RNA-seq Reveals Differentially Expressed Genes between Two indica Inbred Rice Genotypes Associated with Drought-Yield QTLs

 ,
,
Abstract
:1. Introduction
2. Materials and Method
2.1. Dry-Down Experiment
2.2. RNA Extraction
2.3. Pre-Processing
2.4. Read Count Quantification
2.5. Data Filtering and Normalization
2.6. Differential Expression Analysis
2.7. Co-Localization Analysis
3. Results and Discussion
3.1. Read Mapping
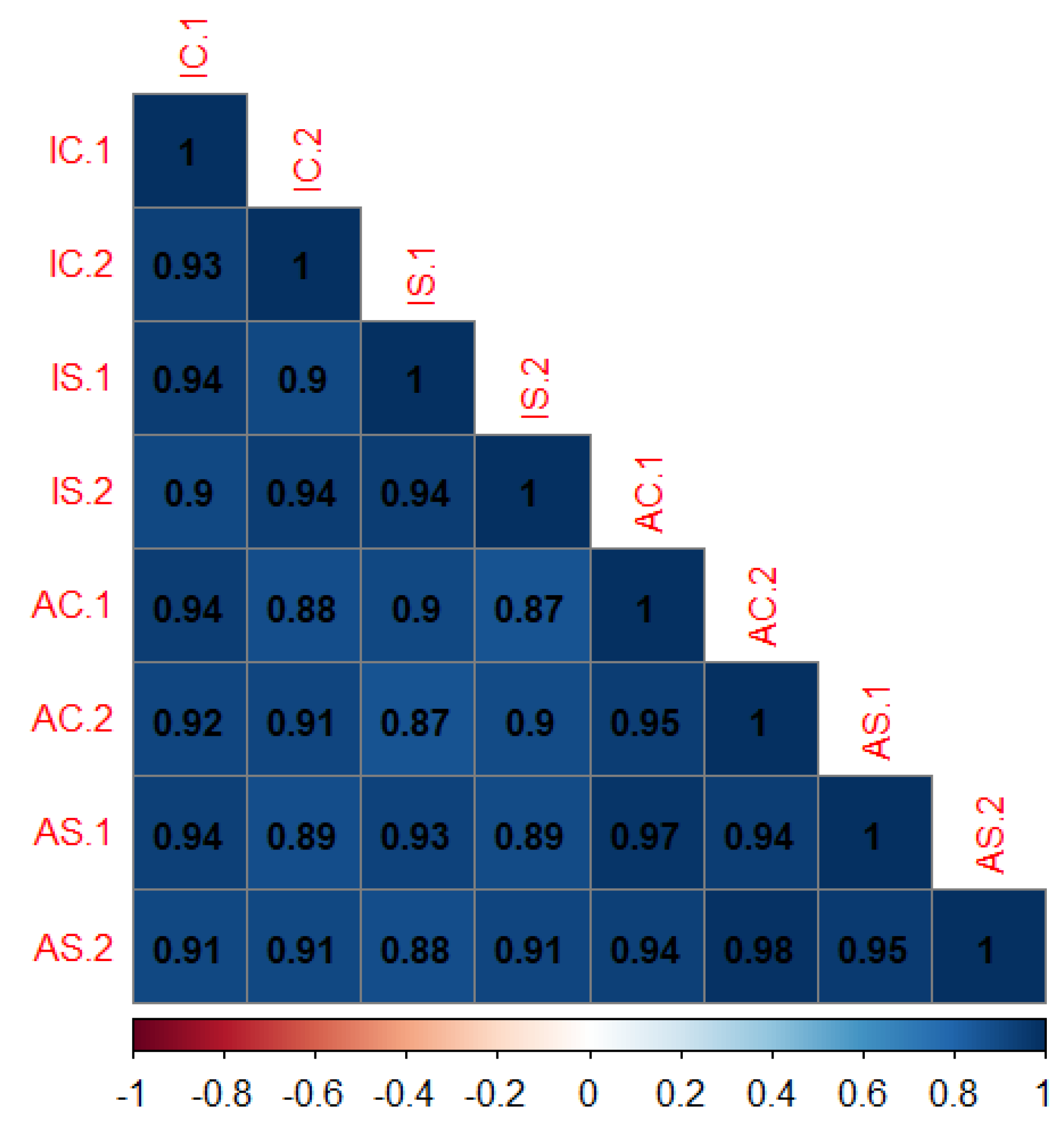
3.2. MA Plots and Spearman’s Coefficient of Correlations
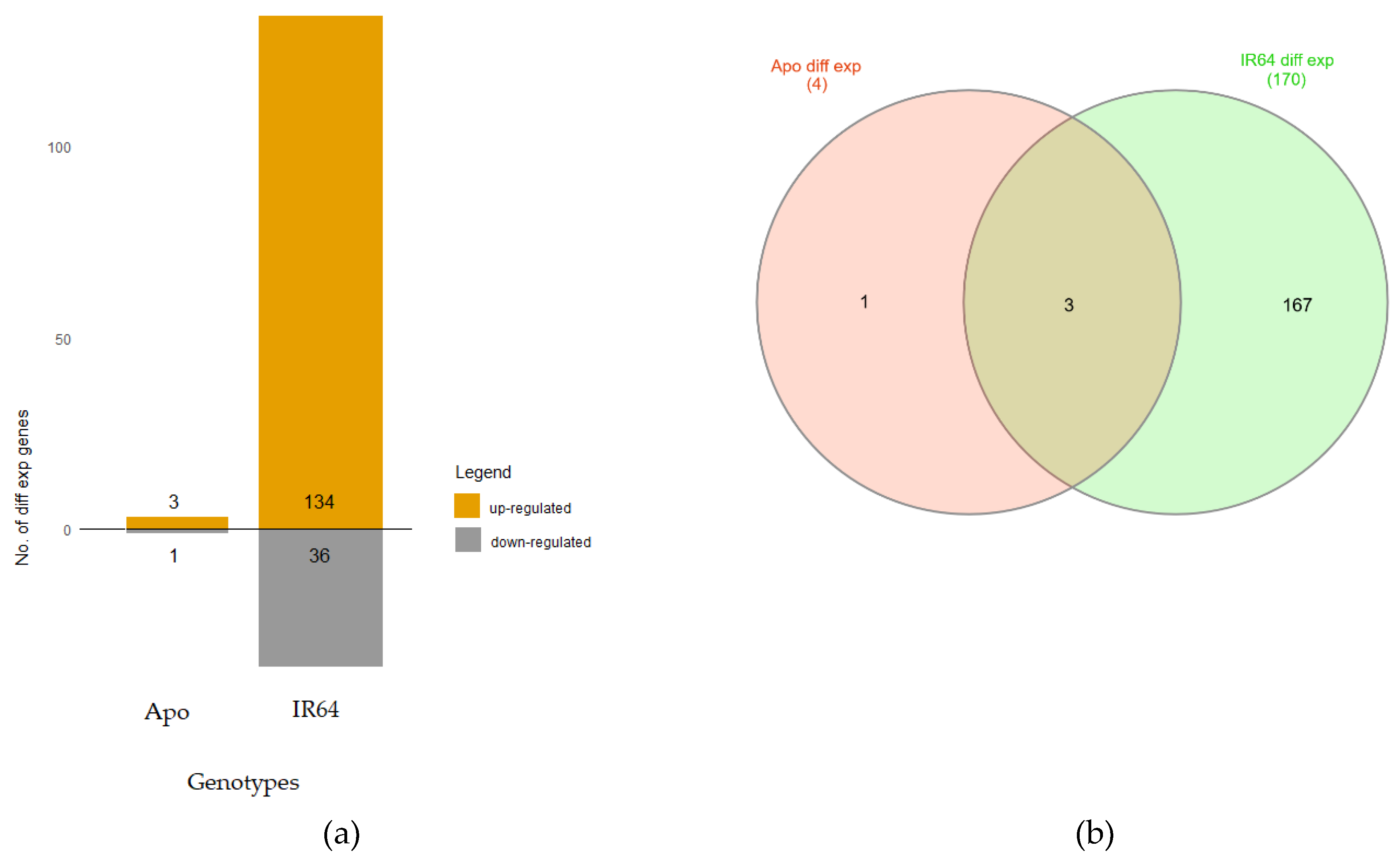
3.3. Pairwise DE (PDE) Between Treatments of the Same Genotype
3.4. Differences Due to Genotypic Background across Treatments
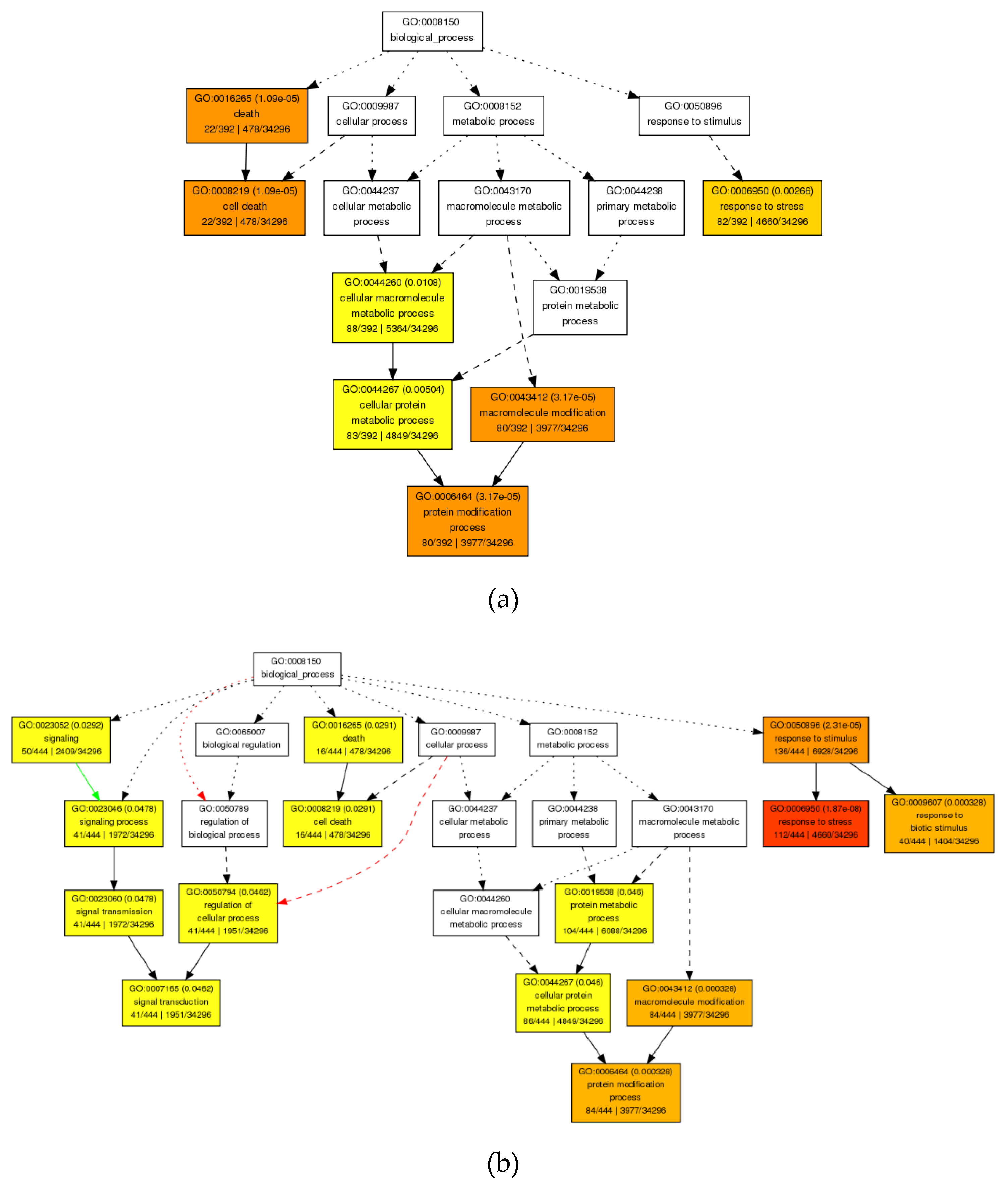
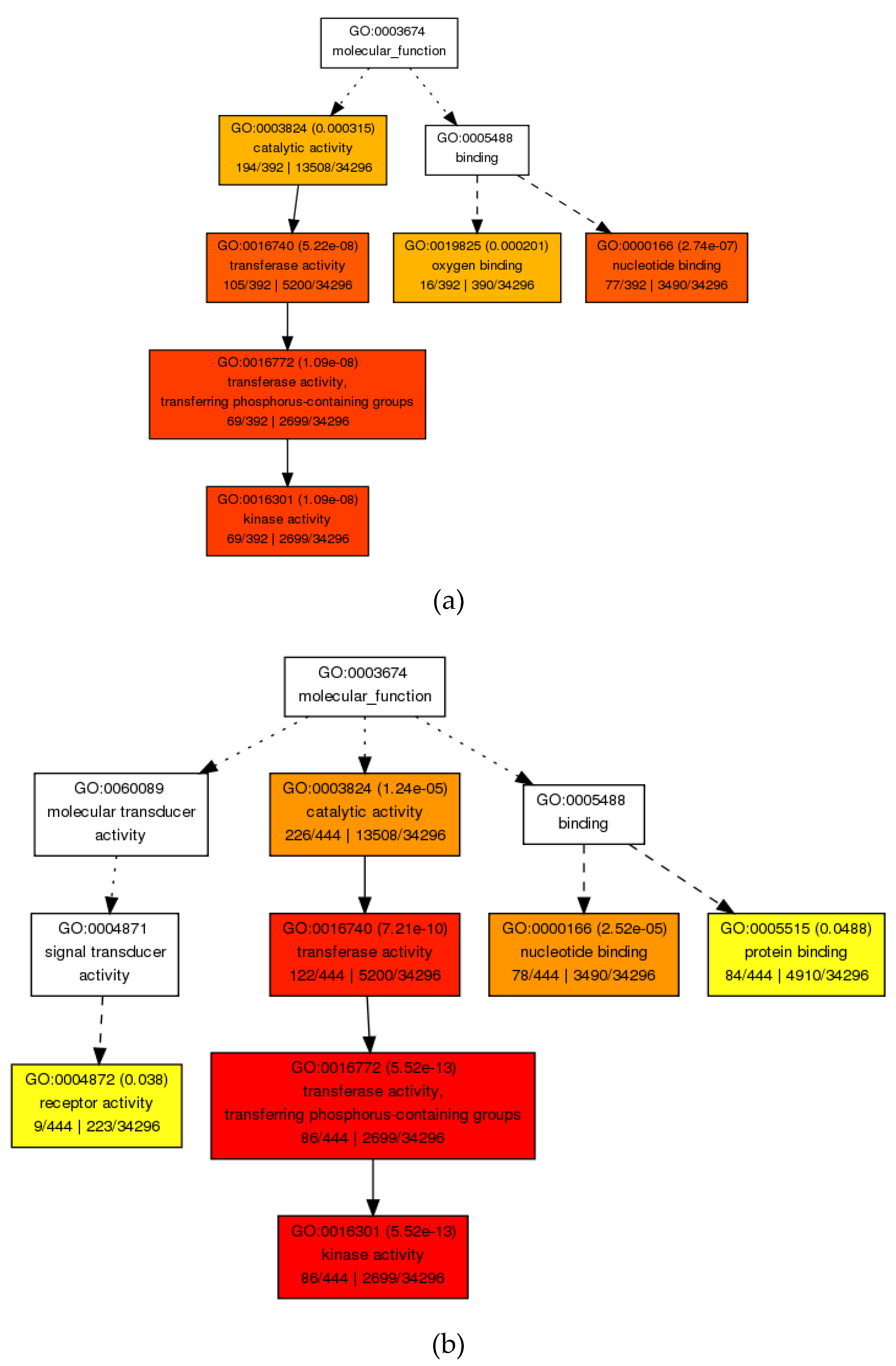
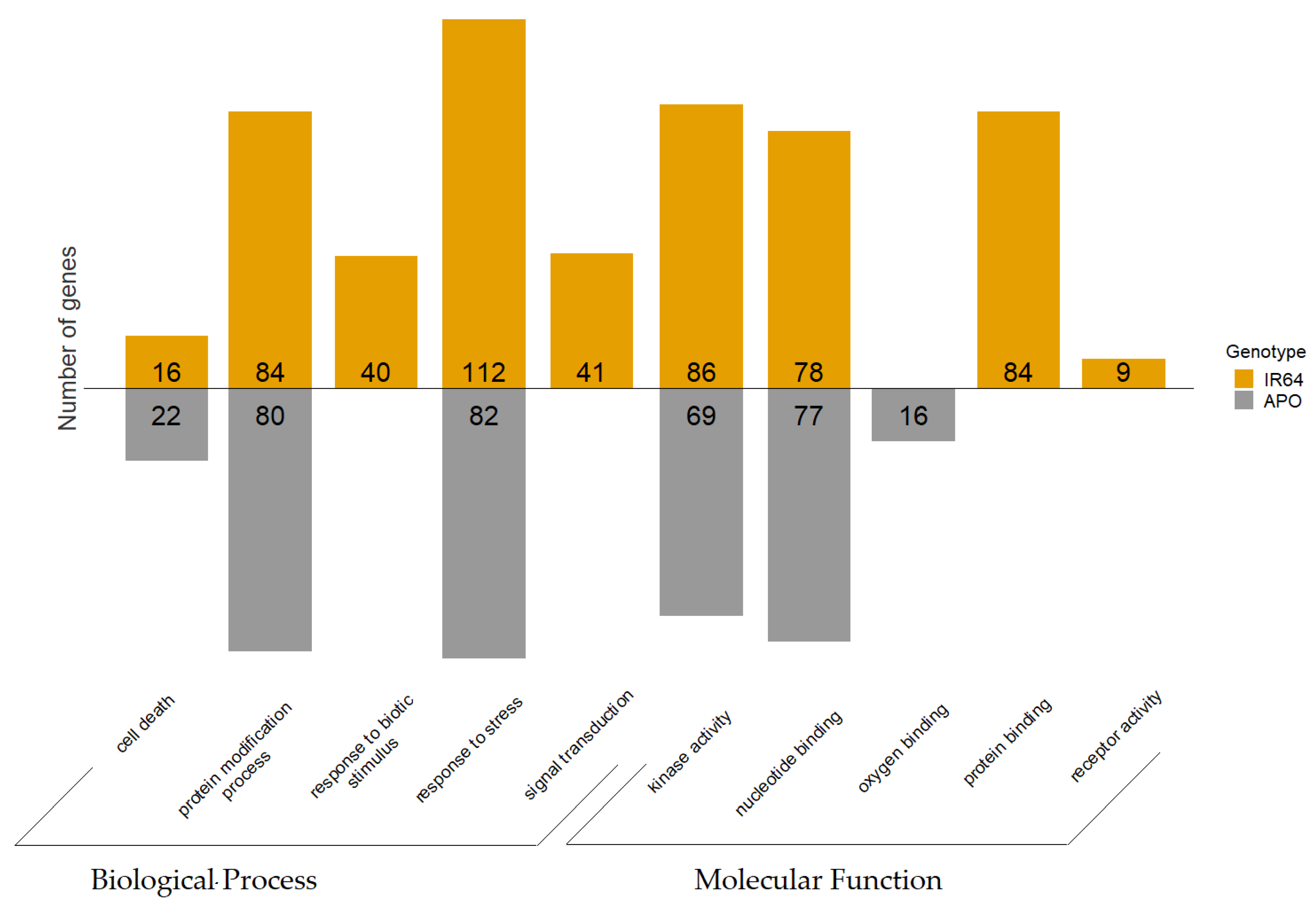
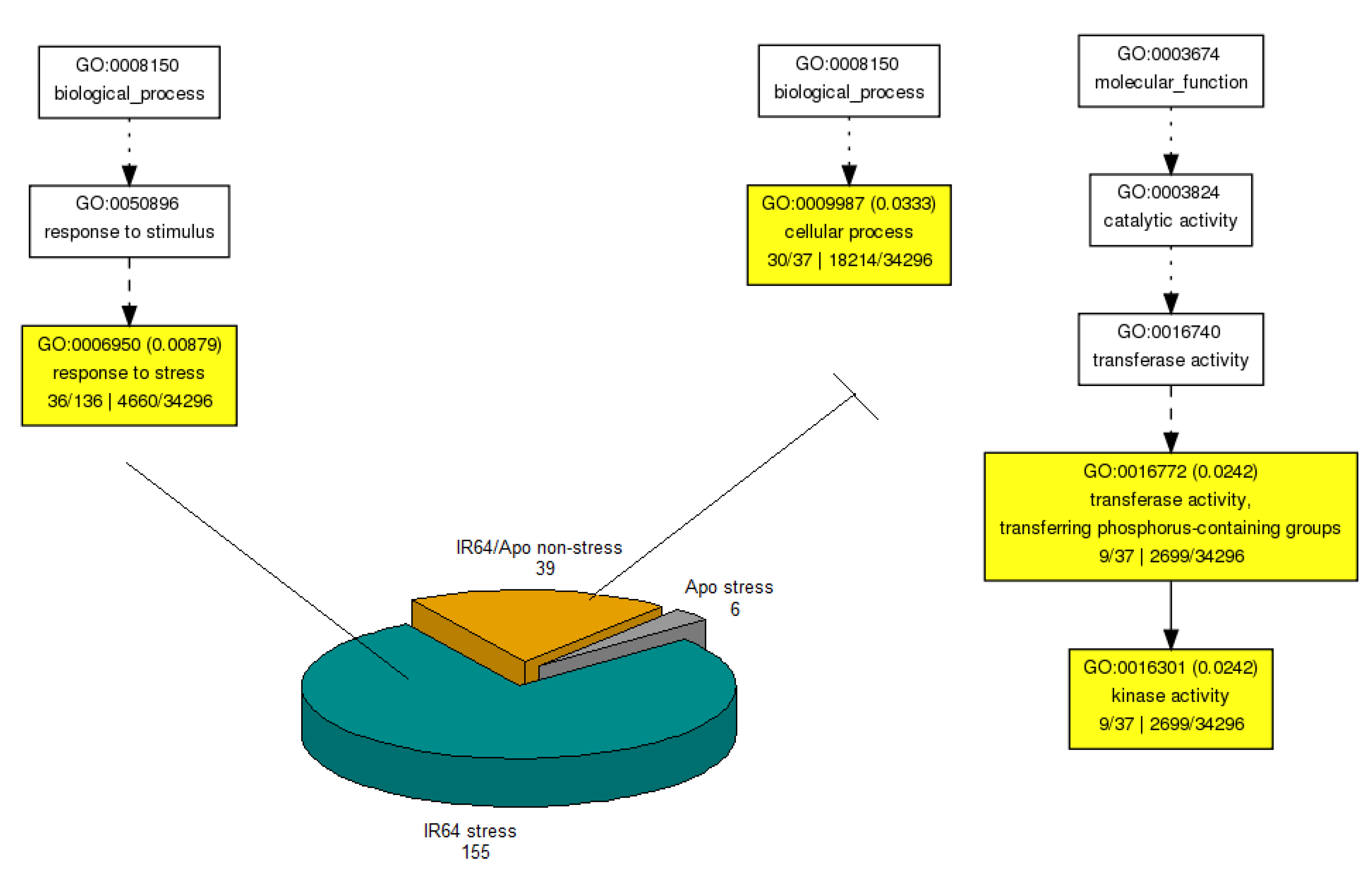
3.4.1. GO Enrichment Analysis
3.4.2. KEGG Pathway Analysis of GDE Genes
3.5. Differences Due to Drought (G × E) Using 3-Way DE Model
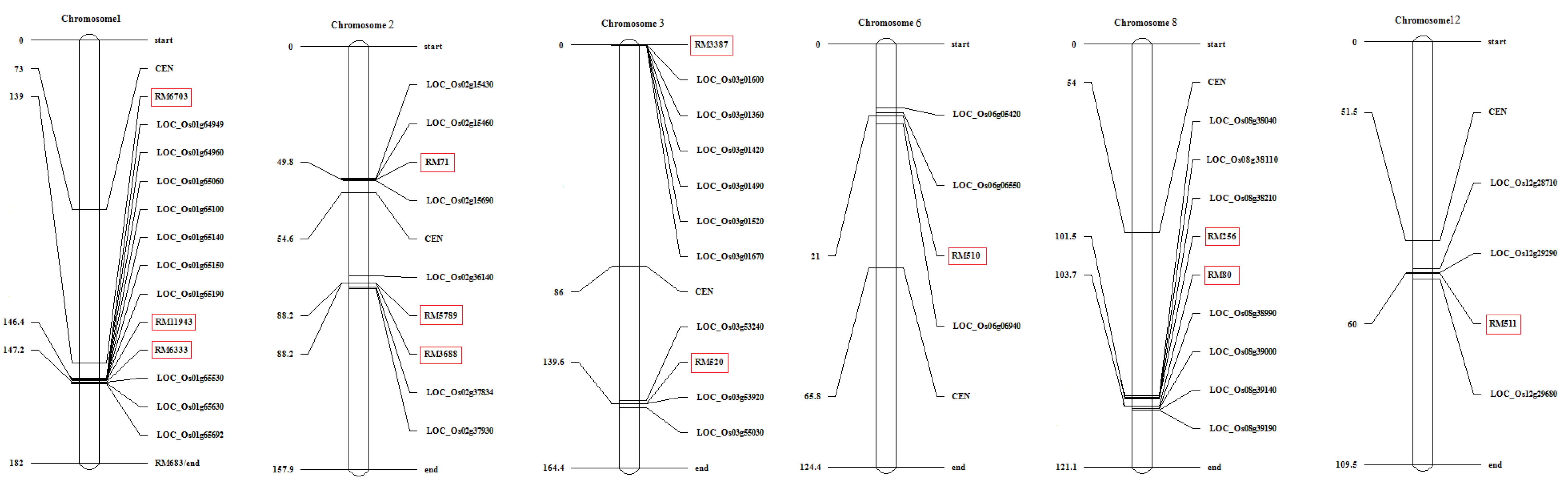
3.6. Several DEGs Co-Localize with Drought-Yield QTLs
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Availability of Data and Materials
Conflicts of Interest
References
- Li, Z.K.; Xu, J.L. Breeding for drought and salt tolerant rice (Oryza sativa L.): Progress and perspectives. In Advances in Molecular Breeding toward Drought and Salt Tolerant Crops; Jenks, M.A., Hasegawa, P.M., Jain, S.M., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 531–564. [Google Scholar]
- Pandey, S.; Bhandari, H. Drought: Economic costs and research implications. In Drought Frontiers in Rice: Crop Improvement for Increased Rainfed Production; Bennett, J., Hardy, B., Serraj, R., Eds.; World Scientific Publishing Co. International Rice Research Institute (IRRI): Los Baños, Philippines, 2009; pp. 3–17. [Google Scholar]
- IRRI (International Rice Research Institute). IRRI Rice Facts; IRRI (International Rice Research Institute): Los Baños, Philippines, 1995. [Google Scholar]
- Sandhu, N.; Jain, S.; Kumar, A.; Mehla, B.S.; Jain, R. Genetic variation, linkage mapping of QTL and correlation studies for yield, root, and agronomic traits for aerobic adaptation. BMC Genet. 2013, 14, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serraj, R.; McNally, K.L.; Slamet-Loedin, I.; Kohli, A.; Haefele, S.M.; Atlin, G.; Kumar, A. Drought Resistance Improvement in Rice: An Integrated Genetic and Resource Management Strategy. Plant Prod. Sci. 2011, 14, 1–14. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring Expression Profiles of Rice Genes under Cold, Drought, and High-Salinity Stresses and Abscisic Acid Application Using cDNA Microarray and RNA Gel-Blot Analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.; Jiao, Y.; Qin, Y.; Liu, X.; He, K.; Chen, C.; Ma, L.; Wang, J.; Xiong, L.; et al. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant Mol. Biol. 2007, 63, 591–608. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Lafitte, H.R.; Gao, Y.M.; Fu, B.Y.; Torres, R.; Li, Z.K. QTLs for drought escape and tolerance identified in a set of random introgression lines of rice. Theor. Appl. Genet. 2005, 111, 1642–1650. [Google Scholar] [CrossRef]
- Bernier, J.; Kumar, A.; Venuprasad, R.; Spaner, D.; Atlin, G.N. A large-effect QTL for grain yield under reproductive stage drought stress in upland rice. Crop Sci. 2007, 47, 505–516. [Google Scholar] [CrossRef]
- Venuprasad, R.; Dalid, C.O.; Del Valle, M.; Zhao, D.; Espiritu, M.; Cruz, M.S.; Amante, M.; Kumar, A.; Atlin, G.N. Identification and characterization of large-effect quantitative trait loci for grain yield under lowland drought stress in rice using bulk-segregant analysis. Theor. Appl. Genet. 2009, 120, 177–190. [Google Scholar] [CrossRef]
- Liu, G.; Mei, H.; Liu, H.; Yu, X.; Zou, G.; Luo, L. Sensitivities of rice grain yield and other panicle characters to late-stage drought stress revealed by phenotypic correlation and QTL analysis. Mol. Breed. 2010, 25, 603–613. [Google Scholar] [CrossRef]
- Gomez, S.M.; Boopathi, N.M.; Kumar, S.S.; Ramasubramanian, T.; Chengsong, Z.; Jeyaprakash, P.; Senthil, A.; Babu, R.C. Molecular mapping and location of QTLs for drought-resistance traits in indica rice (Oryza sativa L.) lines adapted to target environments. Acta Physiol. Plant. 2010, 32, 355–364. [Google Scholar] [CrossRef]
- Kamoshita, A.; Babu, R.C.; Boopathi, N.M.; Fukai, S. Phenotypic and genotypic analysis of drought-resistance traits for development of rice cultivars adapted to rainfed environments. Field Crops Res. 2008, 109, 1–23. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, W.; Chang, Y.; Ma, X.; Tu, H.; Xiong, F.; Jiang, N.; Feng, H.; Huang, C.; Yang, P.; et al. Genome-Wide Association Studies of Image Traits Reveal Genetic Architecture of Drought Resistance in Rice. Mol. Plant 2018, 11, 789–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlin, G.N.; Laza, M.; Amante, M.; Lafitte, H.R. Agronomic performance of tropical aerobic, irrigated and traditional upland rice varieties in three hydrological environments at IRRI. In 4th International Crop Science Congress: New Directions for a Diverse Planet; Fischer, T., Turner, N., Eds.; Regional Institute, Limited: Brisbane, Australia, 2004. [Google Scholar]
- Zhang, J.; Chen, L.L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.M.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. PNAS 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, T.J.; Kelly, K.A. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinform. 2010, 11, 422. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, T.J. Advanced Analysis Using baySeq; Generic Distribution Definitions (Vignette). 2017. Available online: https://www.bioconductor.org (accessed on 31 May 2017).
- Cal, A.J.; Liu, D.; Mauleon, R.; Hsing, Y.C.; Serraj, R. Transcriptome profiling of leaf elongation zone under drought in contrasting rice cultivars. PLoS ONE 2013, 8, e54537. [Google Scholar] [CrossRef]
- Serraj, R.; Dongcheng, L.; Hong, H.; Sellamuthu, R.; Impa, S.; Cairns, J.; Dimayuga, G.; Torres, R. Novel Approaches for Integration of Physiology, Genomics and Breeding for Drought Resistance Improvement in Rice. 2014. Available online: http://www.intlcss.org/ (accessed on 30 June 2016).
- Sinclair, T.; Ludlow, M. Influence of soil water supply on the plant water balance of four tropical grain legumes. Funct. Plant Biol. 1986, 13, 329. [Google Scholar] [CrossRef]
- Rice Plant Biology. Available online: http://rice.plantbiology.msu.edu/ (accessed on 25 May 2017).
- Gordon, A. FASTX-Toolkit: FASTQ/A Short-Reads Pre-Processing Tools. 2009. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 30 June 2017).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. Method 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large Scale Comparison of Gene Expression Levels by Microarrays and RNAseq Using TCGA Data. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, C.; Kechris, K. Differential correlation for sequencing data. BMC Res. Notes 2017, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardcastle, T.J. baySeq: Empirical Bayesian Analysis of Patterns of Differential Expression in Count Data (Vignette). 2017. Available online: https://www.bioconductor.org/ (accessed on 31 May 2017).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chandra, S.; Singh, D.; Pathak, J.; Kumari, S.; Kumar, M.; Poddar, R.; Balyan, H.S.; Gupta, P.K.; Prabhu, K.V.; Mukhopadhyay, K. De Novo Assembled Wheat Transcriptomes Delineate Differentially Expressed Host Genes in Response to Leaf Rust Infection. PLoS ONE 2016, 11, e0148453. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AgriGO. Available online: http://systemsbiology.cau.edu.cn/agriGOv2/ (accessed on 31 July 2017).
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- KEGG. Available online: https://www.genome.jp/kaas-bin/ (accessed on 31 March 2020).
- Ereful, N.C.; Liu, L.Y.; Tsai, E.; Kao, S.M.; Dixit, S.; Mauleon, R.; Malabanan, K.; Thomson, M.; Laurena, A.; Lee, D.; et al. Analysis of Allelic Imbalance in Rice Hybrids Under Water Stress and Association of Asymmetrically Expressed Genes with Drought-Response QTLs. Rice 2016, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Plants Ensembl. Available online: http://plants.ensembl.org (accessed on 31 August 2017).
- Temnykh, S.; Park, W.D.; Ayres, N.; Cartinhour, S.; Hauck, N.; Lipovich, L.; Cho, Y.G.; Ishii, T.; McCouch, S.R. Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 2000, 100, 697–712. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Wade, L.J.; McLaren, C.G.; Quintana, L.; Harnpichitvitaya, D.; Rajatasereekul, S.; Sarawgi, A.K.; Kumar, A.; Ahmed, H.U.; Singh, A.K.; Rodriguez, R.; et al. Genotype by environment interactions across diverse rainfed lowland rice environments. Field Crops Res. 1999, 64, 35–50. [Google Scholar] [CrossRef]
- Lafitte, H.R.; Courtois, B.; Arraudeau, M. Genetic improvement of rice in aerobic systems: Progress from yield to genes. Field Crops Res. 2002, 75, 171–190. [Google Scholar] [CrossRef]
- Hsiao, T.C. The soil plant atmosphere continuum in relation to drought and crop production. In Drought Resistance in Crops with Emphasis on Rice; International Rice Research Institute: Los Baños, Philippines, 1982; pp. 39–52. [Google Scholar]
- O’Toole, J.C. Adaptation of rice to drought-prone environments. In Drought Resistance in Crops with Emphasis on Rice; International Rice Research Institute: Los Baños, Philippines, 1982; pp. 195–213. [Google Scholar]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. Japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [Green Version]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 11. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Ereful, N.C.; Liu, L.Y.; Kao, S.M.; Tsai, E.; Laurena, A.; Thomson, M.; Greenland, A.; Powell, W.; Mackay, I.; Leung, H. cis dominantly explains regulatory divergence between two indica rice genotypes; drought further enhances regulatory differences. bioRxiv 2019. [Google Scholar] [CrossRef]
- Dudoit, S.; Yang, Y.H.; Callow, M.J.; Speed, T.P. Statistical methods for identifying genes with DE in replicated cDNA microarray experiments. Stat. Sin. 2002, 12, 111–139. [Google Scholar]
- Cleveland, W.S.; Devlin, S.J.; Grosse, E. Regression by Local Fitting. J. Econom. 1988, 37, 87–114. [Google Scholar] [CrossRef]
- Cleveland, W.S.; Grosse, E. Computational Methods for Local Regression. Stat. Comput. 1991, 1, 47–62. [Google Scholar] [CrossRef]
- Cheng, X.; Gou, X.; Yin, H.; Mysore, K.S.; Li, J.; Wen, J. Functional characterisation of brassinosteroid receptor MtBRI1 in Medicago truncatula. Sci. Rep. 2017, 7, 9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.; Wani, S.H.; Singh, B.; Bohra, A.; Dar, Z.A.; Lone, A.A.; Pareek, A.; Singla-Pareek, S.L. Transcription Factors and Plants Response to Drought Stress: Current Understanding and Future Directions. Front. Plant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef] [Green Version]
- Kosová, K.; Vítámvás, P.; Prášil, I.T. Wheat and barley dehydrins under cold, drought, and salinity—What can LEA-II proteins tell us about plant stress response? Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Dai, Z.; Lu, R.; Wu, B.; Tang, Q.; Xu, Y.; Cheng, C.; Su, J. Transcriptome Analysis of Two Species of Jute in Response to Polyethylene Glycol (PEG)-induced Drought Stress. Sci. Rep. 2017, 7, 16565. [Google Scholar] [CrossRef] [Green Version]
- Duan, F.; Ding, J.; Lee, D.; Lu, X.; Feng, Y.; Song, W. Overexpression of SoCYP85A1, a Spinach Cytochrome p450 Gene in Transgenic Tobacco Enhances Root Development and Drought Stress Tolerance. Front. Plant Sci. 2017, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, M.H.C. Drought stress and reactive oxygen species: Production, scavenging and signalling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [Green Version]
- Su, T.; Li, W.; Wang, P.; Ma, C. Dynamics of Peroxisome Homeostasis and Its Role in Stress Response and Signaling in Plants. Front. Plant Sci. 2019, 10, 705. [Google Scholar] [CrossRef] [Green Version]
- Ullah, A.; Manghwar, H.; Shaban, M.; Khan, A.H.; Akbar, A.; Ali, U.; Ali, E.; Fahad, S. Phytohormones enhanced drought tolerance in plants: A coping strategy. Environ. Sci. Pollut. Res. 2018, 25, 33103–33118. [Google Scholar] [CrossRef] [PubMed]
- Landi, S.; Hausman, J.F.; Guerriero, G.; Esposito, S. Poaceae vs. Abiotic Stress: Focus on Drought and Salt Stress, Recent Insights and Perspectives. Front. Plant Sci. 2017, 8, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, X.; Zhang, D.; Tang, H.; Sun, B.; Li, C.; Hao, L.; Liu, C.; Li, Y.; Shi, Y.; et al. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS ONE 2017, 12, e0179477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramene. Available online: www.archive.grameme.org (accessed on 31 March 2017).
- Vikram, P.K.; Swamy, M.; Dixit, S.; Uddin, A.H.; Cruz, M.T.; Singh, A.K.; Kumar, A. qDTY 1.1.; a major QTL for rice grain yield under reproductive-stage drought stress with a consistent effect in multiple elite genetic backgrounds. BMC Genet. 2011, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Liu, R.; Yan, L.; Liu, Z.Q.; Jiang, S.C.; Shen, Y.Y.; Wang, X.F.; Zhang, D.P. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J. Exp. Bot. 2012, 63, 1095–1106. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Genotype | Rep 1 | Rep 2 | ||
|---|---|---|---|---|---|
| Number of Reads | % Overall Alignment Rates | Number of Reads | % Overall Alignment Rates | ||
| Control (Non-stressed) | APO | 16,037,800 | 82.77 | 26,626,256 | 89.69 |
| IR64 | 16,083,456 | 83.72 | 25,871,136 | 83.73 | |
| Stressed | APO | 13,338,980 | 84.44 | 26,821,175 | 89.49 |
| IR64 | 13,255,340 | 71.46 | 27,709,000 | 76.82 | |
| Total | 58,715,576 | 107,027,567 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ereful, N.C.; Liu, L.-y.; Greenland, A.; Powell, W.; Mackay, I.; Leung, H. RNA-seq Reveals Differentially Expressed Genes between Two indica Inbred Rice Genotypes Associated with Drought-Yield QTLs. Agronomy 2020, 10, 621. https://doi.org/10.3390/agronomy10050621
Ereful NC, Liu L-y, Greenland A, Powell W, Mackay I, Leung H. RNA-seq Reveals Differentially Expressed Genes between Two indica Inbred Rice Genotypes Associated with Drought-Yield QTLs. Agronomy. 2020; 10(5):621. https://doi.org/10.3390/agronomy10050621
Chicago/Turabian StyleEreful, Nelzo C., Li-yu Liu, Andy Greenland, Wayne Powell, Ian Mackay, and Hei Leung. 2020. "RNA-seq Reveals Differentially Expressed Genes between Two indica Inbred Rice Genotypes Associated with Drought-Yield QTLs" Agronomy 10, no. 5: 621. https://doi.org/10.3390/agronomy10050621