3.2. Chemistry
3.2.1. General Procedure for Compounds 4a,b
A mixture of
3 (0.35 mmol) [
35], a catalytic amount of Et
3N (0.1 mL) and SOCl
2 (9.35 mmol) was stirred at room temperature for 30 min. Then the excess of SOCl
2 was removed in vacuo and the residue oil was dissolved in cold anhydrous THF (1 mL). To this suspension, the appropriate amine (0.75 mmol) was added and the mixture was stirred at room temperature for 2 h. After cooling, cold water was added (2–5 mL) and the suspension was extracted with CH
2Cl
2 (3 × 15 mL); the solvent was evaporated under vacuum to afford the desired final compounds, which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 as eluent (4a), or by crystallization from ethanol (4b).
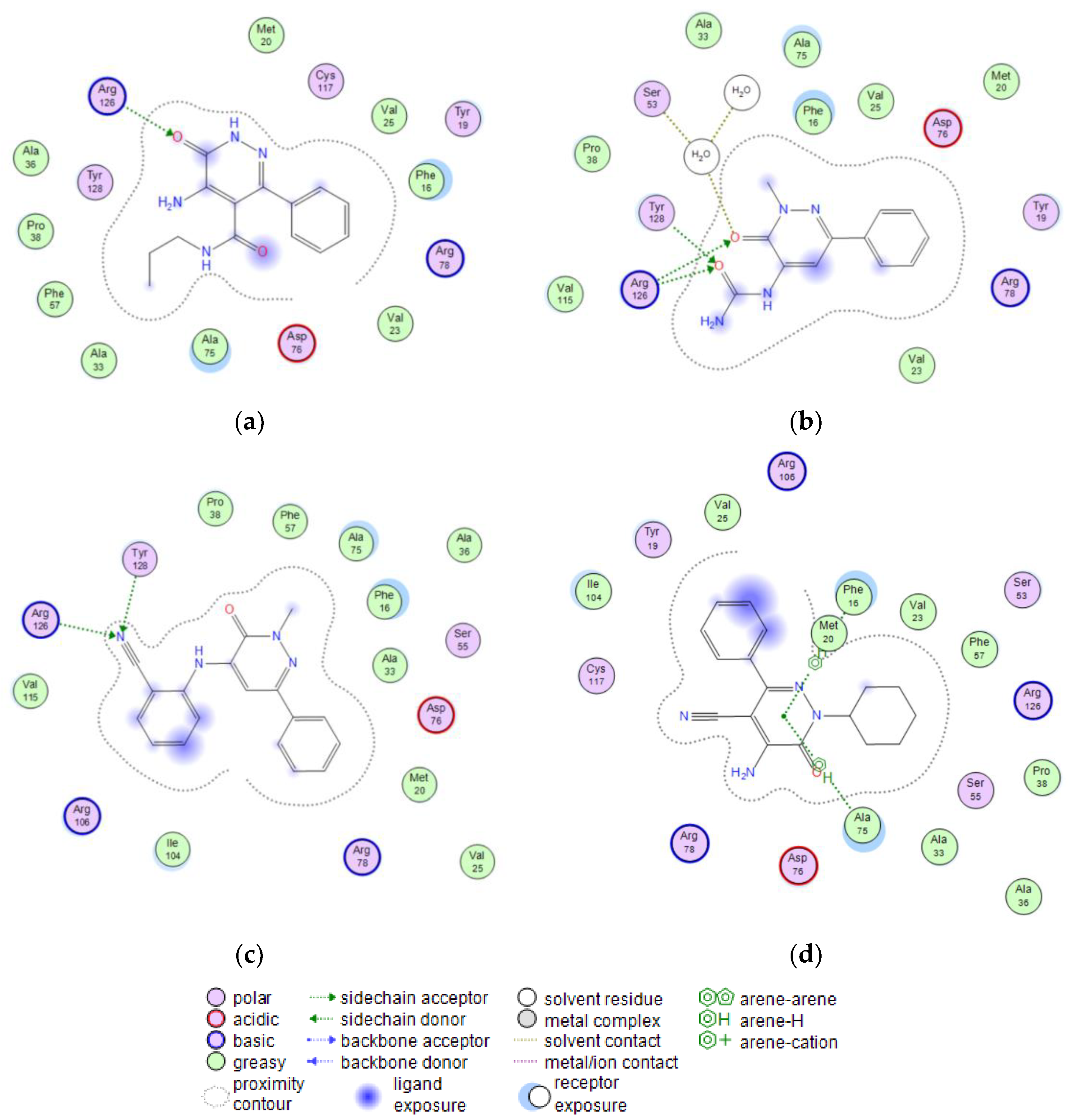
5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid phenylamide (4a)
Yield = 40%; mp = 228–229 °C (EtOH). Light brown solid, 1H NMR (400 MHz, DMSO-d6) δ 6.57 (s, 2H, NH2), 7.02 (t, 1H, J = 7.4 Hz, ArCONH), 7.23 (t, 2H, J = 7.8 Hz, Ar), 7.32 (d, 2H, J = 7.3 Hz, Ar), 7.39 (d, 2H, J = 8.0 Hz, Ar), 7.47–7.49 (m, 2H, Ar), 10.04 (s, 1H, CONH2), 12.88 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ163.75, 156.35, 155.43, 145.54, 141.87, 138.61, 128.43, 128.24, 127.93, 123.86, 119.95, 109.88. MS-ESI for C17H14N4O2 (Calcd, 306.11), [M + H]+ at m/z 306.96, tR = 11.825. Anal. Calcd for C17H14N4O2: C, 66.66; H, 4.61; N, 18.29. Found C, 66.92; H, 4.63; N, 18.36.
5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid propylamide (4b)
Yield = 35%; mp = 228–230 °C (EtOH). Yellow coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 0.57–0.61 (m, 2H, CH3), 1.18 (dp, 2H, J = 14.2, 7.2 Hz, CH2), 2.94 (q, 2H, J = 6.8 Hz, NHCH2), 6.31 (s, 2H, NH2), 7.36 (dt, 2H, J = 4.5, 1.6 Hz, Ar), 7.44 (dq, 2H, J = 6.4, 1.7 Hz, Ar), 7.98 (t, 1H, J = 5.8 Hz, Ar), 12.79 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ 164.94, 156.30, 145.52, 141.61, 137.07, 128.17, 127.90, 127.80, 110.26, 40.55, 21.51, 11.24. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 273.02, tR = 9.970. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
3.2.2. General Procedure for Compounds 5a,b
A mixture of 4a,b (0.43 mmol), K2CO3 (0.86 mmol) and 0.50 mmol of ethyl bromide in anhydrous DMF (2 mL) was refluxed for 30–90 min. After cooling, the mixture was diluted with cold water (15 mL) and compound 5a was recovered by filtration under vacuum. For compound 5b the suspension was extracted with CH2Cl2 (3 × 15 mL) and the solvent was evaporated in vacuo. The crude products were purified by crystallization from ethanol.
5-Amino-1-ethyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid phenylamide (5a)
Yield = 90%; mp = 172–173 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.42 (t, 3H, CH3CH2, J = 7.2 Hz), 4.26 (q, 2H, CH3CH2, J = 7.2 Hz), 6.70 (exch br s, 1H, CONH), 6.93 (d, 2H, Ar, J = 8.0 Hz), 7.04 (t, 1H, Ar, J = 8.0 Hz), 7.20 (t, 2H, Ar, J = 8.0 Hz), 7.49–7.54 (m, 3H, Ar), 7.55–7.60 (m, 2H, Ar). Anal. Calcd for C19H18N4O2: C, 68.25; H, 5.43; N, 16.76. Found C, 68.41; H, 5.44; N, 16.72.
5-Amino-1-ethyl-6-oxo-3-phenyl-1,6-dihydro-pyridazine-4-carboxylic acid propylamide (5b)
Yield = 80%; mp = 141–143 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 0.64 (t, 3H, NH-CH2CH2CH3, J = 7.2 Hz), 1.15 (sex, 2H, NH-CH3CH2CH2, J = 7.6 Hz), 1.43 (t, 3H, N-CH2CH3, J = 7.2 Hz), 3.05 (q, 2H, NH-CH2CH2CH3, J = 7.2 Hz), 4.25 (q, 2H, N-CH2CH3, J = 7.2 Hz), 5.02 (exch br s, 1H, CONHCH2), 6.95 (exch br s, 2H, NH2), 7.45–7.51 (m, 5H, Ar). Anal. Calcd for C16H20N4O2: C, 63.98; H, 6.71; N, 18.65. Found C, 63.83; H, 6.70; N, 18.70.
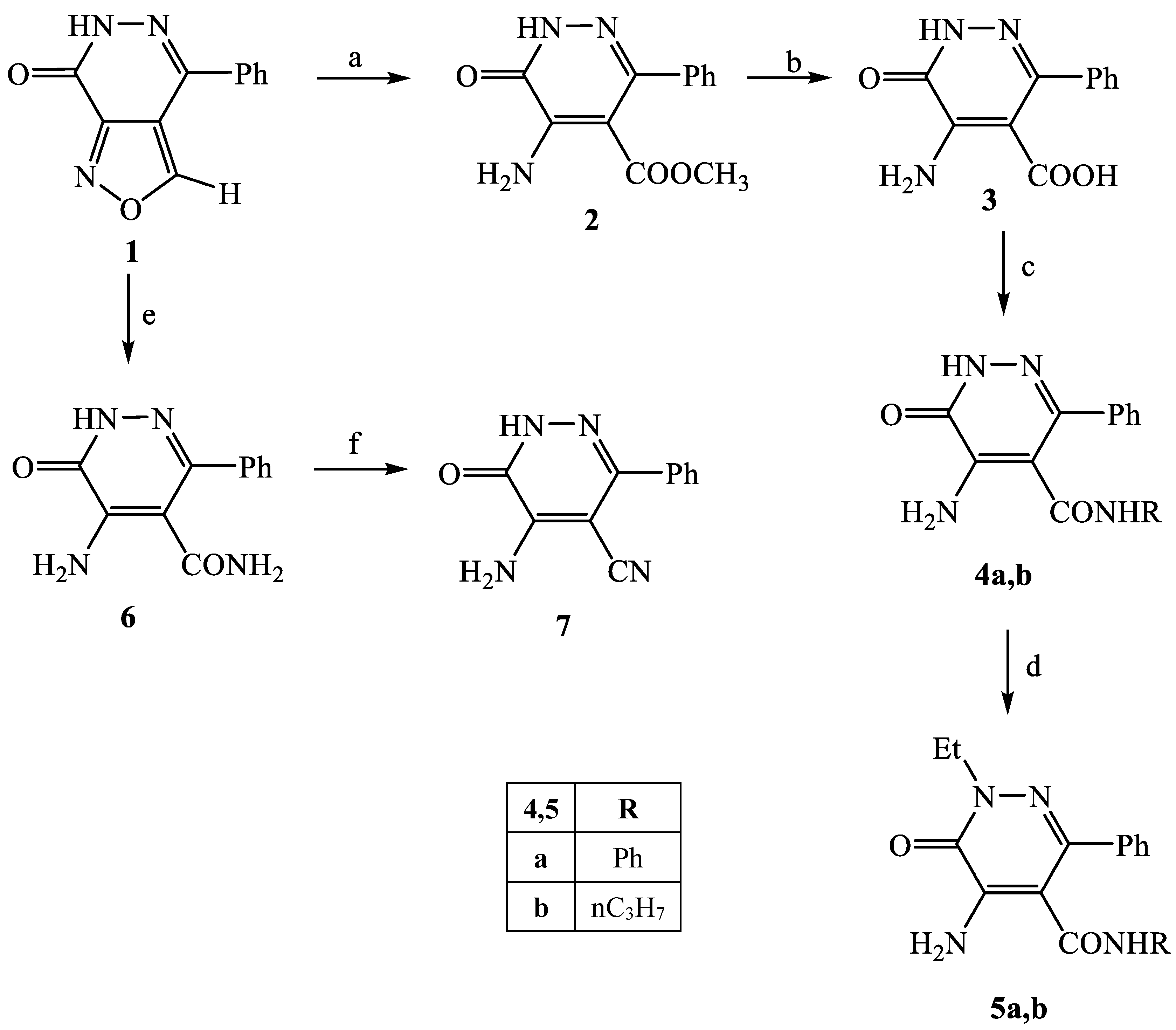
3.2.3. 5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (6)
A mixture of isoxazolopyridazinone
1 (0.94 mmol) [
29], 2 mL of 33% NH
3 and a catalytic amount of piperidine was stirred at 60 °C for 90 min in a sealed/pressure vessel. After cooling the precipitate was recovered by suction and recrystallized with diethyl ether. Yield = 46%; mp > 300 °C (Et
2O). Light brown solid,
1H NMR (400 MHz, DMSO-d
6) δ 6.38 (s, 2H, NH
2), 7.38 (dd, 3H, Ar, J = 5.0, 2.1 Hz), 7.47–7.49 (m, 2H, Ar), 12.79 (s, 1H, ArNH).
13C NMR (100 MHz, DMSO-d
6) δ 167.25, 156.28, 145.36, 141.66, 137.17, 128.17, 127.97, 127.83, 109.75. MS-ESI for C
11H
10N
4O
2 (Calcd, 230.08), [M + H]
+ at
m/z 230.95, t
R = 6.091. Anal. Calcd for C
11H
10N
4O
2: C, 57.39; H, 4.38; N, 24.34. Found C, 57.16; H, 4.36; N, 24.24.
3.2.4. 5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carbonitrile (7)
A suspension of 6 (0.40 mmol) in POCl3 (8 mmol) was stirred at 60 °C for 1–2 h. After cooling, the reaction mixture was treated with cold water (15 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL). The organic solvent was evaporated to afford the desired final compound which was purified by crystallized from diethyl ether. Yield = 48%; mp = 287–289 °C (Et2O). Yellow coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 6.78 (s, 1H, NH2), 7.48 (tt, 3H, J = 3.9, 2.4 Hz, Ar), 7.57–7.61 (m, 2H, Ar), 12.98 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ154.47, 154.06, 149.95, 145.72, 135.21, 129.28, 128.29, 128.13, 115.68, 113.42. MS-ESI for C11H8N4O (Calcd, 212.07), [M + H]+ at m/z 212.89, tR = 10.234. Anal. Calcd for C11H8N4O: C, 62.26; H, 3.80; N, 26.40. Found C, 62.01; H, 3.78; N, 26.29.
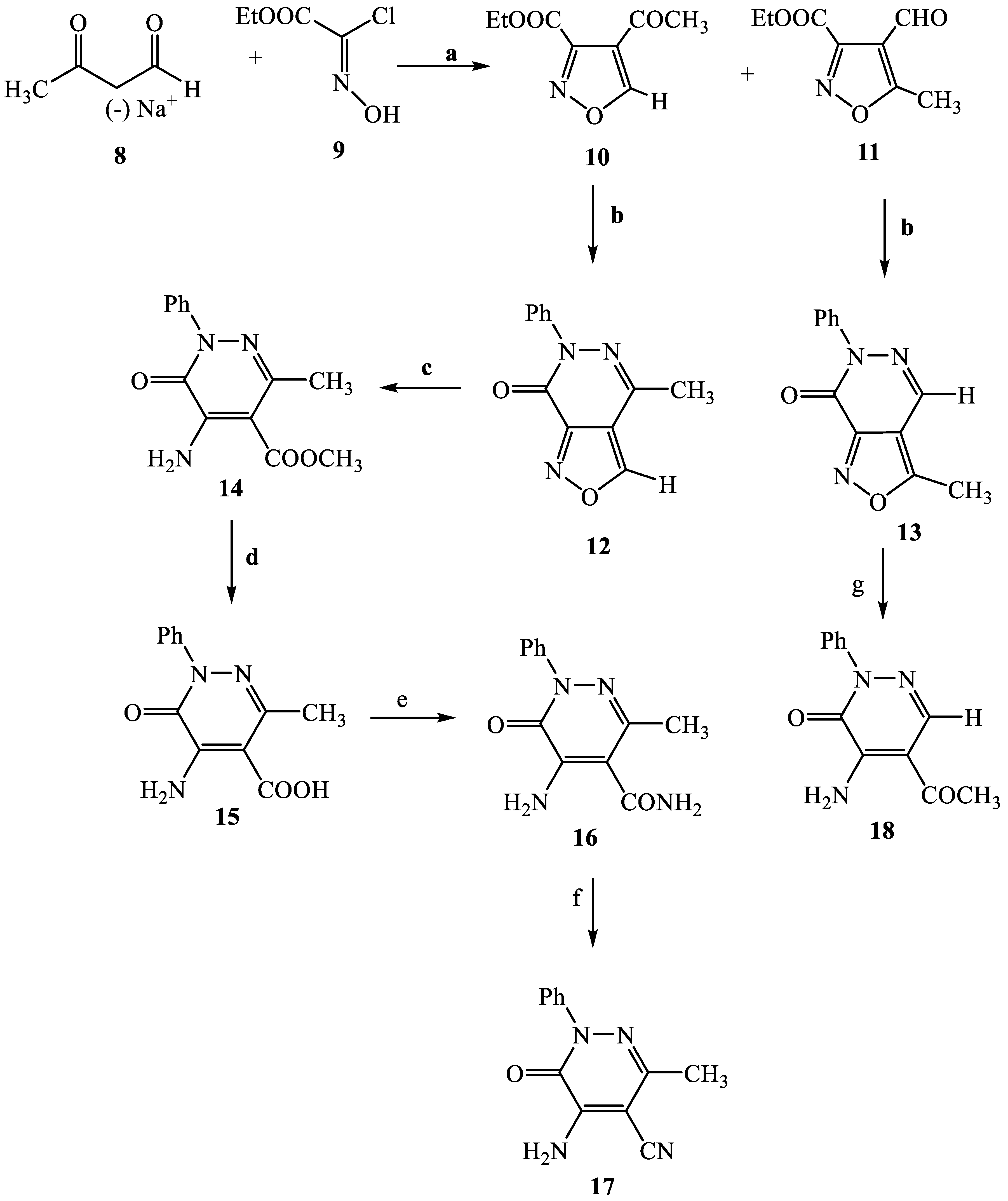
3.2.5. 4-Acetyl-isoxazole-3-carboxylic acid ethyl ester (10)
To a cooled (−5 °C) and stirred suspension of
8 (9.9 mmol) in anhydrous ethanol, a solution of ethyl chloro(hydroximino)acetate
9 (6.6 mmol) in the same solvent (11 mL) was added dropwise. The solvent was evaporated in vacuo, cold water was added (10 mL) and the suspension was extracted with CH
2Cl
2 (3 × 15 mL). A mixture of isoxazoles
10 and
11 [
36] was obtained and they were separated by flash column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 15%; oil.
1H NMR (400 MHz, CDCl
3) δ 1.45 (t, 3H, CH
2CH
3, J = 7.2 Hz), 2.55 (s, 3H, COCH
3), 4.51 (q, 2H, CH
2CH
3, J = 7.2 Hz), 8.95 (s, 1H, Ar). Anal. Calcd for C
8H
9NO
4: C, 52.46; H, 4.95; N, 7.65. Found C, 52.33; H, 4.94; N, 7.67.
3.2.6. General Procedure for Compounds 12 and 13
To a cooled and stirred mixture of isoxazoles 10 or 11 (6.56 mmol) and 2.5 g of PPA (25 mmol) in 2 mL of anhydrous EtOH, 7.87 mmol of phenylhydrazine were added. The reaction was carried out at 70 °C for 30 min. After cooling the solvent was evaporated under vacuum, cold water was added (10 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded the desired compounds.
4-Methyl-6-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (12)
Yield = 90%; mp = 200–201 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.54 (s, 3H, CH3), 7.38 (t, 1H, Ar, J = 7.6 Hz), 7.45–7.50 (m, 2H, Ar), 7.55–7.60 (m, 2H, Ar), 9.22 (s, 1H, C=CH). Anal. Calcd for C12H9N3O2: C, 63.43; H, 3.99; N, 18.49. Found C, 63.58; H, 4.00; N, 18.44.
3-Methyl-6-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (13)
Yield = 80%; mp = 188–190 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.88 (s, 3H, CH3), 7.42 (t, 1H, Ar, J = 7.4 Hz), 7.51 (t, 2H, Ar, J = 7.8 Hz), 7.60 (d, 2H, Ar, J = 7.6 Hz), 8.16 (s, 1H, N=CH). Anal. Calcd for C12H9N3O2: C, 63.43; H, 3.99; N, 18.49. Found C, 63.55; H, 3.99; N, 18.46.
5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid methyl ester (14)
A mixture of 12 (6.21 mmol) and Et3N (0.8 mL) in 2 mL of CH3OH was heated at 60 °C for 2 h. After cooling, ice water (20 mL) was added and the suspension was extracted with CH2Cl2 (3 × 15 mL). Then the solvent was evaporated in vacuo to afford compound 14 which was purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 80%; mp = 91–93 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.53 (s, 3H, CH3), 3.95 (s, 3H, COOCH3), 7.39 (t, 1H, Ar, J = 7.4 Hz), 7.49 (t, 2H, Ar, J = 8.4 Hz), 7.65 (d, 2H, Ar, J = 8.4 Hz), 8.16 (exch br s, 2H, NH2). Anal. Calcd for C13H13N3O3: C, 60.22; H, 5.05; N, 16.21. Found C, 60.08; H, 5.06; N, 16.26.
3.2.7. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid (15)
A mixture of 14 (2.12 mmol), ethanol (3 mL) and 6N NaOH (2 mL) was stirred at reflux for 30 min. After cooling, the solvent was evaporated under vacuum, cold water was added (2–3 mL) and the mixture was acidified with 6N HCl. The precipitate was recovered by vacuum filtration and crystallized from cyclohexane. Yield = 90%; mp = 214–216 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 2.37 (s, 3H, CH3), 7.39 (t, 1H, Ar, J = 7.2 Hz), 7.46 (t, 2H, Ar, J = 8.0 Hz), 7.54 (d, 2H, Ar, J = 8.0 Hz), 8.25 (exch br s, 2H, NH2). Anal. Calcd for C12H11N3O3: C, 58.77; H, 4.52; N, 17.13. Found C, 58.61; H, 4.51; N, 17.16.
3.2.8. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (16)
A mixture of 15 (1.88 mmol), a catalytic amount of Et3N (0.1 mL) and SOCl2 (51 mmol) was refluxed for 30 min. After cooling, the excess of SOCl2 was removed in vacuo and the residue oil was dissolved in cold dry THF (1 mL). To this suspension a solution of 33% NH3 (2 mL) in 1.5 mL of dry THF was added and the mixture was stirred at room temperature for 15 min. After evaporation of the solvent, the mixture was diluted with cold water (20 mL) and the precipitate obtained was filtered and crystallized from ethanol. Yield = 80%; mp = 247–249 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 2.21 (s, 3H, CH3), 7.35–7.40 (m, 1H, Ar), 7.45–7.51 (m, 5H, Ar), 7.65 (s, 1H, NH2), 7.88 (s, 1H, CONH2). 13C NMR (100 MHz, DMSO-d6) δ 167.39, 155.53, 143.12, 141.99, 141.63, 128.82, 127.99, 125.85, 110.42, 20.24. MS-ESI for C12H12N4O2 (Calcd, 244.10), [M + H]+ at m/z 244.95, tR = 7.921. Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.24; H, 4.97; N, 23.03.
3.2.9. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carbonitrile (17)
Compound 17 was obtained starting from compound 16, through the same procedure described for 7. After dilution with cold water, the precipitate was recovered by filtration under vacuum and the solid obtained was purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 20%; mp = 201–203 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 3H, CH3), 7.38 (ddd, 1H, J = 7.7, 5.5, 3.6 Hz, Ar), 7.42–7.51 (m, 4H, Ar). 13C NMR (100 MHz, DMSO-d6) δ 153.37, 149.50, 144.17, 141.60, 130.03, 127.68, 125.90, 115.14, 100.83, 20.47. MS-ESI for C12H10N4O (Calcd, 226.08), 226.96 m/z [M + H]+, 435.11 m/z [2M+H-H2O]+, 451.07 m/z [2M-H2+H]+. tR =11.385. Anal. Calcd for C12H10N4O: C, 63.71; H, 4.46; N, 24.76. Found C, 63.96; H, 4.48; N, 24.85.
3.2.10. 5-Acetyl-4-amino-2-phenylpyridazin-3(2H)-one (18)
Intermediate 13 (1.01 mmol) was suspended in 3.5 mL of EtOH, then 6.08 mmol of HCOONH4 and 40 mg of 10% Pd/C were added. The mixture was refluxed for 2 h and after cooling, CH2Cl2 (5 mL) was added. The solution was stirred for 5 min, then the catalyst was filtered off and the solvent was evaporated in vacuo to furnish desiderd compound 18. Yield = 98%; mp = 181–183 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.60 (s, 3H, COCH3), 6.95 (exch br s, 1H, NH2), 7.42 (t, 1H, Ar, J = 7.6 Hz), 7.51 (t, 2H, Ar, J = 7.6 Hz), 7.64 (d, 2H, Ar, J = 7.6 Hz), 8.13 (s, 1H, C6-H), 9.15 (exch br s, 1H, NH2). Anal. Calcd for C12H11N3O2: C, 62.87; H, 4.84; N, 18.33. Found C, 62.69; H, 4.83; N, 18.28.
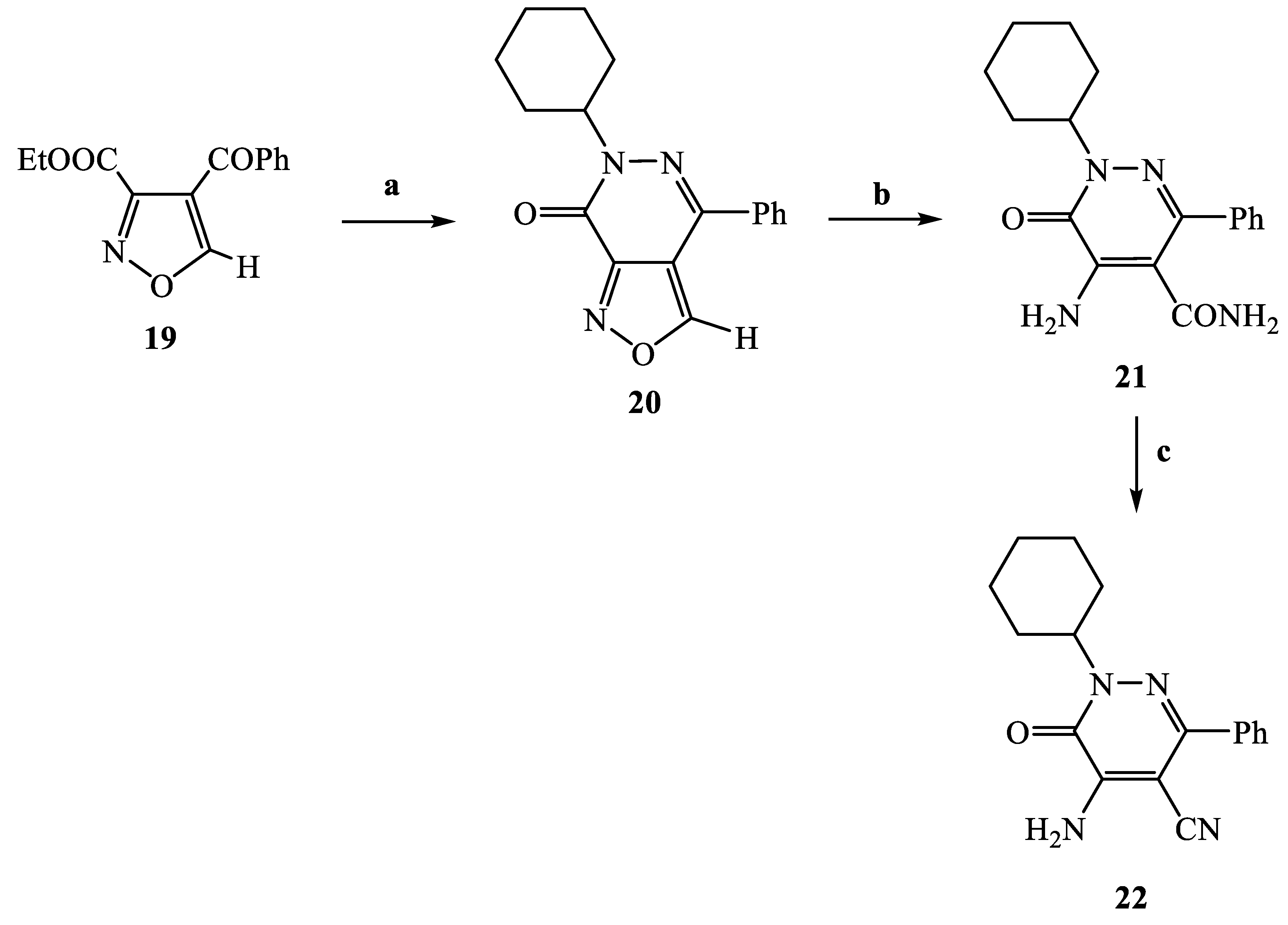
3.2.11. 6-Cyclohexyl-4-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (20)
Compound
20 was obtained starting from
19 [
34] adopting the general procedure described for compounds
12 and
13, but using cyclohexyl hydrazine as reagent. The mixture was heated at 70 °C for 5 h. After dilution with ice-water, the precipitate was recovered by filtration under vacuum and crystallized from ethanol. Yield = 45%; mp = 211–213 °C (EtOH).
1H NMR (400 MHz, CDCl
3) δ 1.27–1.33 (m, 1H, C
6H
11), 1.45–1.51 (m, 3H, C
6H
11), 1.75–1.80 (m, 1H, C
6H
11), 1.85–1.95 (m, 5H, C
6H
11), 5.05–5.10 (m, 1H, C
6H
11), 7.50–7.60 (m, 3H, Ar), 7.85 (d, 2H, Ar, J = 7.2 Hz), 9.30 (s, 1H, isoxazole). Anal. Calcd for C
17H
17N
3O
2: C, 69.14; H, 5.80; N, 14.23. Found C, 69.33; H, 4.82; N, 18.28.
3.2.12. 5-Amino-1-cyclohexyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (21)
A mixture of 20 (0.64 mmol) and 33% NH3 was stirred at 120 °C for 3 h in a sealed/pressure vessel. After cooling, ice-water was added and the suspension was extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded the desired final compound. Yield = 20%; mp = 125–128 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 1.20–1.25 (m, 1H, C6H11), 1.35–1.48 (m, 2H, C6H11), 1.60–1.65 (m, 2H, C6H11), 1.70–1.88 (m, 5H, C6H11), 4.77–4.82 (m, 1H, C6H11), 6.55 (exch br s, 2H, NH2), 7.25–7.31 (m, 3H, Ar), 7.44 (d, 2H, Ar, J = 7.6 Hz), 8.50 (exch br s, 2H, CONH2). Anal. Calcd for C17H20N4O2: C, 65.37; H, 6.45; N, 17.94. Found C, 65.52; H, 6.46; N, 17.99.
3.2.13. 5-Amino-1-cyclohexyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carbonitrile (22)
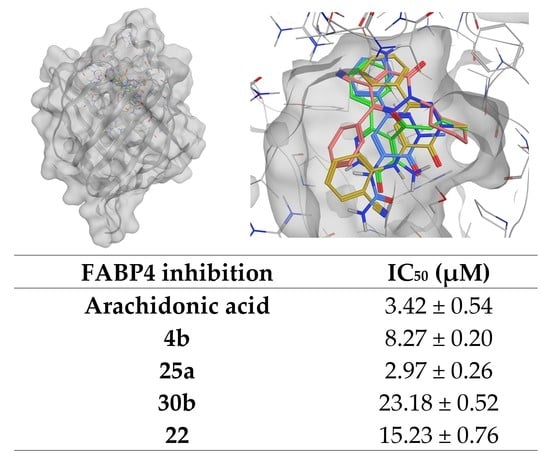
Compound 22 was obtained starting from compound 21, through the same procedure described for 7 and 17. After dilution with cold water, the precipitate was recovered by suction and the solid was purified by crystallization from etanol. Yield = 90%; mp = 170–172 °C (EtOH). Yellow coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.23 (qt, 1H, J = 13.2, 3.3 Hz, CH2-cyclohexane), 1.46 (ttt, 2H, J = 13.8, 7.8, 3.0 Hz, CH2-cyclohexane), 1.71 (dt, 1H, J = 13.3, 3.4 Hz, CH2-cyclohexane), 1.84–1.91 (m, 6H, J = 4.4, 3.5 Hz, CH2-cyclohexane), 4.84–4.93 (m, 1H, CH2-cyclohexane), 7.45–7.50 (m, 3H, Ar), 7.72–7.74 (m, 2H, Ar). 13C NMR (100 MHz, CDCl3) δ 152.65, 148.93, 144.34, 135.02, 129.91, 128.75, 128.20, 114.83, 85.25, 58.19, 30.99, 25.60. MS-ESI for C17H18N4O (Calcd, 294.15), [M + H]+ at m/z 294.99, [M + ACN + H]+ at m/z 336.01, tR = 17.509. Anal. Calcd for C17H18N4O: C, 69.37; H, 6.16; N, 19.03. Found C, 69.09; H, 6.13; N, 18.95.
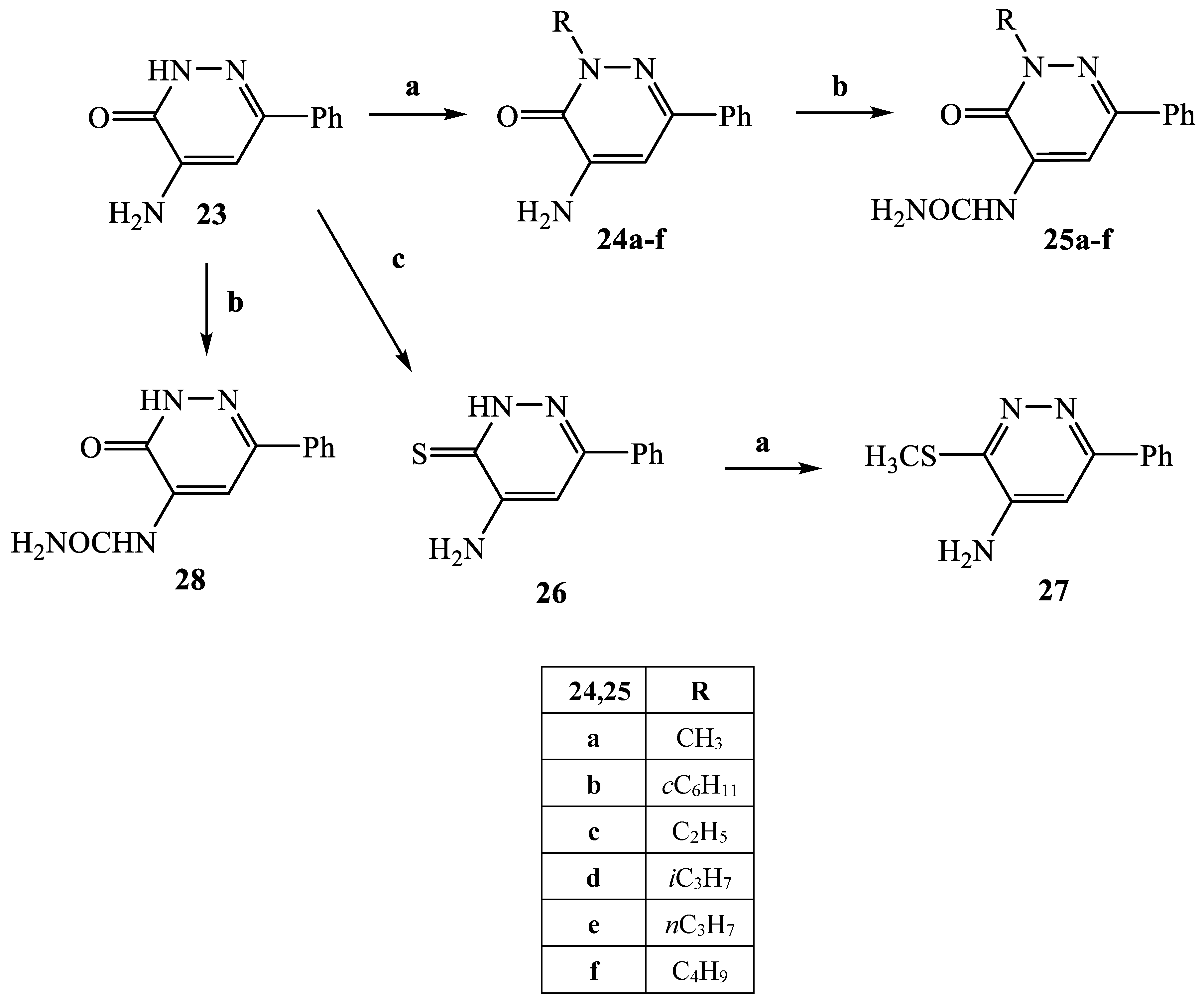
3.2.14. General Procedure for 24b, 24d-f
A mixture of
23 [
37] (0.80 mmol), K
2CO
3 (1.60 mmol) and 0.96–1.44 mmol of the appropriate alkyl or cycloalkyl bromide in anhydrous DMF (1 mL) was refluxed for 2–4 h. After cooling, the mixture was diluted with cold water (20 mL) and extracted with CH
2Cl
2 (3 × 15 mL). Evaporation of the solvent afforded the desired final compounds which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 (for
24b,e,f) or 1:2 (for
24d) as eluent.
4-Amino-2-cyclohexyl-6-phenylpyridazin-3(2H)-one (24b)
Yield = 21%; mp = 120–124 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.20–1.35 (m, 1H, C6H11), 1.40–1.50 (m, 2H, C6H11), 1.70–1.80 (m, 1H, C6H11), 1.90–2.05 (m, 6H, C6H11), 4.85–5.10 (m, 3H, 1H C6H11 + 2H NH2), 6.75 (s, 1H, -CH pyridaz.), 7.35–7.50 (m, 3H, Ar), 7.70 (d, 2H, Ar, J = 7.6 Hz). Anal. Calcd for C16H19N3O: C, 71.35; H, 7.11; N, 15.60. Found C, 71.52; H, 7.10; N, 15.56.
4-Amino-2-isopropyl-6-phenylpyridazin-3(2H)-one (24d)
Yield = 85%; mp = 122–124 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.45 (d, 6H, CH(CH3)2, J = 6.8 Hz), 4.97 (exch br s, 2H, NH2), 5.41 (quin, 1H, CH(CH3)2, J = 6.8 Hz), 6.75 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 3H, Ar), 7.81 (d, 2H, Ar, J = 7.6 Hz). Anal. Calcd for C13H15N3O: C, 68.10; H, 6.59; N, 18.33 Found C, 68.31; H, 6.60; N, 18.29.
4-Amino-6-phenyl-2-propylpyridazin-3(2H)-one (24e)
Yield = 83%; mp = 79–81 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.02 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.93 (sex, 2H, CH2CH2CH3, J = 7.2 Hz), 4.23 (t, 2H, CH2CH2CH3, J = 7.2 Hz), 4.99 (exch br s, 2H, NH2), 6.73 (s, 1H, -CH pyridaz.), 7.38–7.50 (m, 3H, Ar), 7.78 (d, 2H, Ar, J = 8.0 Hz). Anal. Calcd for C13H15N3O: C, 68.10; H, 6.59; N, 18.33 Found C, 68.28; H, 6.60; N, 18.31.
4-Amino-2-butyl-6-phenylpyridazin-3(2H)-one (24f)
Yield = 94%; mp = 67–69 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.00 (t, 3H, CH2CH2CH2CH3, J = 7.2 Hz), 1.45 (m, 2H, CH2CH2CH2CH3), 1.87 (m, 2H, CH2CH2CH2CH3), 4.26 (t, 2H, CH2CH2CH2CH3, J = 7.2 Hz), 4.99 (exch br s, 2H, NH2), 6.75 (s, 1H, -CH pyridaz.), 7.38–7.50 (m, 3H, Ar), 7.76 (d, 2H, Ar, J = 8.0 Hz). Anal. Calcd for C14H17N3O: C, 69.11; H, 7.04; N, 17.27 Found C, 69.29; H, 7.03; N, 17.32.
3.2.15. General Procedure for Compounds 25a–f
To a cooled (0 °C) and stirred suspension of the appropriate pyridazinone 24a–f (0.65 mmol) in anhydrous THF (1–3 mL), anhydrous sodium acetate (1.55 mmol) and triphosgene (2.26 mmol) were added. The mixture was stirred for 10 min at room temperature and refluxed for 2 h. Then, the suspension was cooled to 0 °C and 1 mL of 33% NH3 was added and the mixture was stirred for 30–90 min at room temperature. After evaporation of the solvent, ice/cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum and purified by crystallization from ethanol to obtain the pure samples of 25a–f.
(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25a)
Yield = 65%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.77 (s, 3H, CH3), 7.42–7.51 (m, 3H, Ar), 7.73–7.76 (m, 2H, Ar), 8.35 (s, 1H, Ar), 8.98 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.69, 155.11, 145.12, 137.92, 135.75, 129.39, 129.09, 126.02, 106.62, 20.93. MS-ESI for C12H12N4O2 (Calcd, 244.10), [M + H]+ at m/z 244.95, tR = 11.531. Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94 Found C, 59.24; H, 4.97; N, 23.03.
(2-Cyclohexyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25b)
Yield = 35%; mp = 261–263 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.18–1.31 (m, 1H, C6H11), 1.40–1.51 (m, 2H, C6H11), 1.64–1.72 (m, 1H, C6H11), 1.70–1.90 (m, 6H, C6H11), 4.87 (m, 1H, C6H11), 6.80 (exch br s, 2H, NH2), 7.45–7.55 (m, 3H, Ar), 7.79 (d, 2H, Ar, J = 7.6 Hz), 8.37 (s, 1H, -CH pyridaz.), 8.96 (exch br s, 1H, NHCONH2). Anal. Calcd for C17H20N4O2: C, 65.37; H, 6.45; N, 17.94. Found C, 65.18; H, 6.46; N, 17.91.
(2-Ethyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25c)
Yield = 25%; mp = 270–271 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 1.33 (t, 3H, J = 7.1 Hz, NCH2CH3), 4.20 (q, 2H, J = 7.2 Hz, NCH2), 6.70 (exch br s, 2H, NHCONH2) 7.47 (dt, 3H, J = 13.1, 7.1 Hz, Ar), 7.73 (dd, 2H, J = 23.5, 7.6 Hz, Ar), 8.34 (s, 1H, Ar), 8.98 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.36, 154.51, 150.89, 145.06, 137.97, 135.75, 128.88, 125.84, 106.29, 47.00, 13.39. MS-ESI for C13H14N4O2 (Calcd, 258.11), [M + H]+ at m/z 259.02, 215.90 m/z [M-CONH2 + H]+. tR = 12.090. Anal. Calcd for C13H14N4O2: C, 60.45; H, 5.46; N, 21.69. Found C, 60.21; H, 5.44; N, 21.60.
(2-Isopropyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25d)
Yield = 68%; mp = 260–263 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 1.36 (d, 6H, J = 6.7 Hz, NCH(CH3)2), 5.24 (q, 1H, J = 6.6 Hz, NCH), 7.47 (dt, 2H, J = 15.9, 7.2 Hz, Ar), 7.67–7.79 (m, 3H, Ar), 8.34 (s, 1H, Ar), 8.95 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.69, 154.31, 144.75, 137.69, 136.13, 132.04, 129.30, 129.08, 127.97, 125.91, 105.93, 49.71, 20.96. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 272.95, tR = 14.248. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
(3-Oxo-6-phenyl-2-propyl-2,3-dihydro-pyridazin-4-yl)urea (25e)
Yield = 60%; mp = 273–275 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 0.89 (td, 3H, J = 7.4, 2.7 Hz, NCH2CH2CH3), 1.79 (q, 2H, J = 7.3 Hz, NCH2CH2), 4.13 (t, 2H, J = 7.1 Hz, NCH2), 7.42–7.51 (m, 2H, Ar), 7.69 (d, 1H, J = 5.6 Hz, Ar), 7.73–7.76 (m, 2H, Ar), 8.33 (s, 1H, Ar), 8.96 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.68, 154.88, 145.06, 137.94, 135.87, 132.03, 129.36, 129.07, 128.06, 126.03, 106.37, 53.19, 21.39, 11.13. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 272.95, 229.90 m/z [M-CONH2 + H]+. tR = 14.037. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
(2-Butyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25f)
Yield = 85%; mp = 265–267 °C (EtOH). White solid, 1H NMR (400 MHz, DMSO-d6 + D2O) δ 0.88 (t, 3H, J = 7.4 Hz, CH3), 1.27 (q, 2H, J = 7.3 Hz, CH2CH3), 1.73 (q, 2H, J = 7.2 Hz, CH2CH2CH3), 4.13–4.16 (m, 2H, CH2ArN), 7.41–7.51 (m, 3H, Ar), 7.73–7.75 (m, 2H, Ar), 8.08 (s, 1H, NHCONH2), 8.36 (s, 1H, Ar), 9.10 (s, 2H, CONH2). 13C NMR (100 MHz, DMSO-d6) δ 154.80, 154.60, 137.56, 135.69, 129.20, 128.94, 127.78, 125.79, 118.03, 106.44, 51.11, 29.92, 19.24, 13.53. MS-ESI for C15H18N4O2 (Calcd, 286.14), [M + H]+ at m/z 286.94, tR = 31.162. Anal. Calcd for C15H18N4O2: C, 62.92; H, 6.34; N, 19.57. Found C, 62.66; H, 6.31; N, 19.49.
3.2.16. 4-Amino-6-phenylpyridazine-3(2H)-thione (26)
A mixture of
23 [
37] (0.86 mmol) and Lawesson’s reagent (1.71 mmol) in anhydrous toluene (2–3 mL) was heated at 90 °C for 5 h. After cooling the solvent was evaporated under vacuum, cold water was added (10 mL) and the mixture was extracted with CH
2Cl
2 (3 × 15 mL). Evaporation of the solvent afforded
26 which was purified by flash column chromatography using CH
2Cl
2/CH
3OH 10:1 as eluent. Yield = 63%; mp = 175–178 °C (Cyclohexane).
1H NMR (400 MHz, CDCl
3) δ 5.77 (exch br s, 2H, NH
2), 6.80 (s, 1H, -CH pyridaz.), 7.45–7.55 (m, 3H, Ar), 7.75–7.81 (m, 2H, Ar). Anal. Calcd for C
10H
9N
3S: C, 59.09; H, 4.46; N, 20.67. Found C, 59.23; H, 4.45; N, 20.62.
3.2.17. 3-Methylsulfanyl-6-phenyl-pyridazin-4-ylamine (27)
Compound 27 was obtained, starting from compound 26, through the general procedure described for 24b and 24d–f. After dilution with cold water, the precipitate was recovered by suction and purified by crystallization. Yield = 40%; mp = 168–170 °C (Cyclohexane). Greenish colour solid, 1H NMR (400 MHz, DMSO-d6) δ 2.64 (s, 3H, SCH3), 6.27 (exch br s, 2H, NH2), 7.00 (s, 1H, Ar), 7.46 (dt, 3H, ArH, J = 12.6, 6.9 Hz), 7.90–7.93 (m, 2H, Ar). 13C NMR (100 MHz, DMSO-d6) δ155.31, 147.71, 144.50, 137.20, 129.38, 129.02, 126.51, 102.80, 12.72. MS-ESI for C11H11N3S (Calcd, 217.07), [M + H]+ at m/z 217.86, tR = 9.922. Anal. Calcd for C11H11N3S: C, 60.80; H, 5.10; N, 19.34. Found C, 60.56; H, 5.08; N, 19.26.
3.2.18. (3-Oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (28)
Compound
28 was obtained, starting from
23 [
37], through the same procedure described for
25a–f. Yield = 85%; mp >300 °C (EtOH).
1H NMR (400 MHz, DMSO-d
6) δ 7.41–7.50 (m, 3H, Ar), 7.72–7.75 (m, 2H, Ar), 8.33 (s, 1H, Ar), 8.94 (s, 1H, NHCONH
2), 13.21 (s, 1H, ArNH).
13C NMR (100 MHz, DMSO-d
6) δ156.00, 155.33, 145.54, 139.57, 138.27, 135.77, 134.50, 128.87, 125.70, 106.97. MS-ESI for C
11H
10N
4O
2 (Calcd, 230.08), [M + H]
+ at
m/z 230.88, t = 10.042. Anal. Calcd for C
11H
10N
4O
2: C, 57.39; H, 4.38; N, 24.34. Found C, 57.62; H, 4.39; N, 24.44.
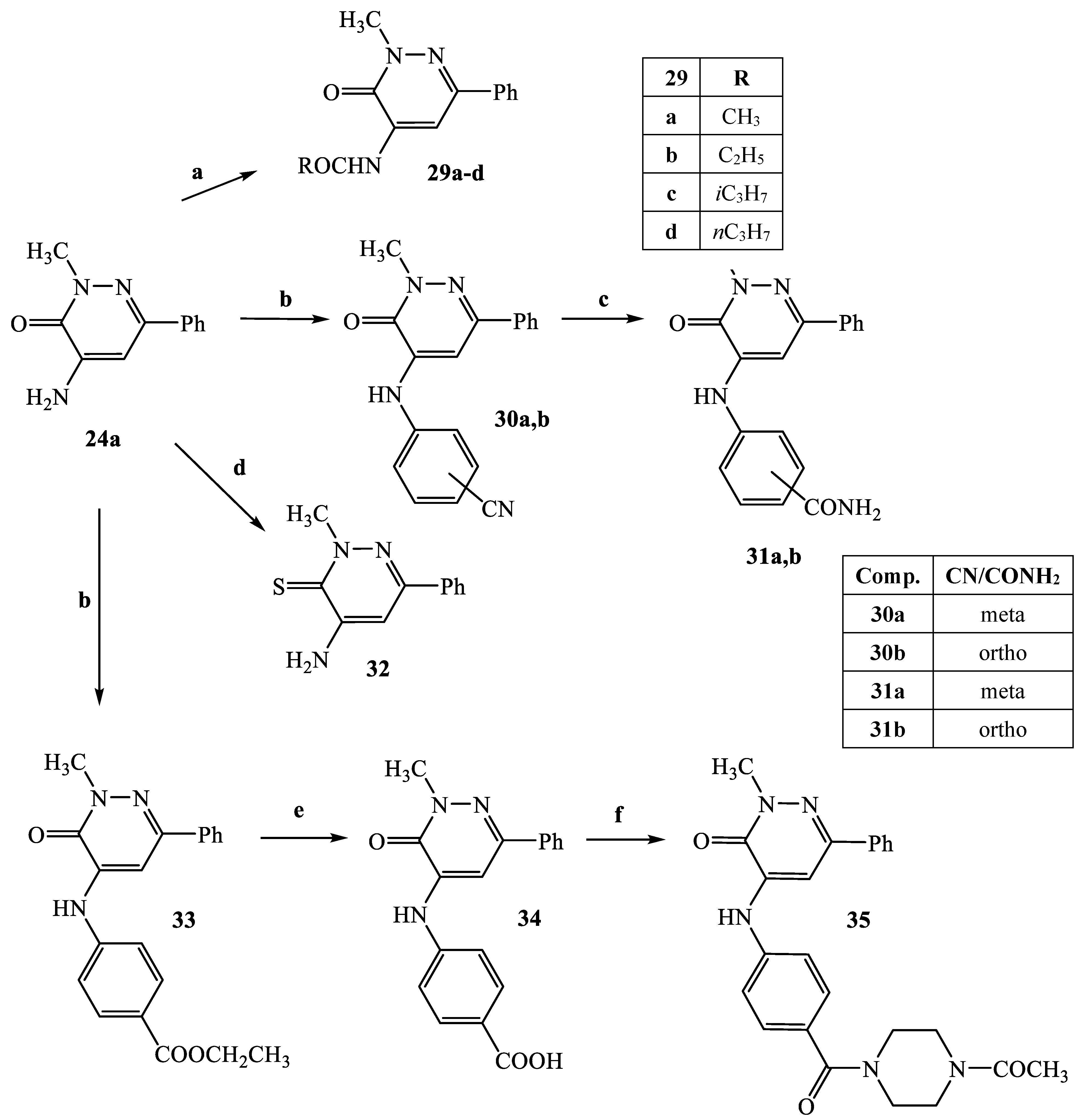
3.2.19. General Procedure for Compounds 29a–d
A mixture of
24a [
38] (0.39 mmol) and the appropriate R-anhydride (13.1 mmol) in 1 mL of pyridine was heated at 140 °C for 5 h in a sealed/pressure vessel. After cooling, ice/cold water was added (50 mL), the precipitate was recovered by filtration under vacuum and purified by crystallization from ethanol to obtain the desired compounds.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)acetamide (29a)
Yield = 90%; mp = 211–212 °C (EtOH). Brownish black coloured solid, 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H, CH3CONH), 3.91 (s, 3H, CH3ArN), 7.42–7.48 (m, 3H, Ar), 7.80–7.83 (m, 2H, Ar), 8.61 (s, 1H, ArH). 13C NMR (100 MHz, CDCl3) δ 196.96, 155.66, 146.61, 135.61, 135.59, 129.59, 128.97, 126.46, 110.82, 40.91, 24.98. MS-ESI for C13H13N3O2 (Calcd, 243.10), [M + H]+ at m/z 243.90, tR = 13.311. Anal. Calcd for C13H13N3O2: C, 64.19; H, 5.39; N, 17.27. Found C, 64.45; H, 5.41; N, 17.34.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)propionamide (29b)
Yield = 93%; mp = 210–211 °C (EtOH). Ash coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.26 (td, 3H, J = 7.5, 1.1 Hz, CH3CH2CONH), 2.49–2.55 (m, 2H, CH2CONH), 3.92 (s, 3H, CH3ArN), 7.41–7.47 (m, 3H, Ar), 7.81–7.84 (m, 2H, Ar), 8.65 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 173.77, 155.73, 146.62, 135.63, 135.60, 129.58, 128.95, 126.43, 110.76, 40.90, 31.00, 9.24. MS-ESI for C14H15N3O2 (Calcd, 257.12), [M + H]+ at m/z 257.90, tR = 14.604. Anal. Calcd for C14H15N3O2: C, 65.36; H, 5.88; N, 16.33. Found C, 65.10; H, 5.90; N, 16.39.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)isobutyramide (29c)
Yield = 95%; mp = 146–148 °C (EtOH). Brown coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.28 (dd, 6H, J = 6.9, 1.2 Hz, (CH3)2CHCONH), 2.64–2.71 (m, 1H, CHCONH), 3.92 (s, 3H, CH3ArN), 7.41–7.46 (m, 3H, Ar), 7.81–7.85 (m, 2H, Ar), 8.66 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 177.10, 155.82, 146.61, 135.71, 135.59, 129.57, 128.94, 126.42, 110.83, 40.87, 36.95, 19.45. MS-ESI for C15H17N3O2 (Calcd, 271.13), [M + H]+ at m/z 272.04, tR = 16.829. Anal. Calcd for C15H17N3O2: C, 66.40; H, 6.32; N, 15.49. Found C, 66.66; H, 6.34; N, 15.55.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)butyramide (29d)
Yield = 92%; mp = 187–189 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.05 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.80 (sex, 2H, CH2CH2CH3, J = 7.2 Hz), 2.49 (q, 2H, CH2CH2CH3, J = 7.2 Hz), 3.94 (s, 3H, N-CH3), 7.45–7.50 (m, 3H, Ar), 7.85 (d, 2H, Ar, J = 7.6 Hz), 8.63 (exch br s, 1H, NH), 8.68 (s, 1H, -CH pyridaz.). Anal. Calcd for C15H17N3O2: C, 66.40; H, 6.32; N, 15.49. Found C, 66.25; H, 6.31; N, 15.44.
3.2.20. General procedure for compounds 30a,b and 33
A mixture of compound
24a [
38] (0.79 mmol), the appropriate R-phenylboronic acid (0.79 mmol), copper acetate (1.19 mmol) and triethylamine (1.59 mmol) in CH
2Cl
2 (5 mL) was stirred at room temperature for 3–12 h. After evaporation of the solvent, ethyl acetate was added (15–20 mL) and the solution was extracted first with 33% NH
3 (3 × 5 mL) and then with water (2 × 5 mL). The organic layer was evaporated under vacuum and the residue was purified by crystallization from ethanol.
3.2.21. 3-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzonitrile (30a)
Yield = 60%; mp = 234–235 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H, CH3), 7.23 (s, 1H, Ar), 7.42–7.48 (m, 3H, Ar), 7.55–7.59 (m, 2H, Ar), 7.80 (dd, 3H, J = 8.0, 1.8 Hz, ArCN), 7.86 (d, 1H, J = 1.8 Hz, ArCN), 9.03 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 193.78, 157.05, 151.81, 140.85, 136.62, 129.18, 126.83, 111.09, 100.21, 23.94. MS-ESI for C18H14N4O (Calcd, 302.12), [M + H]+ at m/z 302.90, [M + ACN + H]+ at m/z 343.92, tR = 16.247. Anal. Calcd for C18H14N4O: C, 71.51; H, 4.67; N, 18.53. Found C, 71.22; H, 4.65; N, 18.45.
3.2.22. 2-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzonitrile (30b)
Yield = 32%; mp = 178–180 °C (EtOH). White coloured solid, 1H NMR (400 MHz, CDCl3) δ 3.95 (s, 3H, CH3), 7.12 (s, 1H, Ar), 7.41–7.47 (m, 3H, ArCN), 7.55 (d, 1H, J = 8.3 Hz, ArCN), 7.65 (td, 1H, J = 7.8, 1.6 Hz, Ar), 7.72 (ddd, 3H, J = 7.6, 3.6, 1.7 Hz, Ar), 7.96 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 147.91, 138.56, 13.22, 129.42, 128.97, 126.44, 124.63, 121.19, 114.68, 100.94, 40.59. MS-ESI for C18H14N4O (Calcd, 302.12), [M + H]+ at m/z 302.97, [M + ACN + H]+ at m/z 344.06, tR = 16.180. Anal. Calcd for C18H14N4O: C, 71.51; H, 4.67; N, 18.53. Found C, 71.22; H, 4.65; N, 18.45.
3.2.23. 4-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzoic acid ethyl ester (33)
Yield = 80%; mp = 171–172 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) 1.42 (t, 3H, CH2CH3, J = 7.2 Hz), 3.96 (s, 3H, CH3), 4.41 (q, 2H, CH2CH3, J = 7.2 Hz), 7.30–7.40 (m, 3H, 2H Ar + CH pyridaz.), 7.45–7.50 (m, 3H, Ar), 7.77 (d, 2H, Ar, J = 8.8 Hz), 7.90 (exch br s, 1H, NH), 8.12 (d, 2H, Ar, J = 8.8 Hz). Anal. Calcd for C20H19N3O3: C, 68.75; H, 5.48; N, 12.03. Found C, 68.58; H, 5.47; N, 12.06.
3.2.24. General Procedure for Compounds 31a,b
A mixture of appropriate pyridazin-benzonitrile 30a or 30b (0.165 mmol) and 80% H2SO4 (2 mL) was stirred at 80 °C for 4 h. After cooling, ice/cold water (2–3 mL) was slowly added, the precipitate obtained was recovered by filtration under vacuum and purified by crystallization.
3.2.25. 3-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzamide (31a)
Yield = 93%; mp = 214–216 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H, CH3), 7.13 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 5H, 4H Ar + 1H CONH2), 7.60 (d, 1H, Ar, J = 9.2 Hz), 7.64 (d, 1H, Ar, J = 7.2 Hz), 7.76 (d, 2H, Ar, J = 8.0 Hz), 7.91 (s, 1H, Ar), 8.02 (exch br s, 1H, CONH2), 8.93 (exch br s, 1H, NH). Anal. Calcd for C18H16N4O2: C, 67.49; H, 5.03; N, 17.49. Found C, 67.36; H, 5.04; N, 17.53.
3.2.26. 2-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzamide (31b)
Yield = 95%; mp = 140–142 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H, CH3), 7.15 (t, 1H, Ar, J = 7.6 Hz), 7.38 (s, 1H, -CH pyridaz.), 7.43–7.50 (m, 3H, Ar), 7.57 (t, 1H, Ar, J = 7.6 Hz), 7.66 (exch br s, 1H, CONH2), 7.77 (t, 2H, Ar, J = 9.2 Hz), 7.84 (d, 2H, Ar, J = 6.8 Hz), 8.17 (exch br s, 1H, CONH2), 10.68 (exch br s, 1H, NH). Anal. Calcd for C18H16N4O2: C, 67.49; H, 5.03; N, 17.49. Found C, 67.36; H, 5.04; N, 17.53.
3.2.27. 4-Amino-2-methyl-6-phenylpyridazine-3(2H)-thione (32)
Compound
32 was obtained, starting from compound
24a [
38], through the same procedure described for 26. In this case, the mixture was refluxed for 10 h. After cooling, ice/cold water was added. The precipitate was recovered by suction and purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 85%; mp = 134–135 °C (EtOH).
1H NMR (400 MHz, CDCl
3) δ 4.38 (s, 3H, CH
3), 5.90 (exch br s, 2H, NH
2), 6.78 (s, 1H, -CH pyridaz.), 7.45–7.50 (m, 3H, Ar), 7.80–7.85 (m, 2H, Ar). Anal. Calcd for C
11H
11N
3S: C, 60.80; H, 5.10; N, 19.34. Found C, 60.97; H, 5.11; N, 19.30.
3.2.28. 4-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzoic acid (34)
Compound 34 was obtained through the general procedure described for 15. After cooling, the mixture was acidified with 6N HCl and the final product was filtered off to obtain the desired compound. Yield = 90%; mp = 280–281 °C (Diethyl ether). 1H NMR (400 MHz, DMSO-d6) δ 3.82 (s, 3H, CH3), 7.37 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 3H, Ar), 7.58 (d, 2H, Ar, J = 8.8 Hz), 7.84 (d, 2H, Ar, J = 8.4 Hz), 7.95 (d, 2H, Ar, J = 8.4 Hz), 9.16 (exch br s, 1H, NH), 12.78 (exch br s, 1H, OH). Anal. Calcd for C18H15N3O2: C, 67.28; H, 4.71; N, 13.08. Found C, 67.44; H, 4.71; N, 13.05.
3.2.29. 4-[4-(4-Acetyl-piperazine-1-carbonyl)-phenylamino]-2-methyl-6-phenylpyridazin-3(2H)-one (35)
Compound 35 was obtained starting from 34 through the same procedure described for 4a,b. In this case the mixture was stirred at room temperature for 40 min. After cooling, THF was removed in vacuo and cold water was added (10 mL). The crude precipitate was recovered by filtration under vacuum and purified by crystallization. Yield = 94%; mp = 213–215 °C (Cyclohexane). Ligrownown solid, 1H NMR (400 MHz, CDCl3) δ 2.14 (s, 3H, CH3CONH), 3.94 (s, 3H, CH3), 3.60 (d, 8H, J = 46.0 Hz, 2 × NCH2CH2N), 7.22 (s, 1H, Ar), 7.33 (d, 2H, J = 7.9 Hz, Ar), 7.47 (dd, 6H, J = 24.3, 7.9 Hz, Ar), 7.71–7.80 (m, 2H, NH + Ar). 13C NMR (100 MHz, CDCl3) δ 170.15, 169.37, 156.24, 146.00, 140.96, 139.58, 136.38, 130.72, 129.39, 129.32, 128.96, 126.42, 120.74, 99.58, 40.75, 21.55. MS-ESI for C24H25N5O3 (Calcd, 431.20), [M + H]+ at m/z 432.10, [M + Na]+ at m/z 454.08, tR = 13.347. Anal. Calcd for C24H25N5O3: C, 66.81; H, 5.84; N, 16.23. Found C, 66.54; H, 5.82; N, 16.16.
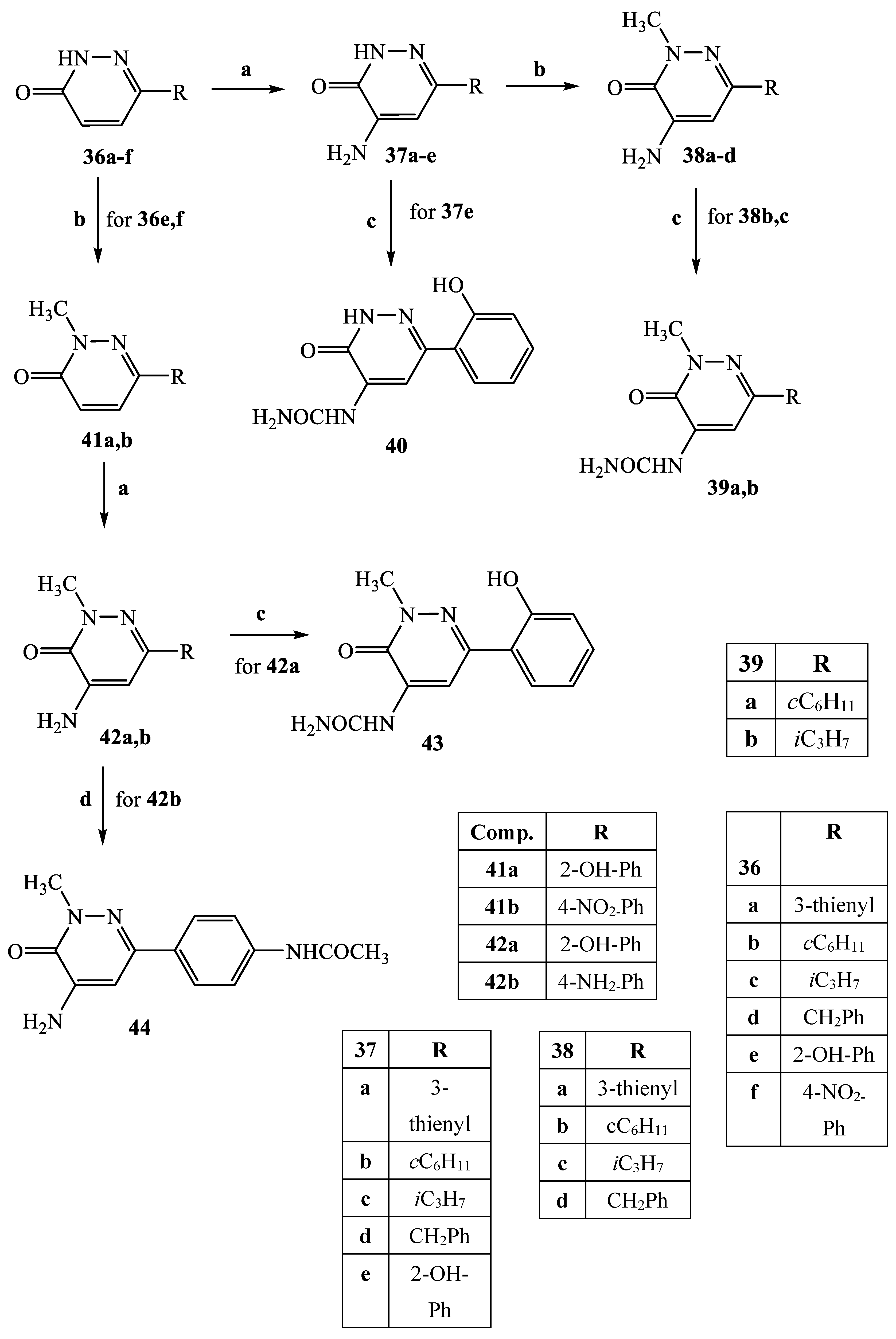
3.2.30. General procedure for compounds 37a–d and 42a,b
A suspension of appropriate pyridazinone
36a–d (1.29 mmol), commercially available, and hydrazine hydrate (48 mmol) was stirred in a sealed/pressure vessel at 180–200 °C for 6–12 h. After cooling, ice-cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum to obtain the desired compounds
37a–d. To obtain compounds
42a,b we adopted the same procedure, using
41a,b (
41a, [
39]) as starting materials.
3.2.31. 4-Amino-6-thiophen-3-yl-pyridazin-3(2H)-one (37a)
Yield = 52%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 6.40 (exch br s, 2H, NH2), 6.68 (s, 1H, -CH pyridaz.), 7.45 (dd, 1H, thiophene, J1 = 1.2 Hz and J2 = 4.8 Hz), 7.60 (dd, 1H, thiophene, J1 = 2.8 Hz and J2 = 4.8 Hz), 7.81 (ds, 1H, thiophene, J = 1.2 Hz), 12.54 (exch br s, 1H, NH). Anal. Calcd for C8H7N3OS: C, 49.73; H, 3.65; N, 21.75. Found C, 49.61; H, 3.65; N, 21.69.
3.2.32. 4-Amino-6-cyclohexylpyridazin-3(2H)-one (37b)
Yield = 46%; mp = 284–287 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.20–1.39 (m, 5H, C6H11), 1.72–1.83 (m, 5H, C6H11), 2.30–2.35 (m, 1H, C6H11), 6.14 (s, 1H, CH pyridaz.), 6.18 (exch br s, 2H, NH2), 12.24 (exch br s, 1H, NH). Anal. Calcd for C10H15N3O: C, 62.15; H, 7.82; N, 21.74. Found C, 62.29; H, 7.80; N, 21.79.
3.2.33. 4-Amino-6-isopropylpyridazin-3(2H)-one (37c)
Yield = 40%; mp = 246–248 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.11 (d, 6H, CH(CH3)2, J = 7.2 Hz), 2.67 (m, 1H, CH(CH3)2), 6.16 (s, 1H, -CH pyridaz.), 6.18 (exch br s, 2H, NH2), 12.23 (exch br s, 1H, NH). Anal. Calcd for C7H11N3O: C, 54.89; H, 7.24; N, 27.43. Found C, 54.76; H, 7.23; N, 27.51.
3.2.34. 4-Amino-6-benzylpyridazin-3(2H)-one (37d)
Yield = 42%; mp = 247–250 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.70 (s, 2H, CH2Ph), 6.04 (s, 1H, -CH pyridaz.), 6.22 (exch br s, 2H, NH2), 7.20–7.40 (m, 5H, Ar), 12.31 (exch br s, 1H, NH). Anal. Calcd for C11H11N3O: C, 65.66; H, 5.51; N, 20.88. Found C, 65.84; H, 5.50; N, 20.83.
3.2.35. 4-Amino-6-(2-hydroxyphenyl)-2-methylpyridazin-3(2H)-one (42a)
Yield = 58%; mp = 212–213 °C (EtOH). White coloured solid, 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H, CH3), 6.84 (s, 1H, Ar), 6.92 (td, 1H, J = 7.7, 1.2 Hz, Ar), 7.00–7.05 (m, 1H, Ar), 7.29 (td, 1H, J = 8.3, 7.8, 1.6 Hz, Ar), 7.58–7.50 (m, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 157.81, 131.24, 126.35, 119.47, 118.30, 98.93, 29.87. MS-ESI for C11H11N3O2 (Calcd, 217.08), [M + H]+ at m/z 217.93, tR = 11.693. Anal. Calcd for C11H11N3O2: C, 60.82; H, 5.10; N, 19.34. Found C, 60.57; H, 5.08; N, 19.26.
3.2.36. 4-Amino-6-(4-aminophenyl)-2-methylpyridazin-3(2H)-one (42b)
Yield = 55%; mp = 208–209 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.65 (s, 3H, N-CH3), 5.35 (exch br s, 2H, NH2), 6.33 (exch br s, 2H, Ph-NH2), 6.59 (d, 2H, Ar, J = 8.0 Hz), 6.63 (s, 1H, -CH pyridaz.), 7.42 (d, 2H, Ar, J = 8.0). Anal. Calcd for C11H11N4O: C, 61.10; H, 5.59; N, 25.91. Found C, C, 61.27; H, 5.58; N, 25.85.
3.2.37. General Procedure for Compounds 38a–d
A mixture of the appropriate pyridazinone 37a–d (0.67 mmol), K2CO3 (1.34 mmol) and CH3I (1.01 mmol) in anhydrous DMF (1.5 mL) was stirred at 80 °C for 1–4 h. After cooling, the mixture was diluted with cold water (15 mL) and compound 38a was recovered by suction and crystallized from ethanol. For compounds 38b–d the suspension was extracted with CH2Cl2 (3 × 15 mL) and the solvent was evaporated in vacuo. The final compounds were purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 (for 38b,d) or CH2Cl2/CH3OH 9.5:0.5 (for 38c) as eluents.
3.2.38. 4-Amino-2-methyl-6-thiophen-3-yl-pyridazin-3(2H)-one (38a)
Yield = 62%; mp = 178–179 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.67 (s, 3H, N-CH3), 6.50 (exch br s, 2H, NH2), 6.68 (s, 1H, -CH pyridaz.), 7.47 (d, 1H, thiophene, J = 4.8 Hz), 7.61 (m, 1H, thiophene), 7.84 (s, 1H, thiophene). Anal. Calcd for C9H9N3OS: C, 52.16; H, 4.38; N, 20.27. Found C, 52.05; H, 4.37; N, 20.22.
3.2.39. 4-Amino-6-cyclohexyl-2-methylpyridazin-3(2H)-one (38b)
Yield = 58%; oil. 1H NMR (400 MHz, CDCl3) δ 1.30–1.43 (m, 5H, C6H11), 1.68–1.92 (m, 5H, C6H11), 2.40 (m, 1H, C6H11), 3.75 (s, 3H, N-CH3), 4.91 (exch br s, 2H, NH2), 6.21 (s, 1H, -CH pyridaz.). Anal. Calcd for C11H17N3O: C, 63.74; H, 8.27; N, 20.27. Found C, 63.87; H, 8.29; N, 20.23.
3.2.40. 4-Amino-6-isopropyl-2-methyl-2H-pyridazin-3-one (38c)
Yield = 49%; oil. 1H NMR (400 MHz, CDCl3) δ 1.18 (d, 6H, CH(CH3)2, J = 7.2 Hz), 2.75 (m, 1H, CH(CH3)2), 3.74 (s, 3H, N-CH3), 4.96 (exch br s, 2H, NH2), 6.21 (s, 1H, -CH pyridaz.). Anal. Calcd for C8H13N3O: C, 57.46; H, 7.84; N, 25.13. Found C, 57.58; H, 7.82; N, 25.07.
3.2.41. 4-Amino-6-benzyl-2-methylpyridazin-3(2H)-one (38d)
Yield = 48%; mp = 104–108 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 3.80 (s, 3H, N-CH3), 3.82 (s, 2H, CH2Ph), 4.81 (exch br s, 2H, NH2), 6.07 (s, 1H, -CH pyridaz.), 7.22–7.35 (m, 5H, Ar). Anal. Calcd for C12H13N3O: C, 66.96; H, 6.09; N, 19.52. Found C, 66.83; H, 5.50; N, 20.83.
3.2.42. General Procedure for Compounds 39a,b, 40 and 43
Compounds 39a,b, 40 and 43 were obtained starting from 38b,c, 37e and 42a, respectively, through the same procedure described for compound 25a–f.
3.2.43. (6-Cyclohexyl-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl)urea (39a)
Yield = 66%; mp = 251–254 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.30–1.40 (m, 5H, C6H11), 1.70–1.85 (m, 5H, C6H11), 2.45 (m, 1H, C6H11), 3.64 (s, 3H, N-CH3), 6.74 (exch br s, 2H, CONH2), 7.79 (s, 1H, -CH pyridaz.), 8.84 (exch br s, 1H, NHCO). Anal. Calcd for C12H18N4O2: C, 57.58; H, 7.25; N, 22.38. Found C, 57.41; H, 7.23; N, 22.43.
3.2.44. (6-Isopropyl-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl)urea (39b)
Yield = 60%; mp = 248–251 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.15 (d, 6H, CH(CH3)2, J = 6.8 Hz), 2.80 (m, 1H, CH(CH3)2), 3.65 (s, 3H, N-CH3), 6.74 (exch br s, 2H, CONH2), 7.81 (s, 1H, -CH pyridaz.), 8.85 (exch br s, 1H, CONH). Anal. Calcd for C9H14N4O2: C, 51.42; H, 6.71; N, 26.65. Found C, 51.31; H, 6,70; N, 26.61.
3.2.45. [6-(2-Hydroxy-phenyl)-3-oxo-2,3-dihydro-pyridazin-4-yl]-urea (40)
Yield = 85%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 6.75 (exch br s, 2H, CONH2), 6.90–6.95 (m, 2H, Ar), 7.27 (t, 1H, Ar, J = 8.4 Hz), 7.45 (dd, 1H, Ar, J1 = 1.2 Hz and J2 = 8.0 Hz), 8.39 (s, 1H, -CH pyridaz.), 8.92 (exch br s, 1H, NHCO), 10.43 (exch br s, 1H, OH), 13.18 (exch br s, 1H, NH). Anal. Calcd for C11H10N4O3: C, 53.66; H, 4.09; N, 22.75. Found C, 53.51; H, 4,08; N, 22.81.
3.2.46. [6-(2-Hydroxyphenyl)-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl]-urea (43)
Yield = 95%; mp = 278–280 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 3.76 (s, 3H, N-CH3), 6.79 (exch br s, 2H, NH2), 6.88–6.95 (m, 2H, Ar), 7.27 (t, 1H, Ar, J = 7.2 Hz), 7.44 (d, 1H, Ar, J = 6.8 Hz), 8.37 (s, 1H, -CH pyridaz.), 8.93 (exch br s, 1H, NHCO), 10.17 (exch br s, 1H, OH). Anal. Calcd for C12H12N4O3: C, 55.38; H, 4.65; N, 21.53. Found C, 55.49; H, 4,65; N, 21,49.
3.2.47. N-[4-(5-Amino-1-methyl-6-oxo-1,6-dihydropyridazin-3-yl)-phenyl]-acetamide (44)
To a cooled (0 °C) and stirred solution of 42b (0.93 mmol) in anhydrous THF (2–3 mL), 1.02 mmol of acetyl chloride was added and the mixture was stirred at room temperature for 20 min. After dilution with cold water (20–30 mL), the precipitate was recovered by filtration under vacuum and purified by crystallization. Yield = 92%; mp = 270–272 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 2.06 (s, 3H, COCH3), 3.69 (s, 3H, N-CH3), 6.48 (exch br s, 2H, NH2), 6.71 (s, 1H, -CH pyridaz.), 7.60–7.70 (m, 4H, Ar), 8.80 (exch br s, 1H, NHCO). Anal. Calcd for C13H14N4O2: C, 60.45; H, 5.46; N, 21.69. Found C, 60.58; H, 5.45; N, 21.63.
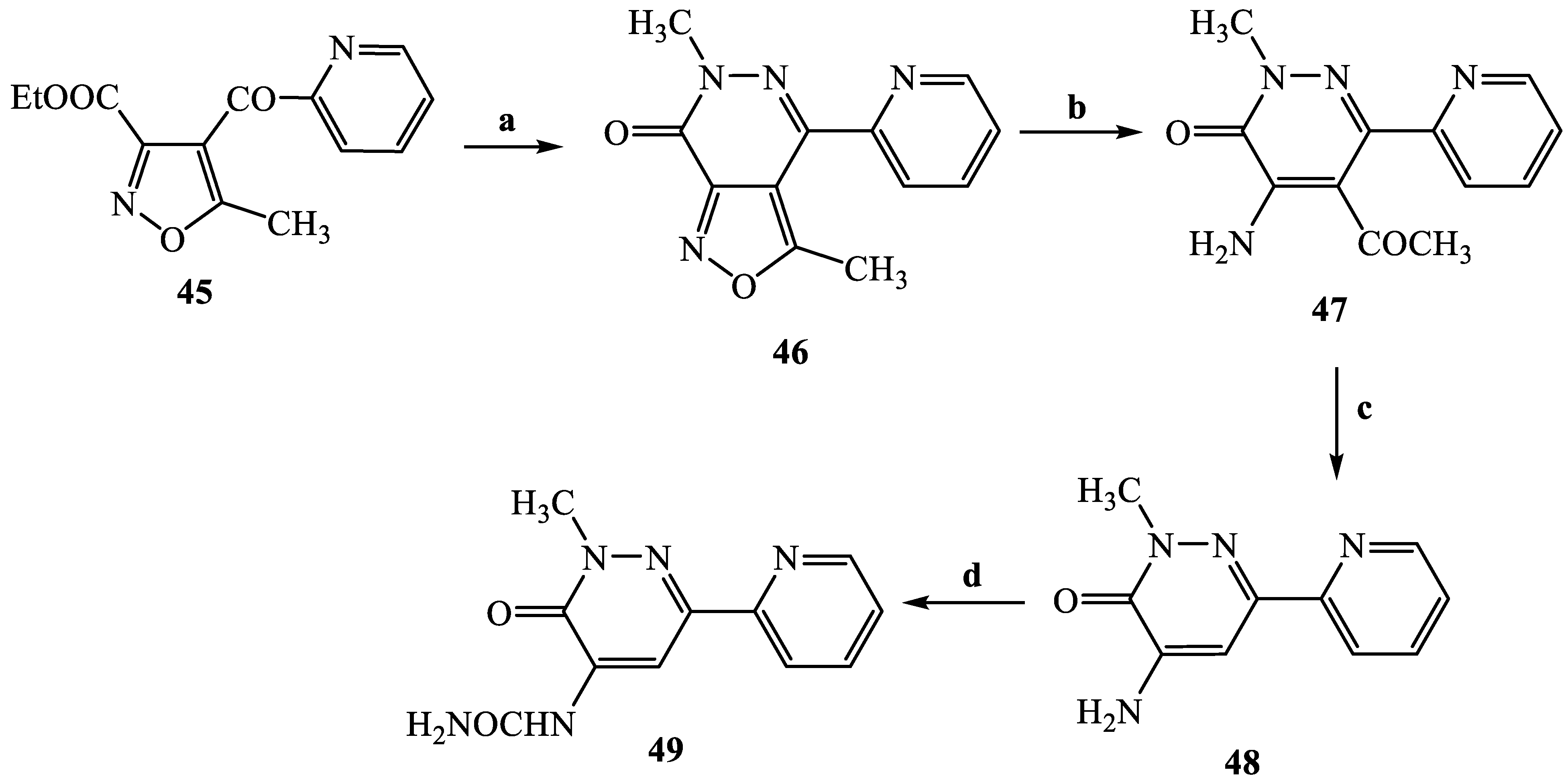
3.2.48. 3,6-Dimethyl-4-pyridin-2-yl-isoxazolo [3,4-d]pyridazin-7(6H)-one (46)
To a cooled (0–4 °C) solution of
45 [
34] (0.38 mmol) in EtOH (2–3 mL), methylhydrazine (1.30 mmol) was added and the mixture was stirred at room temperature for 90 min. The precipitate was recovered by filtration under vacuum to obtain the desired compound. Yield = 84%; mp = 154–155 °C (EtOH).
1H NMR (400 MHz, DMSO-d
6) δ 2.94 (s, 3H, C3-CH
3), 3.74 (s, 3H, N-CH
3), 7.57 (t, 1H, Ar, J = 5.2 Hz), 7.95–8.05 (m, 2H, Ar), 8.76 (d, 1H, Ar, J = 5.2 Hz). Anal. Calcd for C
12H
10N
4O
2: C, 59.50; H, 4.16; N, 23.13. Found C, 59.66; H, 4.16; N, 23.20.
3.2.49. 5-Acetyl-4-amino-2-methyl-6-pyridin-2-yl-pyridazin-3(2H)-one (47)
A mixture of 46 (0.82 mmol), 10% Pd/C (20 mg) and ammonium formate (4.9 mmol) in EtOH (5 mL), was refluxed for 2 h. After addition of CH2Cl2 (4–5 mL) and filtration of charcoal, evaporation of the solvent afforded the product 47. Yield = 65%; mp = 201–203 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.89 (s, 3H, COCH3), 3.73 (s, 3H, N-CH3), 7.23 (exch br s, 2H, NH2), 7.44–7.50 (m, 1H, Ar), 7.89 (d, 1H, Ar, J = 7.2 Hz), 7.96 (t, 1H, Ar, J = 7.2 Hz), 8.57 (d, 1H, Ar, J = 4.4 Hz). Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.18; H, 4.96; N, 22.99.
3.2.50. 4-Amino-2-methyl-6-pyridin-2-yl-pyridazin-3(2H)-one (48)
A suspension of 47 (0.53 mmol) in 1 mL of 48% HBr was stirred in a sealed/pressure vessel at 130 °C for 3 h. After cooling ice-cold water was added and the precipitate was recovered by filtration under vacuum to obtain the desired product 48. Yield = 65%; mp = 294–295 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.75 (s, 3H, N-CH3), 7.24 (s, 1H, -CH pyridaz.), 7.51 (m, 1H, Ar), 8.00 (t, 1H, Ar, J = 7.2 Hz), 8.12 (d, 1H, Ar, J = 8.0 Hz), 8.66 (d, 1H, Ar, J = 4.4 Hz). Anal. Calcd for C10H10N4O: C, 59.40; H, 4.98; N, 27.71. Found C, 59.51; H, 4.99; N, 27.75.
3.2.51. (2-Methyl-3-oxo-6-pyridin-2-yl-2,3-dihydropyridazin-4-yl)-urea (49)
Compound 49 was obtained starting from 48, through the same procedure described for compounds 25a–f, 39a,b, 40 and 43. Yield = 15%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.82 (s, 3H, N-CH3), 6.80 (exch br s, 2H, NH2), 7.46 (m, 1H, Ar), 7.92 (t, 1H, Ar, J = 7.6 Hz), 8.10 (d, 1H, Ar, J = 7.6 Hz), 8.67 (d, 1H, Ar, J = 4.8 Hz), 8.83 (s, 1H, -CH pyridaz.), 8.97 (exch br s, 1H, NHCO). Anal. Calcd for C11H11N5O2: C, 53.87; H, 4.52; N, 28.56. Found C, 53.78; H, 5.00; N, 27.71.
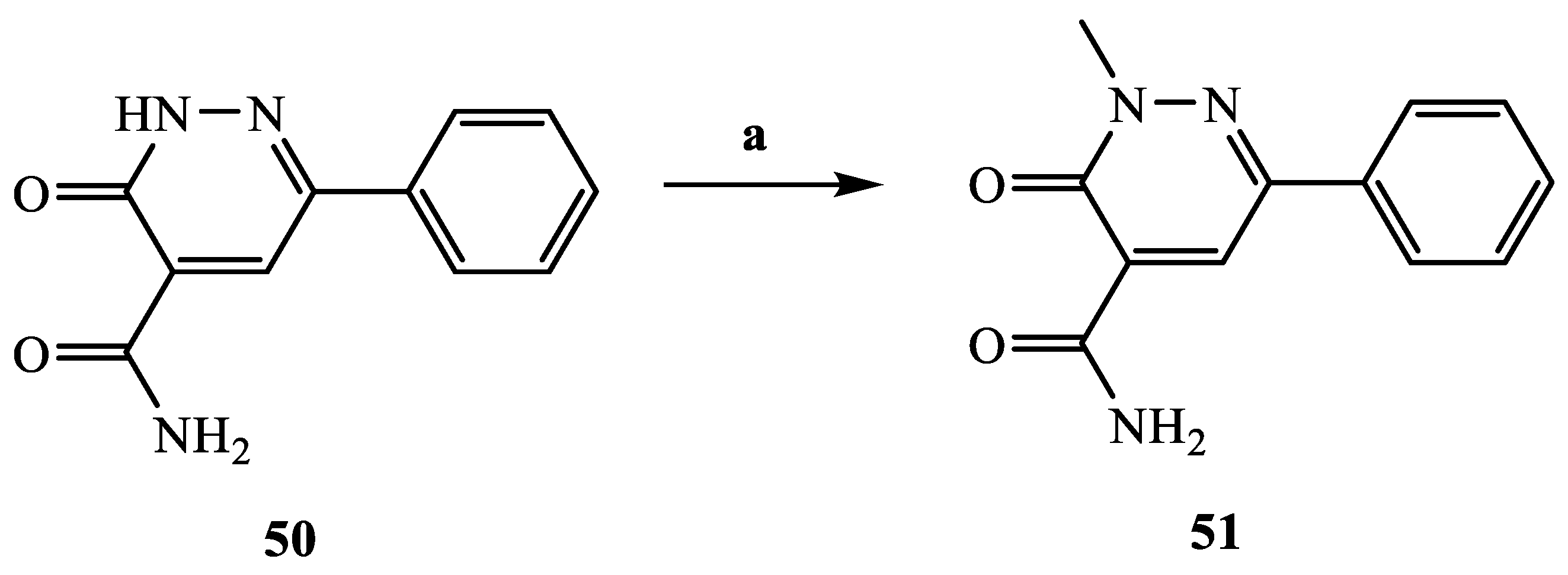
3.2.52. 2-Methyl-3-oxo-6-phenyl-2,3-dihydropyridazine-4-carboxamide (51)
Compound
51 was obtained starting from
50 [
41], through the same procedure described for compounds
38a–d. The compound was purified by crystallization from diethyl ether. Yield = 55%; mp = 215–217 °C (Et
2O). Light brown solid,
1H NMR (400 MHz, CDCl
3) δ 3.99 (s, 3H, CH
3), 5.98 (exch br s, 1H, CONH
2), 7.43–7.51 (m, 3H, Ar), 7.84–7.88 (m, 2H, Ar), 8.72 (s, 1H, Ar), 9.41 (exch br s, 1H, CONH
2).
13C NMR (100 MHz, CDCl
3) δ 163.58, 160.21, 145.33, 134.16, 132.64, 130.02, 129.23, 129.04, 126.15, 41.53. MS-ESI for C
12H
11N
3O
2 (Calcd, 229.08), [M + H]
+ at
m/z 229.90, t
R = 12.205. Anal. Calcd for C
12H
11N
3O
2: C, 62.87; H, 4.84; N, 18.33. Found C, 62.62; H, 4.82; N, 18.26.
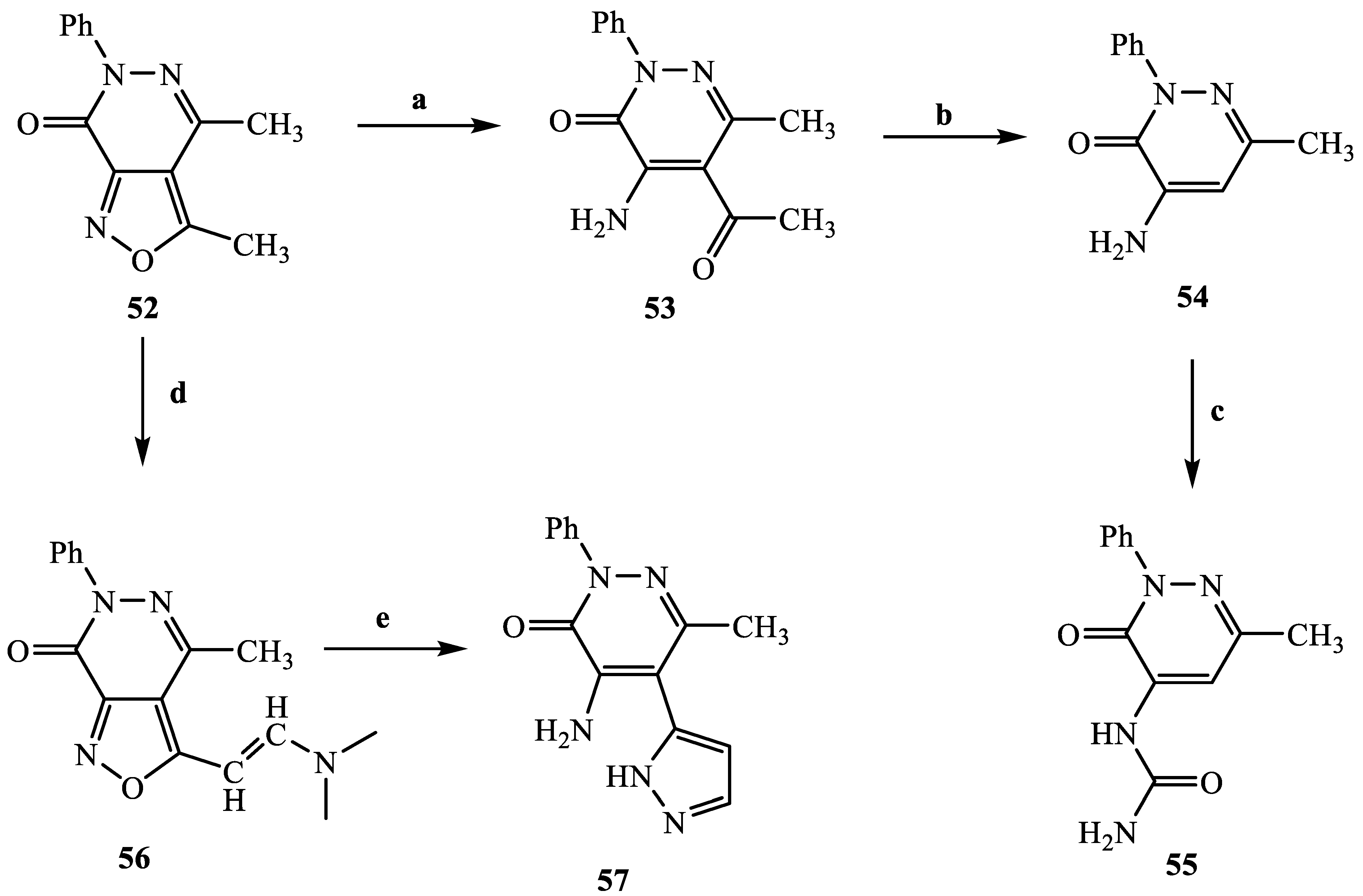
3.2.53. 1-(6-Methyl-3-oxo-2-phenyl-2,3-dihydropyridazin-4-yl)urea (55)
Compound
55 was obtained strating from
54 [
43], through the same procedure for the formation of urea described for compounds
25a–f, 39a,b, 40 and
43. Yield = 95%; mp = 288–290 °C (EtOH). White coloured solid,
1H NMR (400 MHz, DMSO-d
6) δ 2.25 (s, 3H, CH
3), 7.40–7.42 (m, 1H, Ar), 7.47 (d, 2H, J = 8.2 Hz, Ar), 7.50–7.54 (m, 2H, Ar), 7.78 (s, 1H, Ar), 8.91 (s, 1H, NHCONH
2).
13C NMR (100 MHz, DMSO-d
6) δ 155.56, 154.89, 146.52, 141.98, 138.04, 128.86, 128.12, 125.97, 109.55, 21.48. MS-ESI for C
12H
12N
4O
2 (Calcd, 244.10), [M + H]
+ at
m/z 244.88, t
R = 10.100. Anal. Calcd for C
12H
12N
4O
2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.25; H, 4.97; N, 23.03.
3.2.54. (E)-3-(2-(Dimethylamino)vinyl)-4-methyl-6-phenylisoxazolo [3,4-d]pyridazin-7(6H)-one (56)
A mixture of
52 (1.04 mmol) [
42] in 2.5 mL of DMF-DMA was hetaed at 90–100 °C for 1 h. After cooling, ice/cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum to obtained the pure desired compound. Yield = 90%; mp = 224–226 °C dec. (Cyclohexane).
1H NMR (400 MHz, CDCl
3) δ 2.50 (s, 3H, CH
3), 3.00–3.20 (m, 6H, N(CH
3)
2), 5.25 (d, 1H, CH=CH-N, J = 10.0 Hz), 7.30–7.35 (m, 1H, Ar), 7.45–7.50 (m, 2H, Ar), 7.59–7.64 (m, 3H, 1H CH=CH-N + 2H Ar). Anal. Calcd for C
16H
16N
4O
2: C, 64.85; H, 5.44; N, 18.91. Found C, 65.10; H, 5.46; N, 18.98.
3.2.55. 4-Amino-6-methyl-2-phenyl-5-(1H-pyrazol-5-yl)pyridazin-3(2H)-one (57)
A mixture of intermediate 56 (0.81 mmol) and 1 mL of hydrazine hydrate (excess) in 2 mL of abs. EtOH was hetaed at 70 °C for 10 h. After cooling, ice/cold water was added (15 mL). The precipitate obtained was recovered by filtration under vacum and purified by crystallization from ethanol. Yield = 65%; mp = 119–121 °C. (Cyclohexane). Yellow coloured solid, 1H NMR (400 MHz, Methanol-d4) δ 2.31 (s, 3H, CH3), 6.58 (exch br s, 2H, NH2), 7.43 (t, 1H, J = 7.3 Hz, Ar), 7.52 (t, 3H, J = 7.6 Hz, Ar), 7.58 (d, 2H, J = 7.6 Hz, Ar), 7.83 (s, 1H, NH). 13C NMR (100 MHz, Methanol-d4) δ 174.64, 147.92, 143.30, 129.88, 129.32, 127.14, 106.95, 24.30. MS-ESI for C14H13N5O (Calcd, 267.11), [M + H]+ at m/z 267.98. tR = 10.882. Anal. Calcd for C14H13N5O: C, 62.91; H, 4.90; N, 26.20. Found C, 62.66; H, 4.88; N, 26.09.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


