4a,5,9,10,11,12-Hexahydro-6H-benzo[a]cyclohepta[hi]benzofuran - Synthesis of Unnatural Galanthamine Analogs

Institute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163OC, A-1060 Vienna, Austria
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(4), 374-381; https://doi.org/10.3390/70400374
Submission received: 11 November 2001
/
Revised: 29 April 2002
/
Accepted: 30 April 2002
/
Published: 30 April 2002
Abstract
:The synthesis of an unnatural galanthamine analog is reported.
Introduction
Galanthamine (or galantamine, Reminyl®) is a tertiary alkaloid acetylcholinesterase inhibitor (AChEI) which has been approved in several countries for treating symptoms of Alzheimer's type senile dementia [1,2]. In the course of our investigations into the oxidative tandem cyclization [3,4,5] now established as a key step in the industrial galanthamine synthesis [6], we have extended K3[Fe(CN)6] for the preparation of unnatural galanthamine analogs.
![Molecules 07 00374 i001]()

Results and Discussion
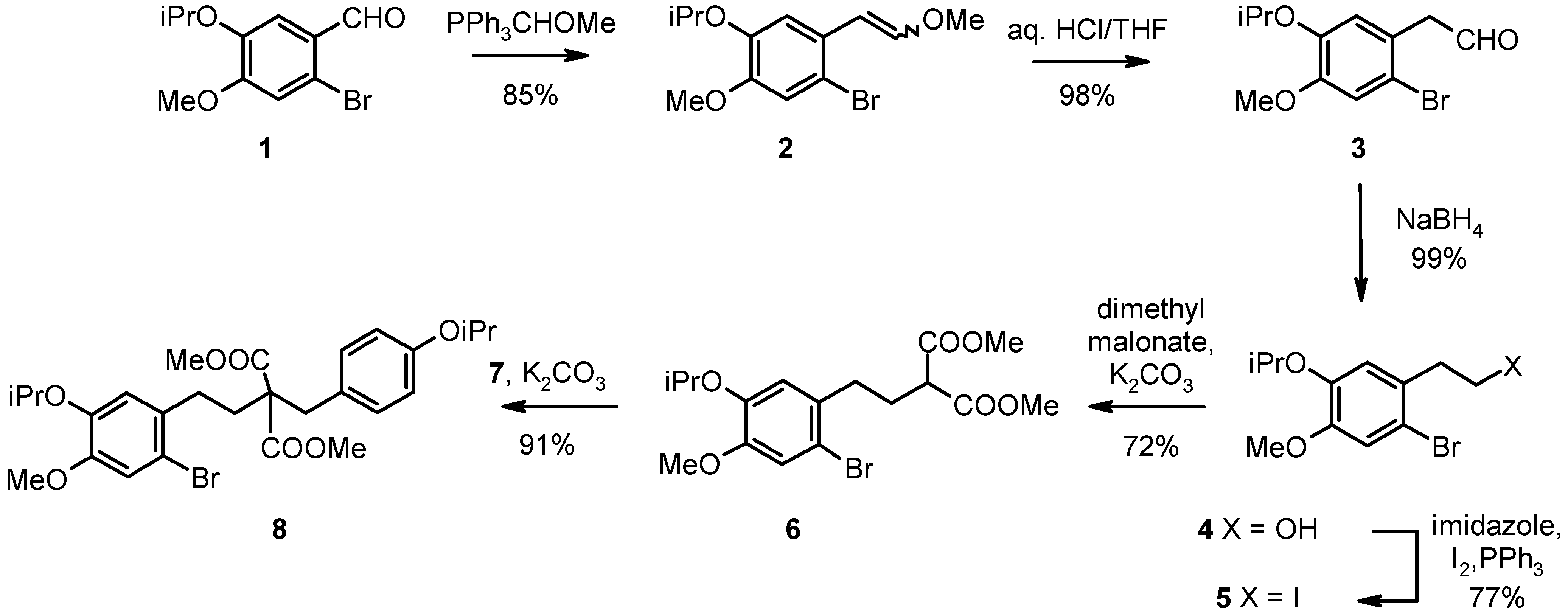
We synthesized the norbelladine analog (11) as intermediate for the oxidative tandem cyclization: 2-bromo-4-methoxy-5-(1-methylethoxy)benzaldehyde (1) [7] was homologized by a Wittig reaction [8] using (methoxymethyl)triphenylphosphonium chloride/potassium-tert.-butoxide and subsequent hydrolysis of the formed enol ether (2), isolated as a mixture of cis/trans isomers, to give the substituted benzene acetaldehyde (3) with 83% yield. This aldehyde was reduced in 99% yield using sodium borohydride in ethanol, and the alcohol (4) was converted to the corresponding alkyl iodide (5) under Appelt conditions with a yield of 77%. Malonate chemistry using 5 and 1-(chloromethyl)-4-(1-methylethoxy)benzene (7) [9] as alkylating agents in presence of anhydrous potassium carbonate gave rise to diester 8 with a yield of 66% for both alkylation steps (see Scheme 1).
Scheme 1.

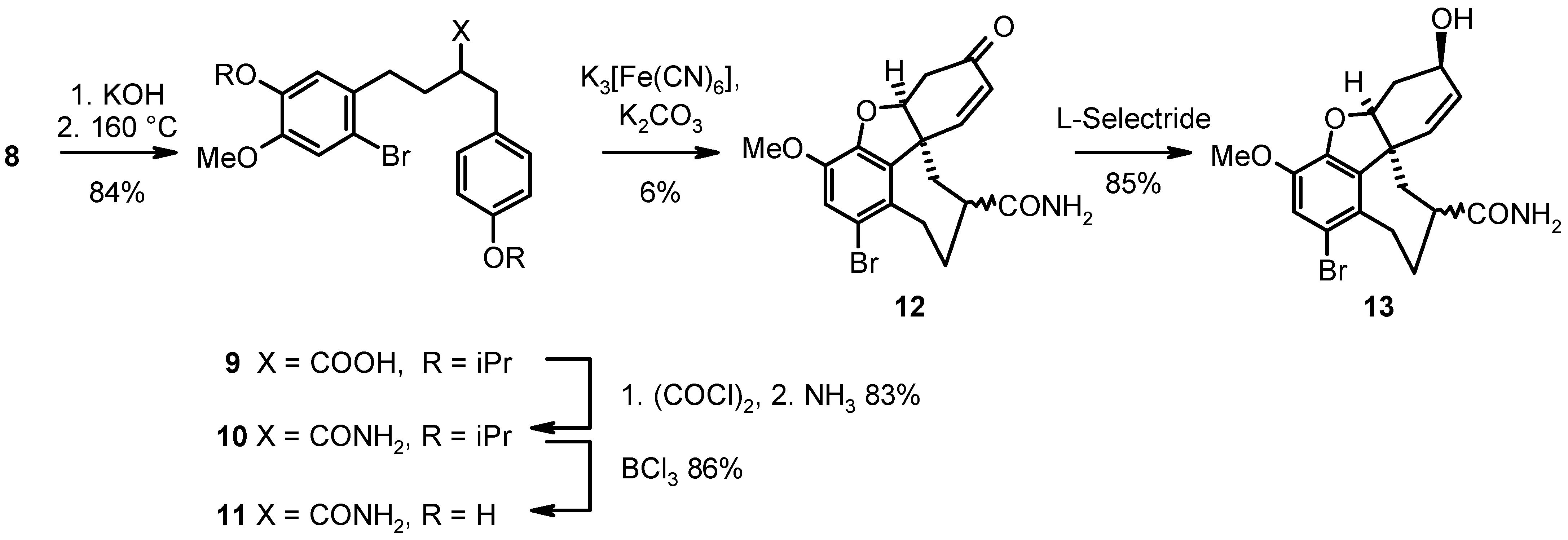
Saponification followed by pyrolytic decarboxylation of the malonic acid intermediate using a kugelrohr apparatus afforded the carboxylic acid (9) with 84% yield, which was converted into the amide (10) with a yield of 83% by subsequent treatment with oxalyl chloride and gaseous ammonia. Cleavage of the isopropyl ether protecting groups in presence of the aryl methyl ether substructure was realized using boron trichloride as Lewis acid with 86% yield. Tandem cyclization of 11 was performed with K3[Fe(CN)6] as oxidant in presence of K2CO3 and afforded the 4a,5,9,10,11,12-hexahydro-6H-benzo[a]cyclohepta[hi]benzofuran derivative (12) with a yield of 6%. Stereoselective reduction of the keto functionality gave rise to the unnatural galanthamine analog (13) with 85% yield (see Scheme 2).
Scheme 2.

Conclusions
We have demonstrated that oxidative tandem cyclization can be successfully developed further to access even carbocyclic galanthamine analogs. Further research in this field is being actively pursued in our laboratory.
Experimental
General
Melting points were determined on a Kofler melting point apparatus. 1H- and 13C-NMR-spectra were recorded on a Bruker AC-200 (200 MHz) pulse FT-NMR spectrometer in CDCl3, DMSO-d6 or MeOH-d4 using TMS as an internal standard. Thin layer chromatography (TLC) was performed on precoated plates (Merck TLC aluminum sheets silica 60 F254) with detection by UV light or with phosphomolybdic acid in aqueous EtOH by heating. All reactions were magnetically stirred under an argon atmosphere. MPLC (medium pressure liquid chromatography) was performed using SiO2 (Baker), a LC-8A pump (Shimadzu), a SPD-6AV UV-detector (Shimadzu) and Büchi glass columns. All the HPLC-MS data were obtained using a HP1100 liquid chromatography system equipped with a diode array detector (Hewlett-Packard) and a 1100 Series MSD with Athmospheric Pressure Chemical Ionization Interface (APCI) in positive and negative ion mode, scanning m/z 200 - 550. Column: Merck Purospher RP18e, 5 µm, solvent A: 97:3 MeCN-H2O, solvent B: 97: 3 20 mM CCl3COOH-MeCN, flow rate: 0.5 mL/min, injection volume: 1 µL, gradient: see Table 1.

{kind=link}
{kind=link}
| Time | 0.00 | 5.00 | 17.00 | 22.00 | 22.50 |
| %A | 20.0 | 20.0 | 60.0 | 60.0 | 20.0 |
| %B | 80.0 | 80.0 | 40.0 | 40.0 | 80.0 |
1-Bromo-5-methoxy-2-(2-methoxyethenyl)-4-(1-methylethoxy)benzene (2). To a suspension of (methoxymethyl)triphenylphosphonium chloride (50.0 g, 152 mmol) in dry THF (330 mL) KOtBu (20.5 g, 183 mmol) was added at 0 °C and stirred for 15 min. Then 2-bromo-4-methoxy-5-(1-methyl-ethoxy)benzaldehyde (1, 33.1 g, 121 mmol) was added and stirred for another 15 min. The reaction mixture was concentrated in vacuo and partitioned between water (300 mL) and Et2O (300 mL). The aqueous layer was extracted with Et2O (2 x 50 mL), the combined organic layer was washed with water (2 x 250 ml) and brine (250 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by MPLC (500 g SiO2, 2:1 petroleum ether-EtOAc). Yield: off-white, rather unstable crystals (32.5 g, 85%), mp. 43 - 45 °C. TLC (2:1 petroleum ether-EtOAc): Rf = 0.75. 1H-NMR (CDCl3) δ 7.00 (s, 1H); 6.90 (s, 1H), 6.83 (d, J = 12.7 Hz, 1Htrans), 6.13 (d, J = 7.6 Hz, 1Hcis), 5.98 (d, J = 12.7 Hz, 1Htrans), 5.50 (d, J = 7.6 Hz, 1Hcis), 4.49 (septet, J = 6.4 Hz, 1H), 3.81 (s, 3H), 3.74 (s, 3Htrans), 3.70 (s, 3Hcis), 1.35 (d, J = 6.4 Hz, 6H); 13C-NMR (CDCl3) δ 149.2 (s), 149.6 (s), 147.6 (s), 148.9 (s), 146.0 (d), 146.7 (d), 127.5 (s), 128.4 (s), 115.7 (d), 117.2 (d), 113.6 (d), 116.2 (d), 113.3 (s), 113.7 (s), 103.8 (d), 104.2 (d), 71.5 (d), 71.9 (d), 56.1 (q), 56.4 (q), 56.0 (q), 60.6 (q), 21.9 (q), 22.0 (q).
2-Bromo-4-methoxy-5-(1-methylethoxy)benzeneacetaldehyde (3). 2 (20.0 g, 66.4 mmol) was stirred in THF (250 mL)/2 N HCl (10 mL) for 3 h under reflux. The mixture was concentrated in vacuo, and the residue was partitioned between water (200 mL) and Et2O (200 mL). The aqueous layer was extracted with Et2O (3 x 50 mL), the combined organic layer was washed with water (2 x 250 ml), satd. NaHCO3 (2 x 250 mL) and brine (250 mL), dried over Na2SO4/charcoal, filtered and concentrated in vacuo. Yield: yellow crystals (18.7 g, 98%), mp. 43 - 45 °C. TLC (4:1 petroleum ether-EtOAc): Rf = 0.8. Anal. Calcd for C12H15BrO3: C, 50.19; H, 5.27. Found: C, 50.49; H, 5.06. 1H-NMR (CDCl3): δ 9.59 (t, J = 2.2 Hz, 1H), 7.06 (s, 1H), 6.73 (s, 1H), 4.46 (septet, J = 7.0 Hz, 1H), 3.81 (s, 3H), 3.73 (d, J = 2.2 Hz, 2H), 1.31 (d, J = 7.0 Hz, 6H); 13C-NMR (CDCl3): δ 198.5 (d), 150.5 (s), 146.9 (s), 124.0 (s), 118.6 (d), 116.2 (d), 115.3 (s), 71.9 (d), 56.1 (q), 49.9 (t), 21.9 (q).
2-Bromo-4-methoxy-5-(1-methylethoxy)benzeneethanol (4). 3 (2.60 g, 9.05 mmol) was added to a suspension of sodium borohydride (0.34 g, 9.1 mmol) in dry ethanol (40 mL) within 30 min at 15 °C and stirred for 2 h at this temperature. The mixture was concentrated in vacuo, quenched with satd. NaHCO3 and partitioned between water (200 mL) and Et2O (200 mL). The aqueous layer was extracted with Et2O (3 x 50 mL), the combined organic layer was washed with water (3 x 200 ml) and brine (250 mL), dried over Na2SO4/charcoal, filtered and concentrated in vacuo. Yield: colorless crystals (2.60 g, 99%) , mp. 65 - 67 °C. TLC (9:1 petroleum ether-EtOAc): Rf = 0.3. Anal. Calcd for C12H17BrO3: C, 49.84; H, 5.93. Found: C, 49.69; H, 5.79. 1H-NMR (CDCl3): δ 6.98 (s, 1H), 6.80 (s, 1H), 4.47 (septet, J = 6.3 Hz, 1H), 3.82 (t, J = 7.0 Hz, 2H), 3.80 (s, 3H), 2.90 (t, J = 7.0 Hz, 2H), 1.32 (d, J = 7.3 Hz, 6H); 13C-NMR (CDCl3): δ 149.7 (s), 146.4 (s), 129.6 (s), 118.5 (d), 116.3 (d), 114.8 (s), 71.8 (d), 62.2 (t), 56.1 (q), 38.8 (t), 21.9 (q).
1-Bromo-2-(2-iodoethyl)-5-methoxy-4-(1-methylethoxy)benzene (5). Triphenylphosphine (24.7 g, 94.0 mmol), imidazole (12.8 g, 188.0 mmol) and iodine (23.06 g, 90.9 mmol) in dry CH2Cl2 (150 mL) were stirred for 1 h at ambient temperature. 4 (18.0 g, 62.2 mmol) in dry CH2Cl2 (100 mL) was added at this temperature within 10 min and stirred for additional 2 h. The mixture was filtered, and the filtrate was washed with water (1 x 200 mL). The aqueous layer was extracted with CH2Cl2 (2 x 50 mL), and the combined organic layer was washed with 10% NaHSO3 (2 x 200 mL), water ( 1 x 200 mL), 10% CuSO4 (2 x 200 mL), water (1 x 200 mL) and brine (1 x 200 mL), dried over Na2SO4/charcoal, filtered and concentrated in vacuo. The residue was purified by MPLC (1000 g SiO2, 96:4 petroleum ether- EtOAc). Yield: colorless needles (19.0 g, 77%), mp. 95 - 97 °C. TLC (9:1 petroleum ether-EtOAc): Rf = 0.9. Anal. Calcd for C12H16BrIO2: C, 36.12; H, 4.04. Found: C, 36.38; H, 3.91. 1H-NMR (CDCl3): δ 7.00 (s, 1H), 6.77 (s, 1H), 4.49 (septet, J = 6.3 Hz, 1H), 3.81 (s, 3H), 3.39 - 3.24 (m, 2H), 3.24 - 3.09 (m, 2H), 1.36 (d, J = 7.3 Hz, 6H); 13C-NMR (CDCl3): δ 150.0 (s), 146.5 (s), 131.7 (s), 118.0 (d), 116.3 (d), 114.3 (s), 71.8 (d), 56.1 (q), 40.0 (t), 22.0 (q), 4.2 (t).
2-[2-[2-Bromo-4-methoxy-5-(1-methylethoxy)phenyl]ethyl]propanediacid dimethyl ester (6). 5 (18.0 g, 45.1 mmol), potassium carbonate (32.0 g, 321 mmol, anhydrous, freshly ground) and dimethyl malonate (50.0 g, 378 mmol) were stirred in dry DMF (200 mL) for 12 h at 80 °C. The mixture was filtered and concentrated in vacuo, and the residue was partitioned between water (300 mL) and Et2O (300 mL). The aqueous layer was extracted with Et2O (3 x 50 mL), the combined organic layer was washed with water (4 x 150 ml) and brine (250 mL), dried over Na2SO4/charcoal, filtered and concentrated in vacuo. Excessive dimethyl malonate was removed by distillation (160 °C/15 mbar), and the residue was purified by kugelrohr distillation (170 °C/0.06 mbar). Yield: colorless oil (18.9 g. 72%). TLC (9:1 petroleum ether-EtOAc): Rf = 0.45. Anal. Calcd for C17H23BrO6: C, 50.63; H, 5.75. Found: C, 50.87; H, 5.62. 1H-NMR (CDCl3): δ 6.99 (s, 1H), 6.73 (s, 1H), 4.49 (septet, J = 6.3 Hz, 1H), 3.81 (s, 3H), 3.76 (s, 6H), 3.39 (t, J = 7.9 Hz, 1H), 2.68 (t, J = 7.9 Hz, 2H), 2.18 (q, J = 7.9 Hz, 2H), 1.34 (d, J = 6.3 Hz, 6H); 13C-NMR (CDCl3): δ 169.6 (s), 149.7 (s), 146.6 (s), 131.7 (s), 117.8 (d), 116.3 (d), 114.6 (s), 71.8 (d), 56.2 (q), 52.5 (q), 50.9 (d), 33.1 (t), 29.0 (t), 22.0 (q).
2-[2-[2-Bromo-4-methoxy-5-(1-methylethoxy)phenyl]ethyl]-2-[4-(1-methylethoxy)-phenylmethyl]propanedicarboxylic acid dimethyl ester (8). 6 (10.0 g, 24.8 mmol), 1-(chloromethyl)-4-(1-methylethoxy)benzene (7, 4.58 g, 24.8 mmol) and potassium carbonate (10.3 g, 74.4 mmol, anhydrous, freshly ground) in dry DMF (200 mL) were stirred for 12 h at 80 °C. The suspension was filtered and concentrated, and the residue was partitioned between water (300 mL) and Et2O (300 mL). The aqueous layer was extracted with Et2O (3 x 50 mL), the combined organic layer was washed with water (4 x 150 mL) and brine (1 x 200 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by kugelrohr distillation (170 °C/0.005 mbar). Yield: colorless crystals (12.5 g, 91%), mp. 93 - 95 °C. TLC (4:1 petroleum ether-EtOAc): Rf = 0.5. Anal. Calcd for C27H35BrO7: C, 58.81; H, 6.40. Found: C, 59.03; H, 6.24. 1H-NMR (CDCl3): δ 7.06 (d, J = 7.5 Hz, 2H), 7.00 (s, 1H), 6.78 (d, J = 7.5 Hz, 2H), 6.69 (s, 1H), 4.62 - 4.31 (m, 2H), 3.82 (s, 3H), 3.78 (s, 6H), 3.29 (s, 2H), 2.70 - 2.52 (m, 2H), 2.12 - 1.97 (m, 2H), 1.31 (d, J = 6.3 Hz, 6H), 1.36 (d, J = 6.3 Hz, 6H); 13C-NMR (CDCl3): δ 171.5 (s), 156.9 (s), 149.5 (s), 146.7 (s), 132.4 (s), 130.8 (d), 127.5 (s), 117.7 (d), 116.2 (d), 115.6 (d), 114.4 (s), 71.8 (d), 69.7 (d), 58.8 (s), 56.1 (q), 52.3 (q), 37.8 (t), 32.8 (t), 30.6 (t), 22.0 (q).
2-Bromo-4-methoxy-5-(1-methylethoxy)-α-[4-(1-methylethoxy)phenylmethyl]benzenebutanoic acid (9). 8 (18.1 g, 32.8 mmol) and KOH (17.5 g, 312 mmol) in ethanol (100 mL)/water (20 mL) were refluxed for 12 h. The solution was concentrated to 30 mL in vacuo, a pH < 1 was adjusted by dropwise addition conc. HCl, and the mixture was partitioned between water (250 mL) and Et2O (250 mL). The aqueous layer was extracted with Et2O (2 x 100 mL), the combined organic layer was washed with water (3 x 200 mL) and brine (1 x 150 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was decarboxylated using a kugelrohr apparatus (30 min, 160 °C/0.005 mbar), and the formed carboxylic acid was purified by subsequent distillation (210 °C/0.008 mbar). Yield: colorless oil (13.3 g, 84%). TLC (EtOAc): Rf = 0.7. 1H-NMR (CDCl3): δ 7.04 (d, J = 9.5 Hz, 2H), 6.99 (s, 1H), 6.80 (d, J = 9.5 Hz, 2H), 6.77 (s, 1H), 4.60 - 4.39 (m, 2H), 3.79 (s, 3H), 3.09 - 2.58 (m, 5H), 2.09 - 1.72 (m, 2H), 1.43 - 1.29 (m, 12H); 13C-NMR (CDCl3): δ 181.0 (s), 156.2 (s), 149.3 (s), 146.3 (s), 132.3 (s), 130.7 (s), 129.6 (d), 117.6 (d), 116.1 (d), 115.7 (d), 114.3 (s), 71.6 (d), 69.6 (d), 55.9 (q), 46.7 (d), 37.0 (t), 33.1 (t), 31.7 (t), 21.8 (q).
2-Bromo-4-methoxy-5-(1-methylethoxy)-α-[4-(1-methylethoxy)phenylmethyl]benzenebutanamide (10). To 9 (24.0 g, 50.1 mmol) in dry CH2Cl2 (200 mL) oxalyl chloride (15 mL) was added within 15 min at 0 °C and stirred for 2 h at this temperature. The mixture was concentrated in vacuo, and the residue was dissolved in dry THF (100 mL). NH3 was passed through the solution for 2 h at this temperature. The solution was poured into water (1500 mL), and the precipitate was collected by filtration and triturated with water (4 x 500 mL). Yield: colorless crystals (19.9 g, 83%); mp. 112 - 114 °C. TLC (EtOAc): Rf = 0.8. Anal. Calcd for C24H32BrNO4: C, 60.25; H, 6.74; N, 2.93. Found: C, 60.15; H, 6.55; N, 2.77. 1H-NMR (CDCl3): δ 7.04 (d, J = 9.5 Hz, 2H), 6.96 (s, 1H), 6.72 (d, J = 9.5 Hz, 2H), 6.70 (s, 1H), 6.00 (b, 1H), 5.55 (b, 1H); 4.60 - 4.30 (m, 2H), 3.77 (s, 3H), 2.96 - 2.52 (m, 4H), 2.51 - 2.28 (m, 1H), 2.03 - 1.60 (m, 2H), 1.36 - 1.20 (m, 12H); 13C-NMR (CDCl3): δ 177.4 (s), 156.2 (s), 149.2 (s), 146.4 (s), 132.7 (s), 131.1 (d), 129.7 (d), 117.5 (s), 116.1 (d), 115.7 (d), 114.4 (d), 71.6 (d), 69.6 (d), 56.0 (q), 48.6 (d), 38.0 (t), 33.3 (t), 32.5 (t), 21.9 (q).
2-Bromo-5-hydroxy-α-(4-hydroxyphenylmethyl)-4-methoxybenzenebutanamide (11). To 10 (10.0 g, 20.9 mmol) in dry CH2Cl2 (150 mL) BCl3 (45 mL, 1.6 M in dry CH2Cl2) was added at – 78 °C and stirred for 1 h at this temperature. The mixture was warmed up to ambient temperature and stirred for additional 2 h. Water (400 mL) was added, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 x 50 mL). The combined organic layer was washed with water (2 x 200 mL) and brine (1 x 200 mL), dried over Na2SO4, filtered and concentrated in vacuo. Yield: colorless foam (7.11 g, 86%). TLC (EtOAc): Rf = 0.7. Anal. Calcd for C18H20BrNO4*0.25 H2O: C, 54.22; H, 5.18; N, 3.51. Found: C, 54.05; H, 4.95; N, 3.54. 1H-MR (DMSO-d6): δ 9.25 (s, 1H), 9.13 (s, 1H), 6.92 (d, J = 9.5 Hz, 2H), 6.90 (s, 1H), 6.67 (d, J = 9.5 Hz, 2H), 6.59 (s, 1H), 3.77 (s, 3H), 2.93 - 2.50 (m, 4H), 2.46 - 2.21 (m, 1H), 2.03 - 1.60 (m, 2H); 13C-NMR (DMSO-d6): δ 177.6 (s), 156.2 (s), 151.3 (s), 146.4 (s), 134.9 (s), 132.1 (d), 129.8 (s), 117.7 (d), 116.1 (d), 115.8 (d), 114.9 (s), 58.2 (q), 48.4 (d), 35.8 (t), 29.9 (t), 27.6 (t).
(4aR*,8aR*)-1-Bromo-4a,5,9,10,11,12-hexahydro-3-methoxy-6-oxa-6H-benzo[a]cyclohepta[hi]benzofuran-10-carboxamide (12). To a suspension of 11 (3.00 g, 7.61 mmol) in CHCl3 (300 mL) K3[Fe(CN)6] (13.2 g, 40.0 mmol) and K2CO3 (7.50 g, 54 mmol) in water (75 mL) were added at once and stirred vigorously using a mechanical stirrer for 45 min at ambient temperature. The mixture was filtered using diatomeous earth, and the filtrate was washed with water (3 x 100 mL) and brine (100 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (50 g SiO2, EtOAc). Yield: colorless foam (179 mg, 6%). TLC (EtOAc): Rf = 0.6. 1H-NMR (CDCl3): δ 6.95 (s, 1H), 6.71 (dd, J = 12.1 Hz, J = 2.0 Hz, 1H), 6.02 (d, J = 12.1 Hz, 1H), 5.70 (b, 2H), 4.82 (s, 1H), 3.81(s, 3H), 3.58 (dd, J = 16.5 Hz, J = 6.0 Hz, 1H), 3.13 (dd, J = 6.0 Hz, J = 16.5 Hz, 1H), 2.82 - 2.57 (m, 3H), 2.48 - 2.15 (m, 2H), 2.12 - 1.62 (m, 2H); 13C-NMR (DMSO-d6): δ 196.7 (s), 178.2 (s), 147.3 (d), 145.6 (s), 143.9 (s), 132.5 (s), 131.4 (s), 127.5 (d), 117.0 (s), 114.8 (d), 88.3 (d), 53.5 (q), 49.7 (s), 43.7 (d), 40.9 (t), 39.7 (t), 38.0 (t), 32.1 (t); 13C-NMR (CDCl3): δ 193.8 (s), 176.7 (s), 146.7 (d), 143.5 (s), 143.2 (s), 131.0 (s), 129.9 (s), 127.7 (d), 116.5 (s), 115.1 (d), 87.6 (d), 56.1 (q), 49.1 (s), 44.2 (d), 39.4 (t), 37.0 (t), 32.0 (t), 31.7 (t). HPLC/MS m/z (relative intensity): NI: 393.1 (19%), 392.1 (96%), 391.1 (16%), 390.1 (100%); PI: 394.8 (17%), 393.9 (100%), 392.8 (28%), 374.8 (11%), 348.9 (11%), 268.0 (10%).
(4aα,6β,8aR*)-1-Bromo-4a,5,9,10,11,12-hexahydro-3-methoxy-6-hydroxy-6H-benzo[a]cyclohepta-[hi]-benzofuran-10-carboxamide (13). To a suspension of 12 (160 mg, 0.41 mmol) in dry THF (5 mL) L-Selectride® (2.0 mL, 2.0 mmol, 1 M in THF) was added dropwise at - 5 °C within 15 min and stirred für 4 h at room temperature. The mixture was hydrolyzed with water (3 mL) and concentrated in vacuo. The residue was partitioned between EtOAc (30 mL) and 2 N HCl (20 mL). The aqueous layer was extracted with EtOAc (3 x 5 mL), the combined organic layer was washed with 2 N HCl (2 x 25 mL), water (1 x 25 mL), satd. NaHCO3 (1 x 25 mL) and brine (1 x 25 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (10 g SiO2, EtOAc). Yield: colorless solid (137 mg, 85%). TlC (EtOAc): Rf = 0.5. Anal. Calcd for C18H20BrNO4: C, 54.84; H, 5.11; N, 3.55. Found: C, 54.55; H, 5.22; N, 3.34. 1H-NMR (MeOH-d4): δ 6.99 (s, 1H), 6.03 (d, J = 16.5 Hz, 1H), 5.94 (dd, J = 16.5 Hz, J = 5.9 Hz, 1H), 4.52 (s, 1H), 4.16 (s, 1H), 3.76 (s, 3H), 3.49 (dd, J = 19.8 Hz, J = 5.9 Hz, 1H), 2.90 (t, J = 17.5 Hz, 1H), 2.77 (t, J = 17.5 Hz, 1H), 2.46 (d, J = 17.6 Hz, 1H), 2.29 - 2.10 (m, 2H), 1.98 - 1.53 (m, 3H); 13C-NMR (MeOH-d4): δ 181.6 (s), 148.1 (s), 145.4 (s), 135.4 (s), 132.3 (s), 129.5 (d), 128.7 (d), 117.8 (d), 115.4 (s), 89.0 (d), 70.3 (d), 62.6 (d), 57.2 (q), 45.7 (s), 42.5 (t), 33.5 (t), 33.1 (t), 31.9 (t). HPLC/MS m/z (relative intensity): NI: 395.1 (18%), 394.1 (89%), 393.1 (25%), 392.1 (100%), 390.1 (8%); PI: 392.9 (7%), 376.9 (14%), 375.9 (62%), 359.8 (100%), 358.8 (21%), 330.8 (5%).
References and Notes
- For a recent compilation of galanthamine syntheses see: Trost, B. M.; Toste, F. D. Enantioselective Total Synthesis of (-)-Galanthamine. J. Am. Chem. Soc. 2000, 122, 11262–11263. [Google Scholar]
- For a review of the acetylcholinesterase activity see: Sramek, J. J.; Frackiewicz, E. J.; Cutler, N. R. Review of the acetylcholinesterase inhibitor galanthamine. Expert Opin. Invest. Drugs 2000, 9, 2393–2402. [Google Scholar]
- Kametani, T.; Yamaki, K.; Yagi, H.; Fukumoto, K. Studies on the syntheses of heterocyclic compounds. CCCXV. Modified total synthesis of (-)-galanthamine through phenol oxidation. J. Chem. Soc. C 1969, 2602–2605. [Google Scholar]
- Barton, D. H. R.; Kirby, G. W. Phenol oxidation and biosynthesis. V. Synthesis of galanthamine. J. Chem. Soc. 1962, 806–817. [Google Scholar]
- Kametani, T.; Seino, C.; Yamaki, K.; Shibuya, S.; Fukumoto, K.; Kigasawa, K.; Satoh, F.; Hiiragi, M.; Hayasaka, T. Syntheses of heterocyclic compounds. CCCLXXXVI. Alternative total syntheses of galanthamine and N-benzylgalanthamine iodide. J. Chem. Soc. C 1971, 1043–1047. [Google Scholar]
- Kueenburg, B.; Czollner, L.; Froehlich, J.; Jordis, U. Development of a pilot scale process for the anti-Alzheimer drug (-)-galanthamine using large-scale phenolic oxidative coupling and crystallisation-induced chiral conversion. Org. Process Res. Dev. 1999, 3, 425–431. [Google Scholar]
- Treu, M.; Jordis, U.; Mereiter, K. 12H-[2]-Benzothiepino[6,5a,5-bc]benzofuran: Synthesis of a Sulfur-Analog of Galanthamine. Heterocycles 2001, 55, 1727–1735. [Google Scholar]
- Poschalko, A.; Welzig, S.; Treu, M.; Nerdinger, S.; Mereiter, K.; Jordis, U. Synthesis of (±)-6H-Benzofuro[3a,3,2,ef][3]benzazepine, an Unnatural Analog of (-)-Galanthamine. Tetrahedron 2002, 58, 1513–1518. [Google Scholar]
- Miwa, T.; Yamamoto, M.; Doi, T.; Tarui, N. Preparation process and effect of pyrimidine-5-carboxamides as cGMP phosphodiesterase inhibitors for circulatory and allergic diseases and sexual dysfunction remedies. WO 0127105, 2001. [Chem. Abstr 2000, 134, 311217]. [Google Scholar]
- Sample Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Treu, M.; Jordis, U. 4a,5,9,10,11,12-Hexahydro-6H-benzo[a]cyclohepta[hi]benzofuran - Synthesis of Unnatural Galanthamine Analogs. Molecules 2002, 7, 374-381. https://doi.org/10.3390/70400374
AMA Style
Treu M, Jordis U. 4a,5,9,10,11,12-Hexahydro-6H-benzo[a]cyclohepta[hi]benzofuran - Synthesis of Unnatural Galanthamine Analogs. Molecules. 2002; 7(4):374-381. https://doi.org/10.3390/70400374
Chicago/Turabian StyleTreu, Matthias, and Ulrich Jordis. 2002. "4a,5,9,10,11,12-Hexahydro-6H-benzo[a]cyclohepta[hi]benzofuran - Synthesis of Unnatural Galanthamine Analogs" Molecules 7, no. 4: 374-381. https://doi.org/10.3390/70400374