
Production of Recombinant Human Ceruloplasmin: Improvements and Perspectives

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Expression of Recombinant CP
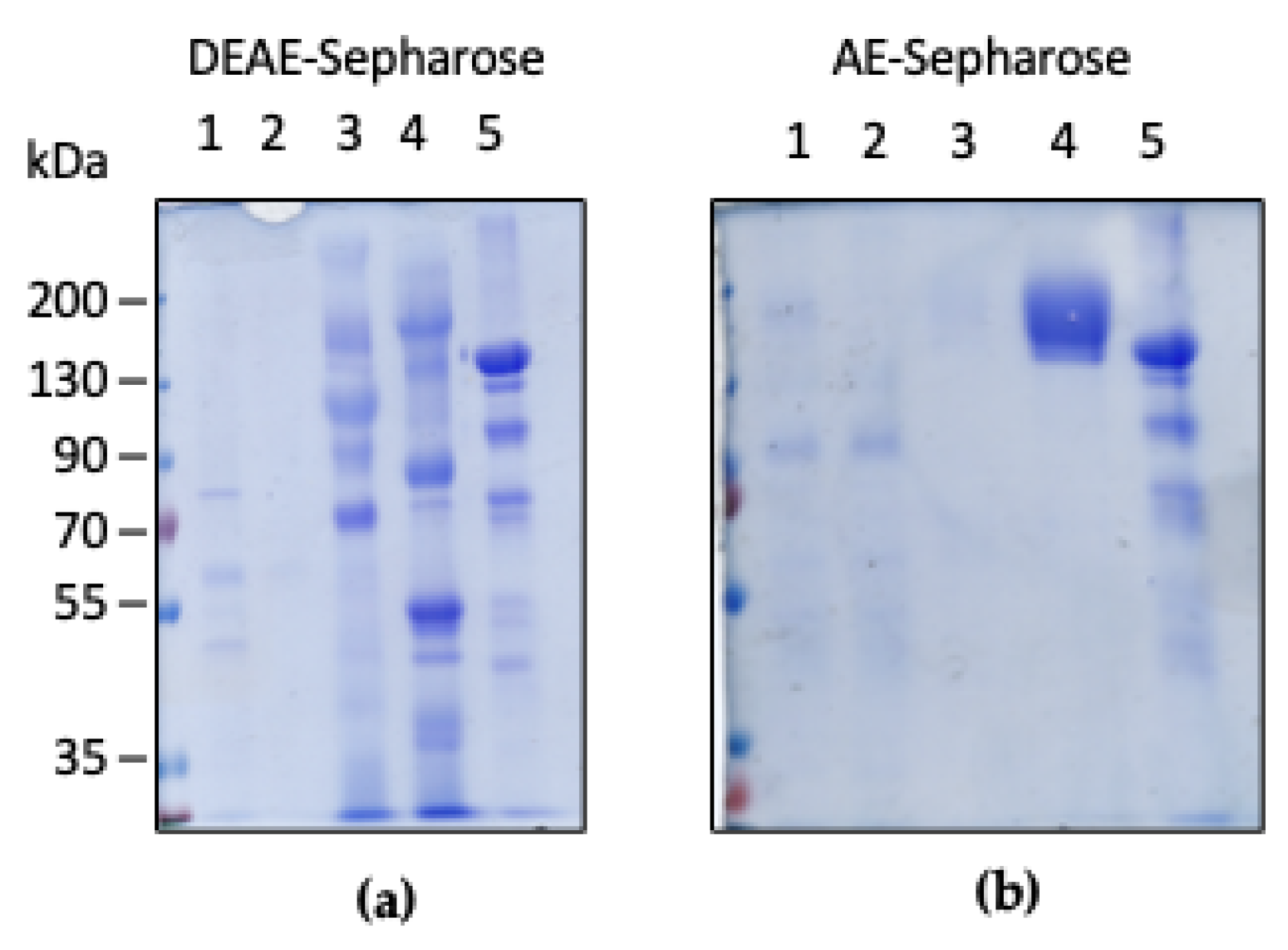
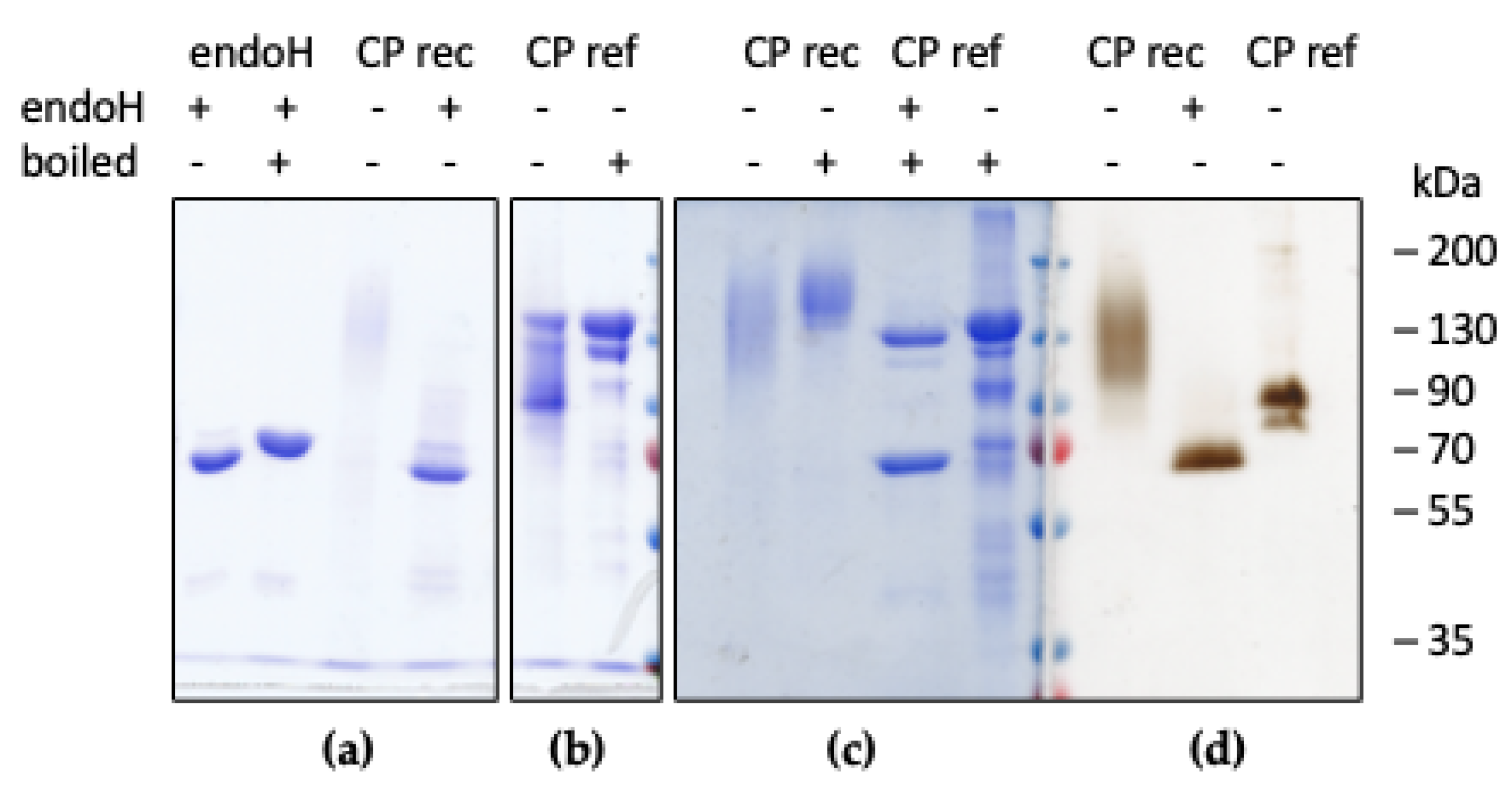
2.2. Purification and Characterization of Recombinant CP
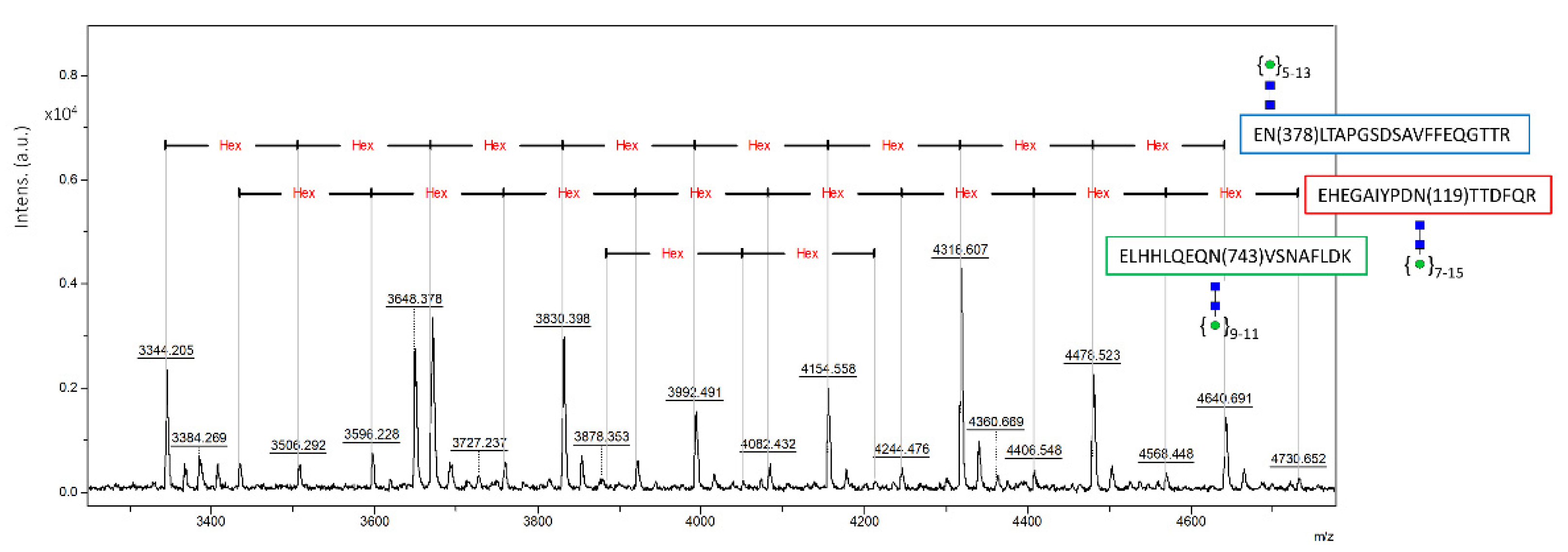
2.3. Glycan Analysis of Recombinant CP
3. Materials and Methods
3.1. Yeast Strains
3.2. Expression and Purification of Recombinant CP
3.3. Characterization of Recombinant CP
3.4. Glycan Analysis
3.4.1. Analysis of N-Glycoprofile
3.4.2. Analysis of N-Glycopeptides and Occupancy of Glycosylation Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Solomon, E.I.; Sundaram, U.; Machonkin, T.E. Multicopper oxidases and oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef]
- Kosman, D.J. Multicopper oxidases: A workshop on copper coordination chemistry, electron transfer, and metallophysiology. J. Biol. Inorg. Chem. 2010, 15, 15–28. [Google Scholar] [CrossRef]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar]
- Vasilyev, V.B. Looking for a partner: Ceruloplasmin in protein-protein interactions. Biometals 2019, 32, 195–210. [Google Scholar] [CrossRef]
- Pietrangelo, A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty years of ferroportin disease: A review or an update of published clinical, biochemical, molecular, and functional features. Pharmaceuticals 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishi, G.; Subramaniam, N.V. Biology of the iron efflux transporter, ferroportin. Adv. Prot. Chem. Struct. Biol. 2021, 123, 1–16. [Google Scholar]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Ji, C.; Steimle, B.L.; Bailey, D.K.; Kosman, D.J. The ferroxidase hephaestin but not amyloid precursor protein is required for ferroportin-supported iron efflux in primary hippocampal neurons. Cell. Mol. Neurobiol. 2018, 38, 941–954. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey Ward, D.; Bonaccorsi di Patti, M.C.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef]
- Dlouhy, A.C.; Bailey, D.K.; Steimle, B.L.; Parker, H.V.; Kosman, D.J. Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. J. Biol. Chem. 2019, 294, 4202–4214. [Google Scholar] [CrossRef]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; McGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Furihata, K.; Takeda, S.; Nakamura, A.; Yamamoto, K.; Morita, H.; Hiyamuta, S.; Ikeda, S.; Shimizu, N.; Yanagisawa, N. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet. 1995, 9, 267–272. [Google Scholar] [CrossRef]
- Kono, S. Aceruloplasminemia. Curr. Drug Targ. 2012, 13, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Pandolfo, M.; Kuhn, J.; Shang, H.; Miyajima, H. The neurological presentation of ceruloplasmin gene mutations. Eur. Neurol. 2008, 60, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; Marchi, G.; Barqué, A.; Esteban-Jurado, C.; Marchetto, A.; Giorgetti, A.; Chelban, V.; Houlden, H.; Wood, N.W.; Piubelli, C.; et al. Genetic and clinical heterogeneity in thirteen new cases with aceruloplasminemia. Atypical anemia as a clue for an early diagnosis. Int. J. Mol. Sci. 2020, 21, 2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyajima, H. Aceruloplasminemia. Neuropathology 2015, 35, 83–90. [Google Scholar] [CrossRef]
- Hellman, N.E.; Kono, S.; Miyajima, H.; Gitlin, J.D. Biochemical analysis of a missense mutation in aceruloplasminemia. J. Biol. Chem. 2002, 277, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Kono, S.; Miyajima, H. Molecular and pathological basis of aceruloplasminemia. Biol. Res. 2006, 39, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorsi di Patti, M.C.; Maio, N.; Rizzo, G.; De Francesco, G.; Persichini, T.; Colasanti, M.; Polticelli, F.; Musci, G. Dominant mutants of ceruloplasmin impair the copper loading machinery in aceruloplasminemia. J. Biol. Chem. 2009, 284, 4545–4554. [Google Scholar] [CrossRef] [Green Version]
- Kono, S.; Yoshida, K.; Tomosugi, N.; Terada, T.; Hamaya, Y.; Kanaoka, S.; Miyajima, H. Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 2010, 1802, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Alessio, M. Aceruloplasminemia: Waiting for an efficient therapy. Front. Neurosci. 2018, 12, 903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanardi, A.; Conti, A.; Cremonesi, M.; D’Adamo, P.; Gilberti, E.; Apostoli, P.; Cannistraci, C.V.; Piperno, A.; David, S.; Alessio, M. Ceruloplasmin replacement therapy ameliorates neurological symptoms in a preclinical model of aceruloplasminemia. EMBO Mol. Med. 2018, 10, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Naylor, S.L.; Lum, J.B.; Cutshaw, S.; McCombs, J.L.; Naberhaus, K.H.; McGill, J.R.; Adrian, G.S.; Moore, C.M.; Barnett, D.R.; et al. Characterization, mapping, and expression of the human ceruloplasmin gene. Proc. Natl. Acad. Sci. USA 1986, 83, 3257–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maio, N.; Polticelli, F.; De Francesco, G.; Rizzo, G.; Bonaccorsi di Patti, M.C.; Musci, G. Role of external loops of human ceruloplasmin in copper loading by ATP7B and Ccc2p. J. Biol. Chem. 2010, 285, 20507–20513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielli, P.; Bellenchi, G.C.; Calabrese, L. Site-directed mutagenesis of human ceruloplasmin: Production of a proteolytically stable protein and structure-activity relationships of type 1 sites. J. Biol. Chem. 2001, 276, 2678–2685. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Stenberg, L.M.; Mauk, A.G. Identification of catalytically important amino acids in human ceruloplasmin by site-directed mutagenesis. FEBS Lett. 2002, 520, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Hellman, N.E.; Kono, S.; Mancini, G.M.; Hoogeboom, A.J.; de Jong, G.J.; Gitlin, J.D. Mechanisms of copper incorporation into human ceruloplasmin. J. Biol. Chem. 2002, 277, 46632–46638. [Google Scholar] [CrossRef] [Green Version]
- Vervecken, W.; Kaigorodov, V.; Callewaert, N.; Geysens, S.; De Vusser, K.; Contreras, R. In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl. Environ. Microbiol. 2004, 70, 2639–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, P.P.; Geysens, S.; Vervecken, W.; Contreras, R.; Callewaert, N. Engineering complex N-type glycosylation in Pichia pastoris using GlycoSwitch technology. Nat. Protoc. 2009, 4, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Ortel, L.T.; Putnam, W.F. Single-chain structure of human ceruloplasmin: The complete amino acid sequence of the whole molecule. Proc. Natl. Acad. Sci. USA 1984, 81, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Barrero, J.J.; Casler, J.C.; Valero, F.; Ferrer, P.; Glick, B.S. An improved secretion signal enhances the secretion of model proteins from Pichia pastoris. Microb. Cell Fact. 2018, 17, 161. [Google Scholar] [CrossRef] [PubMed]
- Musci, G.; Bonaccorsi di Patti, M.C.; Fagiolo, U.; Calabrese, L. Age-related changes in human ceruloplasmin. Evidence for oxidative modifications. J. Biol. Chem. 1993, 268, 13388–13395. [Google Scholar] [CrossRef]
- Stern, R.V.; Caffrey, J.M.; Frieden, E. A tentacle gel simplifies the purification of ceruloplasmin. Biochem. Int. 1992, 27, 281–289. [Google Scholar]
- Yuan, D.S.; Dancis, A.; Klausner, R.D. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J. Biol. Chem. 1997, 272, 25787–25793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerenfaenger, M.; Moritz, M.; Meyer, B. Quantitation of glycopeptides by ESI/MS—Size of the peptide part strongly affects the relative proportions and allows discovery of new glycan compositions of ceruloplasmin. Glycoconj. J. 2019, 36, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Laukens, B.; Jacobs, P.P.; Geysens, K.; Martins, J.; De Wacther, C.; Ameloot, P.; Morelle, W.; Haustraete, J.; Renauld, J.C.; Samyn, B.; et al. Off-target glycans encountered along the synthetic biology route toward humanized N-glycans in Pichia pastoris. Biotechnol. Bioeng. 2020, 117, 2479–2488. [Google Scholar] [CrossRef]
- Looke, M.; Kristjuhan, K.; Kristjuhan, A. Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques 2011, 50, 325–328. [Google Scholar] [CrossRef]
- Pažitná, L.; Nemčovič, M.; Pakanová, Z.; Baráth, P.; Aliev, T.; Dolgikh, D.; Argentova, V.; Katrlík, J. Influence of media composition on recombinant monoclonal IgA1 glycosylation analysed by lectin-based protein microarray and MALDI-MS. J. Biotechnol. 2020, 314–315, 34–40. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | m/z (Permethylated) | Intensity | Relative Intensity (%) |
|---|---|---|---|
| HexNAc2Hex5 | 1579.971 | 764 | 6.1 |
| HexNAc2Hex7 | 1988.437 | 1199 | 9.5 |
| HexNAc2Hex8 | 2192.73 | 2211 | 17.6 |
| HexNAc2Hex9 | 2397.024 | 1045 | 8.3 |
| HexNAc2Hex10 | 2601.325 | 1443 | 11.5 |
| HexNAc2Hex11 | 2805.58 | 1821 | 14.5 |
| HexNAc2Hex12 | 3009.771 | 969 | 7.7 |
| N-Glycosylation Site 1 | Covered by Peptides | Ratio 18O-Asp/Asn after PNGase F 3 | % of Glycosylation |
|---|---|---|---|
| Asn119 * | Yes | 0.61012 | 38 |
| Asn208 | No 2 | - | - |
| Asn339 * | Yes | ND | ND |
| Asn378 * | Yes | 6.2394 | 86 |
| Asn569 | Yes | ND | ND |
| Asn743 * | Yes | 2.2496 | 69 |
| Asn907 | Yes | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaccorsi di Patti, M.C.; Cutone, A.; Nemčovič, M.; Pakanová, Z.; Baráth, P.; Musci, G. Production of Recombinant Human Ceruloplasmin: Improvements and Perspectives. Int. J. Mol. Sci. 2021, 22, 8228. https://doi.org/10.3390/ijms22158228
Bonaccorsi di Patti MC, Cutone A, Nemčovič M, Pakanová Z, Baráth P, Musci G. Production of Recombinant Human Ceruloplasmin: Improvements and Perspectives. International Journal of Molecular Sciences. 2021; 22(15):8228. https://doi.org/10.3390/ijms22158228
Chicago/Turabian StyleBonaccorsi di Patti, Maria Carmela, Antimo Cutone, Marek Nemčovič, Zuzana Pakanová, Peter Baráth, and Giovanni Musci. 2021. "Production of Recombinant Human Ceruloplasmin: Improvements and Perspectives" International Journal of Molecular Sciences 22, no. 15: 8228. https://doi.org/10.3390/ijms22158228