
Emerging Computational Approaches for Antimicrobial Peptide Discovery

 ,
,  ,
,  , , and
, , and
Abstract
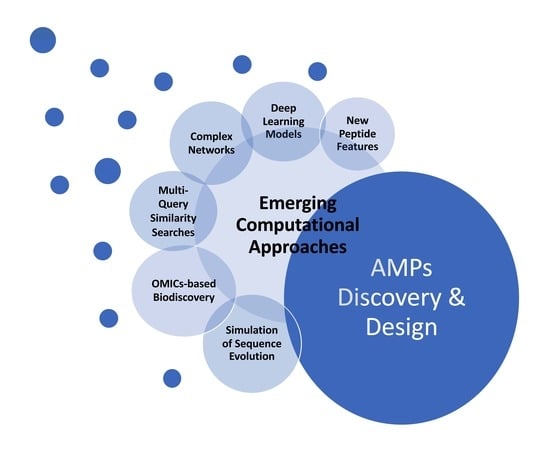
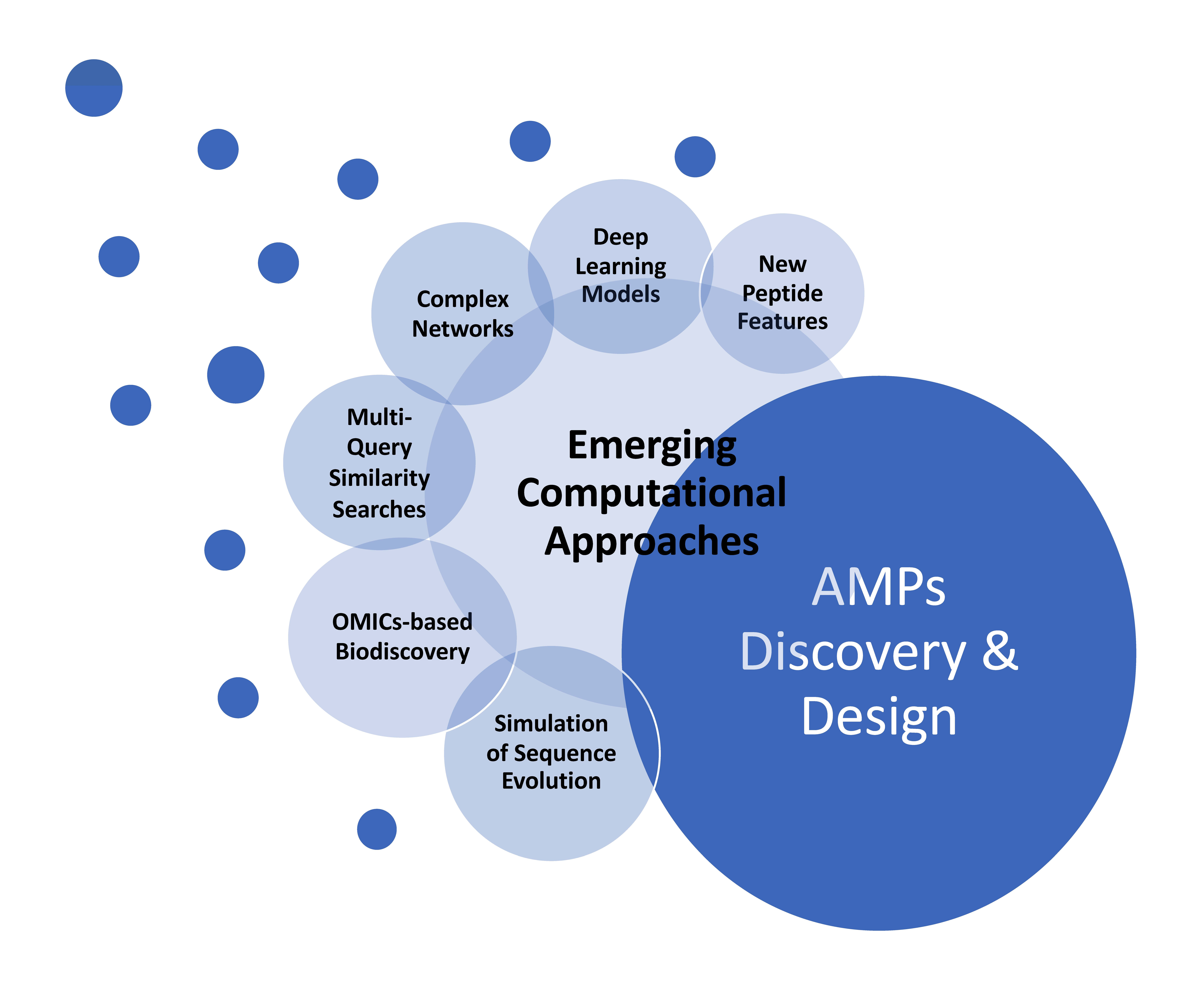
:
1. Introduction
2. Non-Classical Peptide Features for Bioactivity Prediction
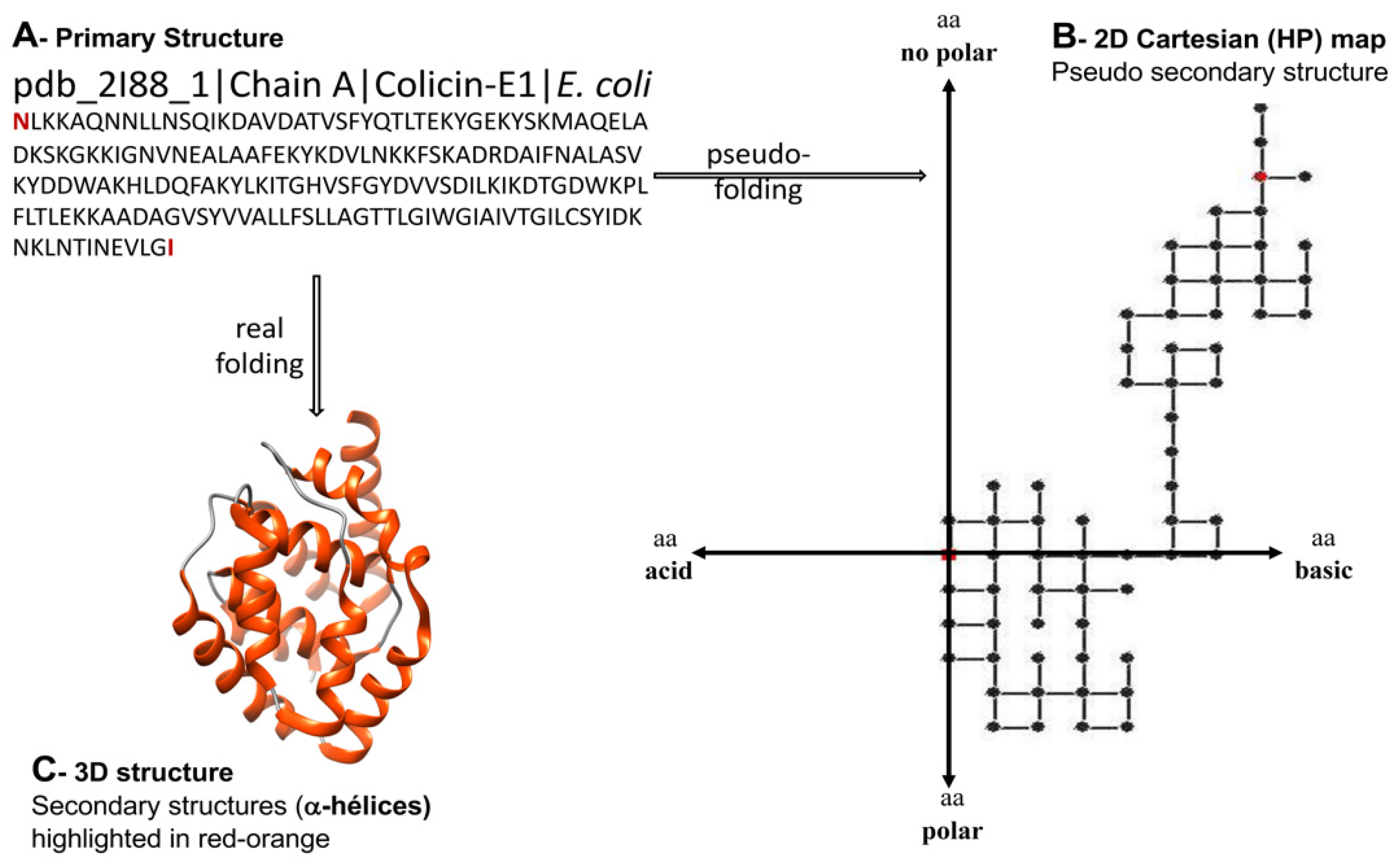
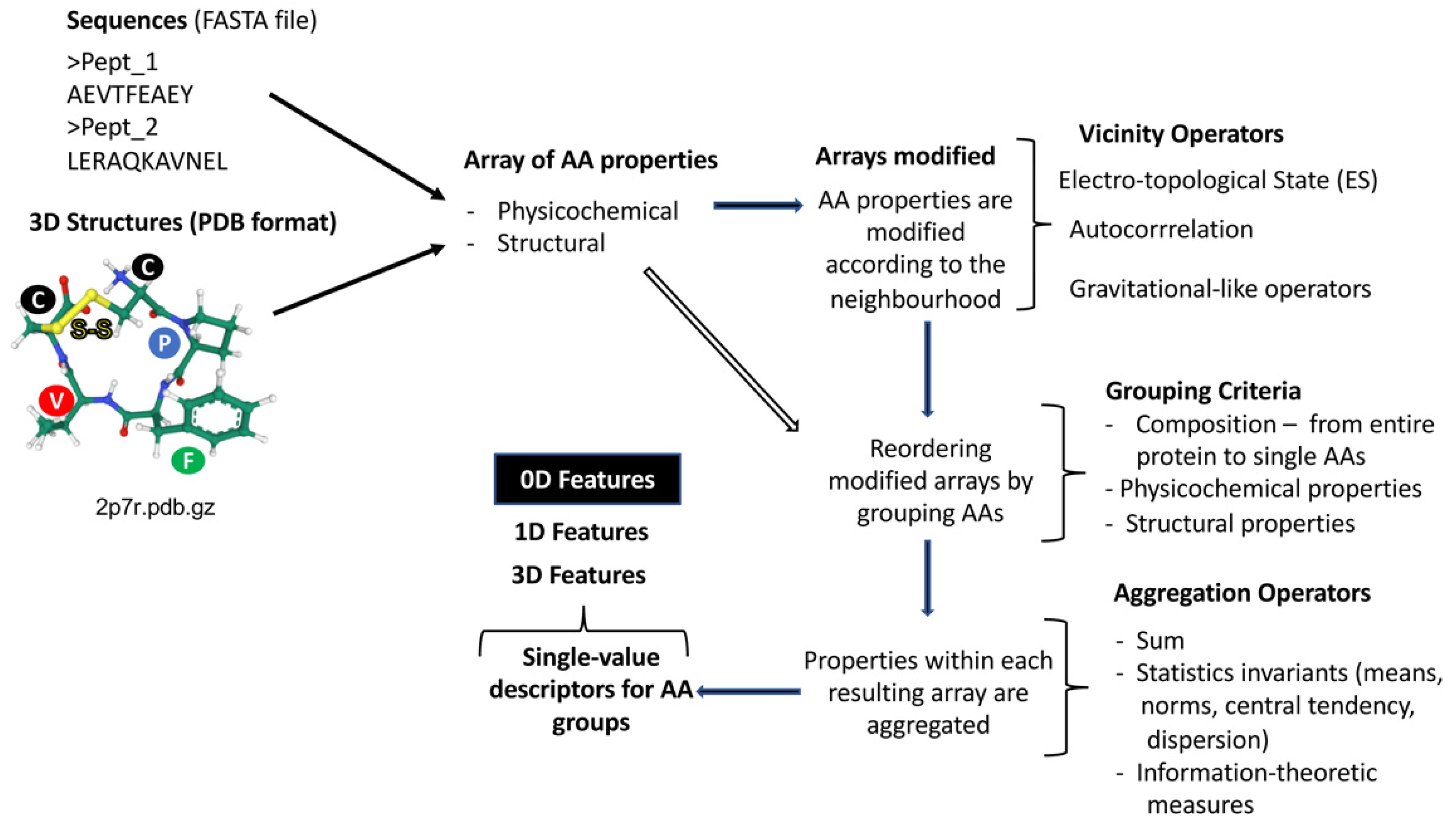
2.1. Peptide Features Inspired in Molecular Descriptors Used in Cheminformatics
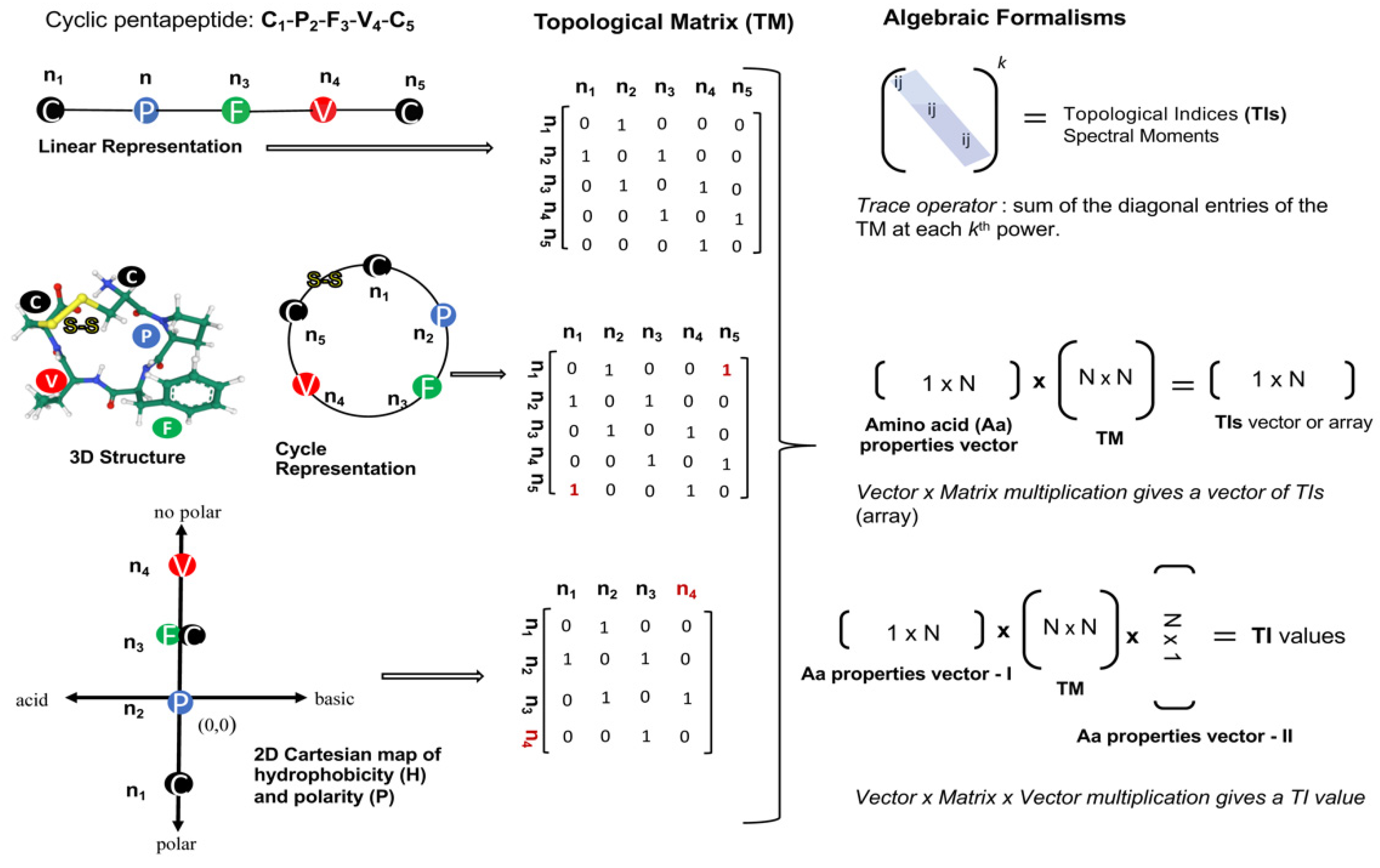
2.1.1. Topological Indices from Algebraic Forms
2.1.2. Topological Indices from Descriptive Statistics
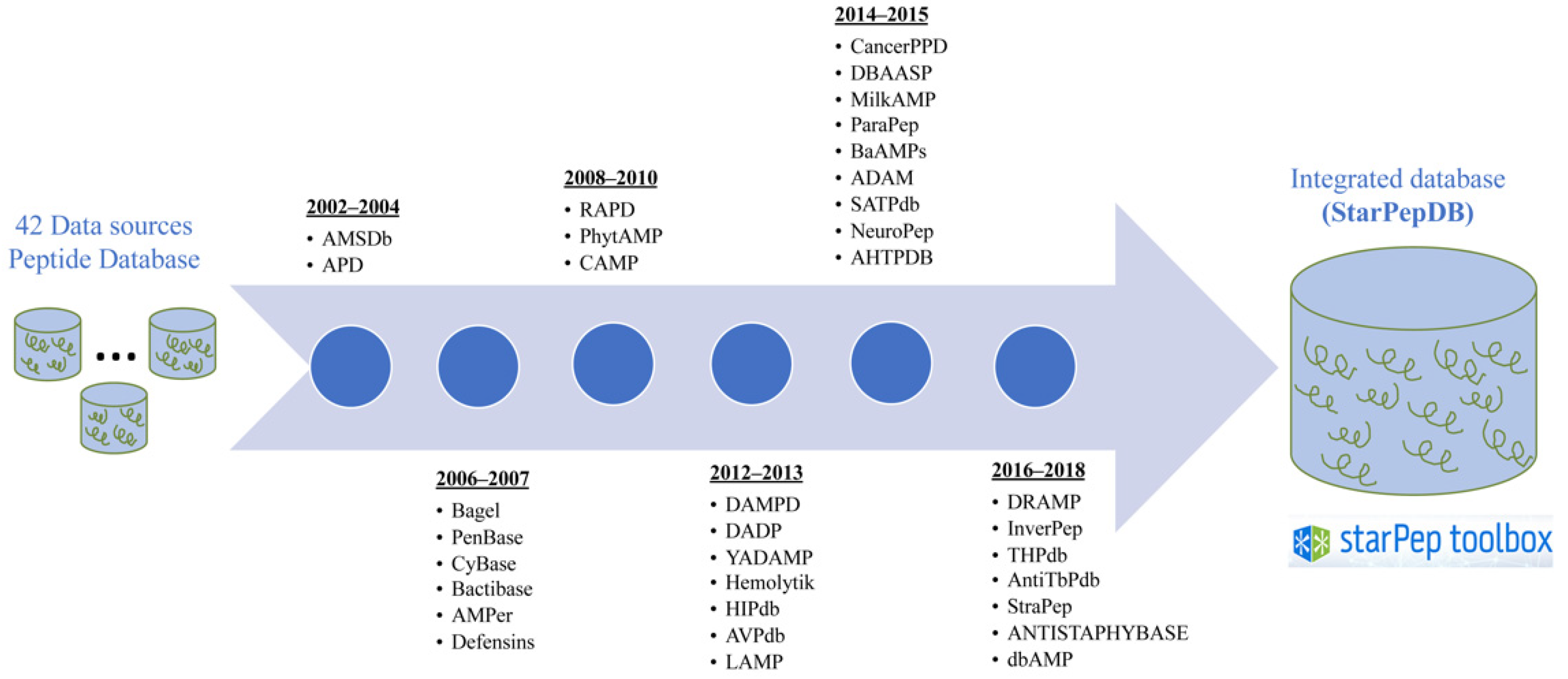
2.2. Integration of Peptide Features from Heterogenous Sources
2.3. NMR-Based Features for Peptides
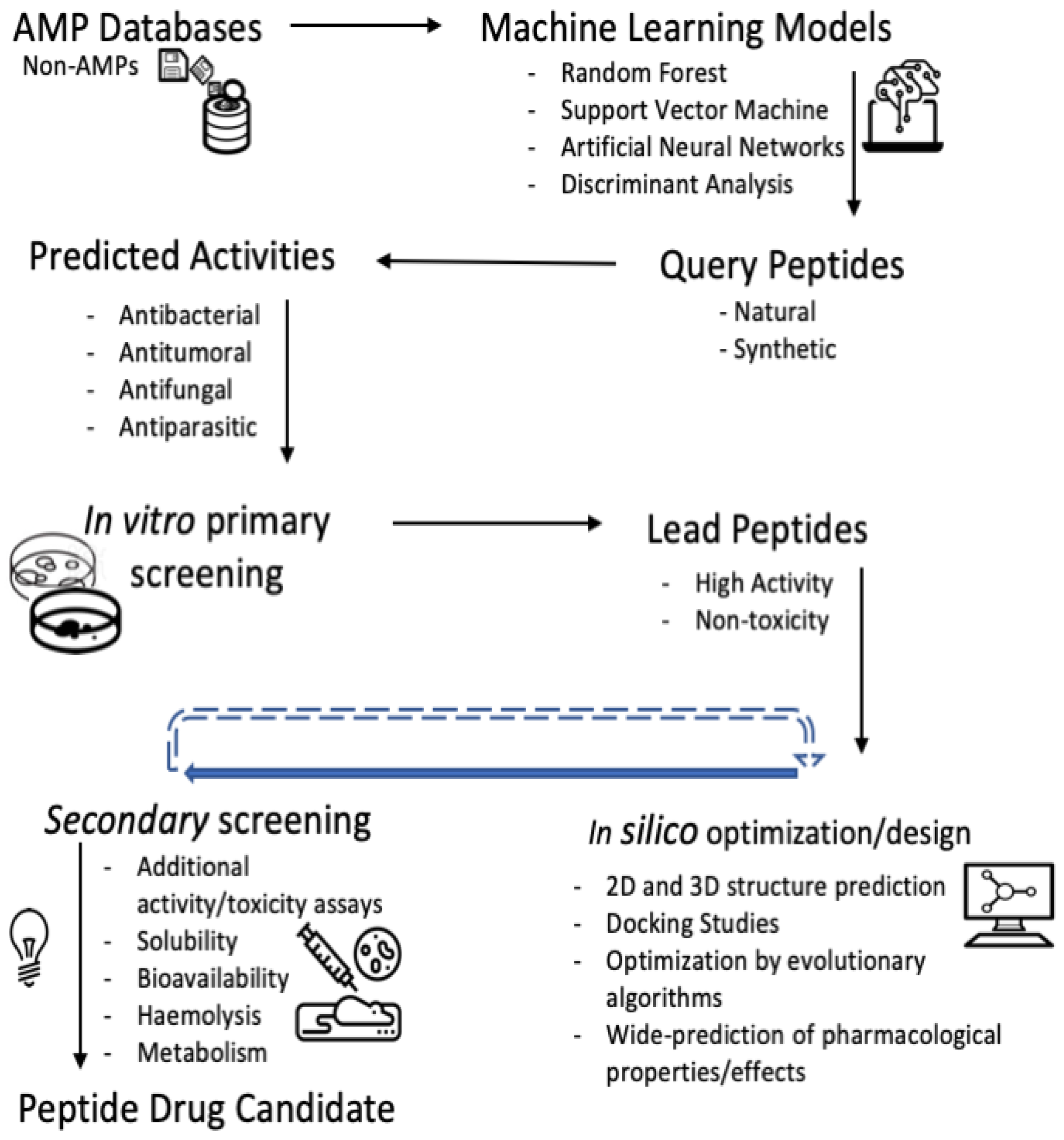
3. Breakthroughs of ML Algorithms in the AMP Prediction
3.1. Data Imbalance and Multi-Label Classification in the Prediction of AMPs—New Algorithm Approaches
3.2. Deep-Learning in the Recognition of AMPs
3.3. Rough Sets Theory in the Classification of AMPs
4. Other Methodologies Than Classical ML for Identifying and Modelling AMPs
4.1. Homology-Based Prediction and Modelling of AMPs
4.2. Emerging ML-Independent Methodologies for AMP Prediction/Design
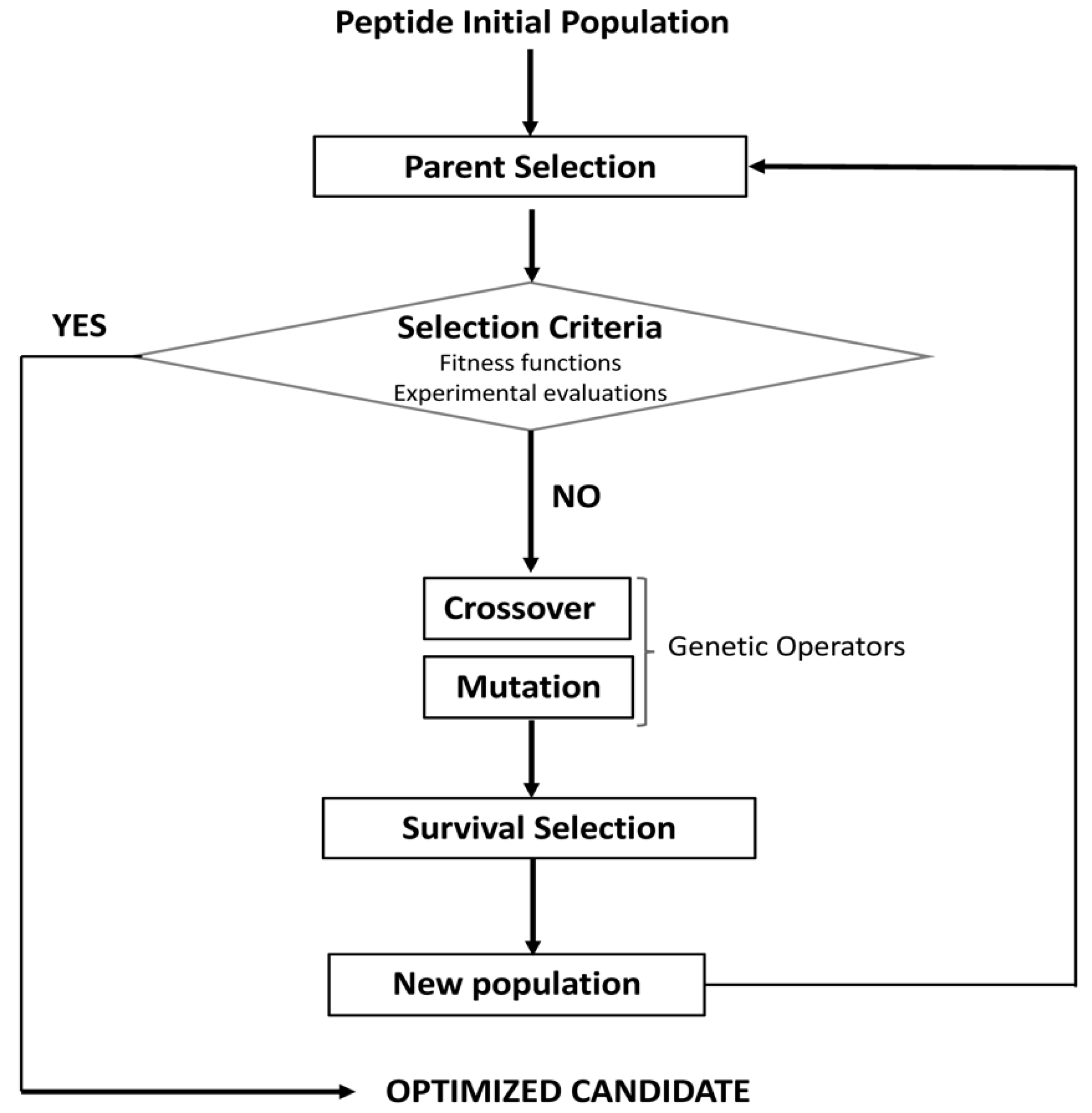
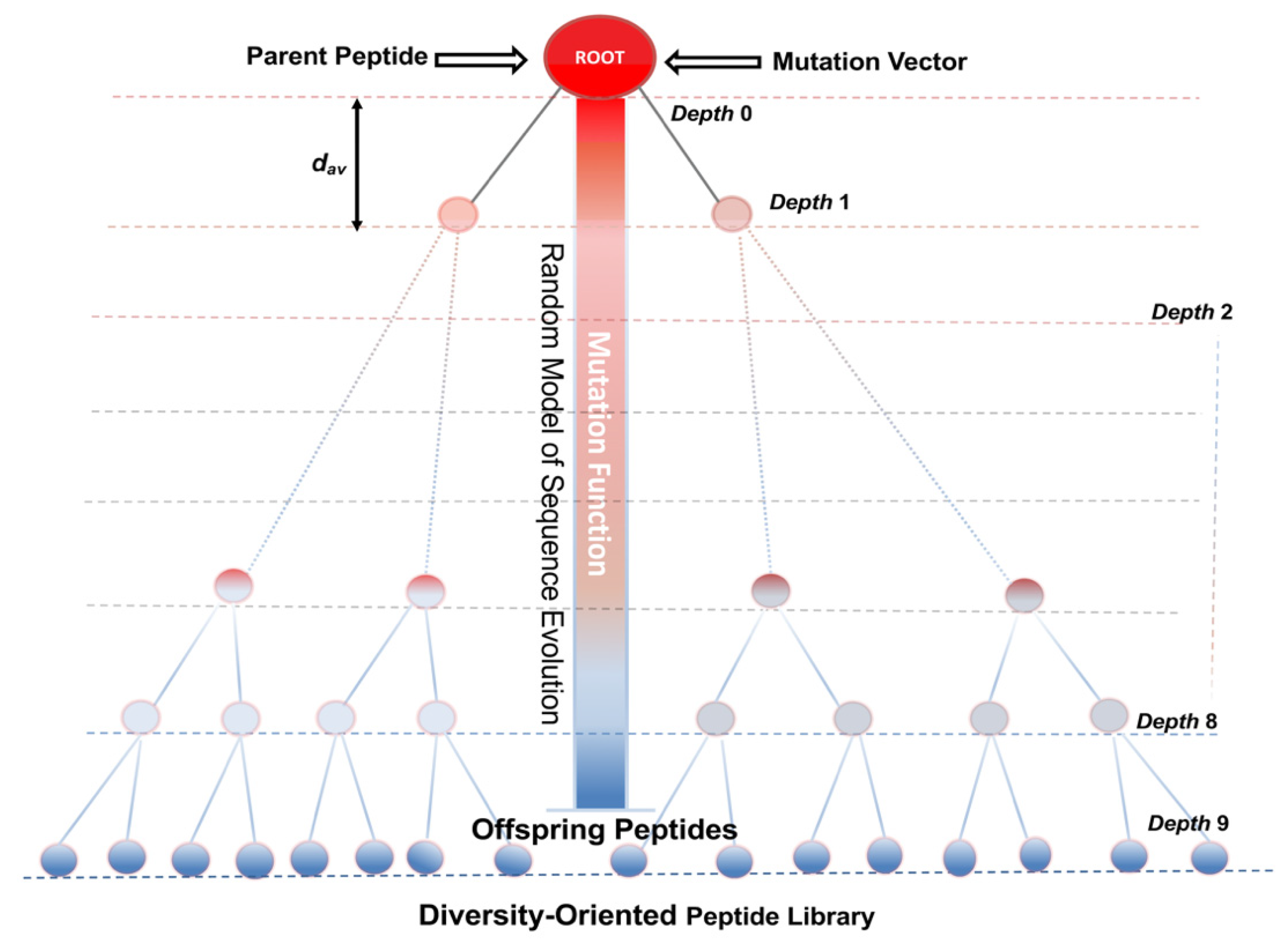
5. Models of Sequence Evolution for the Design and Optimization of Bioactive Peptides
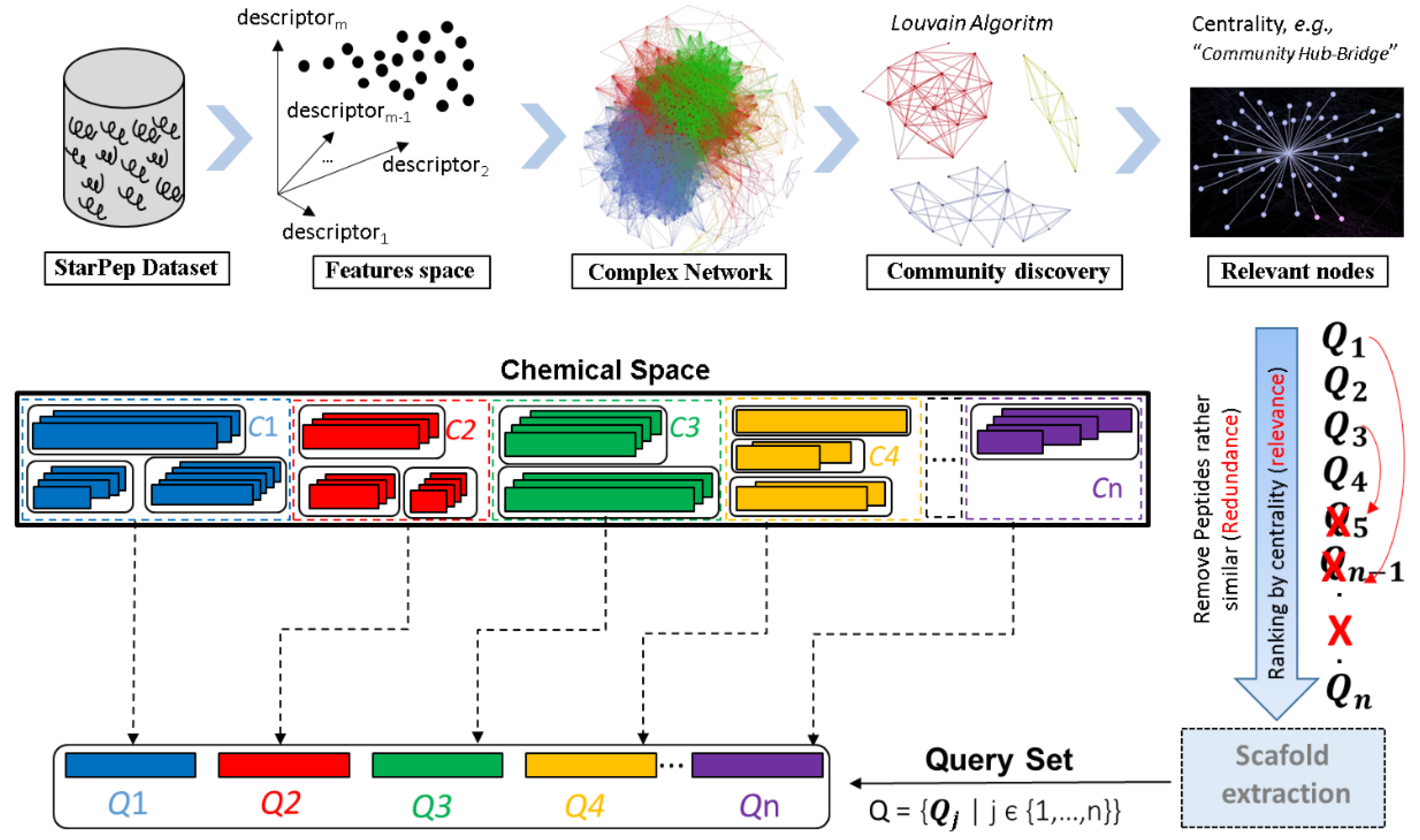
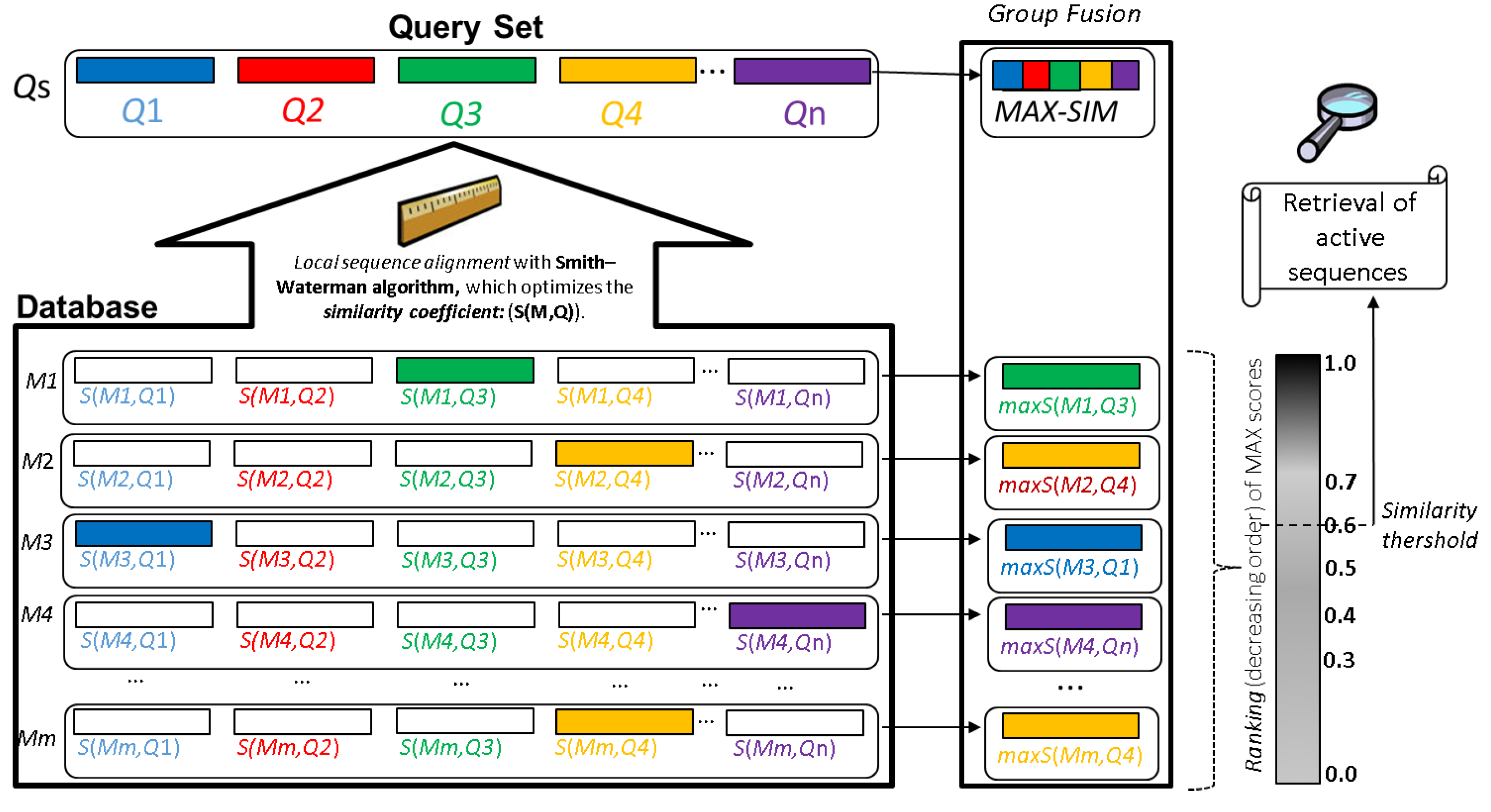
6. Considerations in the Workflow for the High-Throughput Discovering of Bioactive Peptides
6.1. Brief Comparisons between High-Throughput (HT) and Classical Methods
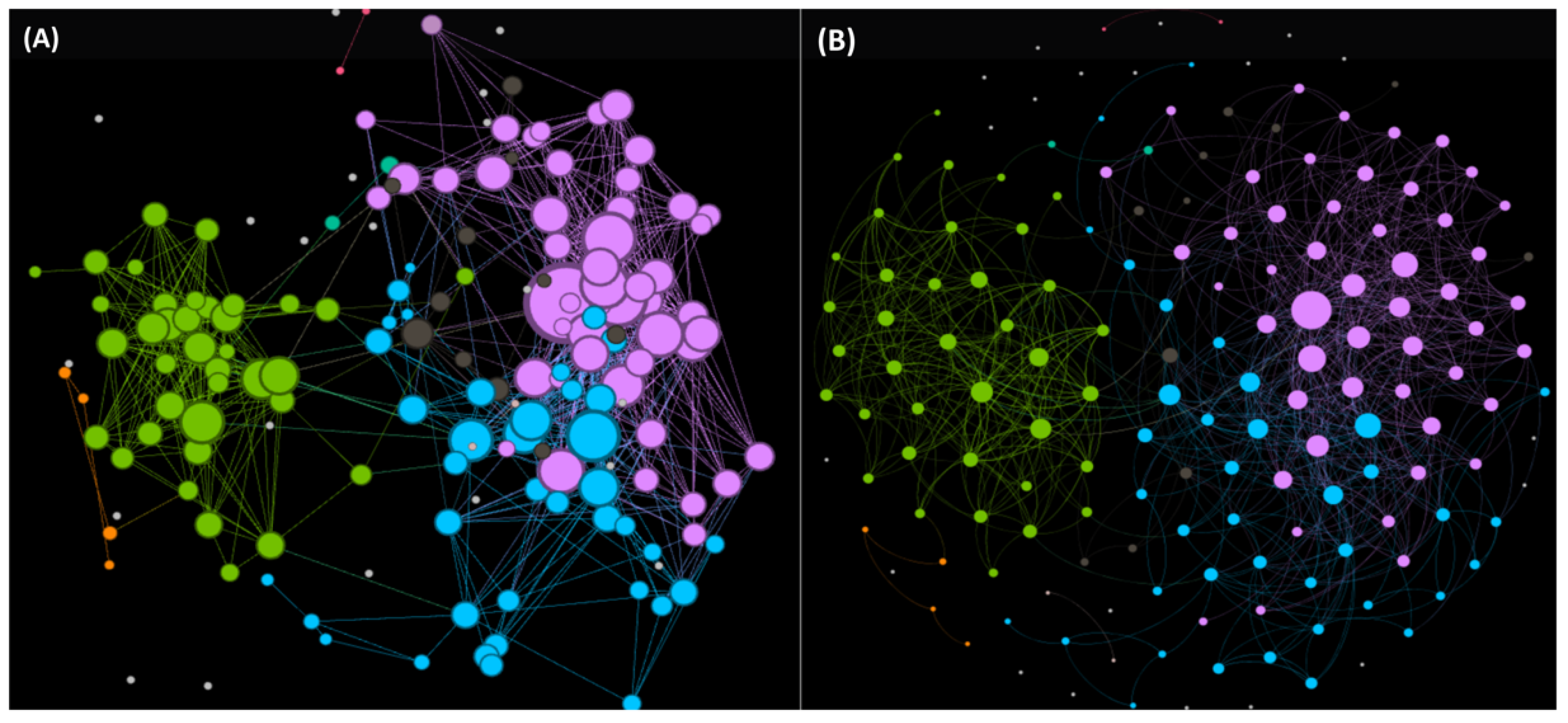
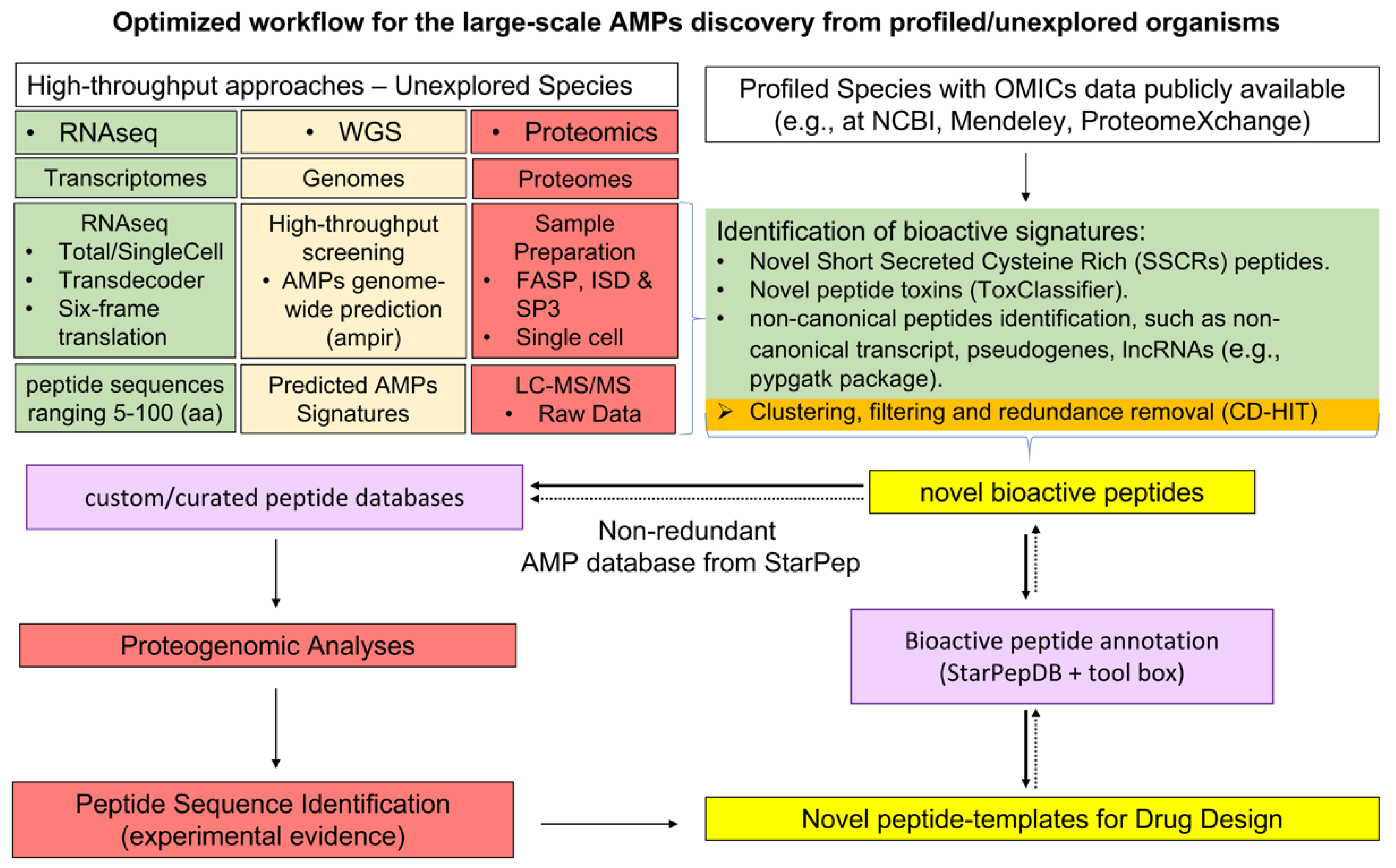
6.2. Optimized Workflow for the Large-Scale AMPs Discovery from Profiled/Unexplored Organisms
7. Concluding Remarks
8. Future Research Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6, S14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara Agudelo, A.; Muñoz Molina, M.; Navarrete Ospina, J.; Salazar Pulido, L.; Castro-Cardozo, B. New Horizons to Survive in a Post-Antibiotics Era. J. Trop Med. Health 2018, 10, JTMH-130. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [Green Version]
- Gohel, V.; Kamal, A. Peptides as Potential Anticancer Agents. Curr. Top. Med. Chem. 2019, 19, 1491–1511. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Porto, W.F.; Pires, A.S.; Franco, O.L. Computational tools for exploring sequence databases as a resource for antimicrobial peptides. Biotechnol. Adv. 2017, 35, 337–349. [Google Scholar] [CrossRef]
- Sundararajan, V.S.; Gabere, M.N.; Pretorius, A.; Adam, S.; Christoffels, A.; Lehväslaiho, M.; Archer, J.A.C.; Bajic, V.B. DAMPD: A manually curated antimicrobial peptide database. Nucleic Acids Res. 2011, 40, D1108–D1112. [Google Scholar] [CrossRef] [Green Version]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMP R3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2016, 44, D1094–D1097. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26467475 (accessed on 23 January 2019). [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-T.; Lee, C.-C.; Yang, J.-R.; Lai, J.Z.C.; Chang, K.Y. A Large-Scale Structural Classification of Antimicrobial Peptides. BioMed Res. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, srep42362. [Google Scholar] [CrossRef]
- Spänig, S.; Heider, D. Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 2019, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, F.; Leier, A.; Xiang, D.; Shen, H.-H.; Lago, T.T.M.; Li, J.; Yu, D.-J.; Song, J. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Briefings Bioinform. 2021, 22, bbab083. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther.-Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cheong, H.H.; Siu, S.W.I. xDeep-AcPEP: Deep Learning Method for Anticancer Peptide Activity Prediction Based on Convolutional Neural Network and Multitask Learning. J. Chem. Inf. Model. 2021, 61, 3789–3803. [Google Scholar] [CrossRef] [PubMed]
- Boone, K.; Camarda, K.; Spencer, P.; Tamerler, C. Antimicrobial peptide similarity and classification through rough set theory using physicochemical boundaries. BMC Bioinform. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chharia, A.; Upadhyay, R.; Kumar, V. Novel fuzzy approach to Antimicrobial Peptide Activity Prediction: A tale of limited and imbalanced data that models won’t hear; 2021. In Proceedings of the NeurIPS 2021 AI for Science Workshop, Vancouver, BC, Canada, 13 December 2021. [Google Scholar]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins: Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xu, D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016, 32, 3745–3752. [Google Scholar] [CrossRef]
- Gull, S.; Shamim, N.; Minhas, F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput. Biol. Med. 2019, 107, 172–181. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Chung, C.-R.; Kuo, T.-R.; Wu, L.-C.; Lee, T.-Y.; Horng, J.-T. Characterization and identification of antimicrobial peptides with different functional activities. Brief. Bioinform. 2019, 21, 1098–1114. [Google Scholar] [CrossRef]
- Pinacho-Castellanos, S.A.; García-Jacas, C.R.; Gilson, M.K.; Brizuela, C.A. Alignment-Free Antimicrobial Peptide Predictors: Improving Performance by a Thorough Analysis of the Largest Available Data Set. J. Chem. Inf. Model. 2021, 61, 3141–3157. [Google Scholar] [CrossRef]
- Ruiz-Blanco, Y.B.; Paz, W.; Green, J.; Marrero-Ponce, Y. ProtDCal: A program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform. 2015, 16, 162. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; García-Jacas, C.R.; Chavez, E.; Beltran, J.A.; Guillen-Ramirez, H.A.; Brizuela, C.A. Automatic construction of molecular similarity networks for visual graph mining in chemical space of bioactive peptides: An unsupervised learning approach. Sci. Rep. 2020, 10, 1–23. [Google Scholar] [CrossRef]
- Kavousi, K.; Bagheri, M.; Behrouzi, S.; Vafadar, S.; Atanaki, F.F.; Lotfabadi, B.T.; Ariaeenejad, S.; Shockravi, A.; Moosavi-Movahedi, A.A. IAMPE: NMR-Assisted Computational Prediction of Antimicrobial Peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. ClassAMP: A Prediction Tool for Classification of Antimicrobial Peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T.J.; Carper, D.L.; Spangler, M.K.; Carrell, A.A.; Rush, T.A.; Minter, S.J.; Weston, D.J.; Labbe, J.L. amPEPpy 1.0: A portable and accurate antimicrobial peptide prediction tool. Bioinformatics 2021, 37, 2058–2060. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.P. A Computational and Statistical Framework for Screening Novel Antimicrobial Peptides. Ph.D. Thesis, George Mason University, Fairfax County, VA, USA, 2015. [Google Scholar]
- García-Jacas, C.R.; Pinacho-Castellanos, S.A.; García-González, L.A.; Brizuela, C.A. Do deep learning models make a difference in the identification of antimicrobial peptides? Brief. Bioinform. 2022, 23, bbac094. [Google Scholar] [CrossRef]
- Wong, K.-C. Evolutionary algorithms: Concepts, designs, and applications in bioinformatics. In Nature-Inspired Computing: Concepts, Methodologies, Tools, and Applications; IGI Global: Hershey, PA, USA, 2017; pp. 111–137. [Google Scholar]
- Bozovičar, K.; Bratkovič, T. Evolving a Peptide: Library Platforms and Diversification Strategies. Int. J. Mol. Sci. 2019, 21, 215. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Reyes, S.G.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Barigye, S.J.; Garcia de la Vega, J.M.; Perez-Castillo, Y.; Castillo-Garit, J.A. Evolutionary algorithm-based generation of optimum peptide sequences with dengue virus inhibitory activity. Future Med. Chem. 2021, 13, 993–1000. [Google Scholar] [CrossRef]
- Fjell, C.D.; Jenssen, H.; Cheung, W.; Hancock, R.; Cherkasov, A. Optimization of Antibacterial Peptides by Genetic Algorithms and Cheminformatics. Chem. Biol. Drug Des. 2010, 77, 48–56. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef]
- Aronica, P.G.; Reid, L.M.; Desai, N.; Li, J.; Fox, S.J.; Yadahalli, S.; Essex, J.W.; Verma, C.S. Computational Methods and Tools in Antimicrobial Peptide Research. J. Chem. Inf. Model. 2021, 61, 3172–3196. [Google Scholar] [CrossRef]
- Ng, X.Y.; Rosdi, B.A.; Shahrudin, S. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Support Vector Machine-Pairwise Algorithm Utilizing LZ-Complexity. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Boone, K.; Wisdom, C.; Camarda, K.; Spencer, P.; Tamerler, C. Combining genetic algorithm with machine learning strategies for designing potent antimicrobial peptides. BMC Bioinform. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Ayala-Ruano, S.; Marrero-Ponce, Y.; Aguilera-Mendoza, L.; Pérez, N.; Agüero-Chapin, G.; Antunes, A.; Aguilar, A.C. Exploring the Chemical Space of Antiparasitic Peptides and Discovery of New Promising Leads through a Novel Approach based on Network Science and Similarity Searching. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Romero, M.; Marrero-Ponce, Y.; Rodríguez, H.; Agüero-Chapin, G.; Antunes, A.; Aguilera-Mendoza, L.; Martinez-Rios, F. A Novel Network Science and Similarity-Searching-Based Approach for Discovering Potential Tumor-Homing Peptides from Antimicrobials. Antibiotics 2022, 11, 401. [Google Scholar] [CrossRef]
- Neuhaus, C.S.; Gabernet, G.; Steuer, C.; Root, K.; Hiss, J.A.; Zenobi, R.; Schneider, G. Simulated Molecular Evolution for Anticancer Peptide Design. Angew. Chem. Int. Ed. 2018, 58, 1674–1678. [Google Scholar] [CrossRef]
- Ruiz-Blanco, Y.B.; Ávila-Barrientos, L.P.; Hernández-García, E.; Antunes, A.; Agüero-Chapin, G.; García-Hernández, E. Engineering protein fragments via evolutionary and protein–protein interaction algorithms: De novo design of peptide inhibitors for F O F 1 -ATP synthase. FEBS Lett. 2020, 595, 183–194. [Google Scholar] [CrossRef]
- Matos, A.; Domínguez-Pérez, D.; Almeida, D.; Agüero-Chapin, G.; Campos, A.; Osório, H.; Vasconcelos, V.; Antunes, A. Shotgun Proteomics of Ascidians Tunic Gives New Insights on Host–Microbe Interactions by Revealing Diverse Antimicrobial Peptides. Mar. Drugs 2020, 18, 362. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.; Domínguez-Pérez, D.; Matos, A.; Agüero-Chapin, G.; Osório, H.; Vasconcelos, V.; Campos, A.; Antunes, A. Putative Antimicrobial Peptides of the Posterior Salivary Glands from the Cephalopod Octopus vulgaris Revealed by Exploring a Composite Protein Database. Antibiotics 2020, 9, 757. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Pérez-Machado, G.; Molina-Ruiz, R.; Pérez-Castillo, Y.; Morales-Helguera, A.; Vasconcelos, V.; Antunes, A. TI2BioP: Topological Indices to BioPolymers. Its practical use to unravel cryptic bacteriocin-like domains. Amino Acids 2010, 40, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Ruso, J.M.; Cordeiro, M.N.D.S. First Multitarget Chemo-Bioinformatic Model To Enable the Discovery of Antibacterial Peptides against Multiple Gram-Positive Pathogens. J. Chem. Inf. Model. 2016, 56, 588–598. [Google Scholar] [CrossRef] [PubMed]
- De Armas, R.R.; Díaz, H.G.; Molina, R.; González, M.P.; Uriarte, E. Stochastic-based descriptors studying peptides biological properties: Modeling the bitter tasting threshold of dipeptides. Bioorganic Med. Chem. 2004, 12, 4815–4822. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Ruso, J.M.; Speck-Planche, A.; Cordeiro, M.N.D.S. Enabling the Discovery and Virtual Screening of Potent and Safe Antimicrobial Peptides. Simultaneous Prediction of Antibacterial Activity and Cytotoxicity. ACS Comb. Sci. 2016, 18, 490–498. [Google Scholar] [CrossRef]
- Estrada, E. Spectral Moments of the Edge Adjacency Matrix in Molecular Graphs. 1. Definition and Applications to the Prediction of Physical Properties of Alkanes. J. Chem. Inf. Comput. Sci. 1996, 36, 844–849. [Google Scholar] [CrossRef]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Agüero-Chapin, G.; Molina-Ruiz, R.; Pérez-Machado, G.; Vasconcelos, V.; Rodríguez-Negrin, Z.; Antunes, A. TI2BioP—Topological Indices to BioPolymers. A Graphical–Numerical Approach for Bioinformatics. In Recent Advances in Biopolymers; IntechOpen: Zagreb, Croatia, 2016. [Google Scholar]
- González-Díaz, H.; González-Díaz, Y.; Santana, L.; Ubeira, F.M.; Uriarte, E.; González-Díaz, H. Proteomics, networks and connectivity indices. Proteomics 2008, 8, 750–778. [Google Scholar] [CrossRef]
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef]
- Randić, M. Graph theoretical approach to structure-activity studies: Search for optimal antitumor compounds. Prog. Clin. Biol. Res. 1985, 172, 309–318. [Google Scholar]
- Moreau, G.; Broto, P. The Autocorrelation of a topological structure. A new molecular descriptor. Nouv. J. Chim. 1980, 4, 359–360. [Google Scholar]
- Balaban, A.T.; Beteringhe, A.; Constantinescu, T.; Filip, P.A.; Ivanciuc, O. Four New Topological Indices Based on the Molecular Path Code. J. Chem. Inf. Model. 2007, 47, 716–731. [Google Scholar] [CrossRef]
- Hall, P.R.; Malone, L.; Sillerud, L.O.; Ye, C.; Hjelle, B.L.; Larson, R.S. Characterization and NMR Solution Structure of a Novel Cyclic Pentapeptide Inhibitor of Pathogenic Hantaviruses. Chem. Biol. Drug Des. 2007, 69, 180–190. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. An Electrotopological-State Index for Atoms in Molecules. Pharm. Res. 1990, 07, 801–807. [Google Scholar] [CrossRef]
- Ivanciuc, O. Building–Block Computation of the Ivanciuc–Balaban Indices for the Virtual Screening of Combinatorial Libraries. Internet Electron. J. Mol. Des. 2002, 1, 1–9. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed.; Wiley-VCH: Mannheim, Germany, 2000; Volume 1, p. 667. [Google Scholar]
- Estrada, E. Characterization of the folding degree of proteins. Bioinformatics 2002, 18, 697–704. [Google Scholar] [CrossRef]
- Estrada, E. Characterization of the amino acid contribution to the folding degree of proteins. Proteins: Struct. Funct. Bioinform. 2004, 54, 727–737. [Google Scholar] [CrossRef]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, A.M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Molina, R.; Agüero-Chapin, G.; Pérez-González, M. TI2BioP (Topological Indices to BioPolymers) Version 2.0; Molecular Simulation and Drug Design (MSDD): Chemical Bioactives Center, Central University of Las Villas, Santa Clara, Cuba, 2011. [Google Scholar]
- Avila-Barrientos, L.P.; Cofas-Vargas, L.F.; Agüero-Chapin, G.; Hernández-García, E.; Ruiz-Carmona, S.; Valdez-Cruz, N.A.; Trujillo-Roldán, M.; Weber, J.; Ruiz-Blanco, Y.B.; Barril, X.; et al. Computational Design of Inhibitors Targeting the Catalytic β Subunit of Escherichia coli FOF1-ATP Synthase. Antibiotics 2022, 11, 557. [Google Scholar] [CrossRef]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Harms, M.; Münch, J.; Sanchez-Garcia, E. PPI-Detect: A support vector machine model for sequence-based prediction of protein-protein interactions. J. Comput. Chem. 2019, 40, 1233–1242. [Google Scholar] [CrossRef]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Beltran, J.A.; Tellez Ibarra, R.; Guillen-Ramirez, H.A.; Brizuela, C.A. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: Toward an organized collection enabling visual network analysis. Bioinformatics 2019, 35, 4739–4747. [Google Scholar] [CrossRef]
- Galpert, D.; Fernández, A.; Herrera, F.; Antunes, A.; Molina-Ruiz, R.; Agüero-Chapin, G. Surveying alignment-free features for Ortholog detection in related yeast proteomes by using supervised big data classifiers. BMC Bioinform. 2018, 19, 166. [Google Scholar] [CrossRef] [Green Version]
- Agüero-Chapin, G.; Molina-Ruiz, R.; Maldonado, E.; de la Riva, G.; Sánchez-Rodríguez, A.; Vasconcelos, V.; Antunes, A. Exploring the adenylation domain repertoire of nonribosomal peptide synthetases using an ensemble of sequence-search methods. PLoS ONE 2013, 8, e65926. [Google Scholar] [CrossRef] [Green Version]
- Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2019, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Borozan, I.; Watt, S.; Ferretti, V. Integrating alignment-based and alignment-free sequence similarity measures for biological sequence classification. Bioinformatics 2015, 31, 1396–1404. [Google Scholar] [CrossRef] [Green Version]
- Empel, A.; Ziv, J. On the complexity of finite sequences. IEEE Trans. Inf. Theory 1976, 22, 75–81. [Google Scholar] [CrossRef]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Feature Selection Methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Kohavi, R.; John, G.H. Wrappers for feature subset selection. Artif. Intell. 1997, 97, 273–324. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Cai, Y.; Liu, J.; Lin, C.; Liu, X. An advanced approach to identify antimicrobial peptides and their function types for penaeus through machine learning strategies. BMC Bioinform. 2019, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Wang, Z.; Jhong, J.-H.; Lee, T.-Y. Identifying anti-coronavirus peptides by incorporating different negative datasets and imbalanced learning strategies. Brief. Bioinform. 2021, 22, 1085–1095. [Google Scholar] [CrossRef]
- Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes 2021, 12, 137. [Google Scholar] [CrossRef]
- Yu, Q.; Dong, Z.; Fan, X.; Zong, L.; Li, Y. HMD-AMP: Protein Language-Powered Hierarchical Multi-label Deep Forest for Annotating Antimicrobial Peptides. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Bernards, M.T.; Shi, Y.; Shao, Q.; He, Y. Sequence-based peptide identification, generation, and property prediction with deep learning: A review. Mol. Syst. Des. Eng. 2021, 6, 406–428. [Google Scholar] [CrossRef]
- Wan, F.; Kontogiorgos-Heintz, D.; de la Fuente-Nunez, C. Deep generative models for peptide design. Digit. Discov. 2022, 1, 195–208. [Google Scholar] [CrossRef]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.-Y.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef]
- Van Oort, C.M.; Ferrell, J.B.; Remington, J.M.; Wshah, S.; Li, J. AMPGAN v2: Machine Learning-Guided Design of Antimicrobial Peptides. J. Chem. Inf. Model. 2021, 61, 2198–2207. [Google Scholar] [CrossRef]
- Das, P.; Wadhawan, K.; Chang, O.; Sercu, T.; Santos, C.N.D.; Riemer, M.; Padhi, I.; Chenthamarakshan, V.; Mojsilovic, A. PepCVAE: Semi-Supervised Targeted Design of Antimicrobial Peptide Sequences. arXiv 2018, arXiv:1810.07743. [Google Scholar]
- Dean, S.N. Variational Autoencoder for the Generation of New Antimicrobial Peptides. ACS Omega 2021, 5, 20746–20754. [Google Scholar] [CrossRef]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Shin, M.K.; Hwang, I.-W.; Jung, J.; Shim, Y.J.; Kim, G.W.; Kim, S.T.; Jang, W.; Sung, J.-S. A Deep Learning Approach with Data Augmentation to Predict Novel Spider Neurotoxic Peptides. Int. J. Mol. Sci. 2021, 22, 12291. [Google Scholar] [CrossRef]
- Wang, C.; Garlick, S.; Zloh, M. Deep Learning for Novel Antimicrobial Peptide Design. Biomolecules 2021, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W. Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther.-Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Babgi, B.A.; Alsayari, J.H.; Davaasuren, B.; Emwas, A.-H.; Jaremko, M.; Abdellattif, M.H.; Hussien, M.A. Synthesis, structural studies, and anticancer properties of [CuBr (PPh3) 2 (4,6-dimethyl-2-thiopyrimidine-S]. Crystals 2021, 11, 688. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Pearson, W.R. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 1990, 183, 63–98. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Ben Hamida, J.; Fliss, I. BACTIBASE: A new web-accessible database for bacteriocin characterization. BMC Microbiol. 2007, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- De Jong, A.; Van Hijum, S.A.F.T.; Bijlsma, J.J.E.; Kok, J.; Kuipers, O.P. BAGEL: A web-based bacteriocin genome mining tool. Nucleic Acids Res. 2006, 34, W273–W279. [Google Scholar] [CrossRef]
- Mulvenna, J.; Mylne, J.; Bharathi, R.; Burton, R.; Shirley, N.; Fincher, G.B.; Anderson, M.; Craik, D.J. Discovery of Cyclotide-Like Protein Sequences in Graminaceous Crop Plants: Ancestral Precursors of Circular Proteins? Plant Cell 2006, 18, 2134–2144. [Google Scholar] [CrossRef] [Green Version]
- Porto, W.F.; Silva, O.N.; Franco, O.L. Prediction and rational design of antimicrobial peptides. In Protein Structure; IntechOpen: Zagreb, Croatia, 2012. [Google Scholar]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Thompson, K. Programming Techniques: Regular expression search algorithm. Commun. ACM 1968, 11, 419–422. [Google Scholar] [CrossRef]
- Jonassen, I. Efficient discovery of conserved patterns using a pattern graph. Comput. Appl. Biosci. 1997, 13, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Fjell, C.D.; Hancock, R.E.W.; Cherkasov, A. AMPer: A database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148–1155. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Gille, C.; Goede, A.; Preißner, R.; Rother, K.; Frömmel, C. Conservation of substructures in proteins: Interfaces of secondary structural elements in proteasomal subunits. J. Mol. Biol. 2000, 299, 1147–1154. [Google Scholar] [CrossRef]
- Lee, J.; Wu, S.; Zhang, Y. Ab Initio Protein Structure Prediction. In From Protein Structure to Function with Bioinformatics; Rigden, D.J., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 3–25. [Google Scholar]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.; Eramian, D.; Shen, M.y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Hammami, R.; Fliss, I. Current trends in antimicrobial agent research: Chemo- and bioinformatics approaches. Drug Discov. Today 2010, 15, 540–546. [Google Scholar] [CrossRef]
- Torrent, M.; Di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2011, 28, 130–131. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C.; Higgins, D.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Tellez-Ibarra, R.; Llorente-Quesada, M.T.; Salgado, J.; Barigye, S.J.; Liu, J. Overlap and diversity in antimicrobial peptide databases: Compiling a non-redundant set of sequences. Bioinformatics 2015, 31, 2553–2559. [Google Scholar] [CrossRef] [Green Version]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of Fingerprint-Based Methods for Virtual Screening Using Multiple Bioactive Reference Structures. J. Chem. Inf. Comput. Sci. 2004, 44, 1177–1185. [Google Scholar] [CrossRef]
- Marasco, D.; Perretta, G.; Sabatella, M.; Ruvo, M. Past and future perspectives of synthetic peptide libraries. Curr. Protein Pept. Sci. 2008, 9, 447–467. [Google Scholar] [CrossRef]
- Irving, M.B.; Pan, O.; Scott, J.K. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr. Opin. Chem. Biol. 2001, 5, 314–324. [Google Scholar] [CrossRef]
- Müller, A.; Gabernet, G.; Hiss, J.A.; Schneider, G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, G.; Wrede, P. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: De novo design of an idealized leader peptidase cleavage site. Biophys. J. 1994, 66, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Schuchhardt, J.; Wrede, P. Peptide design in machina: Development of artificial mitochondrial protein precursor cleavage sites by simulated molecular evolution. Biophys. J. 1995, 68, 434–447. [Google Scholar] [CrossRef] [Green Version]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef]
- Miyata, T.; Miyazawa, S.; Yasunaga, T. Two types of amino acid substitutions in protein evolution. J. Mol. Evol. 1979, 12, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Risler, J.; Delorme, M.; Delacroix, H.; Henaut, A. Amino acid substitutions in structurally related proteins a pattern recognition approach: Determination of a new and efficient scoring matrix. J. Mol. Biol. 1988, 204, 1019–1029. [Google Scholar] [CrossRef]
- Gabernet, G.; Gautschi, D.; Müller, A.T.; Neuhaus, C.S.; Armbrecht, L.; Dittrich, P.S.; Hiss, J.A.; Schneider, G. In silico design and optimization of selective membranolytic anticancer peptides. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Stoye, J.; Evers, D.; Meyer, F. Rose: Generating sequence families. Bioinformatics 1998, 14, 157–163. [Google Scholar] [CrossRef]
- Pang, A.; Smith, A.D.; Nuin, P.A.; Tillier, E.R. SIMPROT: Using an empirically determined indel distribution in simulations of protein evolution. BMC Bioinform. 2005, 6, 236. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, W.; Yang, Z. INDELible: A Flexible Simulator of Biological Sequence Evolution. Mol. Biol. Evol. 2009, 26, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Bosso, M.; Ständker, L.; Kirchhoff, F.; Münch, J. Exploiting the human peptidome for novel antimicrobial and anticancer agents. Bioorganic Med. Chem. 2018, 26, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Pérez, D.; Durban, J.; Agüero-Chapin, G.; López, J.T.; Molina-Ruiz, R.; Almeida, D.; Calvete, J.J.; Vasconcelos, V.; Antunes, A. The Harderian gland transcriptomes of Caraiba andreae, Cubophis cantherigerus and Tretanorhinus variabilis, three colubroid snakes from Cuba. Genomics 2018, 111, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A.; Norton, R.S. Sea Anemones: Quiet Achievers in the Field of Peptide Toxins. Toxins 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 361, 842–844. [Google Scholar] [CrossRef]
- Rodríguez, A.A.; Otero-González, A.; Ghattas, M.; Ständker, L. Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides. Biomedicines 2021, 9, 1381. [Google Scholar] [CrossRef]
- Chevreux, B. MIRA: An Automated Genome and EST Assembler. Ph.D. Thesis, Ruprecht-Karls University, Heidelberg, Germany, 2007. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Sequencing, H. CLC Genomics Workbench. 2011. Available online: https://research.ncsu.edu/gsl/bioinformatic-resources/clc/ (accessed on 17 March 2022).
- Bioinformatics, B.; Valencia, S. OmicsBox-Bioinformatics made easy. March 2019, 3, 2019. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Pérez, D.; Martins, J.C.; Almeida, D.; Costa, P.R.; Vasconcelos, V.; Campos, A. Transcriptomic Profile of the Cockle Cerastoderma edule Exposed to Seasonal Diarrhetic Shellfish Toxin Contamination. Toxins 2021, 13, 784. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, L.C.H.W.; Strugnell, J.M.; Faou, P.; Labiaga, R.; Zhang, J.; Cooke, I.R. Shotgun Proteomics Analysis of Saliva and Salivary Gland Tissue from the Common Octopus Octopus vulgaris. J. Proteome Res. 2018, 17, 3866–3876. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef]
- Fingerhut, L.C.H.W.; Miller, D.J.; Strugnell, J.M.; Daly, N.L.; Cooke, I.R. ampir: An R package for fast genome-wide prediction of antimicrobial peptides. Bioinformatics 2020, 36, 5262–5263. [Google Scholar] [CrossRef]
- Almeida, D.; Domínguez-Pérez, D.; Matos, A.; Agüero-Chapin, G.; Castaño, Y.; Vasconcelos, V.; Campos, A.; Antunes, A. Data Employed in the Construction of a Composite Protein Database for Proteogenomic Analyses of Cephalopods Salivary Apparatus. Data 2020, 5, 110. [Google Scholar] [CrossRef]
- Gacesa, R.; Barlow, D.; Long, P.F. Machine learning can differentiate venom toxins from other proteins having non-toxic physiological functions. PeerJ Comput. Sci. 2016, 2, e90. [Google Scholar] [CrossRef]
- Umer, H.M.; Audain, E.; Zhu, Y.; Pfeuffer, J.; Sachsenberg, T.; Lehtiö, J.; Branca, R.M.; Perez-Riverol, Y. Generation of ENSEMBL-based proteogenomics databases boosts the identification of non-canonical peptides. Bioinformatics 2022, 38, 1470–1472. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- León, I.R.; Schwämmle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative Assessment of In-solution Digestion Efficiency Identifies Optimal Protocols for Unbiased Protein Analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, K.; Kim, S.; Bandeira, N. False discovery rates in spectral identification. BMC Bioinform. 2012, 13, S2. [Google Scholar] [CrossRef] [Green Version]
- The, M.; MacCoss, M.J.; Noble, W.S.; Käll, L. Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J. Am. Soc. Mass Spectrom. 2016, 27, 1719–1727. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Storey, J.D.; MacCoss, M.J.; Noble, W.S. Posterior Error Probabilities and False Discovery Rates: Two Sides of the Same Coin. J. Proteome Res. 2007, 7, 40–44. [Google Scholar] [CrossRef]
- Bhandari, B.K.; Gardner, P.P.; Lim, C.S. Razor: Annotation of signal peptides from toxins. bioRxiv 2021. [Google Scholar] [CrossRef]
- Maxwell, M.; Undheim, E.A.B.; Mobli, M. Secreted Cysteine-Rich Repeat Proteins “SCREPs”: A Novel Multi-Domain Architecture. Front. Pharmacol. 2018, 9, 1333. [Google Scholar] [CrossRef]
- Liu, S.; Bao, J.; Lao, X.; Zheng, H. Novel 3D Structure Based Model for Activity Prediction and Design of Antimicrobial Peptides. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, R.; Agrawal, P.; Patiyal, S.; Raghava, G.P. A Method for Predicting Hemolytic Potency of Chemically Modified Peptides From Its Structure. Front. Pharmacol. 2020, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, S.; Fei, W.; Feng, Y.; Shen, L.; Yang, X.; Wang, M.; Wu, M. Prediction of Anticancer Peptides with High Efficacy and Low Toxicity by Hybrid Model Based on 3D Structure of Peptides. Int. J. Mol. Sci. 2021, 22, 5630. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Su, X.; Wen, M.; Zuo, S.; Hong, L.; Lin, J. Parafold: Paralleling alphafold for large-scale predictions. In Proceedings of the International Conference on High Performance Computing in Asia-Pacific Region Workshops, Kobe, Japan & Online, 12–14 January 2022; pp. 1–9. [Google Scholar]
- Contreras-Torres, E.; Marrero-Ponce, Y.; Terán, J.E.; García-Jacas, C.R.; Brizuela, C.A.; Sánchez-Rodríguez, J.C. MuLiMs-MCoMPAs: A Novel Multiplatform Framework to Compute Tensor Algebra-Based Three-Dimensional Protein Descriptors. J. Chem. Inf. Model. 2019, 60, 1042–1059. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.T.; de la Fuente-Nunez, C. Reprogramming biological peptides to combat infectious diseases. Chem. Commun. 2019, 55, 15020–15032. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classical AMP Prediction Tools | ||||
| Integrated to Database | ML Algorithm | Peptide features | Implementation | Ref. |
| CAMPR3 | RF, SVM, ANN, DA | AAC, net charge, hydrophobicity | http://www.camp3.bicnirrh.res.in/prediction.php | [11] |
| DRAMP 3.0 | ANN, SVM, RF | Secondary structure features | http://shicrazy.pythonanywhere.com/ | [13] |
| ADAM | SVM | AAC | http://bioinformatics.cs.ntou.edu.tw/adam/tool.html | [14] |
| DBAASPv3.0 | Threshold value-based discrimination | Physicochemical properties acconuting for the interaction with membrane | https://dbaasp.org/tools?page=general-prediction | [15] |
| Independent Tools | ||||
| ClasssAMP * | RF, SVM | Sequence-based features | http://www.bicnirrh.res.in/classamp/predict.php | [35] |
| iAMPpred * | SVM | compositional, physicochemical, and structural features | http://cabgrid.res.in:8080/amppred/server.php | [16] |
| iAMP-2L ** | k-NN | PseAAC | http://www.jci-bioinfo.cn/iAMP-2L | [25] |
| AmPEP | RF | Sequence-based features | https://cbbio.online/software/AmPEP/ | [36] |
| amPEPpy | RF | Global protein sequence descriptors | https://github.com/tlawrence3/amPEPpy | [37] |
| AMPScannerv1 ** | RF | Physicochemical features | https://www.dveltri.com/ascan/v1/index.html | [38] |
| AMPfun ** | RF | AAC-based features, physicochemical features and word frequency-based features | http://fdblab.csie.ncu.edu.tw/AMPfun/index.html | [30] |
| AMAP ** | SVM and XGboost tree | AAC-based features | http://amap.pythonanywhere.com/ | [28] |
| Emerging AMP prediction tools | ||||
| MLAMP ** | RF | Non-classical PSeAAC | http://www.jci-bioinfo.cn/MLAMP | [27] |
| IAMPE | RF, k-NN, SVM, XGboost | NMR-based features | http://cbb1.ut.ac.ir/AMPClassifier/Index | [34] |
| AMPDiscover ** | RF/DNN | Non-classical protein features (ProtDCal) | https://biocom-ampdiscover.cicese.mx/ | [31,39] |
| ABP-Finder ** | RF | Non-classical protein features (ProtDCal) | https://protdcal.zmb.uni-due.de/ABP-Finder/index.php | [Unpub] |
| AMPScannerv2 | DNN | AA alphabet | https://www.dveltri.com/ascan/v2/ascan.html | [19] |
| ACP-DL | DNN | Binary profile feature and K-mer sparce matrix | https://github.com/haichengyi/ACP-DL (Standalone) | [20] |
| xDeep-AcPEP * | DNN | Physicochemical, biochemical, evolutionary and positional | https://app.cbbio.online/acpep/home | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agüero-Chapin, G.; Galpert-Cañizares, D.; Domínguez-Pérez, D.; Marrero-Ponce, Y.; Pérez-Machado, G.; Teijeira, M.; Antunes, A. Emerging Computational Approaches for Antimicrobial Peptide Discovery. Antibiotics 2022, 11, 936. https://doi.org/10.3390/antibiotics11070936
Agüero-Chapin G, Galpert-Cañizares D, Domínguez-Pérez D, Marrero-Ponce Y, Pérez-Machado G, Teijeira M, Antunes A. Emerging Computational Approaches for Antimicrobial Peptide Discovery. Antibiotics. 2022; 11(7):936. https://doi.org/10.3390/antibiotics11070936
Chicago/Turabian StyleAgüero-Chapin, Guillermin, Deborah Galpert-Cañizares, Dany Domínguez-Pérez, Yovani Marrero-Ponce, Gisselle Pérez-Machado, Marta Teijeira, and Agostinho Antunes. 2022. "Emerging Computational Approaches for Antimicrobial Peptide Discovery" Antibiotics 11, no. 7: 936. https://doi.org/10.3390/antibiotics11070936