
Vaccine Adjuvants: from 1920 to 2015 and Beyond

Abstract
:1. The Evolution of Vaccines

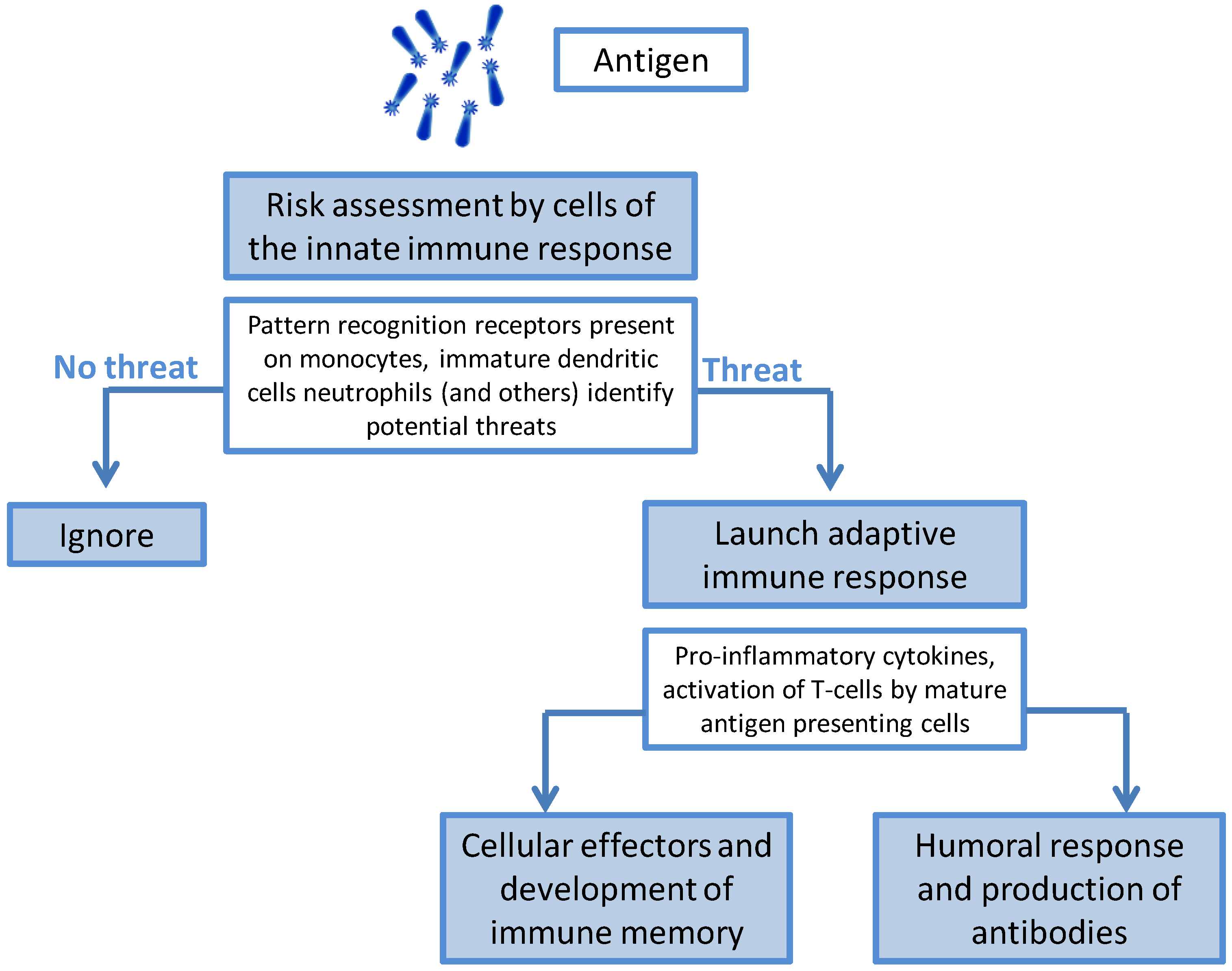
The Immunology of Infection and Immunization

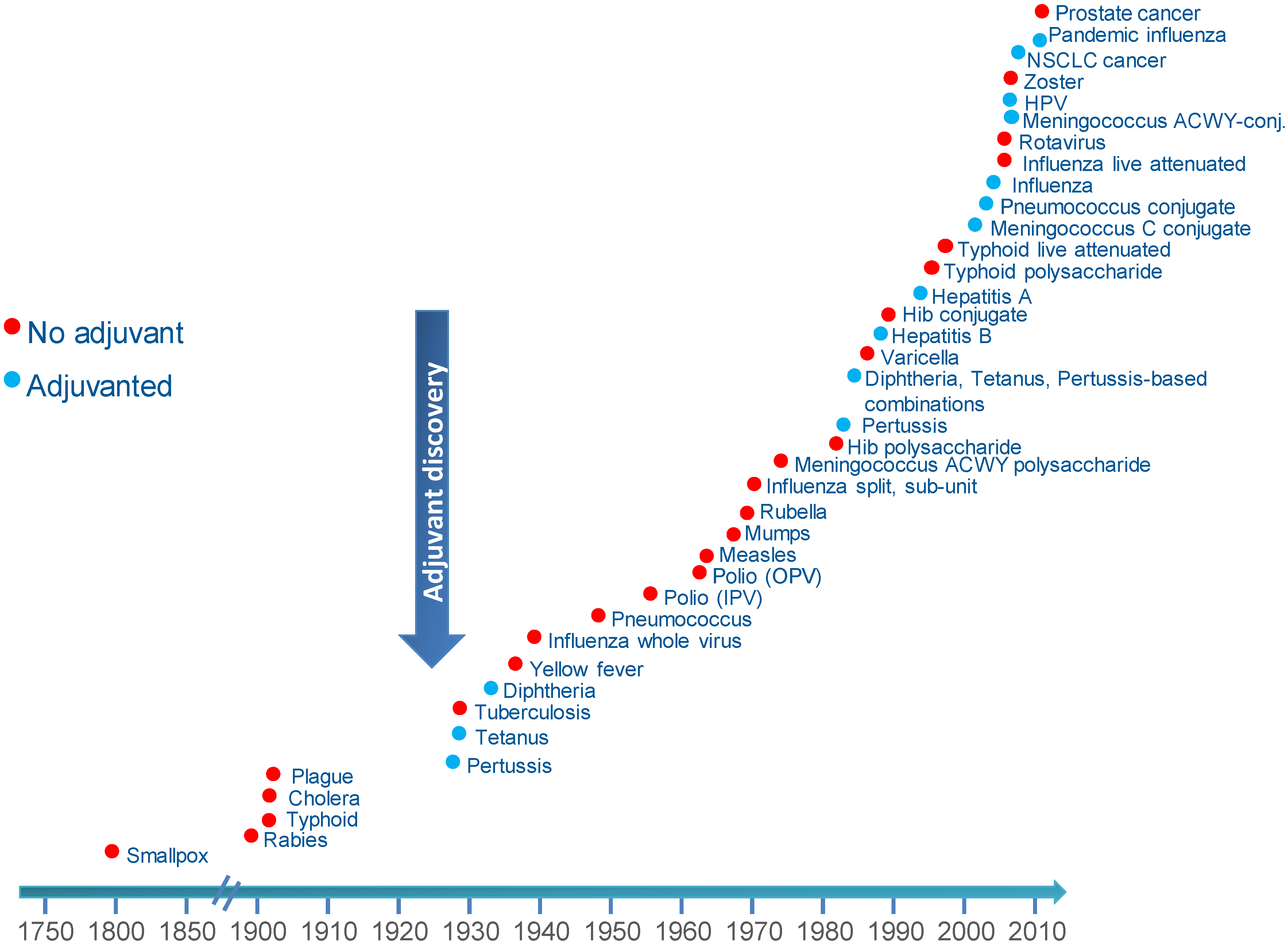
2. The Discovery of Adjuvants

3. New Vaccines: Challenges and Solutions

3.1. Challenging Pathogens
3.2. Challenging Populations

3.3. Potential Solutions for New Vaccines
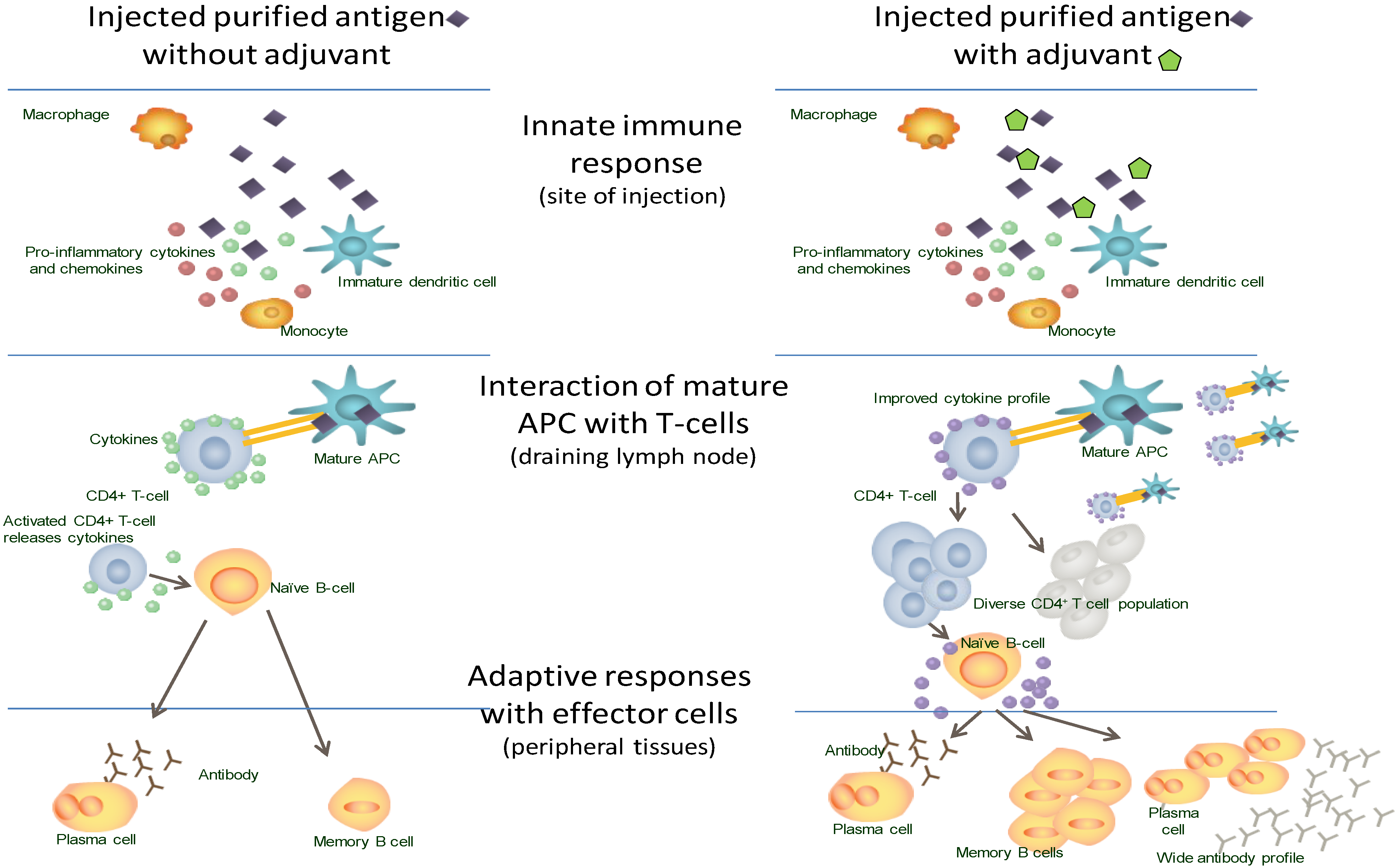
4. Modern Adjuvants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adjuvant | Composition | Major Immune Effects | ||
|---|---|---|---|---|
| (vaccines where used) | Component | Origin | Other Uses | |
| Aluminum (D, T, pertussis, IPV, hepatitis A & B, HPV, meningococcal and pneumococcal) | Aluminum as salts mixed with antigen (adsorption) | Naturally occurring present in soil, water, air | Medicines, cosmetics, food industry | Increases local inflammation, improves antigen update by APCs. Acts to increase antibody production |
| Virosomes (Hepatitis and influenza) | Vesicles where influenza antigens in aqueous volume are enclosed within a standard phospholipid cell membrane bilayer | Natural phospholipids, Seasonal influenza glycoproteins | None | Increases uptake by APCs. May interact with B cells leading to T-cell activation. |
| AS04 (Hepatitis B, HPV) | (3-deacyl-monophosphoryl lipid A) derived from LPS from Salmonella Minnesota, Aluminum salts | Natural exposure to LPS from Gram-negative bacteria occurs frequently | None | Directly stimulates TLR-4 increasing APC maturation and Th1 responses. |
| MF59® (Influenza-seasonal and pandemic) | Squalene | Animal source (shark liver oil). Found naturally in human tissues: adipose tissues, skin, arterial walls, skeleton, muscles, lymph nodes | Cosmetics, moisturizers | Increases APC recruitment and activation. Promotes antigen uptake and migration of cells to lymph nodes. |
| AS03 (Influenza-pandemic) |
|
|
| Promotes local production of cytokines and recruitment of innate cells. |
| Thermo-reversible oil-in-water (Influenza-pandemic) | Squalene | Animal source (shark liver oil). See above | Naturally occurring. See above | Not reported |
| ISA51 (therapeutic vaccine NSCLC) | Mineral oil DRAKEOL 6 VR Surfactant mannide-mono-oleate | Refined mineral oil of vegetable origin | Food industry | Strongly immunogenic |
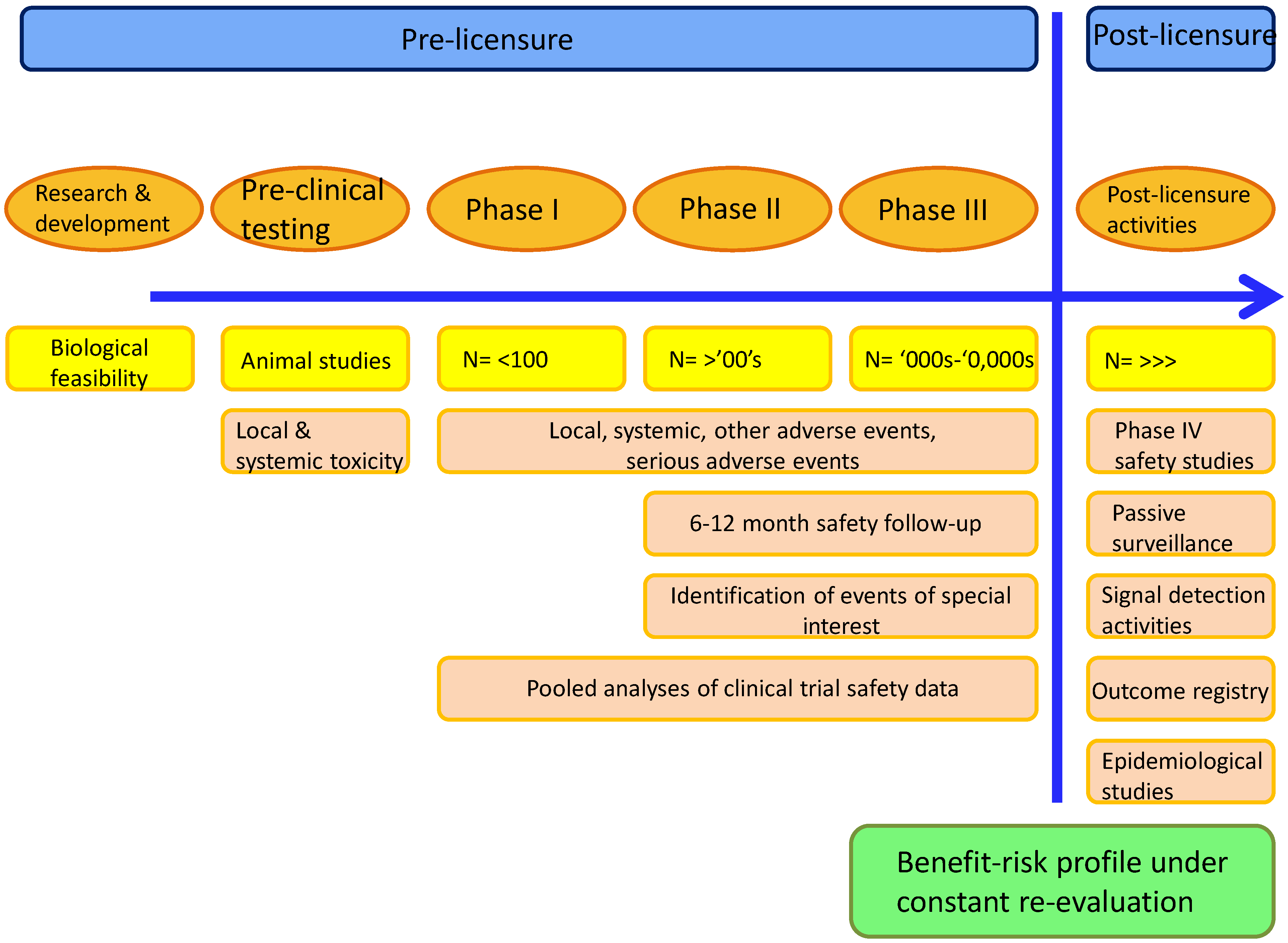
5. Determining the Safety of Adjuvanted Vaccines

Assessment of the Benefit-Risk Ratio
6. Potential Safety Concerns around Adjuvanted Vaccines
6.1. Reactogenicity
6.2. Immune-Mediated Diseases
6.3. Gulf War Syndrome
6.4. Myofasciitis
7. Challenges around Implementing Vaccination Programs with Novel Vaccines
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gross, C.P.; Sepkowitz, K.A. The myth of the medical breakthrough: Smallpox, vaccination, and Jenner reconsidered. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 1998, 3, 54–60. [Google Scholar]
- Strugnell, R.; Zepp, F.; Cunningham, A.L.; Tantawhichien, T. Vaccine antigens. Underst. Mod. Vaccines Perspect. Vaccinol. 2011, 1, 61–88. [Google Scholar] [CrossRef]
- WHO. Global Vaccine Action Plan 2011–2020. Available online: http://www.who.int/immunization/global_vaccine_action_plan/GVAP_doc_2011_2020/en/ (accessed on 23 October 2014).
- Zepp, F. Principles of vaccine design-Lessons from nature. Vaccine 2010, 28, C14–C24. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, P.; Santos, J. Vaccine evolution. Underst. Mod. Vaccines Perspect. Vaccinol. 2011, 1, 1–24. [Google Scholar] [CrossRef]
- Cherry, J.D. Historical review of pertussis and the classical vaccine. J. Infect. Dis. 1996, 174, S259–S263. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Vurdien, J.E.; White, J.M. The epidemiology of pertussis in England and Wales. Commun. Dis. Rep. Rev. 1992, 2, R152–R154. [Google Scholar]
- Romanus, V.; Jonsell, R.; Bergquist, S.O. Pertussis in Sweden after the cessation of general immunization in 1979. Pediatr. Infect. Dis. J. 1987, 6, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Sato, H. Development of acellular pertussis vaccines. Biol. J. Int. Assoc. Biol. Stand. 1999, 27, 61–69. [Google Scholar]
- Pichichero, M.E.; Deloria, M.A.; Rennels, M.B.; Anderson, E.L.; Edwards, K.M.; Decker, M.D.; Englund, J.A.; Steinhoff, M.C.; Deforest, A.; Meade, B.D. A safety and immunogenicity comparison of 12 acellular pertussis vaccines and one whole-cell pertussis vaccine given as a fourth dose in 15- to 20-month-old children. Pediatrics 1997, 100, 772–788. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.L.; Ware, R.S.; Grimwood, K.; Lambert, S.B. Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA 2012, 308, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Wendelboe, A.M.; van Rie, A.; Salmaso, S.; Englund, J.A. Duration of immunity against pertussis after natural infection or vaccination. Pediatr. Infect. Dis. J. 2005, 24, S58–S61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Prietsch, S.O.M.; Axelsson, I.; Halperin, S.A. Acellular vaccines for preventing whooping cough in children. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Storsaeter, J.; Wolter, J. Is there a need for a new generation of vaccines against pertussis? Expert Opin. Emerg. Drugs 2006, 11, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Geeraedts, F.; Goutagny, N.; Hornung, V.; Severa, M.; de Haan, A.; Pool, J.; Wilschut, J.; Fitzgerald, K.A.; Huckriede, A. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLOS Pathog. 2008, 4, e1000138. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Leo, O. Key concepts in immunology. Vaccine 2010, 28, C2–C13. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Koyama, S.; Nakagawa, A.; Coban, C.; Akira, S. Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 2008, 3, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+ T cells: Differentiation and functions. Clin. Dev. Immunol. 2012. [Google Scholar] [CrossRef]
- Garlapati, S. Do we know the Th1/Th2/Th17 determinants of vaccine response? Expert Rev. Vaccines 2012, 11, 1307–1310. [Google Scholar] [CrossRef] [PubMed]
- Klein Klouwenberg, P.; Bont, L. Neonatal and infantile immune responses to encapsulated bacteria and conjugate vaccines. Clin. Dev. Immunol. 2008. [Google Scholar] [CrossRef]
- Kapikian, A.Z.; Mitchell, R.H.; Chanock, R.M.; Shvedoff, R.A.; Stewart, C.E. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am. J. Epidemiol. 1969, 89, 405–421. [Google Scholar] [PubMed]
- Moghaddam, A.; Olszewska, W.; Wang, B.; Tregoning, J.S.; Helson, R.; Sattentau, Q.J.; Openshaw, P.J.M. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines. Nat. Med. 2006, 12, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Dormitzer, P.R.; Nokes, D.J.; Rappuoli, R.; Roca, A.; Graham, B.S. Strategic priorities for respiratory syncytial virus (RSV) vaccine development. Vaccine 2013, 31, B209–B215. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.; Hem, S.L. Immunologic adjuvants. In Vaccines; Saunders Elsevier: Philadelphia, PA, USA, 2004; pp. 69–79. [Google Scholar]
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Cousens, S.; Johnson, H.L.; Lawn, J.E.; Rudan, I.; Bassani, D.G.; Jha, P.; Campbell, H.; Walker, C.F.; Cibulskis, R.; et al. Global, regional, and national causes of child mortality in 2008: A systematic analysis. Lancet 2010, 375, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- WHO. The global burden of disease: 2004 update. Available online: http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/ (accessed on 5 January 2014).
- Leroux-Roels, G. Unmet needs in modern vaccinology: Adjuvants to improve the immune response. Vaccine 2010, 28, C25–C36. [Google Scholar] [CrossRef] [PubMed]
- Kovaiou, R.D.; Herndler-Brandstetter, D.; Grubeck-Loebenstein, B. Age-related changes in immunity: Implications for vaccination in the elderly. Expert Rev. Mol. Med. 2007, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gruver, A.L.; Hudson, L.L.; Sempowski, G.D. Immunosenescence of ageing. J. Pathol. 2007, 211, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Welder, J.H.; Torres, M.P.; Kipper, M.J.; Mallapragada, S.K.; Wannemuehler, M.J.; Narasimhan, B. Vaccine adjuvants: Current challenges and future approaches. J. Pharm. Sci. 2009, 98, 1278–1316. [Google Scholar] [CrossRef] [PubMed]
- Marconi, P.; Argnani, R.; Epstein, A.L.; Manservigi, R. HSV as a vector in vaccine development and gene therapy. Adv. Exp. Med. Biol. 2009, 655, 118–144. [Google Scholar] [PubMed]
- Majhen, D.; Calderon, H.; Chandra, N.; Fajardo, C.A.; Rajan, A.; Alemany, R.; Custers, J. Adenovirus-based vaccines for fighting infectious diseases and cancer: Progress in the field. Hum. Gene Ther. 2014, 25, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.C. Clinical development of Modified Vaccinia virus Ankara vaccines. Vaccine 2013, 31, 4241–4246. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H. DNA vaccines: Roles against diseases. Germs 2013, 3, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Stanberry, L.; Strugnell, R. Vaccines of the future. Underst. Mod. Vaccines Perspect. Vaccinol. 2011, 1, 151–199. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. Baltim. Md 1950 2009, 183, 6186–6197. [Google Scholar]
- Morel, S.; Didierlaurent, A.; Bourguignon, P.; Delhaye, S.; Baras, B.; Jacob, V.; Planty, C.; Elouahabi, A.; Harvengt, P.; Carlsen, H.; et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011, 29, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Ballou, W.R.; Cahill, C.P. Two decades of commitment to malaria vaccine development: GlaxoSmithKline Biologicals. Am. J. Trop. Med. Hyg. 2007, 77, 289–295. [Google Scholar] [PubMed]
- Kester, K.E.; Cummings, J.F.; Ofori-Anyinam, O.; Ockenhouse, C.F.; Krzych, U.; Moris, P.; Schwenk, R.; Nielsen, R.A.; Debebe, Z.; Pinelis, E.; et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: Safety, efficacy, and immunologic associates of protection. J. Infect. Dis. 2009, 200, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, G.; Leroux-Roels, I.; Clement, F.; Ofori-Anyinam, O.; Lievens, M.; Jongert, E.; Moris, P.; Ballou, W.R.; Cohen, J. Evaluation of the immune response to RTS,S/AS01 and RTS,S/AS02 adjuvanted vaccines: Randomized, double-blind study in malaria-naïve adults. Hum. Vaccines Immunother. 2014. [Google Scholar] [CrossRef] [Green Version]
- Bejon, P.; Lusingu, J.; Olotu, A.; Leach, A.; Lievens, M.; Vekemans, J.; Mshamu, S.; Lang, T.; Gould, J.; Dubois, M.-C.; et al. Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N. Engl. J. Med. 2008, 359, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Bovier, P.A. Epaxal: A virosomal vaccine to prevent hepatitis A infection. Expert Rev. Vaccines 2008, 7, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Müller, M.; Kaeser, M.D.; Weydemann, U.; Amacker, M. Influenza virosomes as vaccine adjuvant and carrier system. Expert Rev. Vaccines 2013, 12, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Haensler, J. An update on safety and immunogenicity of vaccines containing emulsion-based adjuvants. Expert Rev. Vaccines 2013, 12, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Baz, M.; Luke, C.J.; Cheng, X.; Jin, H.; Subbarao, K. H5N1 vaccines in humans. Virus Res. 2013, 178, 78–98. [Google Scholar] [CrossRef] [PubMed]
- Giannini, S.L.; Hanon, E.; Moris, P.; van Mechelen, M.; Morel, S.; Dessy, F.; Fourneau, M.A.; Colau, B.; Suzich, J.; Losonksy, G.; et al. Enhanced humoral and memory B cellular immunity using HPV16/18 L1 VLP vaccine formulated with the MPL/aluminium salt combination (AS04) compared to aluminium salt only. Vaccine 2006, 24, 5937–5949. [Google Scholar] [CrossRef] [PubMed]
- Paavonen, J.; Naud, P.; Salmerón, J.; Wheeler, C.M.; Chow, S.-N.; Apter, D.; Kitchener, H.; Castellsague, X.; Teixeira, J.C.; Skinner, S.R.; et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): Final analysis of a double-blind, randomised study in young women. Lancet 2009, 374, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; Franco, E.L.; Wheeler, C.M.; Moscicki, A.-B.; Romanowski, B.; Roteli-Martins, C.M.; Jenkins, D.; Schuind, A.; Costa Clemens, S.A.; Dubin, G. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: Follow-up from a randomised control trial. Lancet 2006, 367, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; Franco, E.L.; Wheeler, C.; Ferris, D.G.; Jenkins, D.; Schuind, A.; Zahaf, T.; Innis, B.; Naud, P.; de Carvalho, N.S.; et al. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: A randomised controlled trial. Lancet 2004, 364, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Roteli-Martins, C.; Naud, P.; de Borba, P.; Teixeira, J.; de Carvalho, N.; Zahaf, T.; Sanchez, N.; Geeraerts, B.; Descamps, D. Sustained immunogenicity and efficacy of the HPV-16/18 AS04-adjuvanted vaccine: Up to 8.4 years of follow-up. Hum. Vaccines Immunother. 2012, 8, 390–397. [Google Scholar] [CrossRef]
- De Carvalho, N.; Teixeira, J.; Roteli-Martins, C.M.; Naud, P.; de Borba, P.; Zahaf, T.; Sanchez, N.; Schuind, A. Sustained efficacy and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine up to 7.3 years in young adult women. Vaccine 2010, 28, 6247–6255. [Google Scholar] [CrossRef] [PubMed]
- Tong, N.K.C.; Beran, J.; Kee, S.A.; Miguel, J.L.; Sánchez, C.; Bayas, J.M.; Vilella, A.; de Juanes, J.R.; Arrazola, P.; Calbo-Torrecillas, F.; et al. Immunogenicity and safety of an adjuvanted hepatitis B vaccine in pre-hemodialysis and hemodialysis patients. Kidney Int. 2005, 68, 2298–2303. [Google Scholar] [CrossRef] [PubMed]
- Kundi, M. New hepatitis B vaccine formulated with an improved adjuvant system. Expert Rev. Vaccines 2007, 6, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Plotkin, S.A.; Black, S.; Coffman, R.L. Assessing the safety of adjuvanted vaccines. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef]
- Lopalco, P.L.; Johansen, K.; Ciancio, B.; de Carvalho Gomes, H.; Kramarz, P.; Giesecke, J. Monitoring and assessing vaccine safety: A European perspective. Expert Rev. Vaccines 2010, 9, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.J.; Kaplanski, C.V.; Lebron, J.A. Nonclinical safety assessment of vaccines and adjuvants. Methods Mol. Biol. 2010, 626, 29–40. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention (CDC). Withdrawal of rotavirus vaccine recommendation. Available online: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm4843a5.htm (accessed on 14 April 2015).
- European Medicines Agency. Questions and answers on the suspension of Hexavac. Doc. Ref. EMEA/304888/2005; EMA: London, UK, 2005. [Google Scholar]
- Garçon, N.; Leroux-Roels, G.; Cheng, W. Vaccine adjuvants. In Understanding Modern Vaccines: Perspectives in Vaccinology; Elsevier: Amsterdam, The Netherlands, 2011; pp. 89–113. [Google Scholar]
- Garçon, N.; Segal, L.; Tavares, F.; van Mechelen, M. The safety evaluation of adjuvants during vaccine development: The AS04 experience. Vaccine 2011, 29, 4453–4459. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.; Granström, M. The role of aluminium for adverse reactions and immunogenicity of diphtheria-tetanus booster vaccine. Acta Paediatr. Oslo Nor. 1992 1994, 83, 159–163. [Google Scholar]
- Waddington, C.S.; Walker, W.T.; Oeser, C.; Reiner, A.; John, T.; Wilkins, S.; Casey, M.; Eccleston, P.E.; Allen, R.J.; Okike, I.; et al. Safety and immunogenicity of AS03B adjuvanted split virion versus non-adjuvanted whole virion H1N1 influenza vaccine in UK children aged 6 months-12 years: Open label, randomised, parallel group, multicentre study. Br. Med. J. 2010. [Google Scholar] [CrossRef]
- Kosalaraksa, P.; Jeanfrau, R.; Frenette, L.; Dramé, M.; Madariaga, M.; Innis, B.L.; Godeaux, O.; Izurieta, P.; Vaughn, D. AS03B-adjuvanted H5N1 influenza vaccine in children 6 months through 17 years of age: A Phase II/III randomized, placebo-controlled, observer-blind trial. J. Infect. Dis. 2014. [Google Scholar] [CrossRef]
- Levie, K.; Gjorup, I.; Skinhøj, P.; Stoffel, M. A 2-dose regimen of a recombinant hepatitis B vaccine with the immune stimulant AS04 compared with the standard 3-dose regimen of Engerix-B in healthy young adults. Scand. J. Infect. Dis. 2002, 34, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Tavares Da Silva, F.; de Keyser, F.; Lambert, P.-H.; Robinson, W.H.; Westhovens, R.; Sindic, C. Optimal approaches to data collection and analysis of potential immune mediated disorders in clinical trials of new vaccines. Vaccine 2013, 31, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, P.; Cohet, C.; Kjaer, S.K.; Latham, N.B.; Lambert, P.-H.; Reisinger, K.; Haupt, R.M. A summary of the post-licensure surveillance initiatives for GARDASIL/SILGARD. Vaccine 2010, 28, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.; Zima, J.; Tavares, F.; Baril, L.; Arellano, F. Post-licensure safety surveillance for AS04-adjuvanted Human Papillomavirus vaccine: More than 4 years of experience. Pharmacoepidemiol. Drug Saf. 2014, 23, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.-G.; David, M.P.P.; Baril, L.; Struyf, F.; Zima, J.; Arellano, F. Safety of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine (CervarixTM): A pooled analysis of clinical trial data. Hum. Vaccin. 2009, 5, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, T.; Descamps, D.; David, M.-P.; Zahaf, T.; Hardt, K.; Izurieta, P.; Dubin, G.; Breuer, T. Analysis of adverse events of potential autoimmune aetiology in a large integrated safety database of AS04 adjuvanted vaccines. Vaccine 2008, 26, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi-Bensouda, L.; Guillemot, D.; Godeau, B.; Bénichou, J.; Lebrun-Frenay, C.; Papeix, C.; Labauge, P.; Berquin, P.; Penfornis, A.; Benhamou, P.-Y.; et al. Autoimmune disorders and quadrivalent human papillomavirus vaccination of young female subjects. J. Intern. Med. 2013, 275, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Langmuir, A.D. Guillain-Barré syndrome: The swine influenza virus vaccine incident in the United States of America, 1976–77: Preliminary communication. J. R. Soc. Med. 1979, 72, 660–669. [Google Scholar] [PubMed]
- Schonberger, L.B.; Bregman, D.J.; Sullivan-Bolyai, J.Z.; Keenlyside, R.A.; Ziegler, D.W.; Retailliau, H.F.; Eddins, D.L.; Bryan, J.A. Guillain-Barre syndrome following vaccination in the National Influenza Immunization Program, United States, 1976–1977. Am. J. Epidemiol. 1979, 110, 105–123. [Google Scholar] [PubMed]
- Greene, S.K.; Rett, M.; Weintraub, E.S.; Li, L.; Yin, R.; Amato, A.A.; Ho, D.T.; Sheikh, S.I.; Fireman, B.H.; Daley, M.F.; et al. Risk of confirmed Guillain-Barre syndrome following receipt of monovalent inactivated influenza A (H1N1) and seasonal influenza vaccines in the Vaccine Safety Datalink Project, 2009–2010. Am. J. Epidemiol. 2012, 175, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Wise, M.E.; Viray, M.; Sejvar, J.J.; Lewis, P.; Baughman, A.L.; Connor, W.; Danila, R.; Giambrone, G.P.; Hale, C.; Hogan, B.C.; et al. Guillain-Barre syndrome during the 2009–2010 H1N1 influenza vaccination campaign: Population-based surveillance among 45 million Americans. Am. J. Epidemiol. 2012, 175, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Dodd, C.N.; Romio, S.A.; Black, S.; Vellozzi, C.; Andrews, N.; Sturkenboom, M.; Zuber, P.; Hua, W.; Bonhoeffer, J.; Buttery, J.; et al. International collaboration to assess the risk of Guillain Barré Syndrome following Influenza A (H1N1) 2009 monovalent vaccines. Vaccine 2013, 31, 4448–4458. [Google Scholar] [CrossRef] [PubMed]
- Isai, A.; Durand, J.; le Meur, S.; Hidalgo-Simon, A.; Kurz, X. Autoimmune disorders after immunisation with Influenza A/H1N1 vaccines with and without adjuvant: EudraVigilance data and literature review. Vaccine 2012, 30, 7123–7129. [Google Scholar] [CrossRef] [PubMed]
- Romio, S.; Weibel, D.; Dieleman, J.P.; Olberg, H.K.; de Vries, C.S.; Sammon, C.; Andrews, N.; Svanström, H.; Mølgaard-Nielsen, D.; Hviid, A.; et al. Guillain-Barré syndrome and adjuvanted pandemic influenza A (H1N1) 2009 vaccines: A multinational self-controlled case series in Europe. PLOS ONE 2014, 9, e82222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Assessment report. Pandemrix. EMA/691037/2013; EMA: London, UK, 2013. [Google Scholar]
- Asa, P.B.; Cao, Y.; Garry, R.F. Antibodies to squalene in Gulf War syndrome. Exp. Mol. Pathol. 2000, 68, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Targher, G.; Franchini, M. Vaccination, squalene and anti-squalene antibodies: Facts or fiction? Eur. J. Intern. Med. 2010, 21, 70–73. [Google Scholar] [CrossRef] [PubMed]
- WHO. Safety of squalene. Available online: http://www.who.int/vaccine_safety/committee/topics/adjuvants/squalene/Jun_2006/en/ (accessed on 23 October 2014).
- Israeli, E.; Agmon-Levin, N.; Blank, M.; Shoenfeld, Y. Macrophagic myofaciitis a vaccine (alum) autoimmune-related disease. Clin. Rev. Allergy Immunol. 2011, 41, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, C.-A. Vaccine adjuvants and macrophagic myofasciitis. Arch. Pédiatrie Organe Off. Sociéte Fr. Pédiatrie 2005, 12, 96–101. [Google Scholar] [CrossRef]
- Verdier, F.; Burnett, R.; Michelet-Habchi, C.; Moretto, P.; Fievet-Groyne, F.; Sauzeat, E. Aluminium assay and evaluation of the local reaction at several time points after intramuscular administration of aluminium containing vaccines in the Cynomolgus monkey. Vaccine 2005, 23, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- WHO. Statement from the Global Advisory Committee on Vaccine Safety on aluminium-containing vaccines. Available online: http://www.who.int/vaccine_safety/committee/topics/aluminium/statement_112002/en/ (accessed on 20 March 2014).
- Expanding immunization coverage. Available online: http://www.unicef.org/immunization/index_coverage.html (accessed 30 June 2014).
- Begg, N. Trust in vaccines: Why it takes more than good faith. Vaccines 2013, 1, 343–347. [Google Scholar] [CrossRef]
- Hardt, K.; Schmidt-Ott, R.; Glismann, S.; Adegbola, R.A.; Meurice, F. Sustaining vaccine confidence in the 21st century. Vaccines 2013, 1, 204–224. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquale, A.D.; Preiss, S.; Silva, F.T.D.; Garçon, N. Vaccine Adjuvants: from 1920 to 2015 and Beyond. Vaccines 2015, 3, 320-343. https://doi.org/10.3390/vaccines3020320
Pasquale AD, Preiss S, Silva FTD, Garçon N. Vaccine Adjuvants: from 1920 to 2015 and Beyond. Vaccines. 2015; 3(2):320-343. https://doi.org/10.3390/vaccines3020320
Chicago/Turabian StylePasquale, Alberta Di, Scott Preiss, Fernanda Tavares Da Silva, and Nathalie Garçon. 2015. "Vaccine Adjuvants: from 1920 to 2015 and Beyond" Vaccines 3, no. 2: 320-343. https://doi.org/10.3390/vaccines3020320