
HCV Drug Resistance Challenges in Japan: The Role of Pre-Existing Variants and Emerging Resistant Strains in Direct Acting Antiviral Therapy

Abstract
:1. Introduction
1.1. Hepatitis C Virus
1.2. HCV Variability
1.3. HCV Treatment
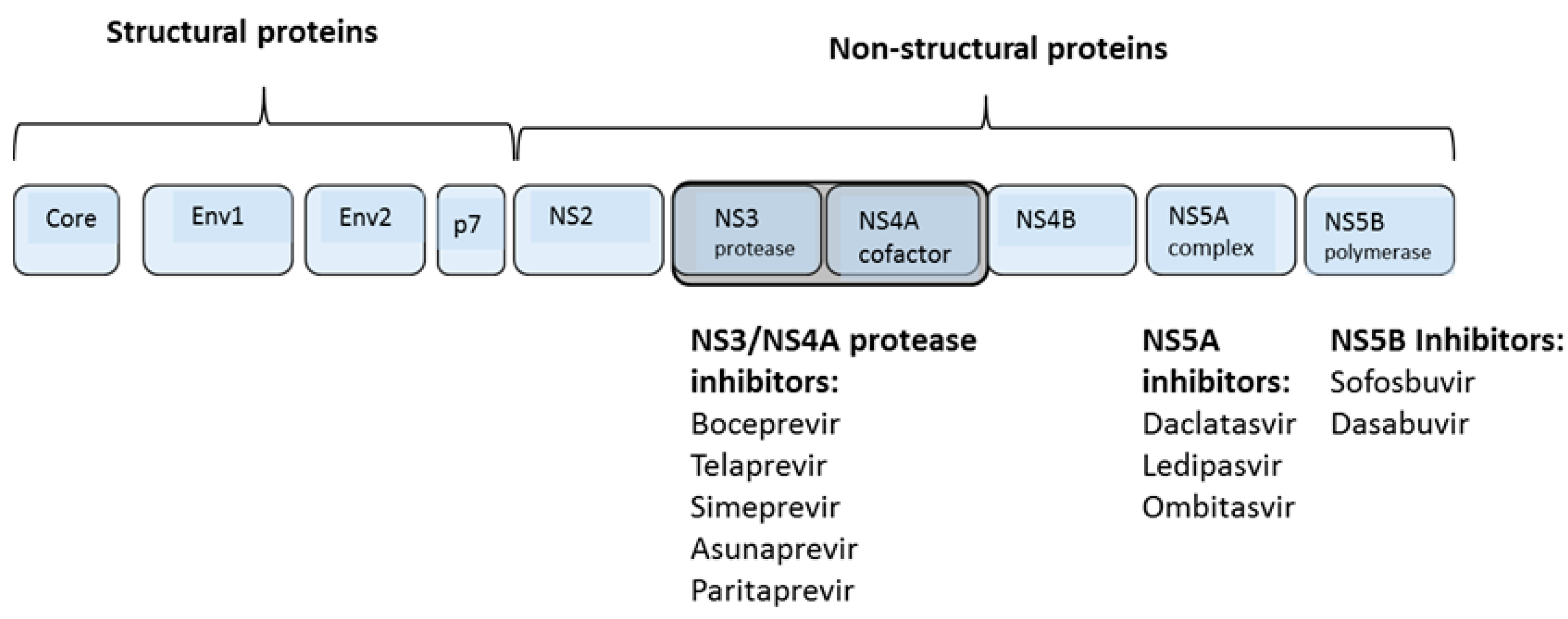
1.4. Direct Acting Antiviral Agents

{kind=link}
{kind=link}
| Class | Drug | Manufacturer | Approved for gt 1 | |
| NS3/4A Protease Inhibitors | ||||
| First-Generation, First-Wave | ||||
| Boceprevir (SCH503034) | Merck | US (2011) | ||
| Telaprevir (VX-950) | Janssen | US (2011); Japan (2011) | ||
| First-Generation, Second-Wave | ||||
| Simeprevir (TMC-435) | Tibotec | US (2013); Japan (2013) | ||
| Faldaprevir (BI-201335) | BI | withdrawn (2014) | ||
| Asunaprevir (BMS-650032) | BMS | Japan (2014) | ||
| Paritaprevir (ABT-450/r) | AbbVie | US (2014) | ||
| Danoprevir (ITMN-191, RG 7227) | Roche | |||
| Sovaprevir (ACH-1625) | Achillion | |||
| Vedroprevir (GS-9451) | Gilead | |||
| Vaniprevir (MK-7009) | Merck | |||
| Second Generation | ||||
| Grazoprevir (MK-5172) | Merck | |||
| ACH-2684 | Achillion | |||
| NS5B Polymerase Inhibitors | ||||
| Nucleoside Inhibitors | ||||
| Sofosbuvir (GS-7977) | Gilead | US (2014); Japan (2015) | ||
| Mericitabine (RG-7218) | Roche | |||
| Non-Nucleoside Inhibitors | ||||
| Thumb II Inhibitors | ||||
| GS-9669 | Gilead | |||
| VX-222 | Vertex | |||
| BMS-791325 | BMS | |||
| Palm I Inhibitors | ||||
| Dasabuvir (ABT-333) | AbbVie | US (2014) | ||
| ABT-072 | AbbVie | |||
| Setrobuvir (ANA-598) | Roche | |||
| NS5A Inhibitors | ||||
| First Generation | ||||
| Daclatasvir (BMS-790052) | BMS | Japan (2014) | ||
| Ledipasvir (GS-5885) | Gilead | US (2014); Japan (2015) | ||
| Ombitasvir (ABT-267) | AbbVie | US (2014) | ||
| PPI-668 | Presidio | |||
| PPI-461 | Presidio | |||
| ACH-2928 | Achillion | |||
| GSK-2336805 | GlaxoSmithKline | |||
| BMS-824393 | BMS | |||
| Samatasvir (IDX719) | Idenix | |||
| Second Generation | ||||
| Elbasavir (MK-8742) | Merck | |||
| ACH-3102 | Achillion | |||
| GS-5816 | Gilead | |||

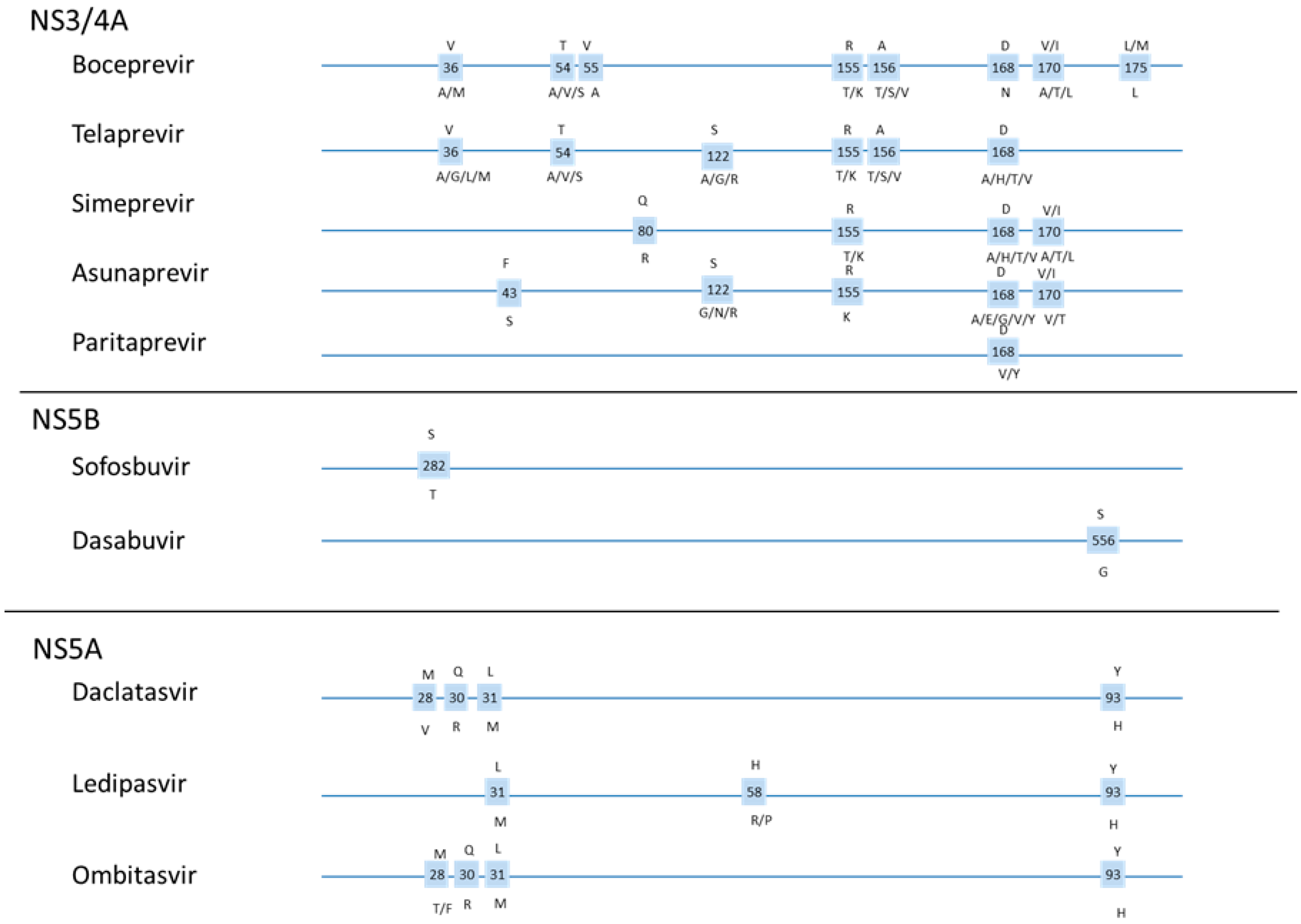
1.5. DAA Resistance

1.6. Resistance Testing
2. NS3/4A Protease Inhibitors
2.1. NS3/4A Protease
2.2. Telaprevir and Boceprevir
2.3. Telaprevir Resistance
2.4. Telaprevir Triple Therapy in Japan
2.5. Second Wave Protease Inhibitors
2.6. Simeprevir
2.7. Asunaprevir (BMS-650032)
2.8. ABT-450/r (Paritaprevir with Ritonovir)
2.9. Second Generation PIs
3. NS5B Polymerase Inhibitors
3.1. NS5B RNA-Dependent RNA Polymerase
3.2. Nucleoside Inhibitors
3.3. Sofosbuvir (GS-7977)
3.4. Non-Nucleoside Inhibitors
3.5. ABT-333 (Dasabuvir)
4. NS5A Inhibitors
4.1. NS5A
4.2. Daclatasvir
4.3. Ledipasvir (GS-5885)
4.4. Ombitasvir (ABT-267)
4.5. Ombitasvir and Paritaprevir/Ritonavir
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29, S74–S81. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N.; Yoshioka, K.; Moriwaki, H.; Okanoue, T.; Sakisaka, S.; Takehara, T.; Oketani, M.; Toyota, J.; Izumi, N.; et al. Accumulation of refractory factors for pegylated interferon plus ribavirin therapy in older female patients with chronic hepatitis C. Hepatol. Res. 2010, 40, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lennon, N.; Berlin, A.M.; Young, S.K.; Lee, B.; Heckerman, D.; Carlson, J.; Reyor, L.L.; et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology 2008, 48, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Mo, H.; Pilot-Matias, T.J.; Molla, A. Evolution of resistant M414T mutants among hepatitis C virus replicon cells treated with polymerase inhibitor A-782759. Antimicrob. Agents Chemother. 2007, 51, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N. Hepatitis C virus: How genetic variability affects pathobiology of disease. J. Gastroenterol. Hepatol. 2011, 26, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, S.; Imamura, M.; Murakami, E.; Hiraga, N.; Tsuge, M.; Kawakami, Y.; Aikata, H.; Abe, H.; Hayes, C.N.; Sasaki, T.; et al. Long term persistence of NS5A inhibitor-resistant hepatitis C virus in patients who failed daclatasvir and asunaprevir therapy. J. Med. Virol. 2015, 87, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N.; Yoshioka, K.; Moriwaki, H.; Okanoue, T.; Sakisaka, S.; Takehara, T.; Oketani, M.; Toyota, J.; Izumi, N.; et al. Factors predictive of sustained virological response following 72 weeks of combination therapy for genotype 1b hepatitis C. J. Gastroenterol. 2011, 46, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Gallego, L. Viral hepatitis: Treating hepatitis C in injection drug users. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, S.; Nelson Hayes, C.; Ochi, H.; Uchida, T.; Kan, H.; Murakami, E.; Abe, H.; Tsuge, M.; Miki, D.; Akiyama, R.; et al. Association between variants in the interferon lambda 4 locus and substitutions in the hepatitis C virus non-structural protein 5A (130 out of 130 characters). J. Hepatol. 2015, 63, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Itakura, J.; Kurosaki, M.; Takada, H.; Nakakuki, N.; Matsuda, S.; Gondou, K.; Asano, Y.; Hattori, N.; Itakura, Y.; Tamaki, N.; et al. Naturally occurring, resistance-associated hepatitis C virus NS5A variants are linked to IL28B genotype and are sensitive to interferon-based therapy. Hepatol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Wyles, D.L.; Mena, A.; Pedreira, J.D.; Castro-Iglesias, A.; Cachay, E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antivir. Res. 2014, 108, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, S.; Ochi, H.; Murakami, E.; Uchida, T.; Kan, H.; Akamatsu, S.; Hayes, C.N.; Abe, H.; Miki, D.; Hiraga, N.; et al. Rapid, sensitive, and accurate evaluation of drug resistant mutant (NS5A-Y93H) strain frequency in genotype 1b HCV by invader assay. PLoS ONE 2015, 10, e0130022. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Kouyama, J.; Naiki, K.; Mochida, S. A novel simple assay system to quantify the percent HCV-RNA levels of NS5A Y93H mutant strains and Y93 wild-type strains relative to the total HCV-RNA levels to determine the indication for antiviral therapy with NS5A inhibitors. PLoS ONE 2014, 9, e112647. [Google Scholar] [CrossRef] [PubMed]
- Panel, A.I.H.G. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology 2015, 62, 932–954. [Google Scholar]
- European Association for Study of the Liver. EASL clinical practice guidelines: Management of hepatitis C virus infection. J. Hepatol. 2014, 60, 392–420. [Google Scholar]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R., Jr.; Ikeda, M.; Lemon, S.M.; Gale, M., Jr. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Reesink, H.W.; Zeuzem, S.; Weegink, C.J.; Forestier, N.; van Vliet, A.; van de Wetering de Rooij, J.; McNair, L.; Purdy, S.; Kauffman, R.; Alam, J.; et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: A phase Ib, placebo-controlled, randomized study. Gastroenterology 2006, 131, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Kieffer, T.L.; Bartels, D.; Hanzelka, B.; Muh, U.; Welker, M.; Wincheringer, D.; Zhou, Y.; Chu, H.M.; Lin, C.; et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 2007, 132, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, L.; Helle, F.; Francois, C.; Castelain, S.; Duverlie, G.; Brochot, E. Simeprevir for the treatment of hepatitis C virus infection. Pharmgenomics Pers. Med. 2014, 7, 241–249. [Google Scholar] [PubMed]
- Hiraga, N.; Imamura, M.; Abe, H.; Hayes, C.N.; Kono, T.; Onishi, M.; Tsuge, M.; Takahashi, S.; Ochi, H.; Iwao, E.; et al. Rapid emergence of telaprevir resistant hepatitis C virus strain from wildtype clone in vivo. Hepatology 2011, 54, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, I.; Akaike, J.; Karino, Y.; Arakawa, T.; Kuwata, Y.; Ohmura, T.; Sato, T.; Kamiya, N.; Yamada, I.; Chayama, K.; et al. Antiviral effects of peginterferon alpha-2b and ribavirin following 24-week monotherapy of telaprevir in Japanese hepatitis C patients. J. Gastroenterol. 2011, 46, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Halfon, P.; Locarnini, S. Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 2011, 55, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Gardiner, D.F.; Lawitz, E.; Martorell, C.; Everson, G.T.; Ghalib, R.; Reindollar, R.; Rustgi, V.; McPhee, F.; Wind-Rotolo, M.; et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 2012, 366, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L. Beyond telaprevir and boceprevir: Resistance and new agents for hepatitis C virus infection. Top. Antivir. Med. 2012, 20, 139–145. [Google Scholar] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Wendt, A.; Adhoute, X.; Castellani, P.; Oules, V.; Ansaldi, C.; Benali, S.; Bourliere, M. Chronic hepatitis C: Future treatment. Clin. Pharmacol. Adv. Appl. 2014, 6, 1–17. [Google Scholar]
- Talwani, R.; Heil, E.L.; Gilliam, B.L.; Temesgen, Z. Simeprevir: A macrocyclic HCV protease inhibitor. Drugs Today 2013, 49, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, S.; Trembling, P.M.; Dusheiko, G.M. TMC435 for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs 2012, 21, 1193–1209. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Daclatasvir + asunaprevir: First global approval. Drugs 2014, 74, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Borgia, F.; Buonomo, A.R.; Zappulo, E.; Castaldo, G.; Borgia, G. ABT-450: A novel protease inhibitor for the treatment of hepatitis C virus infection. Curr. Med. Chem. 2014, 21, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kowdley, K.V.; Coakley, E.; Sigal, S.; Nelson, D.R.; Crawford, D.; Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T.; et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 2014, 370, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Lesburg, C.A.; Cable, M.B.; Ferrari, E.; Hong, Z.; Mannarino, A.F.; Weber, P.C. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999, 6, 937–943. [Google Scholar] [PubMed]
- Gerber, L.; Welzel, T.M.; Zeuzem, S. New therapeutic strategies in HCV: Polymerase inhibitors. Liver int. 2013, 33, S85–S92. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.; Jin, Z.; Dyatkina, N.; Wang, G.; Beigelman, L.; Deval, J. Efficiency of incorporation and chain termination determines the inhibition potency of 2′-modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob. Agents Chemother. 2014, 58, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, S.; Fiorina, L.; Mariani, B.; Gulminetti, R.; Novati, S.; Barbarini, G.; Bruno, R.; Baldanti, F. Naturally occurring resistance mutations to inhibitors of HCV NS5A region and NS5B polymerase in DAA treatment-naive patients. Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M.; et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Lalezari, J.P.; Hassanein, T.; Kowdley, K.V.; Poordad, F.F.; Sheikh, A.M.; Afdhal, N.H.; Bernstein, D.E.; Dejesus, E.; Freilich, B.; et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: A randomised, double-blind, phase 2 trial. Lancet Infect. Dis. 2013, 13, 401–408. [Google Scholar] [CrossRef]
- Lawitz, E.; Mangia, A.; Wyles, D.; Rodriguez-Torres, M.; Hassanein, T.; Gordon, S.C.; Schultz, M.; Davis, M.N.; Kayali, Z.; Reddy, K.R.; et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013, 368, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Noell, B.C.; Besur, S.V.; deLemos, A.S. Changing the face of hepatitis c management—The design and development of sofosbuvir. Drug Des. Dev. Ther. 2015, 9, 2367–2374. [Google Scholar]
- Hedskog, C.; Doehle, B.; Chodavarapu, K.; Gontcharova, V.; Crespo Garcia, J.; de Knegt, R.; Drenth, J.P.; McHutchison, J.G.; Brainard, D.; Stamm, L.M.; et al. Characterization of hepatitis C virus intergenotypic recombinant strains and associated virological response to sofosbuvir/ribavirin. Hepatology 2015, 61, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, O.; Norder, H.; Mukomolov, S.; Magnius, L.O. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J. Virol. 2002, 76, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P.L. Non-nucleoside inhibitors of the HCV NS5B polymerase: Progress in the discovery and development of novel agents for the treatment of HCV infections. Curr. Opin. Investig. Drugs 2007, 8, 614–634. [Google Scholar] [PubMed]
- Nyanguile, O.; Devogelaere, B.; Vijgen, L.; van den Broeck, W.; Pauwels, F.; Cummings, M.D.; de Bondt, H.L.; Vos, A.M.; Berke, J.M.; Lenz, O.; et al. 1a/1b Subtype profiling of nonnucleoside polymerase inhibitors of hepatitis C virus. J. Virol. 2010, 84, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Buonomo, A.R.; Borgia, G. Dasabuvir: A non-nucleoside inhibitor of NS5B for the treatment of hepatitis C virus infection. Rev. Recent Clin. Trials 2014, 9, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, D.R.; Belema, M.; Gao, M.; Goodrich, J.; Kakarla, R.; Knipe, J.O.; Lemm, J.A.; Liu, M.; Lopez, O.D.; Nguyen, V.N.; et al. HCV NS5A replication complex inhibitors. Part 2: Investigation of stilbene prolinamides. Bioorganic Med. Chem. Lett. 2012, 22, 6063–6066. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.W. Drugs for hepatitis C: Unlocking a new mechanism of action. ChemMedChem 2010, 5, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Nettles, R.E.; Gao, M.; Bifano, M.; Chung, E.; Persson, A.; Marbury, T.C.; Goldwater, R.; DeMicco, M.P.; Rodriguez-Torres, M.; Vutikullird, A.; et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology 2011, 54, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Ghalib, R.H.; Rustgi, V.K.; Martorell, C.; Everson, G.T.; Tatum, H.A.; Hezode, C.; Lim, J.K.; Bronowicki, J.P.; Abrams, G.A.; et al. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: A randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect. Dis. 2012, 12, 671–677. [Google Scholar] [CrossRef]
- Chayama, K.; Takahashi, S.; Toyota, J.; Karino, Y.; Ikeda, K.; Ishikawa, H.; Watanabe, H.; McPhee, F.; Hughes, E.; Kumada, H. Dual therapy with the nonstructural protein 5A inhibitor, daclatasvir, and the nonstructural protein 3 protease inhibitor, asunaprevir, in hepatitis C virus genotype 1b-infected null responders. Hepatology 2012, 55, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ikeda, K.; Suzuki, F.; Toyota, J.; Karino, Y.; Chayama, K.; Kawakami, Y.; Ishikawa, H.; Watanabe, H.; Hu, W.; et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J. Hepatol. 2013, 58, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Kumada, H.; Suzuki, Y.; Ikeda, K.; Toyota, J.; Karino, Y.; Chayama, K.; Kawakami, Y.; Ido, A.; Yamamoto, K.; Takaguchi, K.; et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 2014, 59, 2083–2089. [Google Scholar] [CrossRef] [PubMed]
- Karino, Y.; Toyota, J.; Ikeda, K.; Suzuki, F.; Chayama, K.; Kawakami, Y.; Ishikawa, H.; Watanabe, H.; Hernandez, D.; Yu, F.; et al. Characterization of virologic escape in hepatitis C virus genotype 1b patients treated with the direct-acting antivirals daclatasvir and asunaprevir. J. Hepatol. 2013, 58, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Yusim, K.; Boykin, L.; Richardson, R. The Los Alamos hepatitis C sequence database. Bioinformatics 2005, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J.; Levine, S.; Chen, C.; Sheaffer, A.K.; Chaniewski, S.; Voss, S.; Lemm, J.A.; McPhee, F. Combinations of lambda interferon with direct-acting antiviral agents are highly efficient in suppressing hepatitis C virus replication. Antimicrob. Agents Chemother. 2013, 57, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, M.; Yokosuka, O.; Takehara, T.; Sakamoto, N.; Korenaga, M.; Mochizuki, H.; Nakane, K.; Enomoto, H.; Ikeda, F.; Yanase, M.; et al. Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: An open-label, randomised, phase 3 trial. Lancet Infect. Dis. 2015, 15, 645–653. [Google Scholar] [CrossRef]
- Chayama, K.; Notsumata, K.; Kurosaki, M.; Sato, K.; Rodrigues, L., Jr.; Setze, C.; Badri, P.; Pilot-Matias, T.; Vilchez, R.A.; Kumada, H. Randomized trial of interferon- and ribavirin-free ombitasvir/paritaprevir/ritonavir in treatment-experienced hepatitis C virus-infected patients. Hepatology 2015, 61, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chayama, K.; Hayes, C.N. HCV Drug Resistance Challenges in Japan: The Role of Pre-Existing Variants and Emerging Resistant Strains in Direct Acting Antiviral Therapy. Viruses 2015, 7, 5328-5342. https://doi.org/10.3390/v7102876
Chayama K, Hayes CN. HCV Drug Resistance Challenges in Japan: The Role of Pre-Existing Variants and Emerging Resistant Strains in Direct Acting Antiviral Therapy. Viruses. 2015; 7(10):5328-5342. https://doi.org/10.3390/v7102876
Chicago/Turabian StyleChayama, Kazuaki, and C. Nelson Hayes. 2015. "HCV Drug Resistance Challenges in Japan: The Role of Pre-Existing Variants and Emerging Resistant Strains in Direct Acting Antiviral Therapy" Viruses 7, no. 10: 5328-5342. https://doi.org/10.3390/v7102876