. A brief review. Org. Prep. Proced. Int. 1997, 29, 499–539. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bézivin, C.; Tomasi, S.; Dévéhat, F.L.-L.; Boustie, J. Cytotoxic activity of some lichen extracts on murine and human cancer cell lines. Phytomedicine 2003, 10, 499–503. [Google Scholar] [CrossRef]
- Cortés-Benítez, F.; Roy, J.; Maltais, R.; Poirier, D. Impact of androstane A- and D-ring inversion on 17β-hydroxysteroid dehydrogenase type 3 inhibitory activity, androgenic effect and metabolic stability. Bioorg. Med. Chem. 2017, 25, 2065–2073. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, L.; Hernández-Linares, M.G.; Escobar, M.L.; López-Muñoz, H.; Zenteno, E.; Fernández-Herrera, M.A.; Guerrero-Luna, G.; Carrasco-Carballo, A.; Sandoval-Ramírez, J. Antiproliferative, Cytotoxic and Apoptotic Activity of Steroidal Oximes in Cervicouterine Cell Lines. Molecules 2016, 21, 1533. [Google Scholar] [CrossRef] [Green Version]
- Makar, S.; Saha, T.; Swetha, R.; Gutti, G.; Kumar, A.; Singh, S.K. Rational Approaches of Drug Design for the Development of Selective Estrogen Receptor Modulators (SERMs), Implicated in Breast Cancer. Bioorg. Chem. 2020, 94, 103380. [Google Scholar] [CrossRef]
- Miki, Y.; Iwabuchi, E.; Ono, K.; Sasano, H.; Ito, K. Exploring Protein-Protein Interaction in the Study of Hormone-Dependent Cancers. Int. J. Mol. Sci. 2018, 19, 3173. [Google Scholar] [CrossRef] [Green Version]
- Cornel, K.M.C.; Krakstad, C.; Delvoux, B.; Xanthoulea, S.; Jori, B.; Bongers, M.Y.; Konings, G.F.J.; Kooreman, L.F.S.; Kruitwagen, R.F.; Salvesen, H.B.; et al. High mRNA levels of 17β-hydroxysteroid dehydrogenase type 1 correlate with poor prognosis in endometrial cancer. Mol. Cell. Endocrinol. 2017, 442, 51–57. [Google Scholar] [CrossRef]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef]
- Day, J.M.; Tutill, H.J.; Purohit, A.; Reed, M.J. Design and validation of specific inhibitors of 17β-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 2008, 15, 665–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daśko, M.; Demkowicz, S.; Biernacki, K.; Ciupak, O.; Kozak, W.; Masłyk, M.; Rachon, J. Recent progress in the development of steroid sulphatase inhibitors–examples of the novel and most promising compounds from the last decade. J. Enzym. Inhib. Med. Chem. 2020, 35, 1163–1184. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, L.E.; Rzheznikov, V.M. Oxidation of estra-1,3,5(10)-triene-3,11α,17β-triol triacetate with ceric ammonium nitrate. Russ. J. Org. Chem. 2007, 43, 1730–1732. [Google Scholar] [CrossRef]
- Batista, V.S.; Crabtree, R.H.; Konezny, S.J.; Luca, O.R.; Praetorius, J.M. Oxidative functionalization of benzylic C-H bonds by DDQ. New J. Chem. 2012, 36, 1141–1144. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Silvestre, S.; Moreira, V.M. Recent Developments in Oxidative Processes in Steroid Chemistry. Curr. Org. Chem. 2012, 16, 1243–1276. [Google Scholar] [CrossRef]
- Golubovskaya, L.E.; Ivanenko, T.I.; Rzheznikov, V.M. Steroidal nitrates. Part III. Synthesis and antiestrogen activity of the 11α-nitroxy analog of ethynylestradiol. Pharm. Chem. J. 2009, 43, 560–562. [Google Scholar] [CrossRef]
- Berényi, Á.; Minorics, R.; Iványi, Z.; Ocsovszki, I.; Ducza, E.; Thole, H.; Mernyák, E.; Frank, É.; Schneider, G.; Zupkó, I. Synthesis and investigation of the anticancer effects of estrone-16-oxime ethers in vitro. Steroids 2013, 78, 69–78. [Google Scholar] [CrossRef]
- Morozkina, S.N.; Shavva, A.G. Estrone Sulfatase Inhibitors as new anticancer agents. In Chemistry and Biological Activity of Steroids; IntechOpen: London, UK, 2016; pp. 1–26. [Google Scholar]
- Bader, A.; Bkhaitan, M.M.; Abdalla, A.N.; Abdallah, Q.M.A.; Ali, H.I.; Sabbah, D.A.; Albadawi, G.; Abushaikha, G.M. Design and Synthesis of 4-O-Podophyllotoxin Sulfamate Derivatives as Potential Cytotoxic Agents. Evid.-Based Complement. Altern. Med. 2021, 2021, 6672807. [Google Scholar] [CrossRef]
- Shen, G.; Wang, C.; Luo, Y.; Wang, J.; Wang, R.; Xu, W.; Zhang, Y.; Zhang, Y.; Zhang, D.; Jin, C. 2-(6-Hydroxyhexylthio)-5,8-dimethoxy-1,4-naphthoquinone Induces Apoptosis through ROS-Mediated MAPK, STAT3, and NF- κ B Signalling Pathways in Lung Cancer A549 Cells. Evid.-Based Complement. Altern. Med. 2020, 2020, 7375862. [Google Scholar] [CrossRef]
- Mirzaei, S.; Qayumov, M.; Gangi, F.; Behravan, J.; Ghodsi, R. Synthesis and biological evaluation of oxazinonaphthalene-3-one derivatives as potential anticancer agents and tubulin inhibitors. Iran. J. Basic Med. Sci. 2020, 23, 1388–1395. [Google Scholar] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Cajal, S.R.Y. P16Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lespérance, M.; Barbeau, X.; Roy, J.; Maltais, R.; Lagüe, P.; Poirier, D. Chemical synthesis of C3-oxiranyl/oxiranylmethyl-estrane derivatives targeted by molecular modeling and tested as potential inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Steroids 2018, 140, 104–113. [Google Scholar] [CrossRef]
- Woo, L.W.L.; Leblond, B.; Purohit, A.; Potter, B.V.L. Synthesis and evaluation of analogues of estrone-3-O-sulfamate as potent steroid sulfatase inhibitors. Bioorg. Med. Chem. 2012, 20, 2506–2519. [Google Scholar]
- El-Kady, D.S.; Abd Rabou, A.A.; Tantawy, M.A.; Abdel-Rahman, A.A.H.; Abdel-Megeed, A.A.S.; AbdElhalim, M.M.; Elmegeed, G.A. Synthesis and Evaluation of Novel Cholestanoheterocyclic Steroids as Anticancer Agents. Appl. Biochem. Biotechnol. 2019, 188, 635–662. [Google Scholar] [CrossRef]
- Palomino, E. Chemical modulation of activity in steroidal estrogens. Crit. Rev. Biochem. Mol. Biol. 1999, 34, 387–398. [Google Scholar] [CrossRef]
- Kerwin, J.F.; Lancaster, J.R.; Feldman, P.L. Nitric Oxide: A New Paradigm for Second Messengers. J. Med. Chem. 1995, 38, 4343–4362. [Google Scholar] [CrossRef]
- Nussbaumer, P.; Billich, A. Steroid sulfatase inhibitors. Med. Res. Rev. 2004, 24, 529–576. [Google Scholar] [CrossRef]
- Maltais, R.; Ayan, D.; Trottier, A.; Barbeau, X.; Lagüe, P.; Bouchard, J.E.; Poirier, D. Discovery of a non-estrogenic irreversible inhibitor of 17β-Hydroxysteroid dehydrogenase type 1 from 3-substituted-16β-(m-carbamoylbenzyl)-estradiol derivatives. J. Med. Chem. 2014, 57, 204–222. [Google Scholar] [CrossRef]
- Maltais, R.; Trottier, A.; Barbeau, X.; Lagüe, P.; Perreault, M.; Thériault, J.; Lin, S.; Poirier, D. Impact of structural modifications at positions 13, 16 and 17 of 16β-(m-carbamoylbenzyl)-estradiol on 17β-hydroxysteroid dehydrogenase type 1 inhibition and estrogenic activity. J. Steroid Biochem. Mol. Biol. 2016, 161, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Deluca, D.; Möller, G.; Rosinus, A.; Elger, W.; Hillisch, A.; Adamski, J. Inhibitory effects of fluorine-substituted estrogens on the activity of 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2006, 248, 218–224. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 | NHDF |
|---|---|---|---|---|---|---|
| E1 | 41.93 | ND | ND | 29.53 | 42.69 | 61.82 |
| 2 | ND | 29.24 | ND | 46.54 | ND | ND |
| 4 | ND | ND | ND | 32.04 | ND | ND |
| 5 | 5.87 | 7.40 | 5.30 | 10.91 | 14.12 | 12.14 |
| 6 | 53.59 | 44.56 | ND | 36.06 | ND | ND |
| 5-FU | 1.71 | 0.54 | 7.79 | 1.78 | 1.31 | 3.61 |
| Compounds | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 |
|---|---|---|---|---|---|
| E1 | 1.47 | ND | ND | 2.09 | 1.45 |
| 5 | 2.07 | 1.64 | 2.29 | 1.11 | 0.86 |
| 5-FU | 2.11 | 0.49 | 0.46 | 2.03 | 2.76 |
| Compounds | Lowest Energy (kcal·mol−1) | ||
|---|---|---|---|
| ERα | ST | 17β-HSD1 | |
| E1 | −10.3 | −6.2 | −8.1 |
| 2 | −7.2 | −6.2 | −8.3 |
| 4 | −7.3 | −5.9 | −8.2 |
| 5 | −7.4 | −6.9 | −8.0 |
| 6 | −7.0 | −6.3 | −8.3 |
| 17β-estradiol (E2) | −9.9 a | − | − |
| N-acetyl-D-glucosamine | − | −7.2 a | − |
| 5α-Dihydrotestosterone (DHT) | − | - | −8.3 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canário, C.; Matias, M.; Brito, V.; Pires, P.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. C-Ring Oxidized Estrone Acetate Derivatives: Assessment of Antiproliferative Activities and Docking Studies. Appl. Sci. 2022, 12, 3579. https://doi.org/10.3390/app12073579
Canário C, Matias M, Brito V, Pires P, Santos AO, Falcão A, Silvestre S, Alves G. C-Ring Oxidized Estrone Acetate Derivatives: Assessment of Antiproliferative Activities and Docking Studies. Applied Sciences. 2022; 12(7):3579. https://doi.org/10.3390/app12073579
Chicago/Turabian StyleCanário, Catarina, Mariana Matias, Vanessa Brito, Patrícia Pires, Adriana O. Santos, Amílcar Falcão, Samuel Silvestre, and Gilberto Alves. 2022. "C-Ring Oxidized Estrone Acetate Derivatives: Assessment of Antiproliferative Activities and Docking Studies" Applied Sciences 12, no. 7: 3579. https://doi.org/10.3390/app12073579