
Molecular Dynamics Simulations Reveal Structural Interconnections within Sec14-PH Bipartite Domain from Human Neurofibromin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
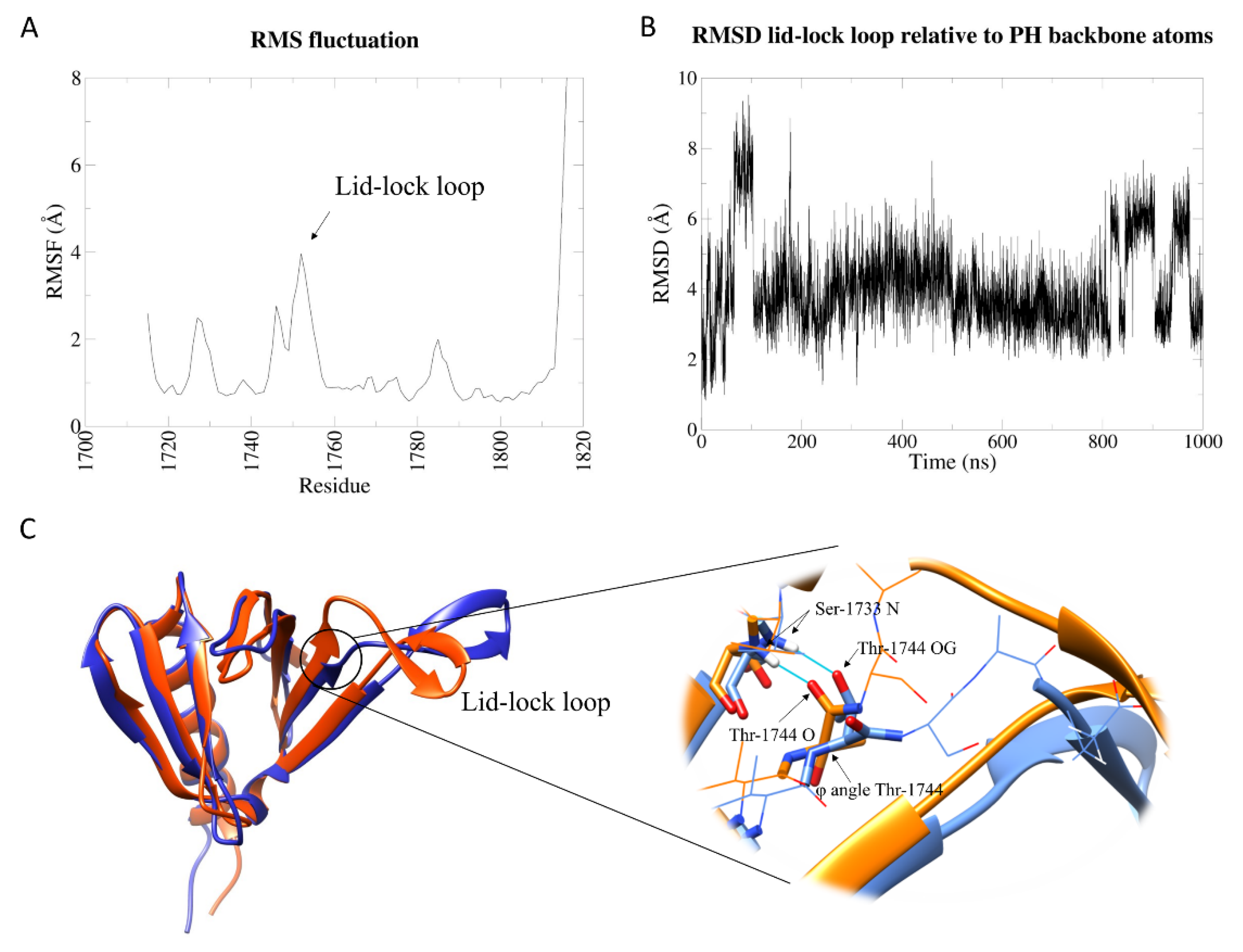
2.1. PH Portion of Sec14-PH Domain Is Independently Stable and Shows Scarce Flexibility
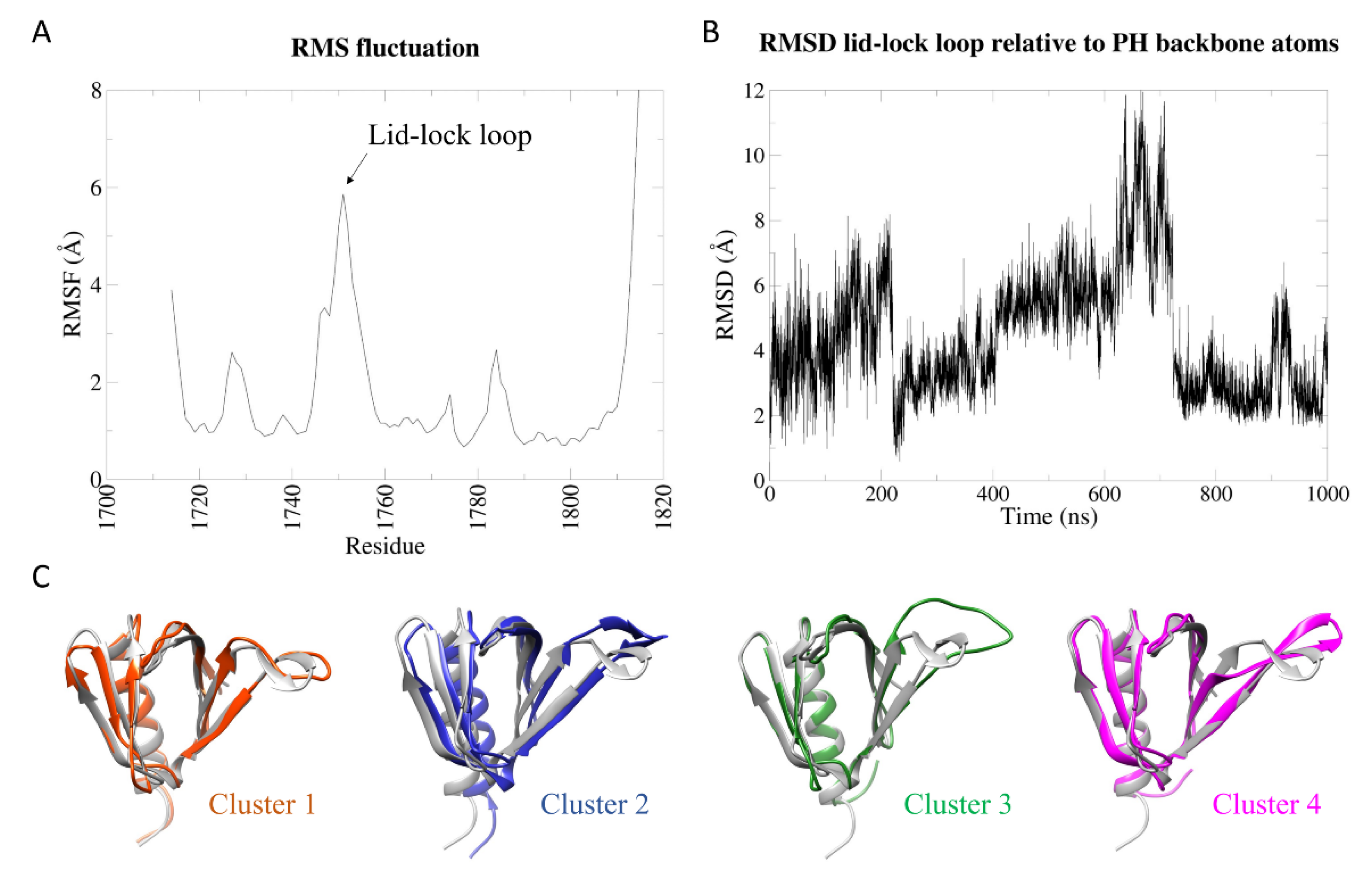
2.2. Dynamic Behaviour of the Whole Wild-Type Sec14-PH Domain
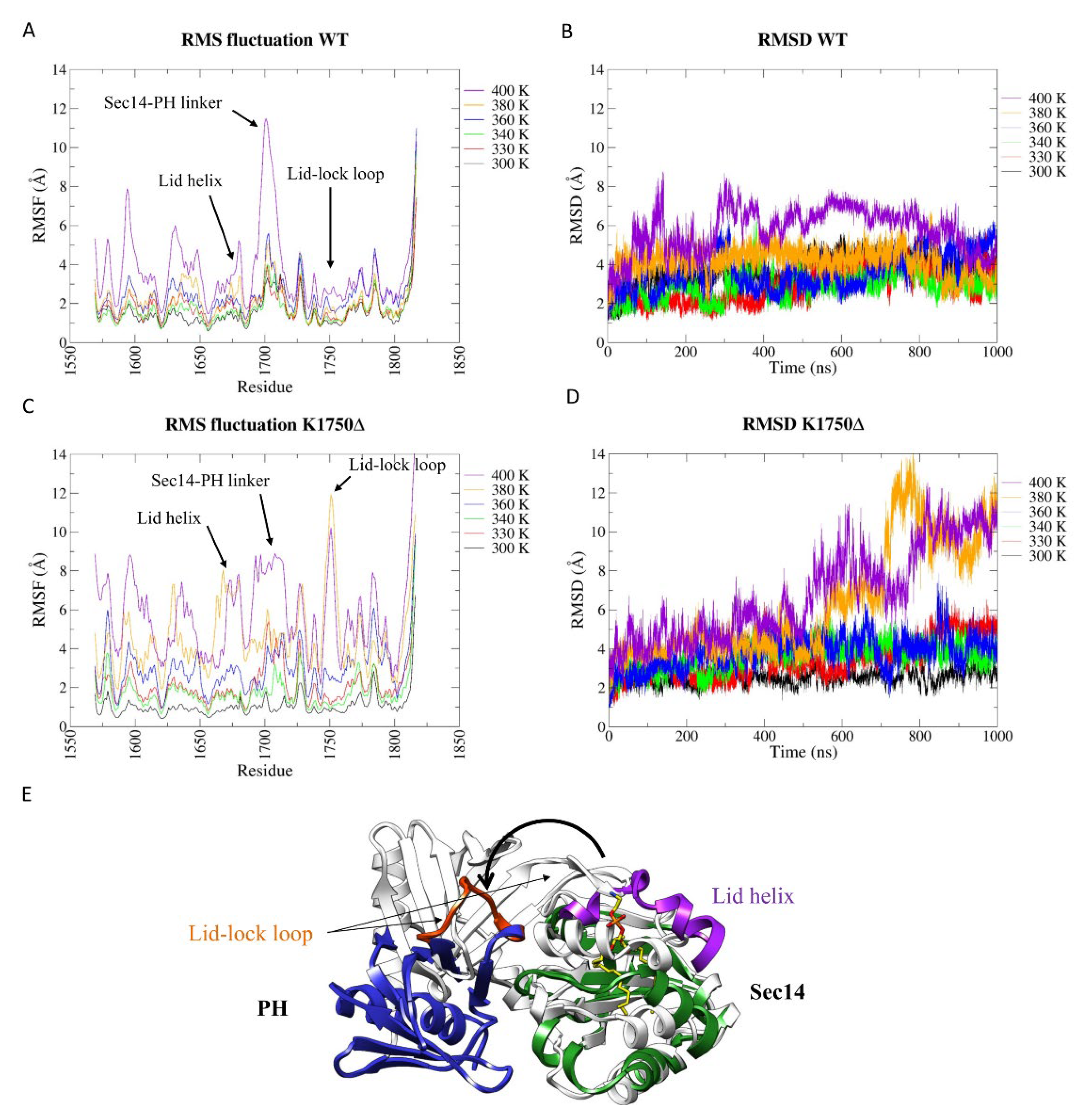
2.3. The K1750Δ Mutation Weakens the Interconnection between the Two Portions of the Bipartite Sec14-PH Domain
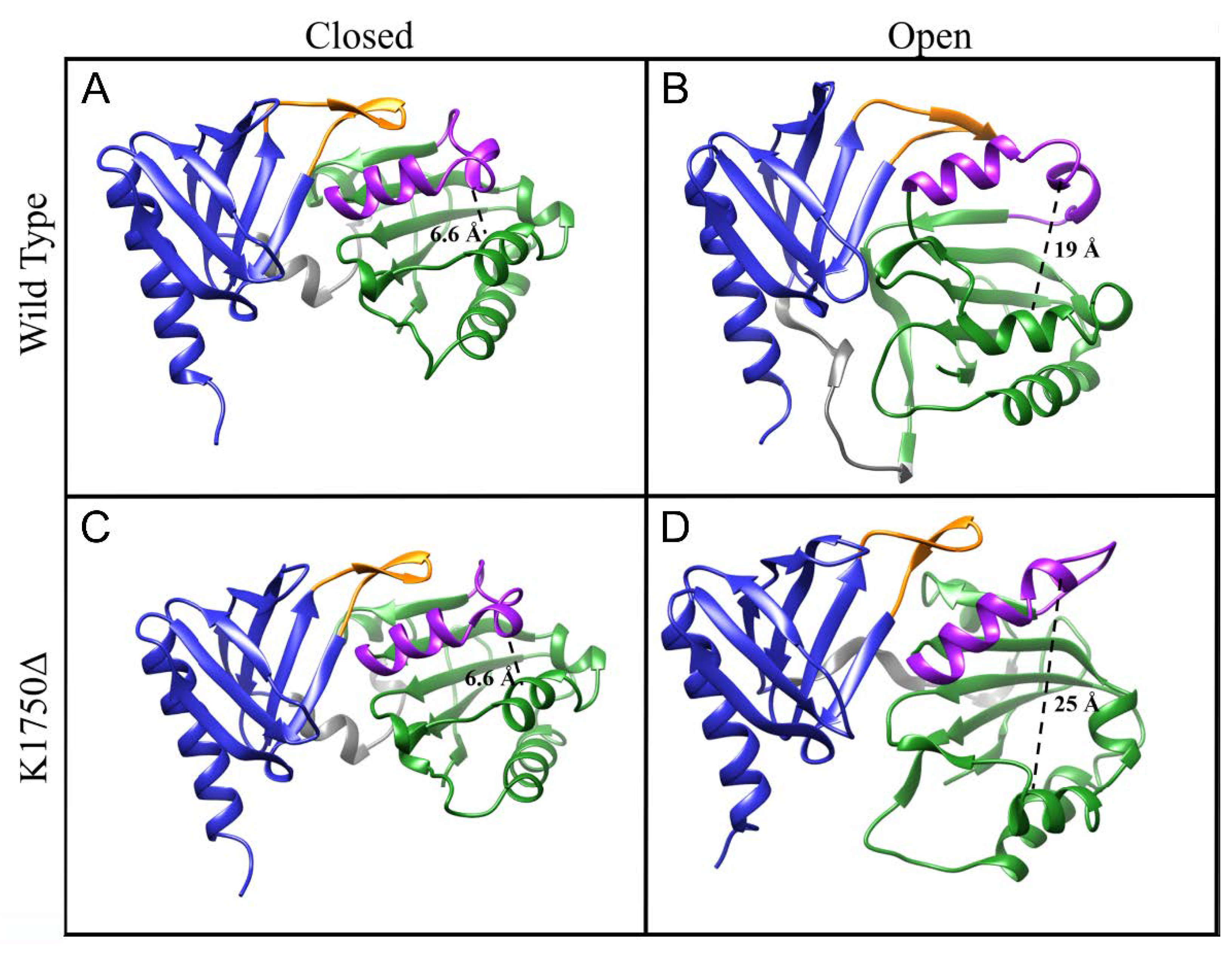
2.4. Opening of the Lipid Pocket
3. Discussion
4. Materials and Methods
4.1. Systems Setup
4.2. Molecular Dynamics Simulations and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brosseau, J.-P.; Liao, C.-P.; Le, L.Q. Translating Current Basic Research into Future Therapies for Neurofibromatosis Type 1. Br. J. Cancer 2020, 123, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M. The GAP-Related Domain of the Neurofibromatosis Type 1 Gene Product Interacts with Ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D Sensitivity to Neurofibromin-Mediated GTP Hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Tokuda, T.; Sato, K.; Okanishi, H.; Nagayama, M.; Hirayama-Kurogi, M.; Ohtsuki, S.; Araki, N. Identification of a Specific Translational Machinery via TCTP-EF1A2 Interaction Regulating NF1-Associated Tumor Growth by Affinity Purification and Data-Independent Mass Spectrometry Acquisition (AP-DIA). Mol. Cell. Proteom. 2019, 18, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.-J.; Zhou, M.-T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-Coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell. Proteom. 2017, 16, 594–607. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The Molecular Landscape of Glioma in Patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef]
- Bonneau, F.; Lenherr, E.D.; Pena, V.; Hart, D.J.; Scheffzek, K. Solubility Survey of Fragments of the Neurofibromatosis Type 1 Protein Neurofibromin. Protein Expr. Purif. 2009, 65, 30–37. [Google Scholar] [CrossRef]
- Welti, S.; Kühn, S.; D’Angelo, I.; Brügger, B.; Kaufmann, D.; Scheffzek, K. Structural and Biochemical Consequences of NF1 Associated Nontruncating Mutations in the Sec14-PH Module of Neurofibromin. Hum. Mutat. 2011, 32, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Tautz, L.; Mustelin, T. The Lipid-Binding SEC14 Domain. Biochim. Biophys. Acta 2007, 1771, 719–726. [Google Scholar] [CrossRef]
- Sha, B.; Phillips, S.E.; Bankaitis, V.A.; Luo, M. Crystal Structure of the Saccharomyces Cerevisiae Phosphatidylinositol-Transfer Protein. Nature 1998, 391, 506–510. [Google Scholar] [CrossRef]
- Ryan, M.M.; Temple, B.R.S.; Phillips, S.E.; Bankaitis, V.A. Conformational Dynamics of the Major Yeast Phosphatidylinositol Transfer Protein sec14p: Insight into the Mechanisms of Phospholipid Exchange and Diseases of sec14p-like Protein Deficiencies. Mol. Biol. Cell 2007, 18, 1928–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, T. Inositol-Lipid Binding Motifs: Signal Integrators through Protein-Lipid and Protein-Protein Interactions. J. Cell Sci. 2005, 118, 2093–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welti, S.; Fraterman, S.; D’Angelo, I.; Wilm, M.; Scheffzek, K. The sec14 Homology Module of Neurofibromin Binds Cellular Glycerophospholipids: Mass Spectrometry and Structure of a Lipid Complex. J. Mol. Biol. 2007, 366, 551–562. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, I.; Welti, S.; Bonneau, F.; Scheffzek, K. A Novel Bipartite Phospholipid-Binding Module in the Neurofibromatosis Type 1 Protein. EMBO Rep. 2006, 7, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Colgan, L.A.; Dao, M.T.; Muntean, B.S.; Sutton, L.P.; Orlandi, C.; Boye, S.L.; Boye, S.E.; Shih, C.-C.; Li, Y.; et al. NF1 is a Direct G Protein Effector Essential for Opioid Signaling to Ras in the Striatum. Curr. Biol. 2016, 26, 2992–3003. [Google Scholar] [CrossRef] [Green Version]
- Nadim, W.D.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.-L.; Zajdel, P.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. Physical Interaction between Neurofibromin and Serotonin 5-HT6 Receptor Promotes Receptor Constitutive Activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef] [Green Version]
- Nix, J.S.; Blakeley, J.; Rodriguez, F.J. An Update on the Central Nervous System Manifestations of Neurofibromatosis Type 1. Acta Neuropathol. 2020, 139, 625–641. [Google Scholar] [CrossRef]
- Chaker-Margot, M.; Werten, S.; Dunzendorfer-Matt, T.; Lechner, S.; Ruepp, A.; Scheffzek, K.; Maier, T. Structural Basis of Activation of the Tumor Suppressor Protein Neurofibromin. Mol. Cell 2022, 82, 1288–1296.e5. [Google Scholar] [CrossRef]
- Lupton, C.J.; Bayly-Jones, C.; D’Andrea, L.; Huang, C.; Schittenhelm, R.B.; Venugopal, H.; Whisstock, J.C.; Halls, M.L.; Ellisdon, A.M. The Cryo-EM Structure of the Human Neurofibromin Dimer Reveals the Molecular Basis for Neurofibromatosis Type 1. Nat. Struct. Mol. Biol. 2021, 28, 982–988. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: AnN⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Johannes, G.E. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Compytational Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizza, F.; Vertemara, J.; Tisi, R. Molecular Dynamics Simulations Reveal Structural Interconnections within Sec14-PH Bipartite Domain from Human Neurofibromin. Int. J. Mol. Sci. 2022, 23, 5707. https://doi.org/10.3390/ijms23105707
Rizza F, Vertemara J, Tisi R. Molecular Dynamics Simulations Reveal Structural Interconnections within Sec14-PH Bipartite Domain from Human Neurofibromin. International Journal of Molecular Sciences. 2022; 23(10):5707. https://doi.org/10.3390/ijms23105707
Chicago/Turabian StyleRizza, Fabio, Jacopo Vertemara, and Renata Tisi. 2022. "Molecular Dynamics Simulations Reveal Structural Interconnections within Sec14-PH Bipartite Domain from Human Neurofibromin" International Journal of Molecular Sciences 23, no. 10: 5707. https://doi.org/10.3390/ijms23105707