Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases


1
Department of Nephrology, Hypertension and Family Medicine, Medical University of Lodz, 90-419 Łódź, Poland
2
Department of Urology, Institute of Medical Sciences, Medical College of Rzeszow University, 35-959 Rzeszow, Poland
*
Author to whom correspondence should be addressed.
Antioxidants 2020, 9(11), 1079; https://doi.org/10.3390/antiox9111079
Submission received: 8 September 2020
/
Revised: 22 October 2020
/
Accepted: 27 October 2020
/
Published: 3 November 2020
(This article belongs to the Special Issue Oxidative Stress and Inflammation in Cardiovascular Diseases)
Abstract
:Chronic kidney disease is highly prevalent worldwide. The decline of renal function is associated with inadequate removal of a variety of uremic toxins that exert detrimental effects on cells functioning, thus affecting the cardiovascular system. The occurrence of cardiovascular aberrations in CKD is related to the impact of traditional risk factors and non-traditional CKD-associated risk factors, including anemia; inflammation; oxidative stress; the presence of some uremic toxins; and factors related to the type, frequency of dialysis and the composition of dialysis fluid. Cardiovascular diseases are the most frequent cause for the deaths of patients with all stages of renal failure. The kidney is one of the vital sources of antioxidant enzymes, therefore, the impairment of this organ is associated with decreased levels of these enzymes as well as increased levels of pro-oxidants. Uremic toxins have been shown to play a vital role in the onset of oxidative stress. Hemodialysis itself also enhances oxidative stress. Elevated oxidative stress has been demonstrated to be strictly related to kidney and cardiac damage as it aggravates kidney dysfunction and induces cardiac hypertrophy. Antioxidant therapies may prove to be beneficial since they can decrease oxidative stress, reduce uremic cardiovascular toxicity and improve survival.
1. Introduction
Chronic kidney disease (CKD) is highly prevalent worldwide (between 11 and 13%), and it is most frequent in developed countries in Europe, USA, Canada, and Australia [1,2]. CKD progresses as GFR decreases and this process results from the deterioration of kidney function, which greatly influences body homeostasis and leads to biological and clinical dysfunctions, including the disturbances in cellular energetic metabolism, protein malnutrition, change in nitrogen input/output, insulin resistance, and significant increase in the synthesis of inflammation/oxidative stress mediators [2]. Finally, it progresses to end-stage renal disease (ESRD) and ends up with the necessity for renal replacement therapy (hemodialysis or peritoneal dialysis) or renal transplantation [3]. The decline of renal function is associated with inadequate removal of a variety of uremic toxins that should be excreted by the kidney. Due to the fact these substances are biologically active, they are both the cause and consequence of CKD [4]. According to studies, uremic toxins exert a detrimental effect on cells involved in the functioning of myocardium and vessels, including smooth muscle cells, endothelial cells (ECs), and platelets leucocytes, thus affecting the cardiovascular system [5]. Cardiovascular diseases (CAD) are the most frequent cause of death for patients with all stages of renal failure, and they are present in >50% of patients undergoing dialysis [4].
2. Cardiovascular Diseases in ESRD Patients
The occurrence of cardiovascular aberrations in CKD is related to the influence of traditional risk factors (hypertension, diabetes mellitus, etc.) and also non-traditional CKD-associated risk factors, including anemia; inflammation; mineral and bone disease abnormalities; oxidative stress; the presence of some uremic toxins; and factors related to the type, frequency of dialysis and the composition of fluid used during the procedure [6,7,8,9]. Increased CAD risk in ESRD patients is also related to the fact that this disease frequently results from hypertension and diabetes mellitus [4]. The prevalence of coronary heart disease and ventricular hypertrophy has been reported to be 40% and 70%, respectively, in renal replacement therapy patients [10]. The likelihood of developing CAD increases linearly in patients whose glomerular filtration rate (eGFR) decreases below ~60–75 mL/min/1.73 m2, and CAD mortality risk in patients with CKD stages G3a to G4 is twice or three times higher compared to patients without CKD [6,11,12]. Also, the prevalence of clinical manifestations of CAD as well as the frequency of arteriosclerosis, LVH, large-vessel coronary disease, myocardial fibrosis, and microvascular disease rises along with the decline of eGFR. The mortality of patients with end-stage renal failure is considerably higher (20 times higher in HD patients) than in the general population [13,14]. Nearly 50% of patients undergoing hemodialysis die of cardiovascular causes [15]. The rate of cardiovascular death is increasing with aggravating kidney impairment even after the adjustment for common CAD risk factors [6,11,16]. Hemodialysis offers a temporary solution for renal dysfunction since it replaces some filtration functions of the kidney, however, it does not diminish morbidity and mortality related to inflammation and its complications, such as cardiovascular disease or oxidative stress [17]. The long-term survival and prognosis of patients undergoing dialysis after acute myocardial events are poor [18]. Numerous studies confirmed that the calcification of the sub-intima and media of large vessels increased the risk of all-cause and cardiovascular mortality in this group [19]. The autopsy studies of patients with chronic kidney disease have revealed the presence of more advanced atherosclerotic plaques, more aggravated medial calcifications of these lesions compared to patients without kidney impairment, as well as inflammation in the coronary plaques [20,21,22,23]. High mortality of dialysis patients may also be ascribed to sudden deaths, which are associated with arrhythmia triggered by shifts in volume, electrolytes and drug concentrations in patients with a myocardial disease (LVH and heart failure) [6]. Mortality related to sudden death and heart failure decreases after kidney transplantation [6]. The impairment of renal clearance in CKD results in the accumulation of toxins, such as p-cresol and indoxyl sulphate, which not only stimulate the expression of intercellular adhesion molecule-1 (ICAM) and monocyte chemotactic protein-1 (MCP-1), but also induce the activation of NADPH oxidase, increasing the production of reactive oxygen species, as well as pose surplus cardiovascular risk in CKD [24,25]. Also, uremic immune dysfunction is considerably associated with high rate of premature mortality in ESRD patients due to the impact on cardiovascular and infectious complications [26]. Malnutrition–inflammation–atherosclerosis (MIA) syndrome is an important complication observed in patients with advanced stages of CKD, and it is accompanied by higher incidence of CVD and rapid progression of atherosclerotic organ damage [27]. The prevalence of MIA in CKD is associated with hypercatabolism, malabsorption due to overhydration and swelling of the gastrointestinal mucous, weakened appetite, loss of protein in the urine and during dialysis, as well as hormonal imbalances. Hormonal disturbances (especially those affecting insulin, insulin-like growth factor (IGF-1), adiponectin, ghrelin, and somatostatin (GH)) contribute to the state of chronic inflammation [26]. In ESRD patients, impaired balance between anti-inflammatory adiponectin and proinflammatory leptin is particularly visible [28]. The presence of proinflammatory cytokines is associated with disturbances of signal transduction and the development of insulin resistance, resulting in lipid metabolism disorders involving abnormal triglyceride metabolism, the rise in lipolysis with an enhanced release of free fatty acids (TNF-α, IL-6), and stimulation of ectopic lipid deposition (leptin) [29]. Moreover, insulin resistance is associated with endothelial dysfunction and an increase in blood pressure, which are both related to increased cardiovascular morbidity. Furthermore, the development of protein–energy wasting (PEW) is one of the strongest predictors of mortality in patients with CKD [26]. Systemic inflammation; uremic toxins; dialysis techniques promoting enhanced catabolism and systemic inflammation; and metabolic dysfunction involving the stimulation of appetite suppression hormones, such as insulin and leptin, which leads to diminished nutrient intake, are all of key importance for the development of PEW [30]. Among adverse effects of PEW there are: poor quality of life, sarcopenia, vascular calcifications, changes in lipid metabolism, increased inflammation and higher prevalence of cardiovascular disease, elevated rates of the number of hospitalizations, and increased mortality [31]. In ESRD, inflammation seems to be further aggravated by uremic immune dysfunction, deficient renal cytokine clearance, as well as inflammatory responses to dialysis. Some authors suggest that the improvement of ESRD patients’ prognosis is possible only as a result of the implementation of several actions targeted at tackling single causes of inflammation in the inflammatory cascade [32]. Apart from chronic inflammation in patients with CKD, especially with ESRD, as well as anemia, the rise in sympathetic tone, uremic toxin activity, aforementioned protein–energy malnutrition, endothelial dysfunction, calcium phosphate disorders, and pro-coagulation contribute to the accelerated development and progression of atherosclerosis [26]. Finally, ESRD is associated with modifications of lipid components, lipoproteins, and proteins. Dyslipidemia observed in ESRD is characterized by the hypertriglyceridemia, increase in IDL-C and chylomicron remnants, and a reduction in HDL-C and apolipoprotein (apo) AI (apoA-I) as well as apoC-II/apoC-III ratio [33,34]. Also, uremia-associated inflammation in ESRD can convert HDL from an antioxidant into a pro-oxidant particle [33]. Again, a vicious circle is observed in uremic patients and it involves reduced catabolism of intermediate-density lipoprotein (IDL) and LDL, which results in their longer residence time in plasma and further in alteration of the apolipoprotein B (apoB) contained in these lipoproteins via carbamylation, oxidation and glycation. Considering changed lipid subfraction turnover, the time of lipoproteins residence in the circulation of CKD patients is prolonged, which translates into higher risk of post-ribosomal modification of lipoproteins (such as glycation, oxidation, and carbamylation). According to studies, extensively modified lipoproteins show decreased affinity towards classic LDL-C receptors, thus they are captured by scavenger receptors (SR) on the surface of abundant macrophages, which in consequence promote atherosclerosis [34,35]. The mechanism underlying the oxidative stress in CKD patients is complex, multifactorial and not fully explained [26]. Due to the importance of oxidative stress in CKD patients, this review purpose is to shed some light on the mechanisms related to enhanced cardiovascular risk in patients with renal insufficiency.
3. Oxidative Stress
The kidney is one of the vital sources of antioxidant enzymes, including glutathione peroxidases, and therefore, the impairment of this organ in the course of CKD is associated with decreased levels of these enzymes as well as increased levels of pro-oxidants [36]. The interplay between oxidants and antioxidants controls crucial pathways and cell metabolism [37,38]. In healthy conditions, reactive species are removed by natural endogenous defense mechanisms. However, in some diseases the defense mechanisms are impaired and/or the production of reactive species is so enhanced that it results in oxidative stress. Numerous studies confirmed the impairment of antioxidant systems (e.g., reduced activity of glutathione peroxidase and copper, zinc superoxide dismutase, and paraoxonase) in patients with CKD [39,40].
Oxidative stress is defined as the imbalance between the production of pro-oxidants and antioxidant defense mechanisms based on reactive oxygen species degradation. Reactive species generation and their cellular localization are usually in equilibrium with the availability of antioxidant enzymes including cystolic catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (Gpx). The normal functioning of cells requires appropriate levels of both elements. In oxidative stress, the production of reactive oxygen species (ROS) exceeds the scavenging capacity of antioxidant systems [3,40]. The enhancement of oxidative stress has been demonstrated already in the early stages of CKD [41,42,43]. Numerous studies indicated that oxidative stress was significantly increased in patients with advanced renal impairment, but this state is exacerbated by hemodialysis [2,7,9,44]. Some authors suggested on the basis of in vivo studies that increased oxidative damage was the result of diminished levels of these enzymes rather than enhanced ROS production [45,46]. However, according to others, oxidative stress is associated with the production of highly reactive intermediates during inflammation; on the other hand, also reactive oxygen species (ROS) are able to stimulate pro-inflammatory mediators, such as NF-κB, thus promoting inflammatory response [41]. Inflammatory cells have been confirmed to be a source of free radicals, such as reactive oxygen and nitrogen species [47]. The upregulation of inflammatory markers observed in CKD patients (including platelet-derived growth factor and tumor necrosis factor-α) results in NADPH oxidase activation and subsequent generation of intracellular O2• and H2O2 [48,49]. The presence of aggravated inflammatory state in CKD can stimulate the activation/recruitment of polymorphonuclear neutrophils and monocytes, which leads to stimulation of myeloperoxidase (MPO) and enhanced ROS production [50,51].
Uremic toxins have been shown to play a vital role in the onset of oxidative stress. Martinon et al. [52] demonstrated that uremic toxins promoted the development of inflammatory state and oxidative stress via priming acute inflammatory polymorphonuclear lymphocytes, stimulating interleukin (IL)-1β and IL-8. In turn, Sakamaki et al. [53] suggested that they stimulated the innate immune response through CD8+ cells. Stockler-Pinto et al. [54] revealed that indoxyl sulphate-related ROS production primarily resulted from the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Also, the synthesis of uric acid can aggravate oxidative stress via the activity of xanthine oxidoreductase, which generates reactive oxygen species [55]. However, some other reports suggest that in the presence of specific components, in various physiochemical circumstances and in different compartments of the human body, uric acid may play an anti-oxidant role in vivo [56]. Uric acid poses strong reducing and antioxidant properties, however, its elevated levels in CKD patients are believed to pose potential risk factors for CVD [57]. Under conditions of oxidative stress, high concentrations of uric acid have been shown to act as a pro-oxidant, particularly when antioxidant systems are impaired. However, it was also found to be cleared by HD as evidenced by a reduction in uric acid levels compared to pre-HD state [58].
The hemodialysis itself also enhances the oxidative stress due to the fact that antioxidant systems, particularly those of low or very-low molecular weight, are filtered during the procedure, and both the dialysis membrane and dialysate can activate leukocytes, leading to the aggravation of inflammation and enhanced ROS production [3]. Increased oxidative stress occurring in HD patients depends on many factors including aging, impairment of the residual renal function and subsequent uremic state, as well as the HD procedure itself [58]. During the initiation of the dialysis process, the membrane and dialysate induce inflammation and promote an important increase in ROS production. Post-dialysis, the levels of oxLDL have been shown to be elevated. However, post-dialysis, the activity of XOD and 8-OHdG levels are considerably diminished, which suggests that markers of oxidative stress are efficiently filtered during the dialysis process [3]. Also, markers of antioxidant defense decreased after HD [3]. Also, Liakopoulos et al. [59] stated that excessive oxidative stress in HD patients was related with the loss of antioxidants during the procedure and the accumulation of oxidative products. The level of oxidative stress was shown to be higher in ESRD patients on peritoneal dialysis (PD) compared to non-dialyzed uremic patients, however, it is lower in comparison to HD patients [59,60]. This observation was confirmed by Chen et al. [61] who demonstrated higher resting levels of superoxide anion in the whole blood after each HD session. Moreover, Granata et al. [62] revealed that patients with CKD and those undergoing hemodialysis show impaired mitochondrial respiration. The aggravation of oxidative stress can also be associated with the impaired activation of nuclear factor erythroid 2-related factor 2 (Nrf2), which is responsible for the regulation of genes encoding detoxifying and antioxidant proteins and enzymes (e.g., CAT, NAD(P)H dehydrogenase [quinone] 1 (NQO1), SOD [63]. Oxidative stress in patients with advanced stages of CKD can also be exacerbated by iron therapy, which is frequently used to treat anemia [36]. This phenomenon is associated with the fact that the administration of intravenous iron and the supersaturation of iron sequestration proteins (e.g., ferritin and transferrin) may result in the formation of free iron showing oxidative properties.
Decreased NO production, the formation ofreactive nitrogen and oxygen species are responsible for the detrimental effects related to oxidative stress
3.1. Nitric Oxide (NO)
The kidney is an important source of L-arginine, which is a precursor for nitric oxide (NO); therefore, the decrease in its mass may result in diminished production of L-arginine and NO activity [47]. Due to the fact that nitric oxide is essential for vascular endothelial cell function, its decreased bioavailability is associated with endothelial dysfunction observed in hypertension, diabetes mellitus, atherosclerosis, and CKD [4]. Asymmetric dimethylarginine (ADMA), which is an endogenous amino acid resembling L-arginine, inhibits endothelial nitric oxide synthase, thus impairing NO synthesis. Elevated ADMA concentrations have been reported in ESRD. Moreover, Ravani et al. [64] suggested that elevated ADMA levels were a strong independent risk factor for the progression of CKD and patient mortality. O2− and H2O2 are the precursors used for the production of even more powerful oxidants. The first of them shows the affinity towards free radical NO and their reaction results in the formation of peroxynitrite (ONOO−). The effects of both ONOO− and hydroxyl (OH−) involve extensive nitrosative and oxidative modifications to proteins, lipids and nucleic acids [41].
3.2. Reactive Oxygen Species (ROS)
Reactive species produced in normal physiological state are inactivated by enzyme systems (e.g., glutathione) as well as other antioxidants (called scavengers) [41]. However, the excessive amount of ROS cannot be neutralized by scavenger systems, and therefore, they cause oxidative damage to proteins, nucleic acids and lipids; impair cellular activity; and hinder enzymatic activity [41]. In the kidneys, ROS are primarily synthesized by the mitochondrial respiratory chain and by enzymes such as NADPH oxidase (NOX) [41]. According to studies, NOX isoforms are vital players in the aggravation of oxidative stress, which results in the worsening of vascular function and promoting fibrosis [65,66]. To a lesser extent, ROS are produced by endoplasmic reticulum, peroxisomes and lysosomes [67]. Nox4 belonging to NADPH oxidase family is expressed in smooth muscle cells, vascular endothelial, as well renal proximal tubules, which explains why renal impairment may influence its expression or activity [68,69]. Also, pro-oxidant enzymes such as xanthine oxidase (XOD), in which activity has been shown to be considerably increased in uremia, is an additional possible source of ROS in CKD [3,70].
Reactive oxygen species (ROS) mainly include (O2•−), the hydroxyl radical (•OH) and hydrogen peroxide (H2O2) [47]. In healthy metabolic cells, their production is counteracted by mitochondrial or cystolic catalase (CAT) or thiol peroxidases, which catalyze H2O2 reduction into water and O2. Mitochondria comprise also other antioxidants, including manganese-SOD (Mn-SOD) and Gpx, which neutralize formed ROS. Mn-SOD converts O2•− to H2O2, which in the next step is decomposed by CAT and Gpx [71]. In peroxisomes, the stabilization of O2•− is related to the activity of copper/zinc-SOD (Cu/Zn-SOD) [72,73]. Also, glutathione homeostasis (Gpx, glutaredoxins, glutathione-S-transferase, peroxiredoxins and thioredoxins) is vital for maintaining cellular redox balance [47,74]. Xanthine oxidase (catalyzes the oxidation of hypoxanthine to uric acid, releasing in consequence ROS (O2•, •OH, and H2O2) are by-products [51]. The formed uric acid accelerates CKD progression to renal failure and enhances the risk of cardiovascular events [75]. Reactive oxygen species are highly reactive and thus damage the variety of cellular structures and functional pathways [47]. Cellular H2O2 is rather stable, however, it still has potential to interact with numerous substances and cause destruction. Ferrous iron (Fe2+) can interact with H2O2, resulting in its cleavage and formation of the most reactive •OH form [76]. Protein tyrosine phosphatases have been shown to be major targets for oxidant signaling due to the fact that they are greatly susceptible to oxidative modification of amino acid residue of cysteine [77].
The presence of oxidative stress and the impairment of antioxidative defense mechanisms in patients with CKD/ESRD have been confirmed in numerous studies. Some products of oxidative metabolism, including advanced glycation end products (such as pentosidine or advanced oxidation protein products), have been demonstrated to accumulate in renal failure [78,79]. Kinugasa E [80] demonstrated increased circulating levels of oxidative stress markers, including advanced glycation end products (AGEs), malondialdehyde (MDA) and 8-hydroxyde-oxyguanosine in blood and/or tissue in CKD patients. Advanced glycation end products (AGEs) acting via a specific receptor (RAGE) activate MAP kinase transduction pathway and in consequence lead to an increase in the level of pro-inflammatory cytokines, enzymes and adhesion molecules [81,82]. Colombo et al. [83] confirmed the existence of a relationship between uremia and oxidative stress, which was assessed on the basis of severe protein oxidative damage (including plasma advanced oxidation protein products) in end-stage renal disease (ESRD) patients on maintenance hemodialysis (HD).
3.3. Consequences of Oxidative Stress
Aggravated oxidative stress has been reported to be involved in the pathomechanisms of several diseases, including cardiovascular disease and chronic kidney disease. Elevated oxidative stress has been demonstrated to be strictly related to kidney and cardiac damage as it aggravates kidney dysfunction and induces cardiac hypertrophy, which is an independent risk factor for heart failure (HF) [43,51]. Oxidative stress has been shown to affect upstream transcriptional gene regulation. Numerous studies provided evidence that proliferator-activated receptors (PPARs), which play key roles in the transcriptional regulation of cell cycle progression, cell differentiation, glucose homeostasis, lipid metabolism, and inflammation, are altered in CKD and CVD [84,85,86]. Oxidative stress results in the damage of nucleic acids, including the modifications of bases (especially guanine in DNA) and covalent crosslinks, leading to single- and double-strand breaks. The oxidation of guanine is associated with the formation of oxidized products including 8-hydroxy-20-deoxyguanosine (8-OH-dG), which are highly prevalent in chronic and degenerative diseases, including CKD [87].
Oxidative stress is responsible for progressive renal damage, which in consequence may lead to renal ischemia, glomeruli damage, cell death and apoptosis, and further worsening of the severe inflammatory processes [41,88]. Fujii et al. correlated oxidative stress with glomerular abnormalities, including glomerular hypertrophy and mesangial proliferation, observed in the course of diabetic nephropathy [89]. It is also an infamous factor responsible for cardiac damage, such as hypertrophy, fibrosis, apoptosis, and remodeling [90]. Numerous mechanisms via which oxidation products promote vascular injury have been suggested [91,92,93,94]. NADPH oxidases, which are the major sources of ROS, participate in the pathogenesis of cardiac remodeling via its impact on redox-sensitive signal transduction [43]. Numerous studies confirmed that both the expression and the activity of NADPH oxidase were elevated in the myocardium of patients with ischemic and non-ischemic heart failure [95,96,97]. Moreover, higher activation of NADPH oxidase was involved in fibrosis and cardiac hypertrophy [98,99]. Uremic toxin, indoxyl sulphate, which promotes the production of ROS through the stimulation of NADPH oxidase or NADPH-like oxidase, has been shown to be involved in vascular disease, as it promotes vascular smooth muscle cell proliferation and vascular calcification; in addition, it is associated with higher mortality observed in CKD patients [100,101,102]. Moreover, this toxin also reduces levels of total glutathione in endothelial cells [43,103].
Oxidative stress-induced endothelial dysfunction and subsequent reduction in NO bioavailability promote the development of atherosclerosis. Peroxynitrite generated from NO is involved in numerous unfavorable vascular actions. The inactivation and the deficiency of NO resulting also from the actions of reactive species decrease the protection of kidney function, which is related to NO-dependent increase in renal blood flow, stimulation of pressure natriuresis, regulation of tubuloglomerular function, and maintenance of fluid and electrolyte homeostasis [41,104].
Oxidative stress is also associated with the formation of oxidized low density lipoprotein (ox- LDL), which play a crucial role in the pathogenesis of atherosclerosis [105]. The accumulation of oxidized low-density lipoproteins in arterial intima is the initial step of atherosclerotic process development [13]. Also, advanced glycation end products, in which production is enhanced in renal failure, exert atherogenic effects [13].
The link between cardiovascular disease and CKD may also involve the actions of the functional mitochondrial angiotensin system, which is regulated by oxidative stress [106]. Angiotensin type II receptors co-localized with angiotensin on the inner mitochondrial membrane of human mononuclear cells were shown to control mitochondrial NO production and respiration. The activation of the renin–angiotensin system (RAAS) in the course of renal impairment is involved in the process of left ventricular (LV) remodeling [107]. Ang II induces vasoconstriction and aldosterone release and it mediates hemodynamic alterations, which in consequence, lead to cardiac and vascular remodeling [108]. Furthermore, both angiotensin II and aldosterone actions involve the activation of mitogen-activated protein kinases (MAPKs), as well as c-Src and Ki-ras2A pathways engaged in the development of inflammation, in the production of O2 and H2O2, endothelial dysfunction, as well as hypertrophic growth [109,110]. Higher O2 levels are associated with enhanced protein kinase C (PKC) activity and NOS uncoupling, as well as consequent loss of vasodilation [51]. ONOO-associated loss of vasodilation and subsequent endothelial dysfunction play a vital role in the development of hypertension and further contributes to hypertrophic remodeling [111]. In CKD patients, oxidative stress leads also to left ventricular hypertrophy (LVH). The role of oxidative stress in the development of cardiac remodeling and heart failure has been summarized at Figure 1.
According to studies, oxidative stress as well as excessive ROS production are important factors mediating osteochondrogenic transdifferentiation of vascular smooth muscle cells (VSMCs) and enhanced vascular calcification [112]. The development of vascular calcifications (VC), which occurs commonly in CKD patients, exerts a direct impact on vessel functions and CVD development. Considerable leukocyte infiltration and the presence of IL-1β and MMP-1 have confirmed that human calcified areas in aortic valves lead to accelerated atherosclerosis, as well as higher rates of cardiovascular and all-cause mortality [113,114,115,116]. Oxidative stress has been demonstrated to contribute to the phenotype switch of vascular smooth muscle cells (VSMCs) even in early CKD [116]. Huang et al. [116] provided evidence for the existence of a kinetic relationship between oxidative stress and vascular calcification and osteoblastic transition. They observed that serum derived from patients with early stage CKD directly induced osteoblastic transition of primary rat VSMCs and calcium deposition in VSCMs, but it did not affect serum phosphorus level. In in vitro studies, hydrogen peroxide (H2O2) and xanthine/xanthine oxidase, which generates superoxide anion, were shown to boost osteochondrogenic transdifferentiation of VSMCs [112,117,118]. Intensified calcification in the presence of H2O2 was associated with higher expression of osteogenic markers, such as osteocalcin (OCN), runt-related transcription factor 2 (Runx2) and alkaline phosphatase (ALP), and lower expression of the contractile VSMCs phenotype markers, such as smooth muscle α-actin (α-SMA) and SM-22α [118]. It has been suggested that Msx2 is an important factor involved not only in transcriptional programming of osteoblastic lineage development but also in BMP-2-mediated vascular calcification through the activation of Wnt catenin signaling and β-catenin-induced activation of Pit1, a type III sodium-dependent phosphate cotransporter [119,120,121]. Cai et al. found that WNT/β-catenin signaling directly elicited osteogenic transdifferentiation and calcification of VSMCs though the modulation of Runx2 gene expression [122]. Huang et al. [116] suggested that the development of vascular calcification could be partly mediated by upregulation of NOX1 as well as ERK kinases as downstream events of NOX1-induced VC. Oxidative stress also indirectly may stimulate vascular calcification. It has been demonstrated that lipid oxidation products present in oxidized low-density lipoprotein rise the activity of ALP and promote calcification of vascular cells, which in consequence may lead to atherosclerosis-associated intimal calcification [123]. Moreover, it seems that oxidative stress in uremia enhances the formation of advanced oxidation protein products (AOPP), but at the same time, the accumulation of AOPP may pose a trigger for enhanced oxidative stress, which gives a positive feedback loop of elevated and maintained oxidative stress in uremic patients [124]. The results of in vitro study revealed that AOPP could directly stimulate osteoblast differentiation and calcification of smooth muscle cells [124]. You et al. [124] demonstrated that AOPP rose the calcium level in human aortic smooth muscle cells (HASMCs) (probably inducing their calcification) and considerably enhanced protein levels and mRNA expression of osteopontin (OPN), which may suggest that AOPP could promote osteoblast differentiation of HASMCs. Moreover, AOPP up-regulated mRNA expression of a transcription factor CBF-α1, which had earlier been found to increase the expression of osteoblast-specific genes, e.g., osteocalcin and alkaline phosphatase [125,126]. Finally, they were shown to considerably lower the expression of SM-α-actin expression [124]. According to in vitro studies, advanced oxidation products can not only trigger the oxidative burst of human monocyte and neutrophil but also induce enhanced production of oxidants by leukocytes [124,127,128]. It has been suggested that the mechanism of AOPP-stimulated smooth muscle cells differentiation may involve the activation of extracellular signal-regulated kinase (ERK), which is a part of MAPK pathway [124]. ERK is able to induce the osteoblast-related gene expression by extracellular matrix-integrin receptor interaction, bone morphogenetic protein 2 (BMP-2) and growth factors, thus leading to osteoblast differentiation [129,130,131]. Moreover, MAPK increases the expression of osteocalcin and AOPP-induced calcium deposition, which results also in the calcification of HASMC [130,132]. The role of oxidative stress in the development of atherosclerosis and other adverse consequences has been presented at Figure 2.
Finally, oxidative stress, especially the exposure to H2O2, has been demonstrated to alter membrane properties of red blood cells (RBC) and accelerate RBC removal in the spleen [51,133]. The increased susceptibility of RBCs to oxidative damage along with the higher risk of ROS production in iron deficiency anemia in CKD create a vicious cycle of enhanced RBC death, anemia and oxidative stress severity [51,133,134]. The lowering of hemoglobin content in iron deficiency-related anemia is associated with the decrease in partial pressure of oxygen, and this hypoxia-resembling state aggravates oxidative stress via auto-oxidation of hemoglobin to met-hemoglobin (metHb) with accompanying generation of O2 [135,136]. Iron deficiency also affects the expression of iron-containing endogenous antioxidant proteins e.g., peroxidase and catalase, as well as concentration of selenium, thus decreasing the activity of selenium-dependent enzyme GPx [137,138,139]. Therefore, it seems that timely intravenous iron replacement and the administration of antioxidants in clinical setting could improve CKD patients’ quality of life and decrease the risk of morbidity [140].
Numerous studies have confirmed that oxidative stress-related oxidation of fatty acid end-products (malondialdehyde) and serum albumin is associated with higher mortality in hemodialysis [141]. According to some authors, the decrease in antioxidant defense in hemodialysis results in enhanced all-cause and cardiovascular mortality in these patients [142,143,144]. Sangeetha Lakshmi et al. [58] revealed that the concentration of malondialdehyde (biomarker of oxidative stress) was considerably increased in patients with CKD and accompanying cardiovascular disease compared to patients with CKD but without cardiovascular disease. Substantial elevation of serum malondialdehyde levels observed in HD patients suffering from CVD, compared with those without CVD, indicated an association between oxidative stress and the development of atherosclerosis in these patients [9]. Juretic et al. [141] observed reduced PON in those patients with uremia who were at higher risk of cardiovascular disease, compared to persons with normal kidney function. It has been suggested that the loss of PON activity may increase the risk for oxidative stress and cardiovascular disease patients with chronic kidney disease, despite the lack of correlation with oxidized LDL [40]. Finally, Russa et al. [2] observed higher values of both oxidative stress and antioxidant barrier in hemodialysis patients with previous acute myocardial infarction compared to patients without cardiovascular events.
4. Treatment Aiming to Decrease the Risk Resulting from Oxidative Stress
According to studies, antioxidant therapies may prove to be beneficial since they can decrease oxidative stress, reduce uremic cardiovascular toxicity and improve survival [104]. Endogenous or dietary antioxidants have also been suggested to exert protective effects against inflammation and kidney damage in patients with CKD [41]. The application of mitochondrial-targeted antioxidant therapy resulted in the improvement of cardiac hypertrophy and diastolic dysfunction through the diminution of oxidative stress, which confirms the role of oxidative stress in the progression of heart failure [145]. Antioxidant supplementation with vitamins A, C, and E; β-carotene; or N-acetyl cysteine (NAC) seems to be beneficial in decreasing cardiovascular risk in hemodialysis patents [3,146]. Vitamin E is a powerful antioxidant exerting anti-inflammatory properties; it has been shown to interfere with cell membrane lipid peroxidation [147]. Observational clinical studies have shown that the intake of vitamin E (more than 100 IU/day), which inhibits oxLDL formation by hindering lipid peroxidation, reduced the rate of coronary events in hemodialysis [41,148,149]. Randomized placebo-controlled Secondary Prevention with Antioxidants of Cardiovascular Disease in End-stage Renal Disease (SPACE) trial revealed that in hemodialysis patients, the supplementation of alpha-tocopherol (800 IU) decreased cardiovascular disease endpoints and resulted in a substantial improvement of cardiovascular complications (myocardial infarction) [149]. Antioxidant therapy with DL-α-tocopherol has been revealed to improve left ventricular hypertrophy (LVH) and to decrease adverse changes within the myocardium in experimental CKD [150].
Vitamin C plays a significant antioxidative role as it can reduce ROS levels, thus providing protection against kidney oxidative damage and helping to maintain vascular and endothelial function [151]. Wang et al. [152] demonstrated that vitamin C (ascorbic acid) diminished oxidative damage, inflammation and renal injury in ischemia nephrotoxic acute kidney injury and rhabdomyolysis-induced renal injury. Deicher et al. observed deficiency of vitamin C (non-enzymatic antioxidant) in hemodialysis patients, which was associated with dietary restrictions and/or its loss during dialysis. Study of HD patients demonstrated that low plasma vitamin C levels predicted fatal and major non-fatal adverse cardiovascular events in this group [153]. Some studies indicate that patients with chronic kidney disease and ESRD patients should be administered a limited dose of daily vitamin C supplement of 75 mg for females and 90 mg for males [154]. However, Jankowska et al. [155] suggested that the supplementation of vitamin C might lead to oxalate accumulation and subsequent elevation in oxidative stress, and therefore, antioxidants administration may not always be the best alternative.
In turn, vitamin D is vital not only for the homeostasis of calcium/phosphorus and skeletal health but also for renal functioning. The deficiency of this vitamin is frequently observed in CKD and ESRD and has been shown to contribute to the deterioration of renal function and increased morbidity and mortality in patients with CKD [56]. Some studies have demonstrated that the intake of vitamin D can reduce kidney injury by suppressing inflammation, fibrosis and apoptosis, via hindering multiple pathways crucial in kidney injury, including renin-angiotensin-aldosterone system (RAAS), NFκ-B, Wnt/β-catenin, and TGF- β/Smad signaling pathways [156,157,158].
Due to the fact that indoxyl sulphate stimulates oxidative stress and hastens the progression of CVD in CKD, the lowering of its concentration may prove beneficial in this group of patients. This suggestion was confirmed in several studies that demonstrated that the use of oral charcoal adsorbent, which decreases the levels of circulating uremic toxins, prevented histological and functional aggravation of CKD and suppressed oxidative stress and the advancement of cardiac damage in CKD [159,160,161]. In other studies, the decrease in heart and left ventricular volumes, cardiac fibrosis, as well as the attenuation of cardiac concentric change were observed in pre-dialysis CKD patients after AST-120 administration [162,163]. Taken together, these results suggest that the administration of AST-120 may become a useful option for improving cardiovascular health in CKD patients [43].
Also, melatonin (N-acetyl-5-methoxytryptamine) has been proven to be highly efficient in many disorders associated with oxidative stress and inflammation in experimental animals [164,165,166]. It is an endogenous neurohormone modulating sleep, immune function, circadian rhythm, and sexual behaviors, which exerts free radical scavenger, antioxidant and anti-inflammatory effects [165,167]. Due to the fact that it scavenges reactive oxygen and nitrogen species and enhances antioxidant defense systems, melatonin prevents tissue damage and hampers transcriptional factors of pro-inflammatory cytokines. Numerous studies have indicated that it indirectly decreases oxidative stress via stimulation of the expression and function of some antioxidant enzymes, enhancing the activities of antioxidative defense systems and glutathione as well as rising the efficacy of the mitochondrial electron transport chain [168,169,170]. CKD patients have been shown to have impaired night-time secretion of melatonin, which is further associated with higher stimulation of intrarenal renin–angiotensin system (RAS), leading to enhanced reactive oxygen species (ROS) production, sodium retention, inflammation, and fibrosis [171]. These pathologies accelerate the progression of CKD to end-stage renal disease (ESRD). The results of studies indicated that the supplementation with exogenous melatonin can reverse adverse changes, slow down the progression of kidney impairment, reduce blood pressure, and also help to maintain the bioavailability of nitric oxide by acting on melatonin receptor MT2 [172]. Studies on animal models indicated that prolonged administration of melatonin enhanced the expression of markers associated with decreased oxidative stress, inflammation and vasoprotection [173]. It has also been shown to improve cardiovascular function as well as renal, cardiac and cerebral damage [174].
The introduction of therapy based on the thiol-containing compound acetylcysteine has been demonstrated to diminish the toxic effects of ischemia reperfusion syndromes of the heart, kidney, liver, and lung and enabled the reduction in the risk of primary cardiovascular endpoint (fatal and non-fatal myocardial infarction) by 40% in hemodialysis patients [13]. Tepel et al. [13] revealed that after adjustment for age, baseline systolic and diastolic blood pressure, medications, smoking, and the duration of hemodialysis, in the study group treated with acetylcysteine, the survival related to the primary end point was higher compared with the control group. Moreover, they observed 30% decrease in cardiac events, 69% reduction in ischemic strokes, and 36% drop in peripheral vascular disease in the acetylcysteine group, however, the differences did not reach the level of statistical significance [13]. Due to the fact that acetylcysteine acts as a free-radical scavenger or as a reactive sulfhydryl compound, it enhances the reductive capacity of the cell ameliorating coronary and peripheral vascular function [175]. However, this antioxidant therapy proved ineffective in patients with heart failure without renal disease [176]. Therefore, it seems that the systemic oxidative stress in uraemic milieu plays a vital role in the development of cardiac disease in renal patients
Some studies indicated that in patients undergoing maintenance, HD plasma concentrations of CoQ10 are reduced, which suggests that CoQ10 supplementation could represent a great antioxidant therapy for these patients [177]. Randomized, double-blind, placebo-controlled study carried out by Rivara et al. [178] demonstrated that administration of CoQ10 (1200 mg daily) as an antioxidant therapy was safe and well tolerated in patients receiving MHD, and it resulted in a substantial, dose-dependent rise in plasma CoQ10 levels compared to placebo and considerably diminished plasma concentrations of F2-isoprostanes, which are considered a robust plasma marker of oxidative stress.
5. Conclusions
Elevated cardiovascular morbidity and mortality in patients with end-stage renal failure remains to be a challenge in medicine. Numerous studies indicate that oxidative stress may play an important role in the development and progression of cardiovascular disease. However, antioxidant therapies seem to exert beneficial effects as they decrease cardiovascular risk and they bring hope for less cardiovascular complications in this group of patients.
Funding
No external funding was received in relation to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Russa, D.; Pellegrino, D.; Montesanto, A.; Gigliotti, P.; Perri, A.; Russa, A.; Bonofiglio, R. Oxidative Balance and Inflammation in Hemodialysis Patients: Biomarkers of Cardiovascular Risk? Oxid. Med. Cell. Longev. 2019, 2019, 8567275. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Garcia, J.A.; Rodriguez-Sanchez, E.; Aceves-Ripoll, J.; Abarca-Zabalia, J.; Susmozas-Sanchez, A.; Gonzalez Lafuente, L.; Bada-Bosch, T.; Hernandez, E.; Merida-Herrero, E.; Praga, M.; et al. Oxidative Status before and after Renal Replacement Therapy: Differences between Conventional High Flux Hemodialysis and on-Line Hemodiafiltration. Nutrients 2019, 11, 2809. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Tepel, M.; Echelmeyer, M.; Orie, N.N.; Zidek, W. Increased intracellular reactive oxygen species in patients with end-stage renal failure: Effect of hemodialysis. Kidney Int. 2000, 58, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.N.; Himmelfarb, J.; Henrich, W.L.; Hakim, R.M. Reassessing the cardiac risk profile in chronic hemodialysis patients: A hypothesis on the role of oxidant stress and other non-traditional cardiac risk factors. J. Am. Soc. Nephrol. 1997, 8, 475–486. [Google Scholar] [PubMed]
- Boaz, M.; Matas, Z.; Biro, A.; Katzir, Z.; Green, M.; Fainaru, M.; Smetana, S. Serum malondialdehyde and prevalent cardiovascular disease in hemodialysis. Kidney Int. 1999, 56, 1078–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzolino, M.; Galassi, A.; Pivari, F.; Ciceri, P.; Conte, F. The Cardiovascular Burden in End-Stage Renal Disease. Contrib. Nephrol. 2017, 191, 44–57. [Google Scholar] [CrossRef]
- Chronic Kidney Disease Prognosis Consortium; Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; de Jong, P.E.; Coresh, J.; Gansevoort, R.T. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: A collaborative meta-analysis. Lancet 2010, 375, 2073–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjunath, G.; Tighiouart, H.; Ibrahim, H.; MacLeod, B.; Salem, D.N.; Griffith, J.L.; Coresh, J.; Levey, A.S.; Sarnak, M.J. Level of kidney function as a risk factor for atherosclerotic cardiovascular outcomes in the community. J. Am. Coll. Cardiol. 2003, 41, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Tepel, M.; van der Giet, M.; Statz, M.; Jankowski, J.; Zidek, W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: A randomized, controlled trial. Circulation 2003, 107, 992–995. [Google Scholar] [CrossRef]
- Parfrey, P.S.; Foley, R.N.; Harnett, J.D.; Kent, G.M.; Murray, D.; Barre, P.E. Outcome and risk factors of ischemic heart disease in chronic uremia. Kidney Int. 1996, 49, 1428–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, D.J.; Covic, A. Coronary artery disease in uremia: Etiology, diagnosis, and therapy. Kidney Int. 2001, 60, 2059–2078. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Qureshi, A.R.; Alvestrand, A.; Divino-Filho, J.C.; Gutierrez, A.; Heimburger, O.; Lindholm, B.; Bergstrom, J. Inflammation, malnutrition, and cardiac disease as predictors of mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 1), S28–S36. [Google Scholar]
- Herzog, C.A.; Ma, J.Z.; Collins, A.J. Poor long-term survival after acute myocardial infarction among patients on long-term dialysis. N. Engl. J. Med. 1998, 339, 799–805. [Google Scholar] [CrossRef] [Green Version]
- London, G.M.; Guerin, A.P.; Marchais, S.J.; Metivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Nakano, T.; Ninomiya, T.; Sumiyoshi, S.; Fujii, H.; Doi, Y.; Hirakata, H.; Tsuruya, K.; Iida, M.; Kiyohara, Y.; Sueishi, K. Association of kidney function with coronary atherosclerosis and calcification in autopsy samples from Japanese elders: The Hisayama study. Am. J. Kidney Dis. 2010, 55, 21–30. [Google Scholar] [CrossRef]
- Nakamura, S.; Ishibashi-Ueda, H.; Niizuma, S.; Yoshihara, F.; Horio, T.; Kawano, Y. Coronary calcification in patients with chronic kidney disease and coronary artery disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.; Buzello, M.; Ritz, E.; Stein, G.; Raabe, G.; Wiest, G.; Mall, G.; Amann, K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol. Dial. Transplant. 2000, 15, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campean, V.; Neureiter, D.; Varga, I.; Runk, F.; Reiman, A.; Garlichs, C.; Achenbach, S.; Nonnast-Daniel, B.; Amann, K. Atherosclerosis and vascular calcification in chronic renal failure. Kidney Blood Press. Res. 2005, 28, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants (Basel) 2020, 9, 752. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Block, G.; Horwich, T.; Fonarow, G.C. Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J. Am. Coll. Cardiol. 2004, 43, 1439–1444. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Silverio, D.; Tsomidou, C.; Salcedo, M.D.; Montan, P.D.; Guzman, E. The Impact of Insulin Resistance and Chronic Kidney Disease on Inflammation and Cardiovascular Disease. Clin. Med. Insights Endocrinol. Diabetes 2018, 11, 1179551418792257. [Google Scholar] [CrossRef]
- Iorember, F.M. Malnutrition in Chronic Kidney Disease. Front. Pediatr. 2018, 6, 161. [Google Scholar] [CrossRef]
- Jadeja, Y.P.; Kher, V. Protein energy wasting in chronic kidney disease: An update with focus on nutritional interventions to improve outcomes. Indian J. Endocrinol. Metab. 2012, 16, 246–251. [Google Scholar] [CrossRef]
- Jankowska, M.; Cobo, G.; Lindholm, B.; Stenvinkel, P. Inflammation and Protein-Energy Wasting in the Uremic Milieu. Contrib. Nephrol. 2017, 191, 58–71. [Google Scholar] [CrossRef]
- Florens, N.; Calzada, C.; Lyasko, E.; Juillard, L.; Soulage, C.O. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins (Basel) 2016, 8, 376. [Google Scholar] [CrossRef] [Green Version]
- Kwan, B.C.; Kronenberg, F.; Beddhu, S.; Cheung, A.K. Lipoprotein metabolism and lipid management in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1246–1261. [Google Scholar] [CrossRef]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.; Rothwell, P.M. Disentangling the multiple links between renal dysfunction and cerebrovascular disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Tavakoli, S.; Asmis, R. Reactive oxygen species and thiol redox signaling in the macrophage biology of atherosclerosis. Antioxid. Redox Signal. 2012, 17, 1785–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thevenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jungers, P.; Descamps-Latscha, B. Glutathione antioxidant system as a marker of oxidative stress in chronic renal failure. Free Radic. Biol. Med. 1996, 21, 845–853. [Google Scholar] [CrossRef]
- Johnson-Davis, K.L.; Fernelius, C.; Eliason, N.B.; Wilson, A.; Beddhu, S.; Roberts, W.L. Blood enzymes and oxidative stress in chronic kidney disease: A cross sectional study. Ann. Clin. Lab. Sci. 2011, 41, 331–339. [Google Scholar] [PubMed]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef] [Green Version]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellstrom, B. Oxidative stress and endothelial function in chronic renal failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar]
- Fujii, H.; Nakai, K.; Fukagawa, M. Role of oxidative stress and indoxyl sulfate in progression of cardiovascular disease in chronic kidney disease. Ther. Apher. Dial. 2011, 15, 125–128. [Google Scholar] [CrossRef]
- Granata, S.; Dalla Gassa, A.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. (Lond.) 2015, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Wong, Y.T.; Chen, J.; Ruan, R. Age-related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mech. Ageing Dev. 2007, 128, 286–292. [Google Scholar] [CrossRef]
- Small, D.G. Glenda Oxidative Stress and Antioxidant Therapy in Chronic Kidney and Cardiovascular Disease. 2013. Available online: https://www.intechopen.com/books/oxidative-stress-and-chronic-degenerative-diseases-a-role-for-antioxidants/oxidative-stress-and-antioxidant-therapy-in-chronic-kidney-and-cardiovascular-disease (accessed on 10 August 2020).
- Imig, J.D.; Ryan, M.J. Immune and inflammatory role in renal disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantar-Zadeh, K.; Brennan, M.L.; Hazen, S.L. Serum myeloperoxidase and mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2006, 48, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Nuhu, F.; Bhandari, S. Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anaemia. Pharmaceuticals (Basel) 2018, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Sakamaki, I.; Inai, K.; Tsutani, Y.; Ueda, T.; Tsutani, H. Binding of monosodium urate crystals with idiotype protein efficiently promote dendritic cells to induce cytotoxic T cells. Cancer Sci. 2008, 99, 2268–2273. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Saldanha, J.F.; Yi, D.; Mafra, D.; Fouque, D.; Soulage, C.O. The uremic toxin indoxyl sulfate exacerbates reactive oxygen species production and inflammation in 3T3-L1 adipose cells. Free Radic. Res. 2016, 50, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Maia, L.; Duarte, R.O.; Ponces-Freire, A.; Moura, J.J.; Mira, L. NADH oxidase activity of rat and human liver xanthine oxidoreductase: Potential role in superoxide production. J. Biol. Inorg. Chem. 2007, 12, 777–787. [Google Scholar] [CrossRef]
- Kim, I.Y.; Lee, D.W.; Lee, S.B.; Kwak, I.S. The role of uric acid in kidney fibrosis: Experimental evidences for the causal relationship. Biomed. Res. Int. 2014, 2014, 638732. [Google Scholar] [CrossRef] [Green Version]
- Reddy, E.P.; Suchitra, M.M.; Bitla, A.R.; Sivakumar, V.; Rao, P.S. Antioxidant Enzymes status in South Indian Hemodialysis patients. Int. J. Biol. Med. Res. 2011, 2, 682–687. [Google Scholar]
- Sangeetha Lakshmi, B.; Harini Devi, N.; Suchitra, M.M.; Srinivasa Rao, P.; Siva Kumar, V. Changes in the inflammatory and oxidative stress markers during a single hemodialysis session in patients with chronic kidney disease. Ren. Fail. 2018, 40, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative Stress in Hemodialysis Patients: A Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017, 3081856. [Google Scholar] [CrossRef]
- Krata, N.; Zagozdzon, R.; Foroncewicz, B.; Mucha, K. Oxidative Stress in Kidney Diseases: The Cause or the Consequence? Arch. Immunol. Ther. Exp. (Warsz.) 2018, 66, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.F.; Chang, C.L.; Liou, S.Y. Increase in resting levels of superoxide anion in the whole blood of uremic patients on chronic hemodialysis. Blood Purif. 1998, 16, 290–300. [Google Scholar] [CrossRef]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Ravani, P.; Tripepi, G.; Malberti, F.; Testa, S.; Mallamaci, F.; Zoccali, C. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: A competing risks modeling approach. J. Am. Soc. Nephrol. 2005, 16, 2449–2455. [Google Scholar] [CrossRef]
- You, Y.H.; Okada, S.; Ly, S.; Jandeleit-Dahm, K.; Barit, D.; Namikoshi, T.; Sharma, K. Role of Nox2 in diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F840–F848. [Google Scholar] [CrossRef] [PubMed]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Biol. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Sachse, A.; Wolf, G. Angiotensin II-induced reactive oxygen species and the kidney. J. Am. Soc. Nephrol. 2007, 18, 2439–2446. [Google Scholar] [CrossRef] [Green Version]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Yoon, Y.J.; Choi, H.J.; Park, S.H.; Kim, C.D.; Kim, I.S.; Kwon, T.H.; Do, J.Y.; Kim, S.H.; Ryu, D.H.; et al. Dialysis modality-dependent changes in serum metabolites: Accumulation of inosine and hypoxanthine in patients on haemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Raha, S.; McEachern, G.E.; Myint, A.T.; Robinson, B.H. Superoxides from mitochondrial complex III: The role of manganese superoxide dismutase. Free Radic. Biol. Med. 2000, 29, 170–180. [Google Scholar] [CrossRef]
- Angermuller, S.; Islinger, M.; Volkl, A. Peroxisomes and reactive oxygen species, a lasting challenge. Histochem. Cell Biol. 2009, 131, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Li, K.W.; Seitz, J.; Volkl, A.; Luers, G.H. Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes—Evidence for a natural piggyback import mechanism in mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef]
- Godoy, J.R.; Oesteritz, S.; Hanschmann, E.M.; Ockenga, W.; Ackermann, W.; Lillig, C.H. Segment-specific overexpression of redoxins after renal ischemia and reperfusion: Protective roles of glutaredoxin 2, peroxiredoxin 3, and peroxiredoxin 6. Free Radic. Biol. Med. 2011, 51, 552–561. [Google Scholar] [CrossRef]
- Su, X.; Xu, B.; Yan, B.; Qiao, X.; Wang, L. Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS ONE 2017, 12, e0187550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, B. Is it oxidative stress or free radical stress and why does it matter? Oxid. Antioxid. Med. Sci. 2012, 1, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.E.; Patel, R.P.; Brookes, P.S.; Darley-Usmar, V.M. Nanotransducers in cellular redox signaling: Modification of thiols by reactive oxygen and nitrogen species. Trends Biochem. Sci. 2002, 27, 489–492. [Google Scholar] [CrossRef]
- Miyata, T.; Taneda, S.; Kawai, R.; Ueda, Y.; Horiuchi, S.; Hara, M.; Maeda, K.; Monnier, V.M. Identification of pentosidine as a native structure for advanced glycation end products in beta-2-microglobulin-containing amyloid fibrils in patients with dialysis-related amyloidosis. Proc. Natl. Acad. Sci. USA 1996, 93, 2353–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Kinugasa, E. Markers and possible uremic toxins: Japanese experiences. Contrib. Nephrol. 2011, 168, 134–138. [Google Scholar] [CrossRef]
- Zill, H.; Gunther, R.; Erbersdobler, H.F.; Folsch, U.R.; Faist, V. RAGE expression and AGE-induced MAP kinase activation in Caco-2 cells. Biochem. Biophys. Res. Commun. 2001, 288, 1108–1111. [Google Scholar] [CrossRef]
- Boulanger, E.; Wautier, M.P.; Wautier, J.L.; Boval, B.; Panis, Y.; Wernert, N.; Danze, P.M.; Dequiedt, P. AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Int. 2002, 61, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, G.; Reggiani, F.; Astori, E.; Altomare, A.; Finazzi, S.; Garavaglia, M.L.; Angelini, C.; Milzani, A.; Badalamenti, S.; Dalle-Donne, I. Advanced oxidation protein products in nondiabetic end stage renal disease patients on maintenance haemodialysis. Free Radic. Res. 2019, 53, 1114–1124. [Google Scholar] [CrossRef]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Lepenies, J.; Hewison, M.; Stewart, P.M.; Quinkler, M. Renal PPARgamma mRNA expression increases with impairment of renal function in patients with chronic kidney disease. Nephrology (Carlton) 2010, 15, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Hongo, M.; Saito, K.; Nagai, R.; Ishizaka, N. Reduction of renal lipid content and proteinuria by a PPAR-gamma agonist in a rat model of angiotensin II-induced hypertension. Eur. J. Pharmacol. 2012, 682, 131–136. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [Green Version]
- Haugen, E.; Nath, K.A. The involvement of oxidative stress in the progression of renal injury. Blood Purif. 1999, 17, 58–65. [Google Scholar] [CrossRef]
- Fujii, H.; Kono, K.; Nakai, K.; Goto, S.; Komaba, H.; Hamada, Y.; Shinohara, M.; Kitazawa, R.; Kitazawa, S.; Fukagawa, M. Oxidative and nitrosative stress and progression of diabetic nephropathy in type 2 diabetes. Am. J. Nephrol. 2010, 31, 342–352. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Murdoch, C.; Shah, A.M. Involvement of NADPH oxidases in cardiac remodelling and heart failure. Am. J. Nephrol. 2007, 27, 649–660. [Google Scholar] [CrossRef]
- Clermont, G.; Lecour, S.; Lahet, J.; Siohan, P.; Vergely, C.; Chevet, D.; Rifle, G.; Rochette, L. Alteration in plasma antioxidant capacities in chronic renal failure and hemodialysis patients: A possible explanation for the increased cardiovascular risk in these patients. Cardiovasc. Res. 2000, 47, 618–623. [Google Scholar] [CrossRef]
- Miyazaki, H.; Matsuoka, H.; Itabe, H.; Usui, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Hemodialysis impairs endothelial function via oxidative stress: Effects of vitamin E-coated dialyzer. Circulation 2000, 101, 1002–1006. [Google Scholar] [CrossRef] [Green Version]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, S.; Santanam, N.; Ramachandran, S.; Meilhac, O. Oxidants and antioxidants in atherogenesis. An appraisal. J. Lipid Res. 1999, 40, 2143–2157. [Google Scholar]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Maack, C.; Kartes, T.; Kilter, H.; Schafers, H.J.; Nickenig, G.; Bohm, M.; Laufs, U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation 2003, 108, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Nediani, C.; Borchi, E.; Giordano, C.; Baruzzo, S.; Ponziani, V.; Sebastiani, M.; Nassi, P.; Mugelli, A.; d’Amati, G.; Cerbai, E. NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and right ventricle. J. Mol. Cell. Cardiol. 2007, 42, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Pagano, P.J. The reactive adventitia: Fibroblast oxidase in vascular function. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; Boyd, R.; Zhu, M.; Chapman, A.; Pimentel, D.R.; Wang, H.D. NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc. Res. 2007, 75, 702–709. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work, G. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran J. Kidney Dis. 2015, 9, 165–179. [Google Scholar] [PubMed]
- Ehara, H.; Yamamoto-Honda, R.; Kitazato, H.; Takahashi, Y.; Kawazu, S.; Akanuma, Y.; Noda, M. ApoE isoforms, treatment of diabetes and the risk of coronary heart disease. World J. Diabetes 2012, 3, 54–59. [Google Scholar] [CrossRef]
- Abadir, P.M.; Foster, D.B.; Crow, M.; Cooke, C.A.; Rucker, J.J.; Jain, A.; Smith, B.J.; Burks, T.N.; Cohn, R.D.; Fedarko, N.S.; et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. USA 2011, 108, 14849–14854. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Kobori, H.; Alper, A.B., Jr.; Shenava, R.; Katsurada, A.; Saito, T.; Ohashi, N.; Urushihara, M.; Miyata, K.; Satou, R.; Hamm, L.L.; et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension 2009, 53, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Morrone, D.; Marzilli, M. Role of RAAS inhibition in preventing left ventricular remodeling in patients post myocardial infarction. Heart Metab. 2010, 47, 9–13. [Google Scholar]
- Cave, A.; Grieve, D.; Johar, S.; Zhang, M.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 1998, 97, 2222–2229. [Google Scholar] [CrossRef] [Green Version]
- Tóth, A.; Balogh, E.; Jeney, V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants 2020, 9, 963. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef] [PubMed]
- Rennenberg, R.J.; Kessels, A.G.; Schurgers, L.J.; van Engelshoven, J.M.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Peeters, M.J.; van den Brand, J.A.J.G.; van Zuilen, A.D.; Koster, Y.; Bots, M.L.; Vervloet, M.G.; Blankestijn, P.J.; Wetzels, J.F.M.; Masterplan Study Group. Abdominal aortic calcification in patients with CKD. J. Nephrol. 2017, 30, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.L.; Shao, J.S.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J. Biol. Chem. 2003, 278, 45969–45977. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.S.; Cheng, S.L.; Pingsterhaus, J.M.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Investig. 2005, 115, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Sun, Y.T.; Sun, W.; Xu, T.H.; Ren, C.; Fan, X.; Sun, L.; Liu, L.L.; Feng, J.M.; Ma, J.F.; et al. High phosphorus level leads to aortic calcification via β-catenin in chronic kidney disease. Am. J. Nephrol. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Cai, T.; Sun, D.; Duan, Y.; Wen, P.; Dai, C.; Yang, J.; He, W. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp. Cell Res. 2016, 345, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [CrossRef]
- You, H.; Yang, H.; Zhu, Q.; Li, M.; Xue, J.; Gu, Y.; Lin, S.; Ding, F. Advanced oxidation protein products induce vascular calcification by promoting osteoblastic trans-differentiation of smooth muscle cells via oxidative stress and ERK pathway. Ren. Fail. 2009, 31, 313–319. [Google Scholar] [CrossRef]
- Banerjee, C.; McCabe, L.R.; Choi, J.Y.; Hiebert, S.W.; Stein, J.L.; Stein, G.S.; Lian, J.B. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J. Cell. Biochem. 1997, 66, 1–8. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Witko-Sarsat, V.; Gausson, V.; Nguyen, A.T.; Touam, M.; Drüeke, T.; Santangelo, F.; Descamps-Latscha, B. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: A potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003, 64, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.F.; Chaudhary, L.; Fausto, A.; Halstead, L.R.; Ory, D.S.; Avioli, L.V.; Cheng, S.L. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J. Biol. Chem. 2001, 276, 14443–14450. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Franceschi, R.T. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem. 2002, 277, 36181–36187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziros, P.G.; Gil, A.P.; Georgakopoulos, T.; Habeos, I.; Kletsas, D.; Basdra, E.K.; Papavassiliou, A.G. The bone-specific transcriptional regulator Cbfa1 is a target of mechanical signals in osteoblastic cells. J. Biol. Chem. 2002, 277, 23934–23941. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Jiang, D.; Thomas, P.; Benson, M.D.; Guan, K.; Karsenty, G.; Franceschi, R.T. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J. Biol. Chem. 2000, 275, 4453–4459. [Google Scholar] [CrossRef] [Green Version]
- Snyder, L.M.; Fortier, N.L.; Trainor, J.; Jacobs, J.; Leb, L.; Lubin, B.; Chiu, D.; Shohet, S.; Mohandas, N. Effect of hydrogen peroxide exposure on normal human erythrocyte deformability, morphology, surface characteristics, and spectrin-hemoglobin cross-linking. J. Clin. Investig. 1985, 76, 1971–1977. [Google Scholar] [CrossRef]
- Vives Corrons, J.L.; Miguel-Garcia, A.; Pujades, M.A.; Miguel-Sosa, A.; Cambiazzo, S.; Linares, M.; Dibarrart, M.T.; Calvo, M.A. Increased susceptibility of microcytic red blood cells to in vitro oxidative stress. Eur. J. Haematol. 1995, 55, 327–331. [Google Scholar] [CrossRef]
- Nagababu, E.; Gulyani, S.; Earley, C.J.; Cutler, R.G.; Mattson, M.P.; Rifkind, J.M. Iron-deficiency anaemia enhances red blood cell oxidative stress. Free Radic. Res. 2008, 42, 824–829. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.C.; Van der Linden, P.; Biro, G.; Hu, L.Q. Physiologic aspects of anemia. Crit. Care Clin. 2004, 20, 187–212. [Google Scholar] [CrossRef]
- Lazarte, S.S.; Monaco, M.E.; Jimenez, C.L.; Ledesma Achem, M.E.; Teran, M.M.; Isse, B.A. Erythrocyte Catalase Activity in More Frequent Microcytic Hypochromic Anemia: Beta-Thalassemia Trait and Iron Deficiency Anemia. Adv. Hematol. 2015, 2015, 343571. [Google Scholar] [CrossRef] [Green Version]
- Rifkind, J.M.; Nagababu, E. Hemoglobin redox reactions and red blood cell aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [Green Version]
- Yetgin, S.; Hincal, F.; Basaran, N.; Ciliv, G. Serum selenium status in children with iron deficiency anemia. Acta Haematol. 1992, 88, 185–188. [Google Scholar] [CrossRef]
- Steinmetz, H.T. The role of intravenous iron in the treatment of anemia in cancer patients. Ther. Adv. Hematol. 2012, 3, 177–191. [Google Scholar] [CrossRef]
- Juretic, D.; Tadijanovic, M.; Rekic, B.; Simeon-Rudolf, V.; Reiner, E.; Baricic, M. Serum paraoxonase activities in hemodialyzed uremic patients: Cohort study. Croat. Med. J. 2001, 42, 146–150. [Google Scholar]
- Chen, Y.H.; Hung, S.C.; Tarng, D.C. Serum bilirubin links UGT1A1*28 polymorphism and predicts long-term cardiovascular events and mortality in chronic hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.S.; Hung, S.C.; Wei, Y.H.; Tarng, D.C. GST M1 polymorphism associates with DNA oxidative damage and mortality among hemodialysis patients. J. Am. Soc. Nephrol. 2009, 20, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Canaud, B.; Cristol, J.; Morena, M.; Leray-Moragues, H.; Bosc, J.; Vaussenat, F. Imbalance of oxidants and antioxidants in haemodialysis patients. Blood Purif. 1999, 17, 99–106. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Jun, M.; Venkataraman, V.; Razavian, M.; Cooper, B.; Zoungas, S.; Ninomiya, T.; Webster, A.C.; Perkovic, V. Antioxidants for chronic kidney disease. Cochrane Database Syst. Rev. 2012, 10, CD008176. [Google Scholar] [CrossRef]
- Singh, U.; Devaraj, S.; Jialal, I. Vitamin E, oxidative stress, and inflammation. Annu. Rev. Nutr. 2005, 25, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218. [Google Scholar] [CrossRef]
- Amann, K.; Tornig, J.; Buzello, M.; Kuhlmann, A.; Gross, M.L.; Adamczak, M.; Buzello, M.; Ritz, E. Effect of antioxidant therapy with dl-alpha-tocopherol on cardiovascular structure in experimental renal failure. Kidney Int. 2002, 62, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Koyner, J.L.; Sher Ali, R.; Murray, P.T. Antioxidants. Do they have a place in the prevention or therapy of acute kidney injury? Nephron Exp. Nephrol. 2008, 109, e109–e117. [Google Scholar] [CrossRef]
- Wang, S.; Eide, T.C.; Sogn, E.M.; Berg, K.J.; Sund, R.B. Plasma ascorbic acid in patients undergoing chronic haemodialysis. Eur. J. Clin. Pharmacol. 1999, 55, 527–532. [Google Scholar] [CrossRef]
- Deicher, R.; Ziai, F.; Bieglmayer, C.; Schillinger, M.; Horl, W.H. Low total vitamin C plasma level is a risk factor for cardiovascular morbidity and mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2005, 16, 1811–1818. [Google Scholar] [CrossRef]
- Chazot, C.; Jean, G.; Kopple, J.D. Can Outcomes be Improved in Dialysis Patients by Optimizing Trace Mineral, Micronutrient, and Antioxidant Status? The Impact of Vitamins and their Supplementation. Semin. Dial. 2016, 29, 39–48. [Google Scholar] [CrossRef]
- Jankowska, M.; Rutkowski, B.; Debska-Slizien, A. Vitamins and Microelement Bioavailability in Different Stages of Chronic Kidney Disease. Nutrients 2017, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Freundlich, M.; Quiroz, Y.; Zhang, Z.; Zhang, Y.; Bravo, Y.; Weisinger, J.R.; Li, Y.C.; Rodriguez-Iturbe, B. Suppression of renin-angiotensin gene expression in the kidney by paricalcitol. Kidney Int. 2008, 74, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Deb, D.K.; Sun, T.; Wong, K.E.; Zhang, Z.; Ning, G.; Zhang, Y.; Kong, J.; Shi, H.; Chang, A.; Li, Y.C. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. 2010, 77, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Kang, Y.S.; Dai, C.; Liu, Y. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J. Am. Soc. Nephrol. 2011, 22, 90–103. [Google Scholar] [CrossRef]
- Akizawa, T.; Asano, Y.; Morita, S.; Wakita, T.; Onishi, Y.; Fukuhara, S.; Gejyo, F.; Matsuo, S.; Yorioka, N.; Kurokawa, K.; et al. Effect of a carbonaceous oral adsorbent on the progression of CKD: A multicenter, randomized, controlled trial. Am. J. Kidney Dis. 2009, 54, 459–467. [Google Scholar] [CrossRef]
- Ishikawa, I.; Araya, M.; Hayama, T.; Sugano, M.; Yamato, H.; Ise, M. Effect of oral adsorbent (AST-120) on renal function, acquired renal cysts and aortic calcification in rats with adriamycin nephropathy. Nephron 2002, 92, 399–406. [Google Scholar] [CrossRef]
- Shimoishi, K.; Anraku, M.; Kitamura, K.; Tasaki, Y.; Taguchi, K.; Hashimoto, M.; Fukunaga, E.; Maruyama, T.; Otagiri, M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 2007, 24, 1283–1289. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Nishijima, F.; Goto, S.; Sugano, M.; Yamato, H.; Kitazawa, R.; Kitazawa, S.; Fukagawa, M. Oral charcoal adsorbent (AST-120) prevents progression of cardiac damage in chronic kidney disease through suppression of oxidative stress. Nephrol. Dial. Transplant. 2009, 24, 2089–2095. [Google Scholar] [CrossRef] [Green Version]
- Nakai, K.F.H.; Kono, K.; Goto, S. Effect of AST-120 on cardiac abnormalities in patients with chronic kidney disease (CKD) stage 4 and 5. In Proceedings of the XLVII ERA-EDTA Congress—II DGfN Congress, Munich, Germany, 25–28 June 2010. [Google Scholar]
- Mayo, J.C.; Sainz, R.M.; Tan, D.X.; Hardeland, R.; Leon, J.; Rodriguez, C.; Reiter, R.J. Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), in macrophages. J. Neuroimmunol. 2005, 165, 139–149. [Google Scholar] [CrossRef]
- Quiroz, Y.; Ferrebuz, A.; Romero, F.; Vaziri, N.D.; Rodriguez-Iturbe, B. Melatonin ameliorates oxidative stress, inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction. Am. J. Physiol.-Ren. Physiol. 2008, 294, F336–F344. [Google Scholar] [CrossRef]
- Carrillo-Vico, A.; Guerrero, J.M.; Lardone, P.J.; Reiter, R.J. A review of the multiple actions of melatonin on the immune system. Endocrine 2005, 27, 189–200. [Google Scholar] [CrossRef]
- Eghbal, M.A.; Eftekhari, A.; Ahmadian, E.; Azarmi, Y.; Parvizpur, A. A review of biological and pharmacological actions of melatonin: Oxidant and prooxidant properties. Pharm. Bioprocess. 2016, 4, 69–81. [Google Scholar]
- Vázquez, J.; González, B.; Sempere, V.; Mas, A.; Torija, M.J.; Beltran, G. Melatonin Reduces Oxidative Stress Damage Induced by Hydrogen Peroxide in Saccharomyces cerevisiae. Front. Microbiol. 2017, 8, 1066. [Google Scholar] [CrossRef] [PubMed]
- Kratz, E.M.; Piwowar, A. Melatonin, advanced oxidation protein products and total antioxidant capacity as seminal parameters of prooxidant-antioxidant balance and their connection with expression of metalloproteinases in context of male fertility. J. Physiol. Pharmacol. 2017, 68, 659–668. [Google Scholar]
- Gitto, E.; Aversa, S.; Reiter, R.J.; Barberi, I.; Pellegrino, S. Update on the use of melatonin in pediatrics. J. Pineal Res. 2011, 50, 21–28. [Google Scholar] [CrossRef]
- Rahman, A.; Hasan, A.U.; Kobori, H. Melatonin in chronic kidney disease: A promising chronotherapy targeting the intrarenal renin–angiotensin system. Hypertens. Res. 2019, 42, 920–923. [Google Scholar] [CrossRef]
- Qiu, F.; Liu, X.; Zhang, Y.; Wu, Y.; Xiao, D.; Shi, L. Aerobic exercise enhanced endothelium-dependent vasorelaxation in mesenteric arteries in spontaneously hypertensive rats: The role of melatonin. Hypertens. Res. 2018, 41, 718–729. [Google Scholar] [CrossRef]
- Agabiti-Rosei, C.; Favero, G.; De Ciuceis, C.; Rossini, C.; Porteri, E.; Rodella, L.F.; Franceschetti, L.; Maria Sarkar, A.; Agabiti-Rosei, E.; Rizzoni, D.; et al. Effect of long-term treatment with melatonin on vascular markers of oxidative stress/inflammation and on the anticontractile activity of perivascular fat in aging mice. Hypertens. Res. 2017, 40, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Erşahin, M.; Sehirli, O.; Toklu, H.Z.; Süleymanoglu, S.; Emekli-Alturfan, E.; Yarat, A.; Tatlidede, E.; Yeğen, B.C.; Sener, G. Melatonin improves cardiovascular function and ameliorates renal, cardiac and cerebral damage in rats with renovascular hypertension. J. Pineal Res. 2009, 47, 97–106. [Google Scholar] [CrossRef]
- Andrews, N.P.; Prasad, A.; Quyyumi, A.A. N-acetylcysteine improves coronary and peripheral vascular function. J. Am. Coll. Cardiol. 2001, 37, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Giam, B.; Kaye, D.M.; Rajapakse, N.W. Role of Renal Oxidative Stress in the Pathogenesis of the Cardiorenal Syndrome. Heart Lung Circ. 2016, 25, 874–880. [Google Scholar] [CrossRef]
- Mehmetoglu, I.; Yerlikaya, F.H.; Kurban, S.; Erdem, S.S.; Tonbul, Z. Oxidative stress markers in hemodialysis and peritoneal dialysis patients, including coenzyme Q10 and ischemia-modified albumin. Int. J. Artif. Organs 2012, 35, 226–232. [Google Scholar] [CrossRef]
- Rivara, M.B.; Yeung, C.K.; Robinson-Cohen, C.; Phillips, B.R.; Ruzinski, J.; Rock, D.; Linke, L.; Shen, D.D.; Ikizler, T.A.; Himmelfarb, J. Effect of Coenzyme Q10 on Biomarkers of Oxidative Stress and Cardiac Function in Hemodialysis Patients: The CoQ10 Biomarker Trial. Am. J. Kidney Dis. 2017, 69, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Nickenig, G.; Harrison, D.G. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part I: Oxidative stress and atherogenesis. Circulation 2002, 105, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Pueyo, M.E.; Gonzalez, W.; Nicoletti, A.; Savoie, F.; Arnal, J.F.; Michel, J.B. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Imaeda, A.; Tanigawa, T.; Aoki, T.; Kondo, Y.; Nakamura, N.; Yoshikawa, T. Antioxidative effects of fluvastatin and its metabolites against oxidative DNA damage in mammalian cultured cells. Free Radic. Res. 2001, 35, 789–801. [Google Scholar] [CrossRef]
- Paoletti, E.; Bellino, D.; Cassottana, P.; Rolla, D.; Cannella, G. Left ventricular hypertrophy in nondiabetic predialysis CKD. Am. J. Kidney Dis. 2005, 46, 320–327. [Google Scholar] [CrossRef]
Figure 1.
The role of oxidative stress in the development of cardiac remodeling and heart failure.

Figure 2.
The role of oxidative stress in the development of atherosclerosis and other adverse consequences.
Figure 2.
The role of oxidative stress in the development of atherosclerosis and other adverse consequences.


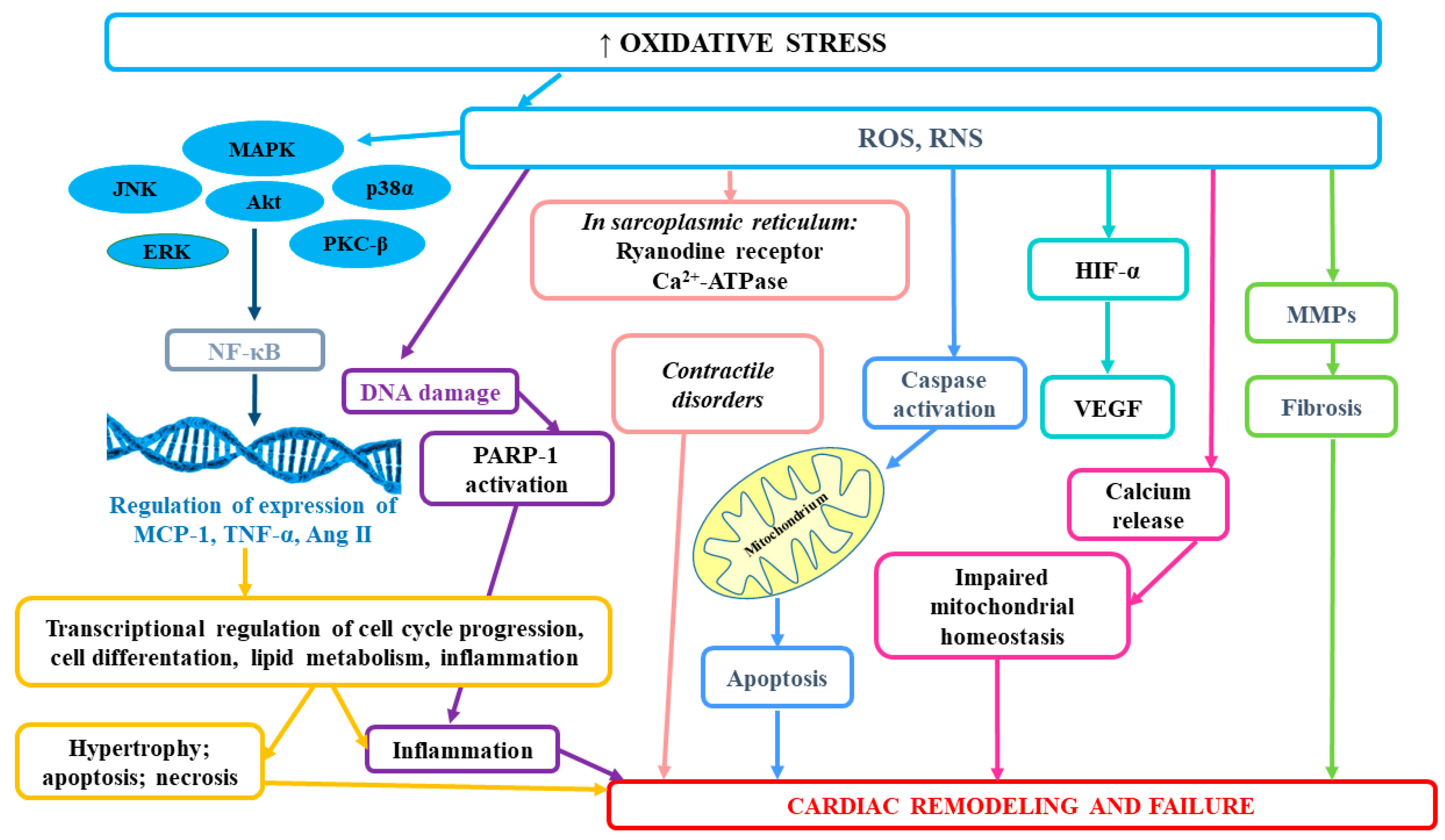{kind=link}
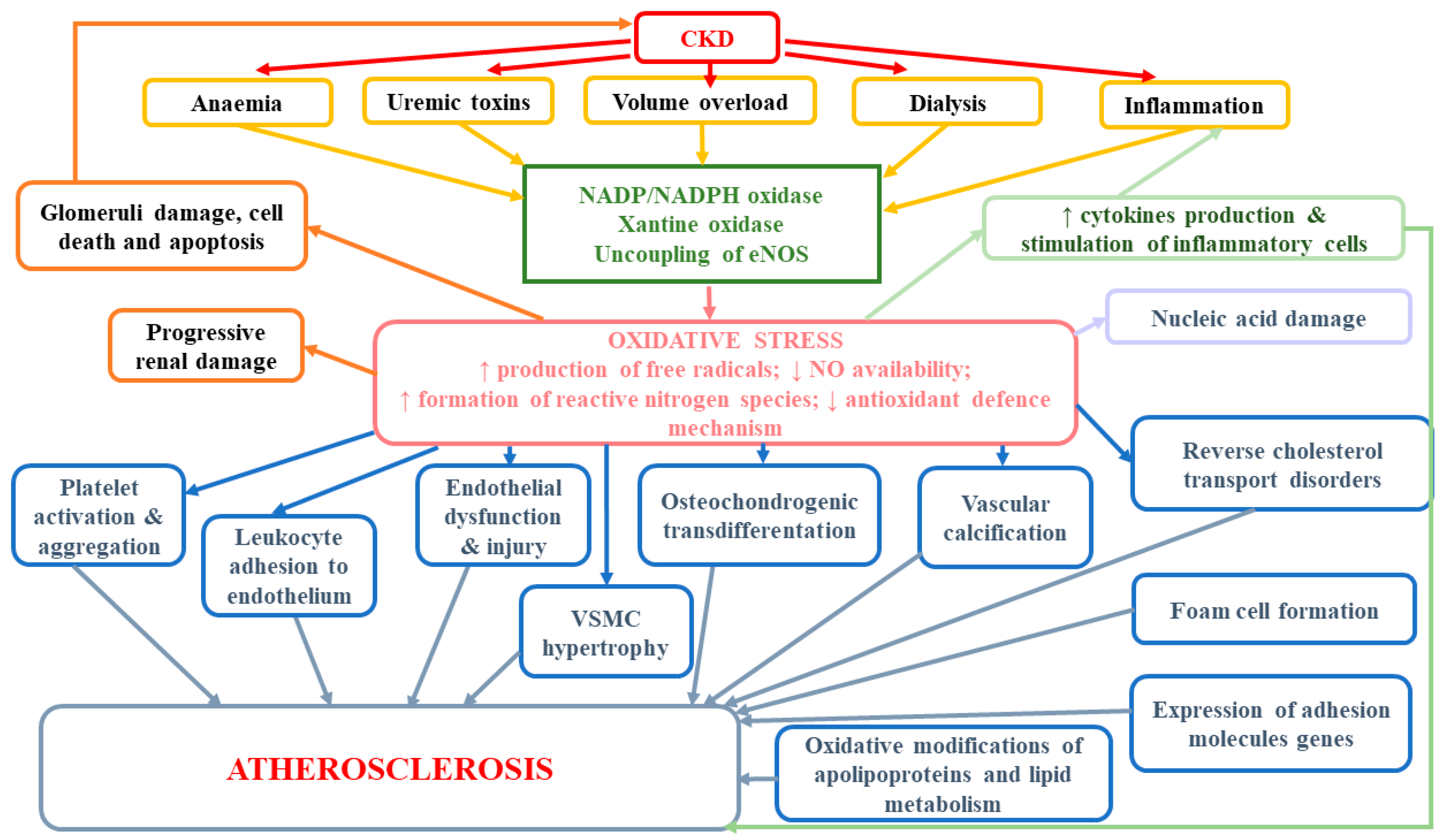{kind=link}
Table 1.
The results of selected articles concerning the adverse impact of oxidative stress.
| Type of Study | Study Group | Most Important Results | Ref |
|---|---|---|---|
| Prospective, randomized, placebo-controlled trial | 134 HD patients randomly assigned either to receive acetylcysteine (600 mg BID) or placebo | Patients in the acetylcysteine group had 40% lower risk of reaching the primary end point (cardiac event, ischemic stroke, peripheral vascular disease) compared with the control group (relative risk, 0.60 [95% CI, 0.38 to 0.95], p = 0.03). Oxidized LDL was significantly lower in the acetylcysteine group compared with the control group (0.13 ± 0.22 arbitrary units vs. 0.55 ± 0.14 arbitrary units, p < 0.01). Conclusions: In hemodialysis patients, treatment with acetylcysteine reduces composite cardiovascular end points. | [13] |
| Case-control | 244 nondiabetic patients with CKD (57 patients with stages 1 to 2 CKD and 187 patients with stages 3 to 5 CKD and 52 normotensive healthy subjects (controls) | LVH is already present in the early stages of renal disease. Strong relationship between elevated pulse pressure and LVH in those with more advanced CKD suggests that increased arterial stiffness might have a role for LVH well before the start of dialysis therapy. | [182] |
| Randomized placebo-controlled Secondary prevention with antioxidants of cardiovascular disease in end-stage renal disease (SPACE) trial | 196 HD patients with pre-existing CAD randomized to receive 800 IU/day vitamin E or matching placebo | Fifteen (16%) patients assigned to vitamin E and 33 (33%) of those assigned to placebo had a primary endpoint (relative risk 0.46 [95% CI 0.27–0.78], p = 0.014); 5.1% patients assigned to vitamin E and 17.2% patients assigned to placebo had myocardial infarction (0.3 [0.11–0.78], p = 0.016). Conclusions: In hemodialysis patients with prevalent cardiovascular disease, supplementation with 800 IU/day vitamin E reduces composite cardiovascular disease endpoints and myocardial infarction. | [149] |
| A meta-analysis of prospective studies | 30 articles reporting calcifications and cardiovascular end-points | The presence of calcifications increased the risk for any cardiovascular event. In a population with renal insufficiency, the event rate for all-cause mortality in patients with calcifications was more than five times higher than in patients without calcifications. The presence of calcifications had the highest predictive power for a cardiovascular or cerebrovascular event in subjects with renal insufficiency. | [114] |
| Randomized controlled trial | 280 patients with CKD not on HD enrolled in the MASTERPLAN study | AAC occur commonly in populations of non-dialysis CKD patients. Calcification score ≥ 4 was associated with cardiovascular events; HR for cardiovascular events in the high calcification score group was 5.5 (95 % confidence interval 1.2–24.8), p = 0.03 | [115] |
| Case-control and animal models | Patients from a clinical trial ChiCTR-OCH-14004447: (a) 24 HD patients vs. 13 healthy individuals (b) patients with CKD stage 2–3 (n = 30), patients with CKD5 (n = 30), and normal adults (n = 15) | A robust elevation in oxidative stress in HD patients vs healthy individuals; the elevation was higher in patients with VC than those without VC. Kinetic relationship among oxidative stress, osteoblastic transition and VC following CKD progression were indicated; the magnitude of osteoblastic transition did not further increase from E4wkCKD to E5wkCKD in rats, suggesting complex contributions of osteoblastic transition to OS-associated VC in early stage CKD. Serum OS levels were increased in both CKD2–3 and CKD5 patients compared to healthy controls, which is consistent with a role of OS in causing osteoblastic transition-mediated VC. This study supported a direct role of NOX1 in the induction of VC in patients with CKD. Conclusions: Oxidative stress plays a role in VC development in HD patients. OS without an increase in serum phosphorus concurrently exists with VC in patients with early stage CKD and in a rat model for early stage CKD. Serum from CDK2–3 patients with OS abnormalities and normal levels of serum phosphorus directly induce calcium deposition in primary VSMCs. Osteoblastic transition of VSMCs contributes to VC in CKD patients; the phenotype switch is in part enhanced by OS, NOX1, and ERK. | [116] |
| VSMC culture; Western Blot Analysis | N/A | H2O2 at concentrations of 0.1 to 0.4 mm induced osteogenic differentiation and calcification of VSMC in a concentration-dependent manner. Runx2, a key transcription factor for osteoblast and chondrocyte differentiation, was required for oxidative stress-induced VSMC calcification. Conclusions: Enhanced expression of Runx2 is sufficient to induce VSMC calcification. Activation of AKT signaling appears to mediate oxidative stress-induced Runx2 expression and activity during VSMC calcification. | [118] |
| Cell culture; animal model | N/A | Runx2 is a direct transcriptional target of WNT/β-catenin signaling pathway. WNT/β-catenin signaling could play a crucial role in promoting VSMCs osteogenic trans-differentiation and the development and progression of vascular calcification. | [122] |
| Human Smooth Muscle Cells (HASMC) Culture | N/A | AOPP increased the calcium content of HASMCs suggesting that AOPP can induce calcification of HASMCs AOPP considerably increased the protein and mRNA expression of OPN in HASMCs, indicating that AOPP can induce osteoblast differentiation of HASMCs; AOPP up-regulated the mRNA expression of CBF-α1 transcription factor. enhancing the expression of osteoblast-specific genes (osteocalcin, alkaline phosphatase). Conclusions: AOPP can trans-differentiate the HASMCs to osteoblast-like cells. | [124] |
CRF—chronic renal failure; HD—maintenance hemodialysis; AAC—abdominal aortic calcification; HR—hazard ratio; AOPP—advanced oxidation protein products.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rysz, J.; Franczyk, B.; Ławiński, J.; Gluba-Brzózka, A. Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases. Antioxidants 2020, 9, 1079. https://doi.org/10.3390/antiox9111079
AMA Style
Rysz J, Franczyk B, Ławiński J, Gluba-Brzózka A. Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases. Antioxidants. 2020; 9(11):1079. https://doi.org/10.3390/antiox9111079
Chicago/Turabian StyleRysz, Jacek, Beata Franczyk, Janusz Ławiński, and Anna Gluba-Brzózka. 2020. "Oxidative Stress in ESRD Patients on Dialysis and the Risk of Cardiovascular Diseases" Antioxidants 9, no. 11: 1079. https://doi.org/10.3390/antiox9111079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.