
Synergic Effect of Phthalide Lactones and Fluconazole and Its New Analogues as a Factor Limiting the Use of Azole Drugs against Candidiasis


 , ,
, ,
Abstract
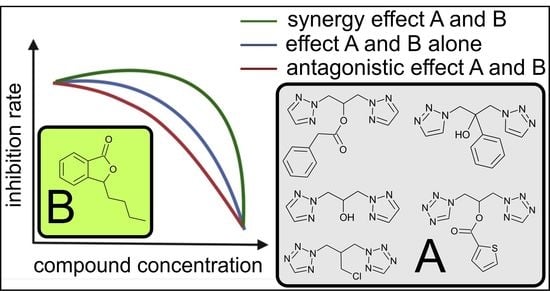
:
1. Introduction
1.1. Candida Albicans as a Potentially Infectious Disease Agent
1.2. Pharmacological Use of Fluconazole and Associated Risks
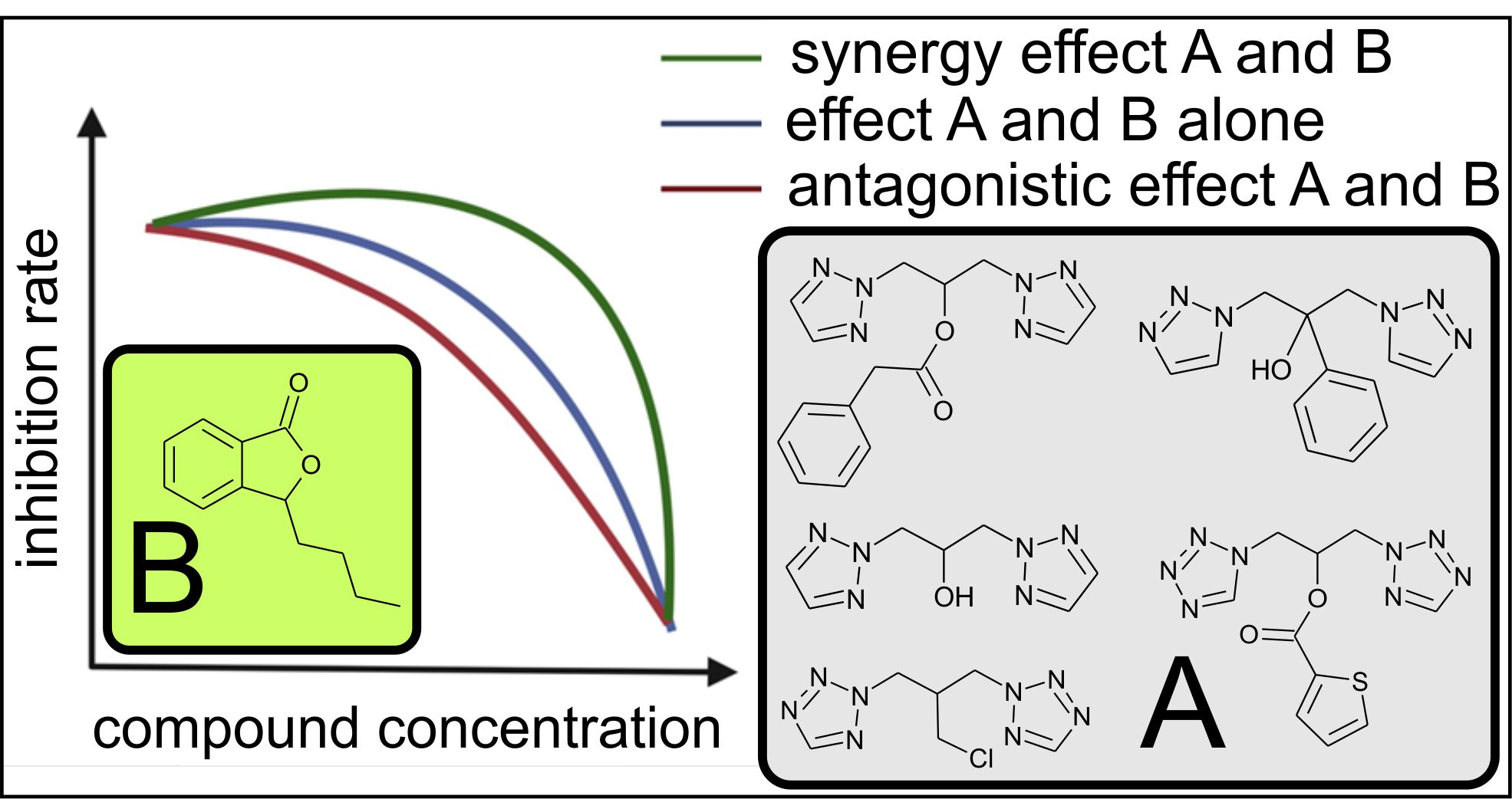
1.3. Mechanism of Action of Fluconazole against Candida Yeast
1.4. Phthalide Lactones, Isolated Plants of the Apiaceae Lindl Family
1.5. Mechanism of Action of 3-n-Butylphthalide on Candida Albicans Cells
2. Results and Discussion
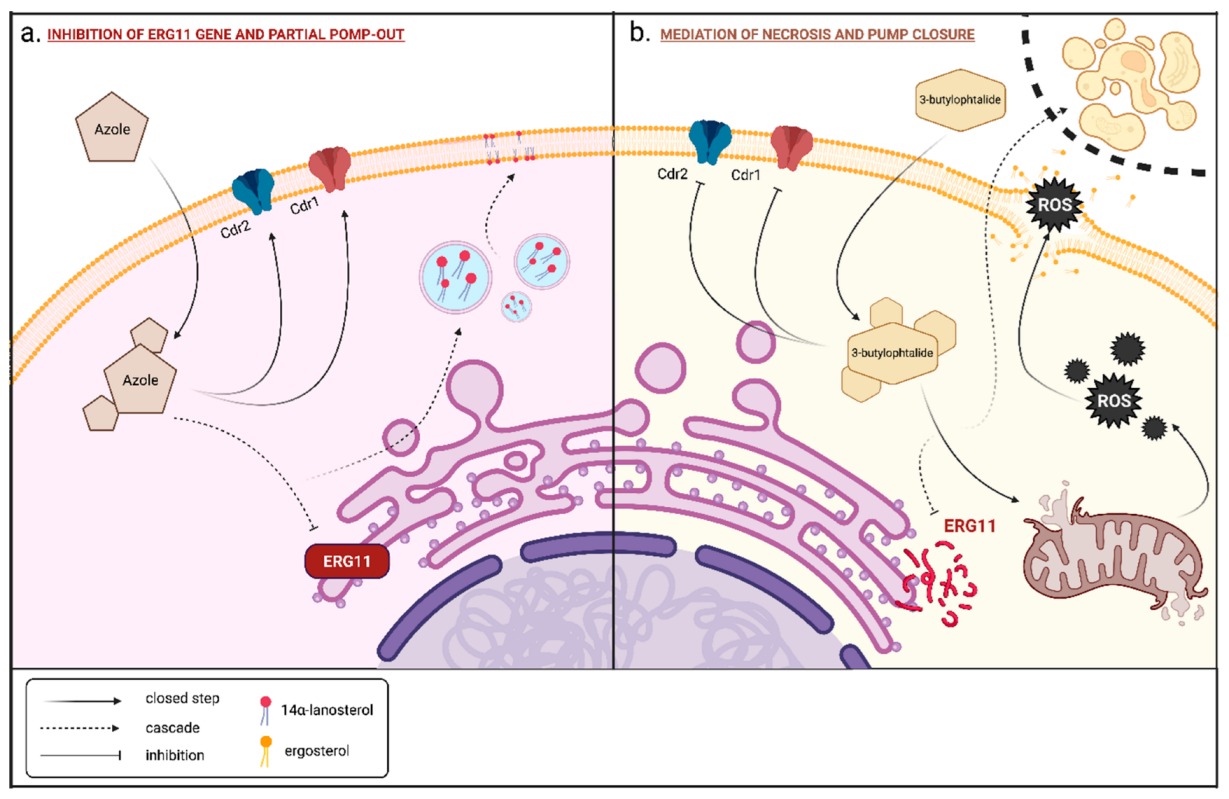
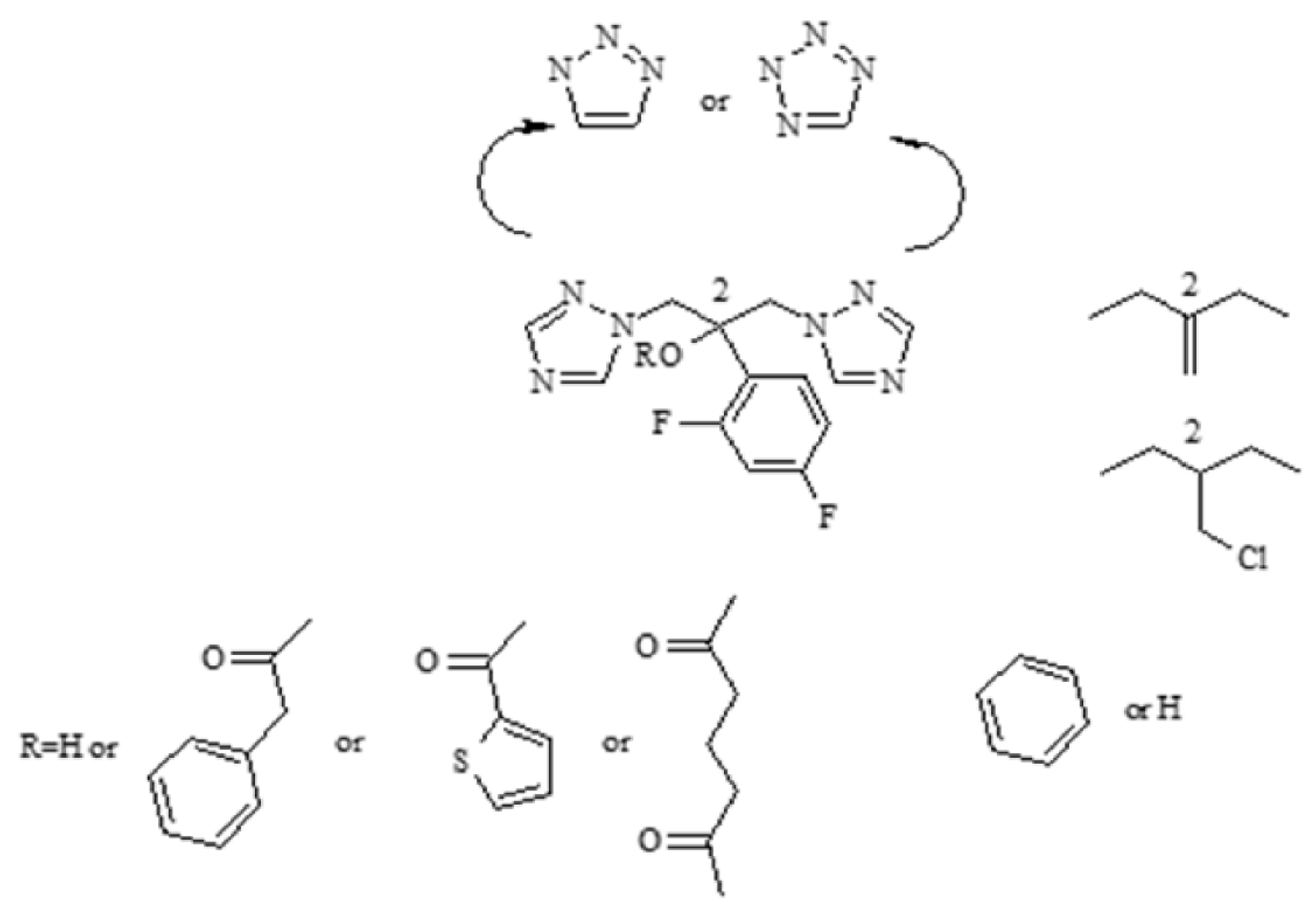
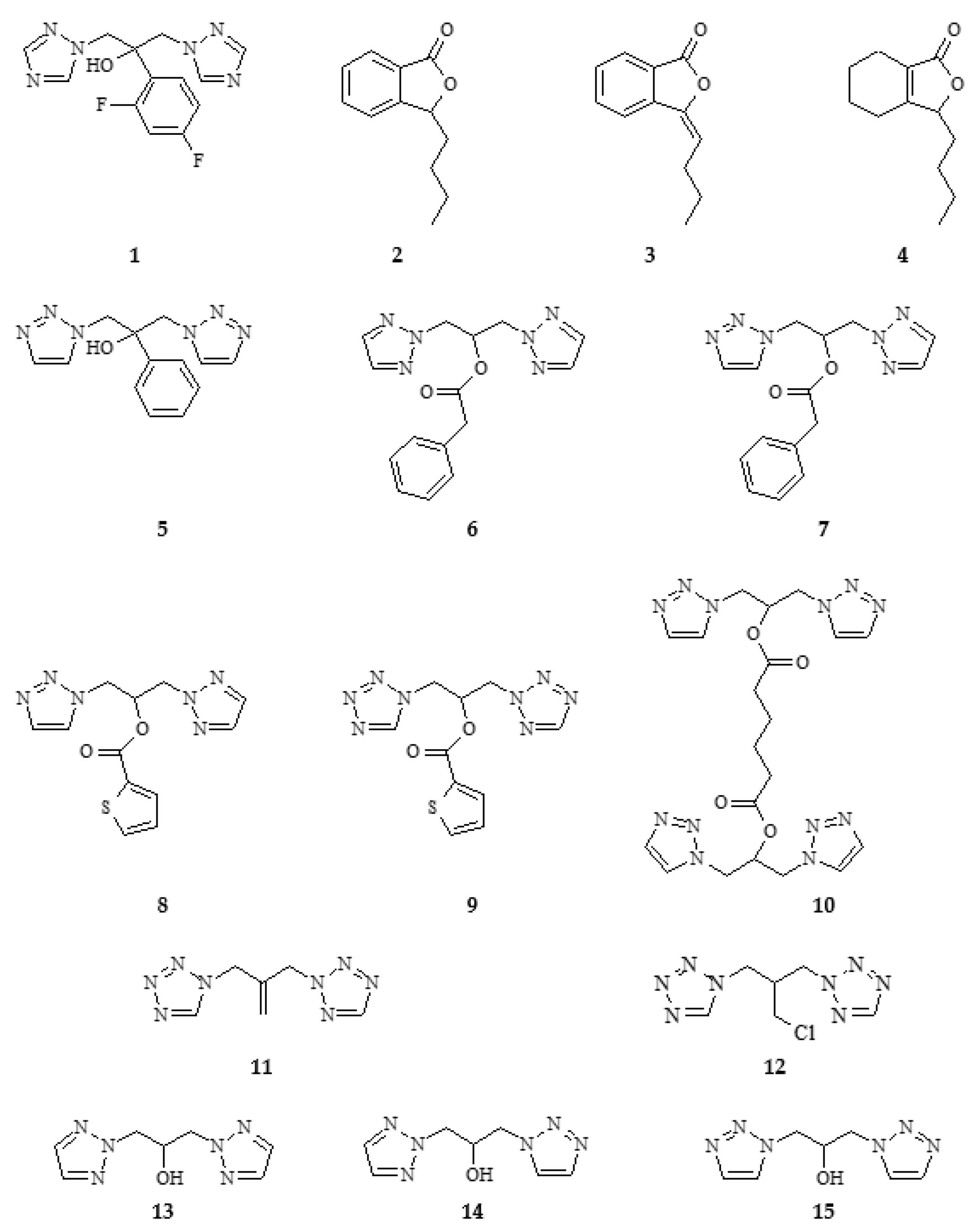
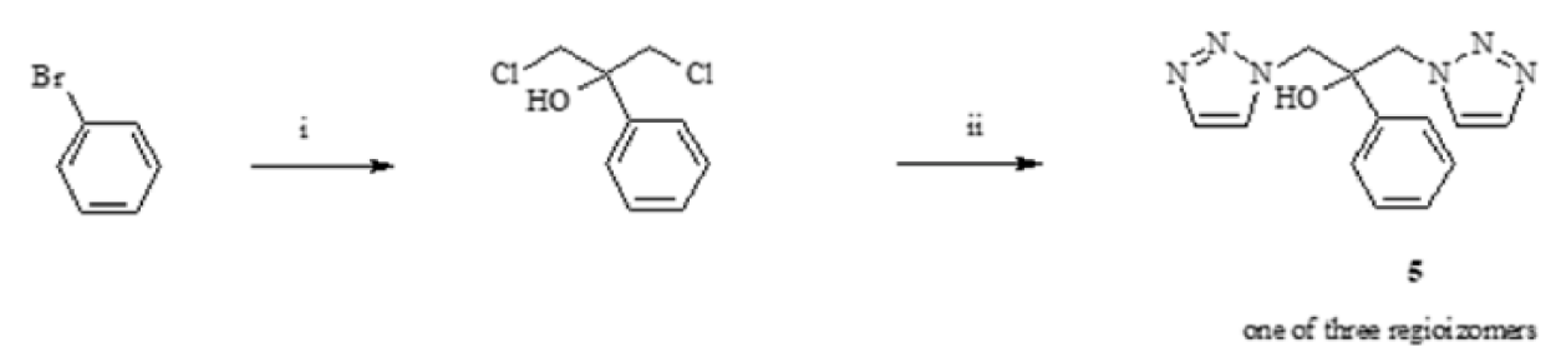
2.1. Synthesis of Phthalide Lactones 2–4 and Analogues of Fluconazole 5–15
Synthesis of Phthalide Lactones 2–4
2.2. In Silico Studies
2.2.1. Phthalide Lactones 2–4
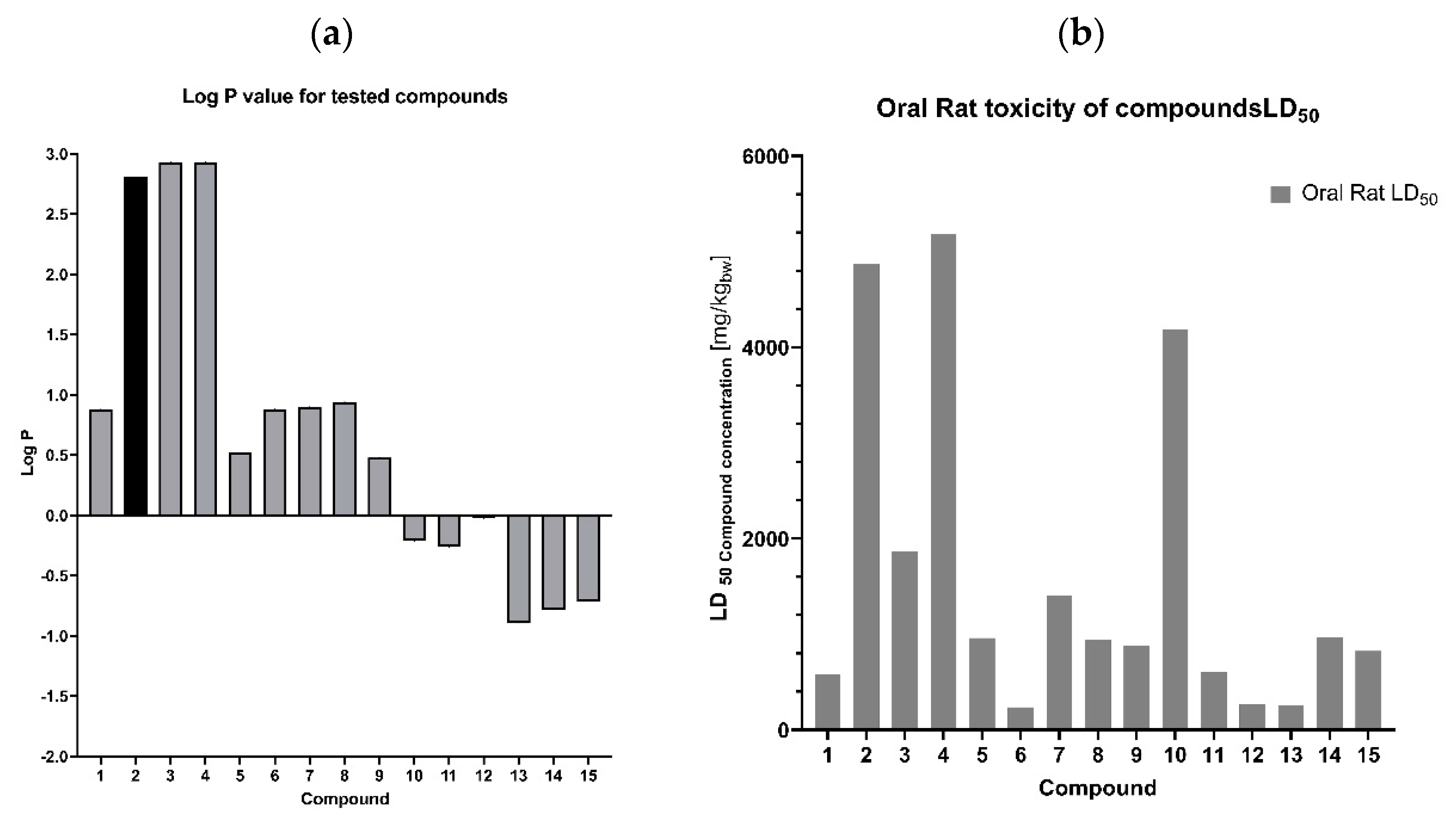
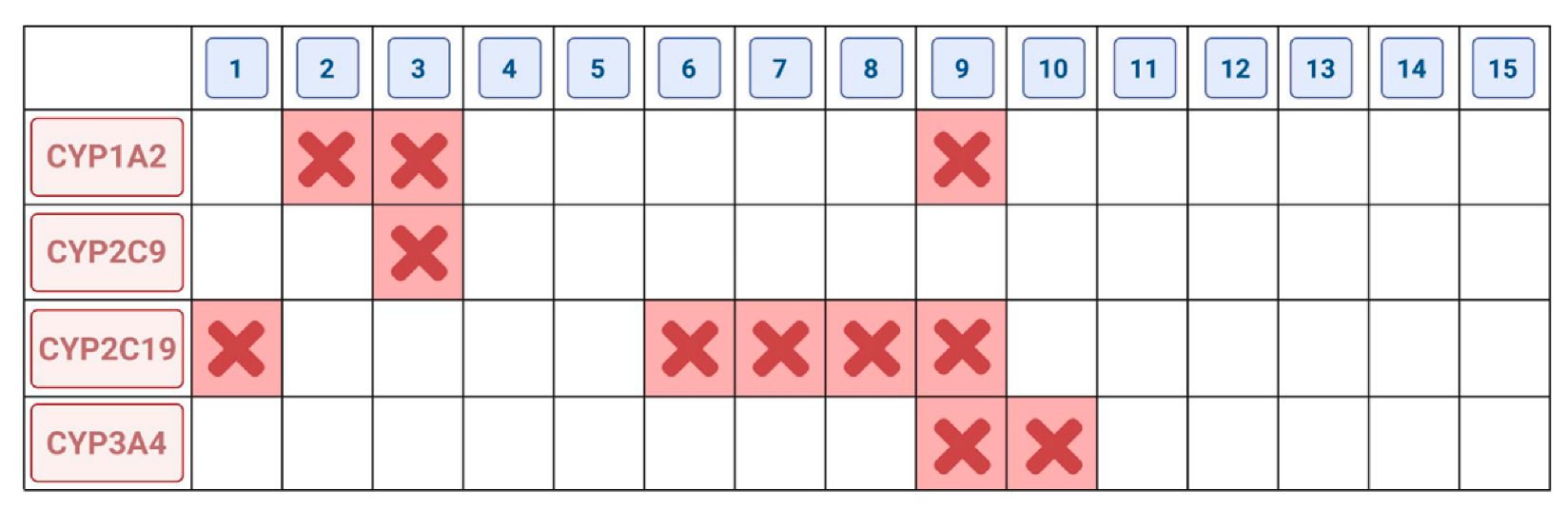
2.2.2. Fluconazole (2) and Its Analogues 5–15
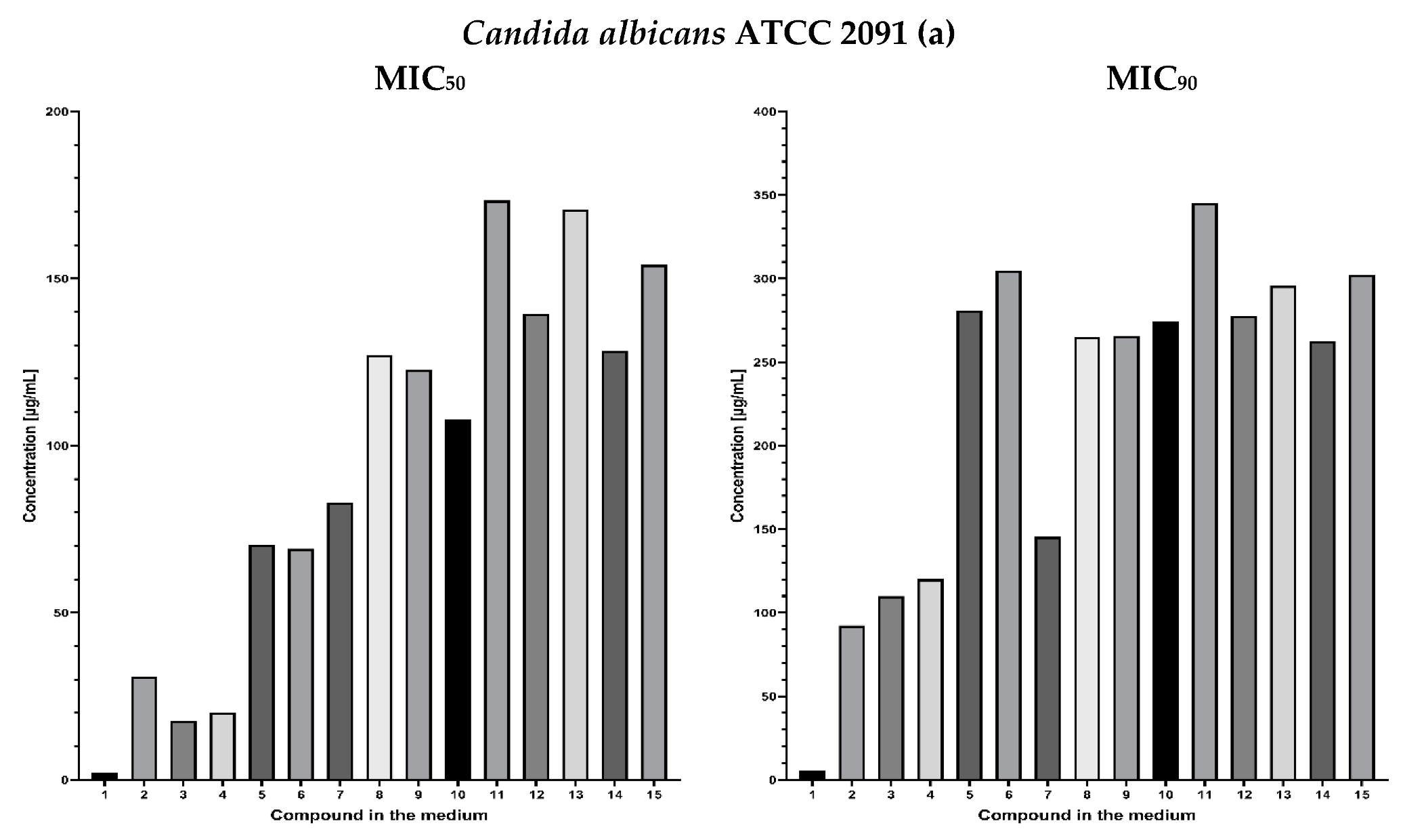
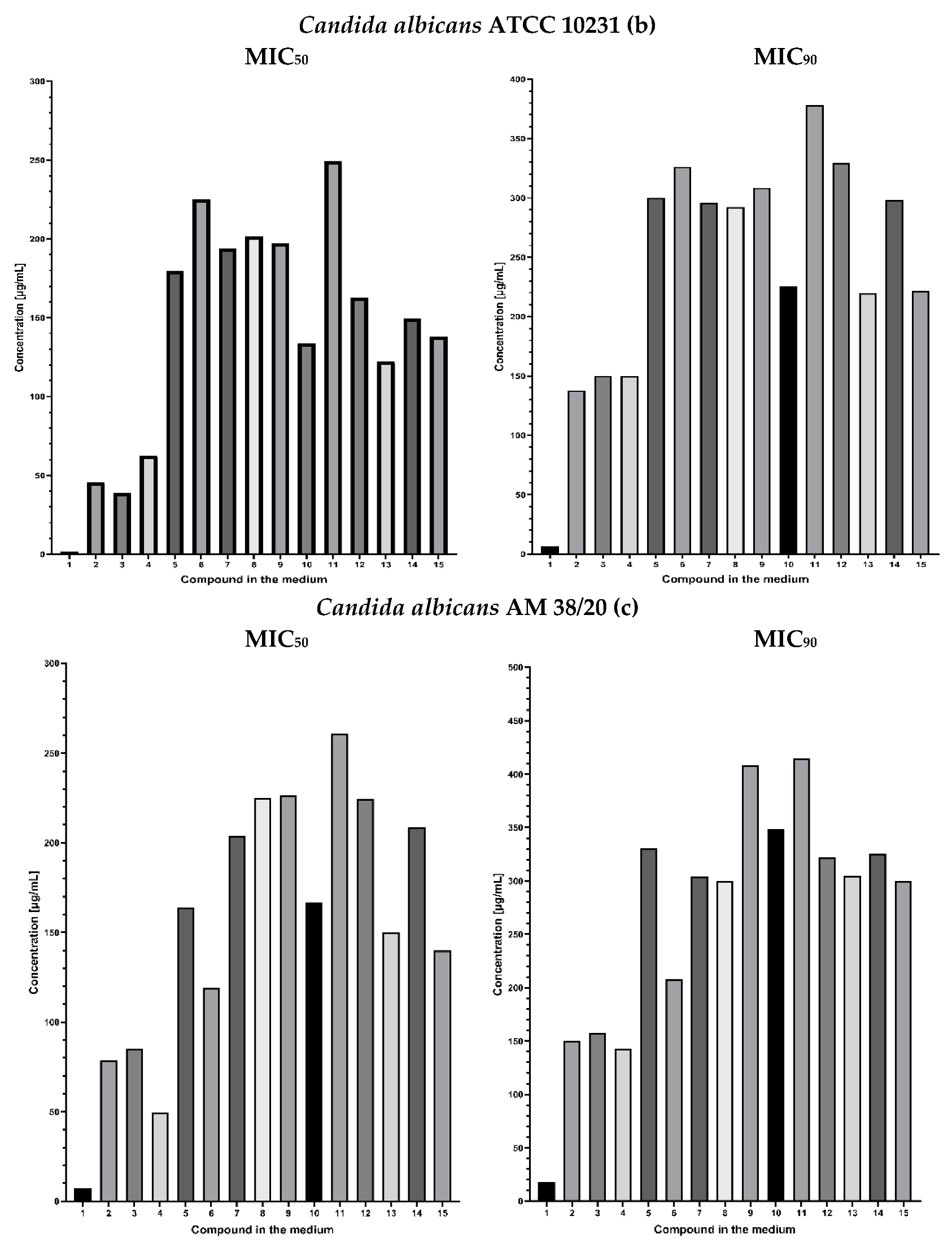
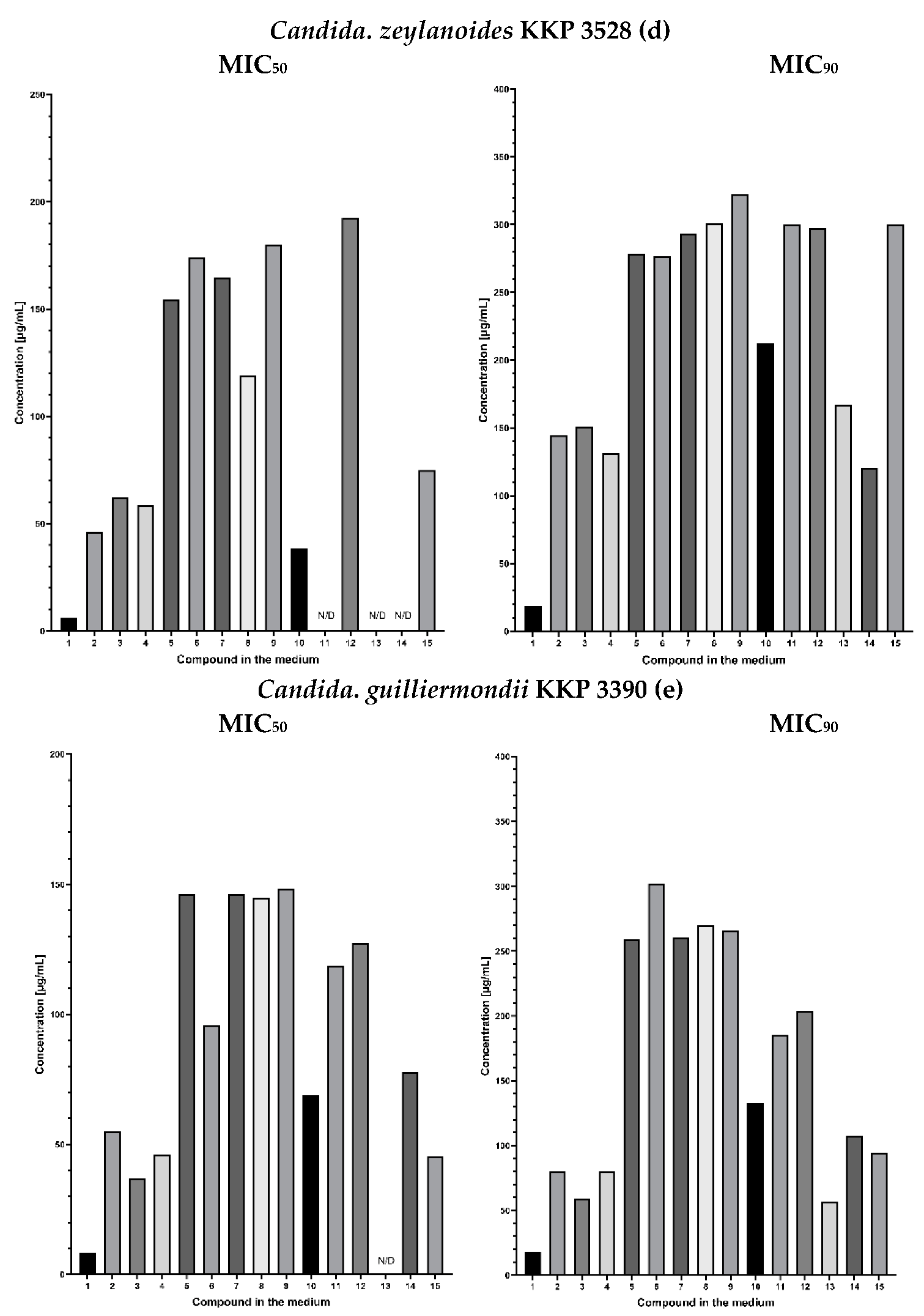
2.3. Fungistatic Activity
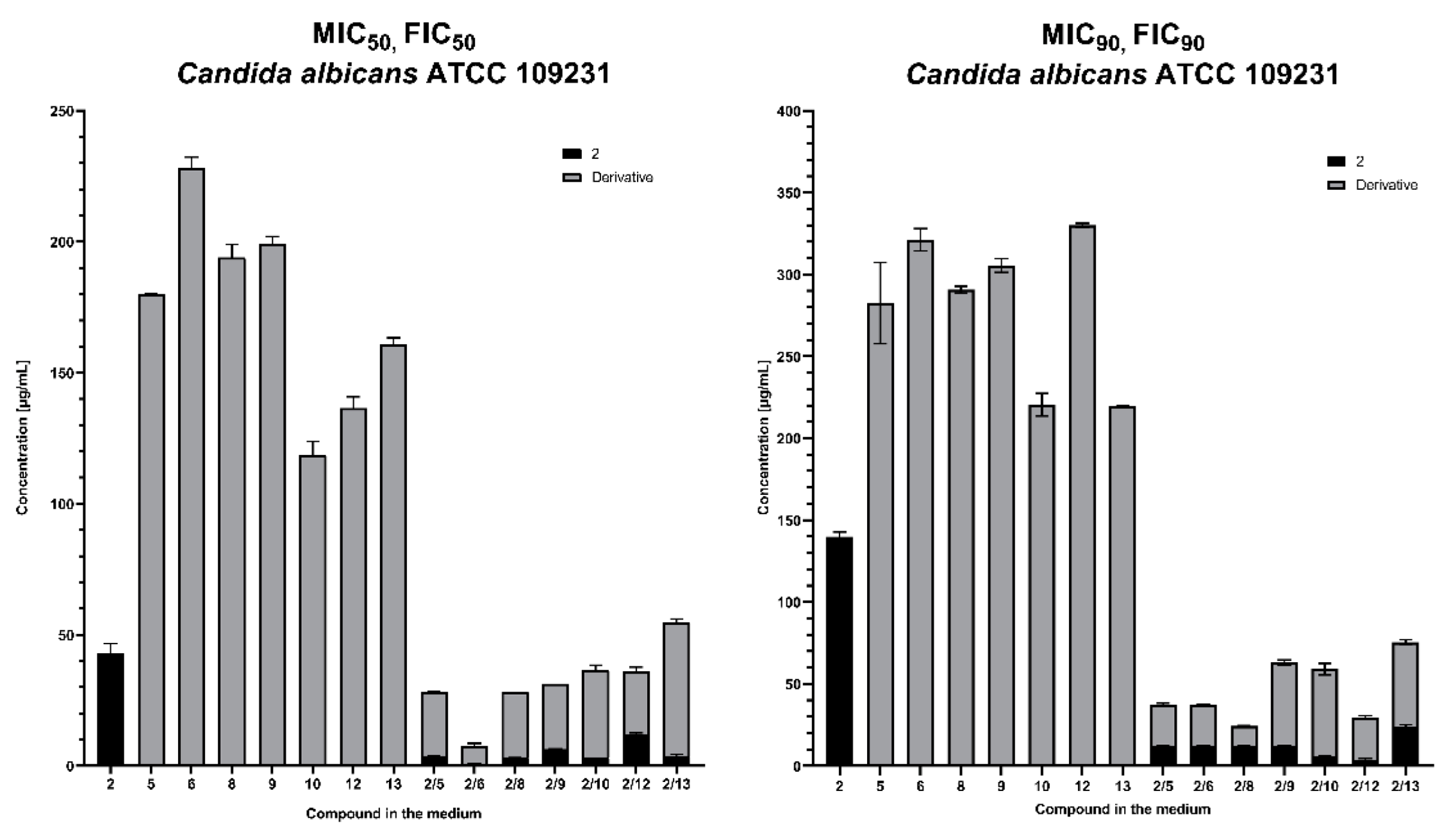
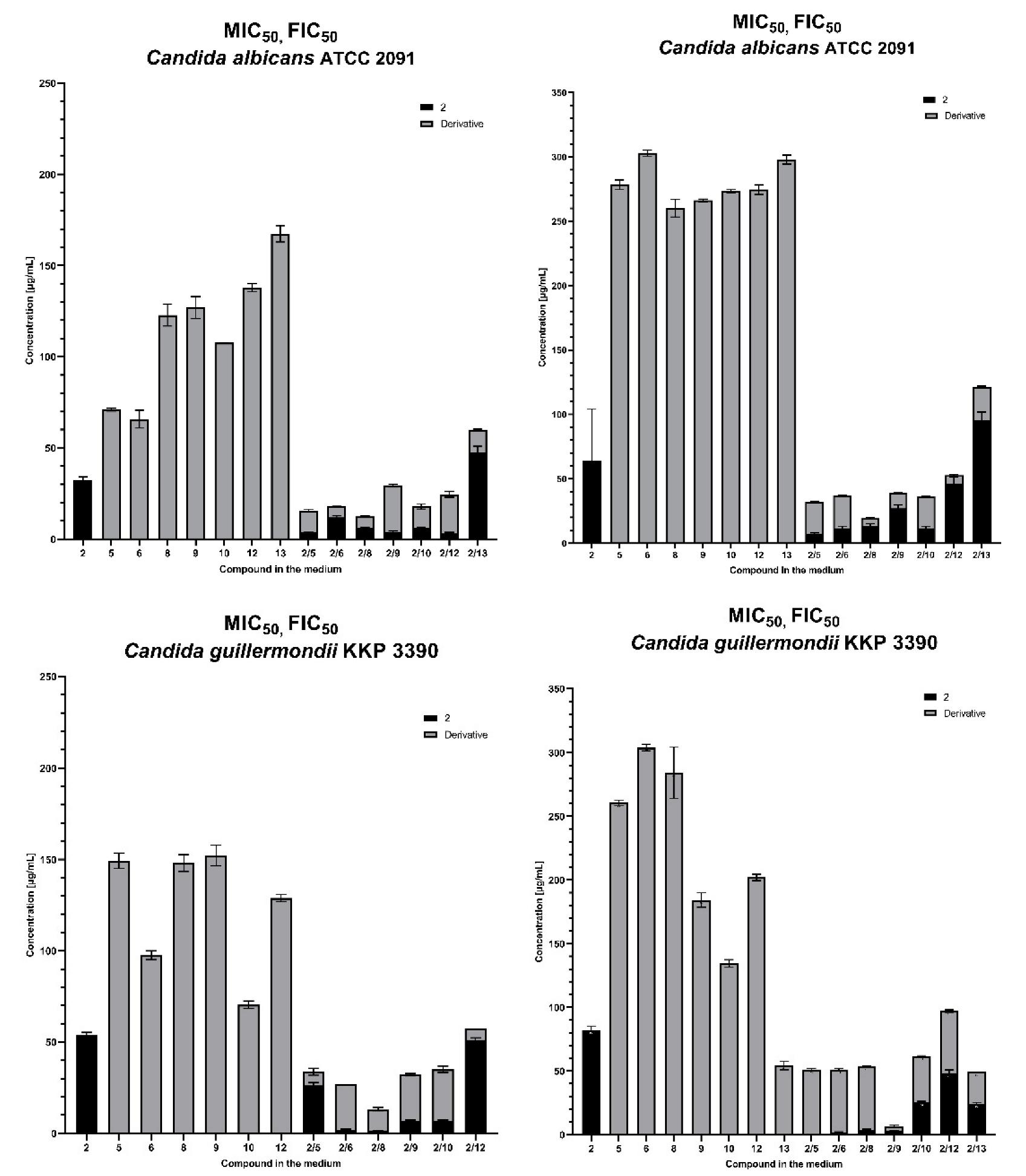
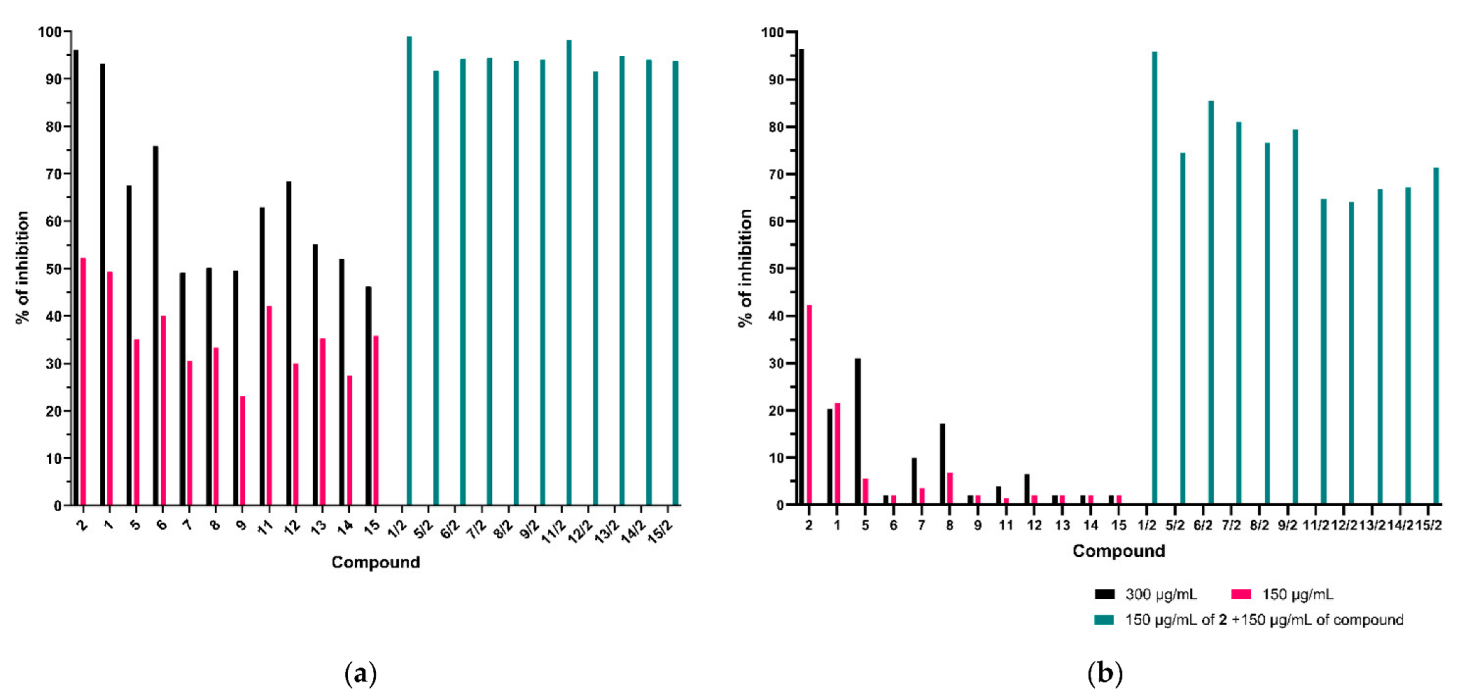
2.3.1. Synergic Effect of Lactones 2–4 and Fluconazole in Combination
2.3.2. Synergistic Effect of 3-Butylophthalide (2) and Azole Derivatives (5, 6, 8, 9, 10, 12, 13) against Selected Candida Strains
3. Materials and Methods
3.1. General
3.2. Chemical Synthesis of Phthalide Lactones 2–4
3.3. Chemical Synthesis of Fluconazole Analogues 5–15
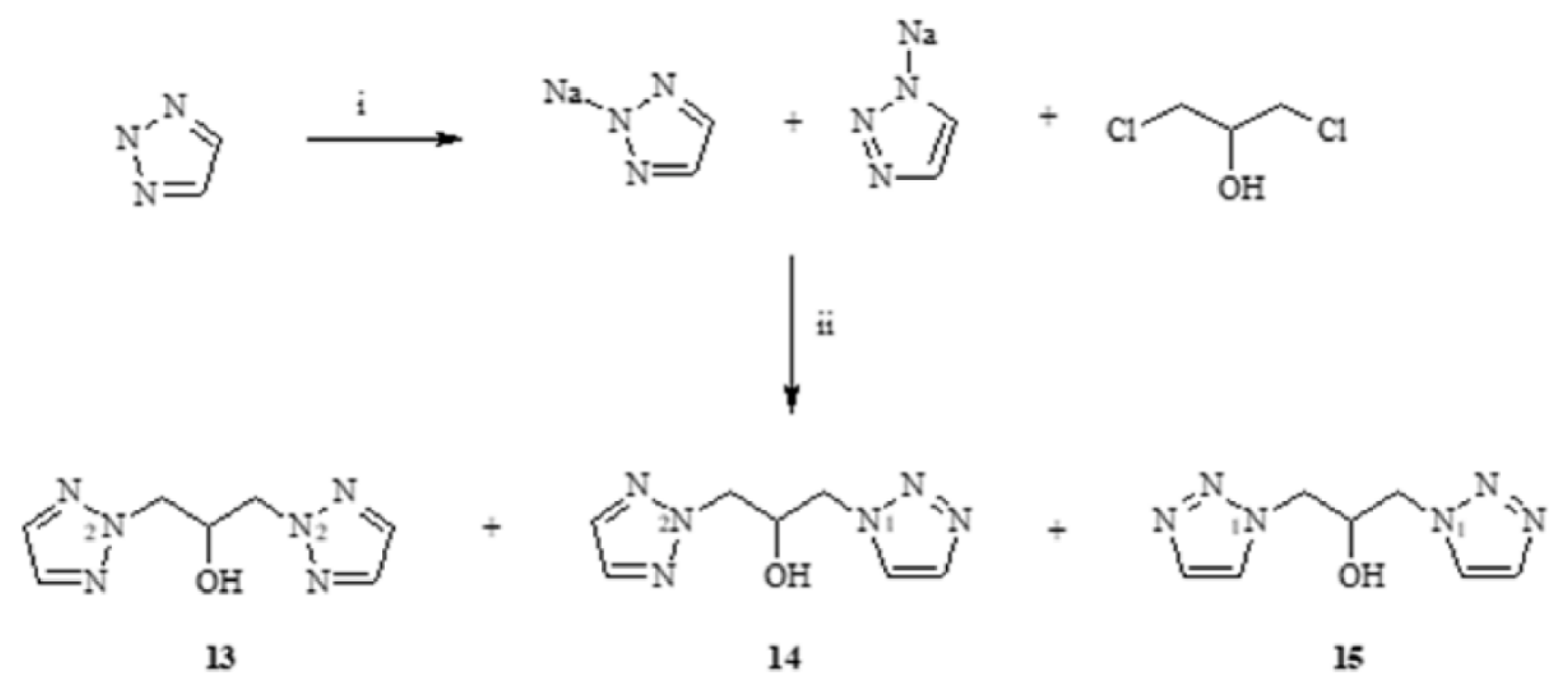
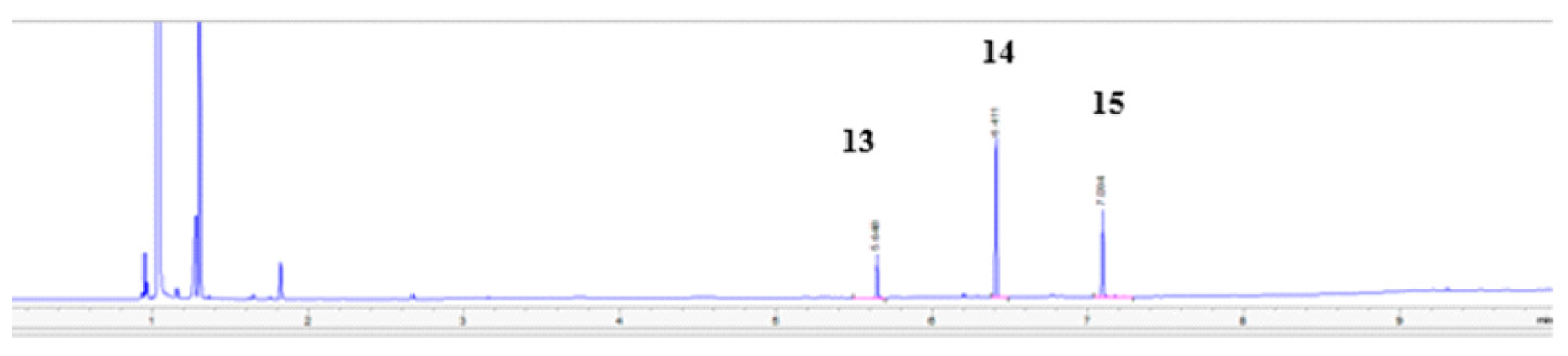
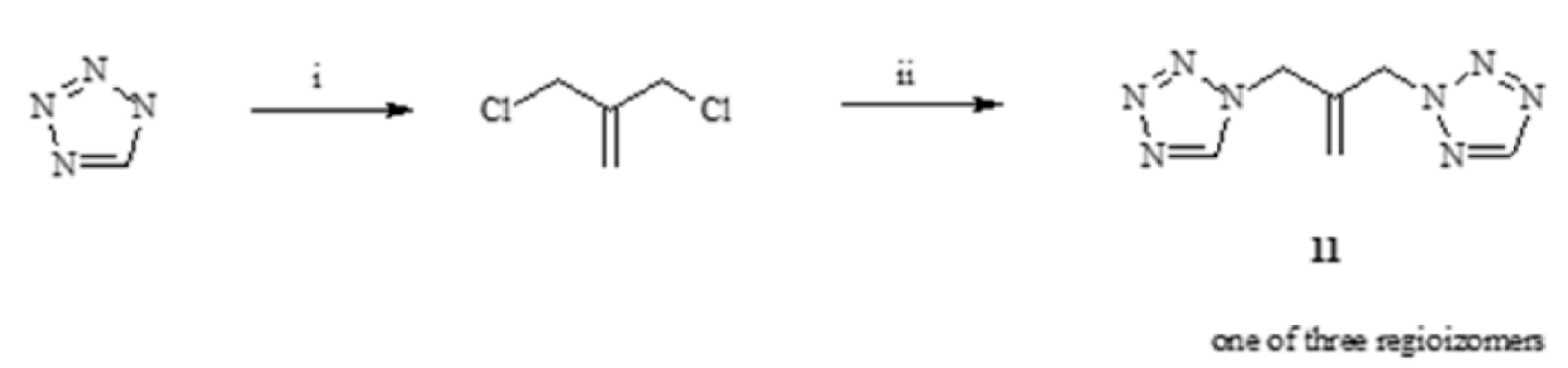
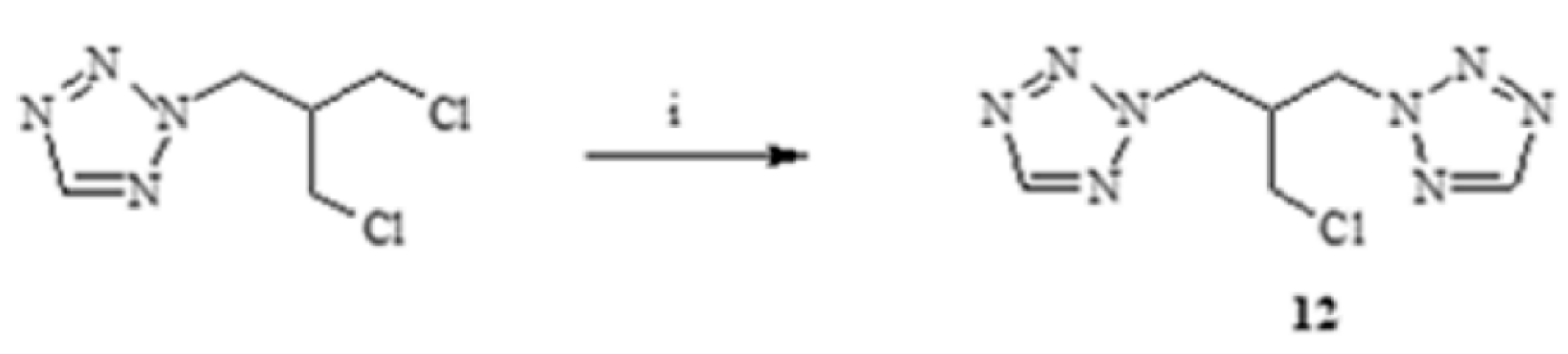
3.3.1. Synthesis of 1,3-(1,2,3-Triazol)propan-2-ol (13–15)
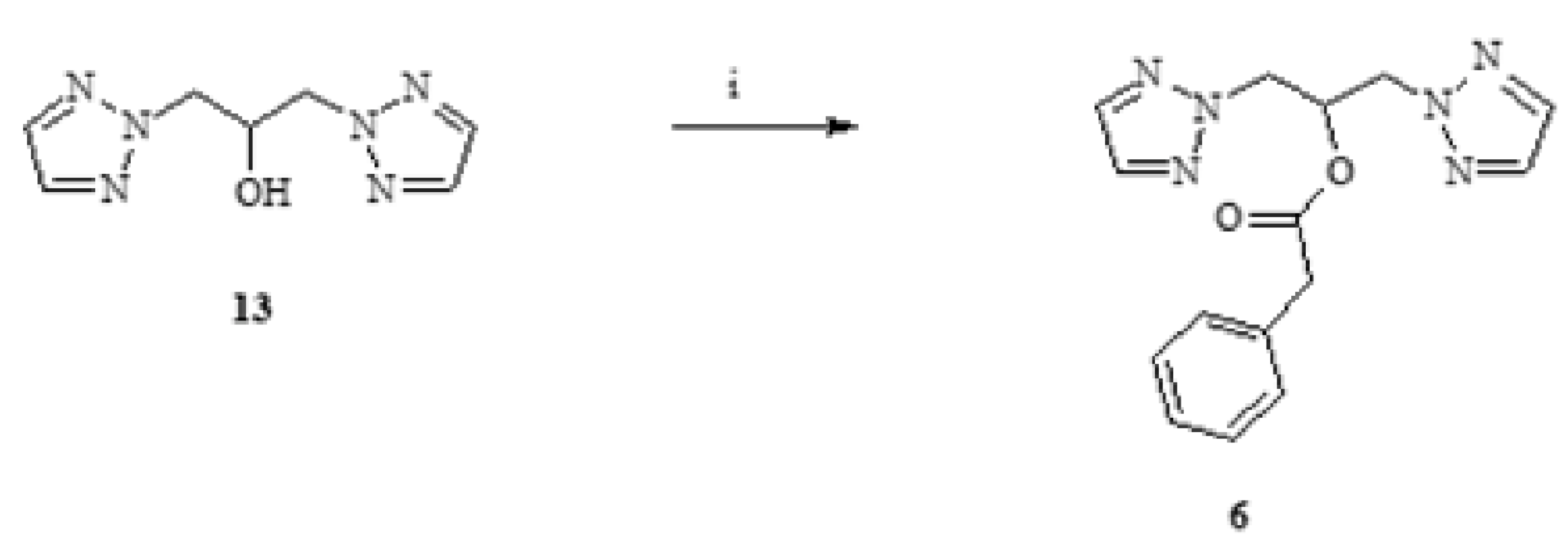
3.3.2. General Methods of Synthesis of Ester: 6–10
3.4. In Silico Methods
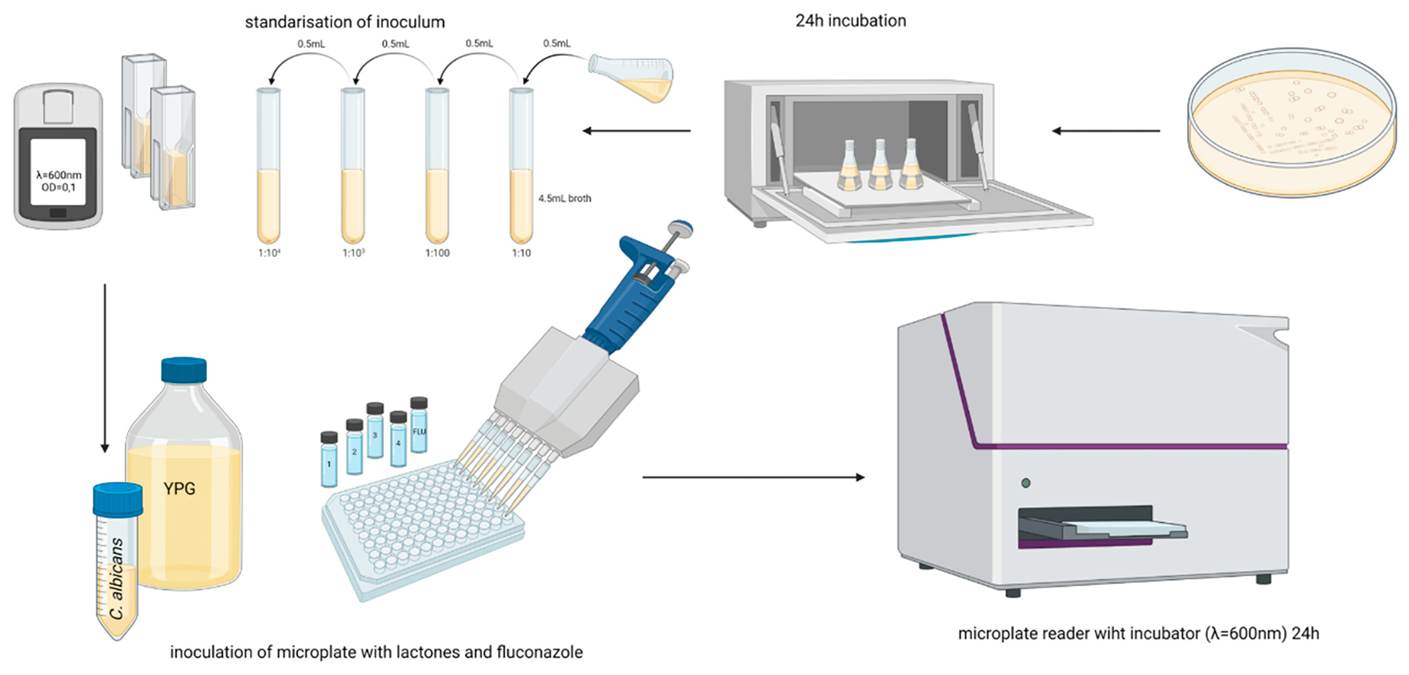
3.5. Method for Determination of Biological Activity
3.5.1. Antifungal Activity
3.5.2. Fractional Inhibitory Concentration
4. Conclusions
- Phthalide lactones in combination with azole compounds were effective in reducing the concentration of fluconazole and its derivatives, due to the synergistic action in the reaction system against fluconazole-resistant strains of Candida albicans isolated from patients and strains that did not develop resistance to fluconazole.
- The synergistic effects of fluconazole (1) and three synthetic lactones identical to naturally occurring celery plants—3-n-butylphthalide (2), 3-n-butylidenephthalide (3), 3-n-butyl-4,5, 6,7—tetrahydrophthalide (4)—against Candida albicans ATCC 10231, C. albicans ATCC 2091, C. guilliermondii KKP 3390 and two strains of C. albicans resistant to fluconazole, were compared with the activity of individual compounds separately. In all cases, these compounds showed high fungistatic activity, which makes them potential agents for use in pharmacology.
- Eleven azole derivatives not previously described in the literature were designed and synthesized. The structures of the compounds obtained were determined by 1HNMR and 13C NMR spectroscopy, and molecular weights were determined by GC-MS or elemental analysis.
- 4.
- High specificity for individual Candida strains was also observed in all tests.
- 5.
- A correlation between log P and fungistatic and synergistic effects has not been established. The drug transporters CDR1 and CDR2 play a key role in the synergistic effect.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, B.; Mishra, A.K.; Kumar, S.; Mandal, S.K.; NSV, L.; Kumar, V.; Baek, K.-H.; Mohanta, Y.K. Antifungal Metabolites as Food Bio-Preservative: Innovation, Outlook, and Challenges. Metabolites 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Tóth, R.; Gácser, A. Mechanisms of Pathogenic Candida Species to Evade the Host Complement Attack. Front. Cell. Infect. Microbiol. 2020, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staniszewska, M.; Kuryk, Ł.; Gryciuk, A.; Kawalec, J.; Rogalska, M.; Baran, J.; Łukowska-Chojnacka, E.; Kowalkowska, A. In Vitro Anti-Candida Activity and Action Mode of Benzoxazole Derivatives. Molecules 2021, 26, 5008. [Google Scholar] [CrossRef] [PubMed]
- Smego, R.A.; Ahmad, H. The Role of Fluconazole in the Treatment of Candida Endocarditis. Medicine 2011, 90, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Akler, M.E.; Vellend, H.; McNeely, D.M.; Walmsley, S.L.; Gold, W.L. Use of Fluconazole in the Treatment of Candidal Endophthalmitis. Clin. Infect. Dis. 1995, 20, 657–664. [Google Scholar] [CrossRef]
- Bavaro, D.F.; Balena, F.; Ronga, L.; Signorile, F.; Romanelli, F.; Stolfa, S.; Sparapano, E.; de Carlo, C.; Mosca, A.; Monno, L.; et al. Emerging Issue of Fluconazole-Resistant Candidemia in a Tertiary Care Hospital of Southern Italy: Time for Antifungal Stewardship Program. J. Med. Mycol. 2022, 32, 101206. [Google Scholar] [CrossRef]
- Corrêa, J.C.R.; Salgado, H.R.N. Review of Fluconazole Properties and Analytical Methods for Its Determination. Crit. Rev. Anal. Chem. 2011, 41, 124–132. [Google Scholar] [CrossRef]
- Fattouh, N.; Hdayed, D.; Geukgeuzian, G.; Tokajian, S.; Khalaf, R.A. Molecular Mechanism of Fluconazole Resistance and Pathogenicity Attributes of Lebanese Candida Albicans Hospital Isolates. Fungal Genet. Biol. 2021, 153, 103575. [Google Scholar] [CrossRef]
- Jacobson, M.A.; Hanks, D.K.; Ferrell, L.D. Fatal Acute Hepatic Necrosis Due to Fluconazole. Am. J. Med. 1994, 96, 188–190. [Google Scholar] [CrossRef]
- Bühler, T.; Medinger, M.; Bouitbir, J.; Krähenbühl, S.; Leuppi-Taegtmeyer, A. Hepatotoxicity Due to Azole Antimycotic Agents in a HLA B*35:02-Positive Patient. Front. Pharmacol. 2019, 10, 645. [Google Scholar] [CrossRef]
- Beck, K.R.; Odermatt, A. Antifungal Therapy with Azoles and the Syndrome of Acquired Mineralocorticoid Excess. Mol. Cell. Endocrinol. 2021, 524, 111168. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics 2020, 9, 312. [Google Scholar] [CrossRef]
- Agrawal, A.; Singh, A.; Verma, R.; Murari, A. Oral Candidiasis: An Overview. J. Oral Maxillofac. Pathol. 2014, 18, 81. [Google Scholar] [CrossRef]
- Spettel, K.; Barousch, W.; Makristathis, A.; Zeller, I.; Nehr, M.; Selitsch, B.; Lackner, M.; Rath, P.-M.; Steinmann, J.; Willinger, B. Analysis of Antifungal Resistance Genes in Candida Albicans and Candida Glabrata Using next Generation Sequencing. PLoS ONE 2019, 14, e0210397. [Google Scholar] [CrossRef] [PubMed]
- Yassin, M.T.; Mostafa, A.A.; Al-Askar, A.A.; Bdeer, R. In Vitro Antifungal Resistance Profile of Candida Strains Isolated from Saudi Women Suffering from Vulvovaginitis. Eur. J. Med. Res. 2020, 25, 1–9. [Google Scholar] [CrossRef]
- Becher, R.; Wirsel, S.G.R. Fungal Cytochrome P450 Sterol 14α-Demethylase (CYP51) and Azole Resistance in Plant and Human Pathogens. Appl. Microbiol. Biotechnol. 2012, 95, 825–840. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Chuang, Y.-C.; Wu, U.-I.; Sun, H.-Y.; Wang, J.-T.; Sheng, W.-H.; Chen, Y.-C.; Chang, S.-C. Mechanisms of Azole Resistance and Trailing in Candida Tropicalis Bloodstream Isolates. J. Fungi 2021, 7, 612. [Google Scholar] [CrossRef]
- Arastehfar, A.; Daneshnia, F.; Hafez, A.; Khodavaisy, S.; Najafzadeh, M.J.; Charsizadeh, A.; Zarrinfar, H.; Salehi, M.; Shahrabadi, Z.Z.; Sasani, E.; et al. Antifungal susceptibility, genotyping, resistance mechanism, and clinical profile of Candida tropicalis blood isolates. Med. Mycol. 2020, 58, 766–773. [Google Scholar] [CrossRef]
- Fan, X.; Xiao, M.; Zhang, D.; Huang, J.J.; Wang, H.; Hou, X.; Zhang, L.; Kong, F.; Chen, S.C.; Tong, Z.H.; et al. Molecular mechanisms of azole resistance in Candida tropicalis isolates causing invasive candidiasis in China. Clin. Microbiol. Infect. 2019, 25, 885–891. [Google Scholar] [CrossRef]
- Flowers, S.A.; Colón, B.; Whaley, S.G.; Schuler, M.A.; Rogers, P.D. Contribution of Clinically Derived Mutations in ERG11 to Azole Resistance in Candida Albicans. Antimicrob. Agents Chemother. 2015, 59, 450–460. [Google Scholar] [CrossRef]
- Pristov, K.E.; Ghannoum, M.A. Resistance of Candida to Azoles and Echinocandins Worldwide. Clin. Microbiol. Infect. 2019, 25, 792–798. [Google Scholar] [CrossRef]
- León, A.; Del-Ángel, M.; Ávila, J.L.; Delgado, G. Phthalides: Distribution in nature, chemical reactivity, synthesis, and biological activity. Prog Chem. Org. Nat. Prod. 2017, 104, 127–246. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Sheng, Y.; Zhang, Y.; Zhang, J.; Du, G. Identification and Comparison of Metabolites After Oral Administration of Essential Oil of Ligusticum Chuanxiong or Its Major Constituent Ligustilide in Rats. Planta Med. 2008, 74, 1684–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spréa, R.M.; Fernandes, Â.; Finimundy, T.C.; Pereira, C.; Alves, M.J.; Calhelha, R.C.; Canan, C.; Barros, L.; Amaral, J.S.; Ferreira, I.C.F.R. Lovage (Levisticum Officinale, W.D.J. Koch) Roots: A Source of Bioactive Compounds towards a Circular Economy. Resources 2020, 9, 81. [Google Scholar] [CrossRef]
- León, A.; Toscano, R.A.; Tortoriello, J.; Delgado, G. Phthalides and Other Constituents from Ligusticum Porteri; Sedative and Spasmolytic Activities of Some Natural Products and Derivatives. Nat. Prod. Res. 2011, 25, 1234–1242. [Google Scholar] [CrossRef]
- Beck, J.J.; Chou, S.-C. The Structural Diversity of Phthalides from the Apiaceae. J. Nat. Prod. 2007, 70, 891–900. [Google Scholar] [CrossRef]
- Momin, R.A.; Nair, M.G. Mosquitocidal, Nematicidal, and Antifungal Compounds from Apium Graveolens, L. Seeds. J. Agric. Food Chem. 2001, 49, 142–145. [Google Scholar] [CrossRef]
- Pannek, J.; Gach, J.; Boratyński, F.; Olejniczak, T. Antimicrobial Activity of Extracts and Phthalides Occurring in Apiaceae Plants. Phytother. Res. 2018, 32, 1459–1487. [Google Scholar] [CrossRef]
- Fan, L.; Luo, B.; Luo, Z.; Zhang, L.; Fan, J.; Xue, W.; Tang, L.; Li, Y. Synthesis and Antifungal Activities of 3-Substituted Phthalide Derivatives. Z. Für Nat. B 2019, 74, 811–818. [Google Scholar] [CrossRef]
- Xiao, B.; Yin, J.; Park, M.; Liu, J.; Li, J.L.; la Kim, E.; Hong, J.; Chung, H.Y.; Jung, J.H. Design and Synthesis of Marine Fungal Phthalide Derivatives as PPAR-γ Agonists. Bioorg. Med. Chem. 2012, 20, 4954–4961. [Google Scholar] [CrossRef]
- Jia, J.; Wei, C.; Liang, J.; Zhou, A.; Zuo, X.; Song, H.; Wu, L.; Chen, X.; Chen, S.; Zhang, J.; et al. The Effects of DL-3-n-butylphthalide in Patients with Vascular Cognitive Impairment without Dementia Caused by Subcortical Ischemic Small Vessel Disease: A Multicentre, Randomized, Double-blind, Placebo-controlled Trial. Alzheimer’s Dement. 2016, 12, 89–99. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Sheng, X.; Huang, Z.; Li, T.; Zhang, M.; Xu, J.; Ji, H.; Yin, J.; Zhang, Y. Design, Synthesis and Biological Evaluation of Hydrogen Sulfide Releasing Derivatives of 3-n-Butylphthalide as Potential Antiplatelet and Antithrombotic Agents. Org. Biomol. Chem. 2014, 12, 5995–6004. [Google Scholar] [CrossRef]
- Diao, X.; Deng, P.; Xie, C.; Li, X.; Zhong, D.; Zhang, Y.; Chen, X. Metabolism and Pharmacokinetics of 3-n -Butylphthalide (NBP) in Humans: The Role of Cytochrome P450s and Alcohol Dehydrogenase in Biotransformation. Drug Metab. Dispos. 2013, 41, 430–444. [Google Scholar] [CrossRef] [Green Version]
- Gach, J.; Olejniczak, T.; Krężel, P.; Boratyński, F. Microbial Synthesis and Evaluation of Fungistatic Activity of 3-butyl-3-hydroxyphthalide, the Mammalian Metabolite of 3-n-butylidenephthalide. Int. J. Mol. Sci. 2021, 22, 7600. [Google Scholar] [CrossRef]
- Chen, X.; Deng, S.; Lei, Q.; He, Q.; Ren, Y.; Zhang, Y.; Nie, J.; Lu, W. MiR-7-5p Affects Brain Edema After Intracerebral Hemorrhage and Its Possible Mechanism. Front. Cell Dev. Biol. 2020, 8, 598020. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Miao, L.; Guo, Y.; Yuan, R.; Tian, H. Design, Synthesis, and Neuroprotective Effects of Novel Hybrid Compounds Containing Edaravone Analogue and 3-n-Butylphthalide Ring-Opened Derivatives. Biochem. Biophys. Res. Commun. 2021, 556, 99–105. [Google Scholar] [CrossRef]
- Liu, X.; Liu, R.; Fu, D.; Wu, H.; Zhao, X.; Sun, Y.; Wang, M.; Pu, X. Dl-3-n-Butylphthalide Inhibits Neuroinflammation by Stimulating Foxp3 and Ki-67 in an Ischemic Stroke Model. Aging 2021, 13, 3763–3778. [Google Scholar] [CrossRef]
- Wang, B.; Wu, C.; Chen, Z.; Zheng, P.; Liu, Y.; Xiong, J.; Xu, J.; Li, P.; Mamun, A.A.; Ye, L.; et al. DL-3-n-Butylphthalide Ameliorates Diabetes-Associated Cognitive Decline by Enhancing PI3K/Akt Signaling and Suppressing Oxidative Stress. Acta Pharmacol. Sin. 2021, 42, 347–360. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, W.; Huang, X.; Hao, L.; Li, Y.; Sun, S. Antifungal Activity and Potential Mechanism of N-Butylphthalide Alone and in Combination With Fluconazole Against Candida Albicans. Front. Microbiol. 2019, 10, 1461. [Google Scholar] [CrossRef]
- Yan, Z.; Hua, H.; Xu, Y.; Samaranayake, L.P. Potent Antifungal Activity of Pure Compounds from Traditional Chinese Medicine Extracts against Six Oral Candida Species and the Synergy with Fluconazole against Azole-Resistant Candida Albicans. Evid.-Based Complement. Altern. Med. 2012, 2012, 106583. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druzhilovskiy, D.S.; Rudik, A.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Dmitriev, A.V.; Pogodin, P.V.; Dubovskaya, V.I.; Ivanov, S.M.; Tarasova, O.A.; et al. Computational platform Way2Drug: From the prediction of biological activity to drug repurposing. Russ. Chem. Bull. 2017, 66, 1832–1841. [Google Scholar] [CrossRef]
- Pugia, M.J.; Knudsen, B.E.; Cason, C.V.; Bartsch, R.A. Synthesis and alkali-metal complexing abilities of crown ether tertiary alcohols. J. Org. Chem. 1987, 52, 541–547. [Google Scholar] [CrossRef]
- Białońska, A.; Bronisz, R. Application of N-(omega-bromoalkyl)tetrazoles for the preparation of bitopic ligands containing pyridylazole chelators or azole rings as building blocks for iron(II) spin crossover polymeric materials. Tetrahedron 2008, 64, 9771–9779. [Google Scholar] [CrossRef]
- Białońska, A.; Bronisz, R.; Rudolf, M.F.; Weselski, M. HS reversible arrow LS Transition in Iron(II) Two-Dimensional Coordination Networks Containing Tris(tetrazol-1-ylmethyl)methane As Triconnected Building Block. Inorg. Chem. 2012, 51, 237–245. [Google Scholar] [CrossRef]
- Tamaian, R.; Moţ, A.; Silaghi-Dumitrescu, R.; Ionuţ, I.; Stana, A.; Oniga, O.; Nastasă, C.; Benedec, D.; Tiperciuc, B. Study of the Relationships between the Structure, Lipophilicity and Biological Activity of Some Thiazolyl-Carbonyl-Thiosemicarbazides and Thiazolyl-Azoles. Molecules 2015, 20, 22188–22201. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Chmiel, T.; Mieszkowska, A.; Kempińska-Kupczyk, D.; Kot-Wasik, A.; Namieśnik, J.; Mazerska, Z. The Impact of Lipophilicity on Environmental Processes, Drug Delivery and Bioavailability of Food Components. Microchem. J. 2019, 146, 393–406. [Google Scholar] [CrossRef]
- Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials. JACC Basic Transl. Sci. 2020, 5, 387–397. [Google Scholar] [CrossRef]
- Kumar, S. Engineering cytochrome P450 biocatalysts for biotechnology, medicine and bioremediation. Expert Opin. Drug Metab. Toxicol. 2010, 6, 115–131. [Google Scholar] [CrossRef]
- Botham, K.M.; Mayes, P.A. Lipids of Physiologic Significance. In Harper’s Illustrated Biochemistry, 31st ed.; Murray, R.K., Granner, D.K., Mayes, P.A., Rodwell, V.W., Eds.; McGraw-Hill Education: New York, NY, USA, 2018; Available online: https://accessmedicine.mhmedical.com/content.aspx?aid=1160192421 (accessed on 10 August 2022).
- Lorian, V. Antibiotics in Labolatory Medicine, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Meletiadis, J.; Pournaras, S.; Roilides, E.; Walsh, T.J. Defining Fractional Inhibitory Concentration Index Cutoffs for Additive Interactions Based on Self-Drug Additive Combinations, Monte Carlo Simulation Analysis, and In Vitro In Vivo Correlation Data for Antifungal Drug Combinations against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2010, 54, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A. CLSI Document M27-A2. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts Approved Standard; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2002. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ATCC 10231 | ATCC 2091 | KKP 3390 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MICsynergy | FIC | MICsynergy | FIC | MICsynergy | FIC | |||||||
| 50% | 90% | 50% | 90% | 50% | 90% | 50% | 90% | 50% | 90% | 50% | 90% | |
| 1 | 0.78 | 1.56 | 1.41 | 0.27 | 0.78 | 1.56 | 1.17 | 0.57 | 1.56 | 0.78 | 0.31 | 0.28 |
| 2 | 0.78 | 3.13 | 25 | 25 | 3.13 | 1.26 | ||||||
| 1 | 0.62 | 1.59 | 0.42 | 0.28 | 0.55 | 1.41 | 0.42 | 0.47 | 1.27 | 0.58 | 0.32 | 0.35 |
| 3 | 1.20 | 4.47 | 3.72 | 22.25 | 4.32 | 20.18 | ||||||
| 1 | 0.42 | 1.43 | 0.41 | 0.34 | 0.70 | 1.48 | 0.55 | 0.49 | 1.22 | 6.73 | 0.43 | 0.65 |
| 4 | 1.32 | 15.27 | 4.45 | 20.27 | 11.58 | 24.86 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krężel, P.; Olejniczak, T.; Tołoczko, A.; Gach, J.; Weselski, M.; Bronisz, R. Synergic Effect of Phthalide Lactones and Fluconazole and Its New Analogues as a Factor Limiting the Use of Azole Drugs against Candidiasis. Antibiotics 2022, 11, 1500. https://doi.org/10.3390/antibiotics11111500
Krężel P, Olejniczak T, Tołoczko A, Gach J, Weselski M, Bronisz R. Synergic Effect of Phthalide Lactones and Fluconazole and Its New Analogues as a Factor Limiting the Use of Azole Drugs against Candidiasis. Antibiotics. 2022; 11(11):1500. https://doi.org/10.3390/antibiotics11111500
Chicago/Turabian StyleKrężel, Piotr, Teresa Olejniczak, Aleksandra Tołoczko, Joanna Gach, Marek Weselski, and Robert Bronisz. 2022. "Synergic Effect of Phthalide Lactones and Fluconazole and Its New Analogues as a Factor Limiting the Use of Azole Drugs against Candidiasis" Antibiotics 11, no. 11: 1500. https://doi.org/10.3390/antibiotics11111500