
Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel


Department of Civil, Chemical, Environmental and Materials Engineering, University of Bologna, via Terracini 28, 40131 Bologna, Italy
*
Author to whom correspondence should be addressed.
Energies 2021, 14(12), 3562; https://doi.org/10.3390/en14123562
Submission received: 7 May 2021
/
Revised: 4 June 2021
/
Accepted: 9 June 2021
/
Published: 15 June 2021
(This article belongs to the Section B2: Clean Energy)
Abstract
:The need for lowering the environmental impacts has incentivized the investigation of biomass and biofuels as possible alternative sources for energy supply. Among the others, oxygenated bio-derived molecules such as alcohols, esters, acids, aldehydes, and furans are attractive substances as chemical feedstock and for sustainable energy production. Indeed, the presence of oxygen atoms limits the production of aromatic compounds, improves combustion efficiency (thus heat production) and alleviates the formation of carbon soot. On the other hand, the variability of their composition has represented one of the major challenges for the complete characterization of combustion behaviour. This work gives an overview of the current understanding of the detailed chemical mechanisms, as well as experimental investigations characterizing the combustion process of these species, with an emphasis on the laminar burning velocity and the ignition delay time. From the review, the common intermediates for the most relevant functional groups and combustion of biofuels were identified. The gathered information can be intended for the sake of core mechanism generation.
1. Introduction
Fossil fuels are still the main feedstock for global energy production [1,2]. However, sustainable sources like biofuels may offer many economic, technological, and environmental advantages due to the significant reduction of particulate matter, soot formation, unburned hydrocarbon, and NOx emissions [3]. On the contrary, their incomplete combustion produces a small amount of harmful chemical components for the environment and human health (e.g., acetic acid, aldehydes, and ketones) [1,2,4]. Recently, the use of oxygenated bio-derived fuels (oxy-biofuels) such as alcohols, esters, acids, aldehydes, and furans have attracted the attention of researchers worldwide [5,6,7,8,9]. This trend is due to their positive answers to the environmental issues and also complying with the strict emission regulations of transportation sectors [2,4]. Indeed, the existence of oxygenated functional groups in the molecular arrangement changes the electronic structure of the fuel, thus limiting the production of aromatic compounds, carbon soot [3,10,11]. Besides, the presence of oxygen reduces the C–H bond strength being bond dissociation energies 80.6 kcal mol−1 and 257.3 kcal mol−1 in the absence and presence of oxygen, respectively [12]. In this framework, the design and optimization of any combustion process based on oxy-biofuels need the definition of a detailed chemical kinetic model. However, many experimental studies are hindered by technical difficulties [13,14,15] related to the different functionality of oxygen-rich biomass, intermediates, and products; to the temperature sensitivity of the products [4,16]; to the short lifetime of intermediate products; and the product dependency on the residence time of volatiles.
This work gathers the available information in the current literature on the progress in experimental and modelling efforts geared towards the combustion of common primary alcohols (methanol, ethanol, and butanol); organic acids (acetic acid and crotonic acid); and other important oxygenated substances like acetaldehyde and furan.
2. Research Metrics of Oxy-Biofuel
The International Energy Agency (IEA) released a report on the current state and trends of renewable energy sources, including the global policies and market distributions. As part of this, an increase in worldwide biofuel production by 9.84 · 106 tons to 1.5 · 108 tons has been reported from 2018 to 2020. Based on the rising energy production, IEA forecasts a consistent increase of up to 25% in the next five years, reaching 1.87 · 108 tons by 2024. Figure 1 shows data collected from the most important scientific and patent database over the past ten years (2009–2020). The following figure contains information on the number of publications devoted to the production or consumption of liquid and solid biofuels (Figure 1a), distinguished in terms of chemical class (Figure 1b), and authors’ affiliations (Figure 1c). Quite clearly, the high number of scientific articles per year is approximately constant in recent years. Similarly, scientific research on oxygenated biofuels (such as alcohols, furans, aldehydes, esters, phenols, and acids) is increasing [5,6,9,17]. Indeed, most of the biofuel research is devoted to the investigation of these chemical classes, as shown in Figure 1b, which reports data referring to 2020. Additional information can be gained by the comparison of the share of published articles for countries, as shown in Figure 1c. It is worth noting that Europe, the United States of America, and China lead the innovation in this field, followed by India and Brazil, confirming the relevance of local policies. In addition to their scientific contributions, the European Union set two new policy directives in 2003 on biofuel [18]. The first policy aimed to make 20% of their automobile fuel; biofuel, hydrogen, natural gas, and other renewable fuels by 2020, as per the agreement by all EU member states in the Renewable energy Directives (RED) 2009/28/EC. The second EU directive was to put tax deductions on biofuels. Besides, under the climate and energy framework of 2030, the EU member countries have agreed to reduce greenhouse gas emissions by 40% by 2030 (compared to 1990), make 27% of their energy from renewable sources, and increase in energy efficiency by at least 27% [18]. To achieve this and realize the EU bio-based economy beyond 2020, the member states urged and prioritized detailed biofuel research, development of safe and environmentally friendly combustors, financial incentives, and upgrading of biorefineries [18]. Given such concerns from the EU Member States, the United States Energy Independence and Security Act (EISA) of 2007 defined the Renewable Fuel Standard program known as RFS2.1 [19]. EISA directed the use of biofuels and established to attain a 50% reduction in greenhouse gas emissions up to 2022. Despite the number of publications, some unsolved problems limit the application of this technological solution yet. In this light, an extensive review focused on the combustion mechanisms of oxygenated fuels could highlight the existing gap of knowledge.
3. Combustion Chemistry of Oxy-Biofuels
Oxygenated species as a potential replacement to conventional fuels must be strictly reviewed from different practical viewpoints. In addition to the sustainability of the source, the compatibility of the fuel within transportation sectors and combustion machinery need to be analyzed [20]. In advance, it is worth knowing the common pyrolysis products and pathways of biomass degradation in general. Besides, the chemical behaviours of flammable mixtures can be estimated by aggregating the kinetic mechanisms of each component, in agreement with the hierarchical approach adopted for mechanism generations [21]. Biomass can be transformed into biofuel by using different processes, as recently reviewed by Cossu et al. [22]. A schematic representation of alternative routes to produce biofuels is given in Figure 2.
Recalling that most of the species included in syngas, alkanes, and alkenes have been widely studied as intermediates of traditional fuels as well, this review will be focused on oxygenated compounds (i.e., alcohols, aldehydes, heterocyclic organic compounds, and acids), which is pertinently a new topic. Due to the oxygenated functional groups, biofuel combustion can result in different reaction sequences and primary reactions from that of conventional fuel chemistry [23]. More specifically, the oxygen atom in the hydrocarbons affects the electronic structure and reactivity of the fuel, because it modifies the bond dissociation energies and increases, hinders, and initiates various reaction pathways as compared to the parent fuel molecule (e.g., alkane) [24]. To make it clear, the bond dissociation energies of methane and methanol at room temperature are presented as 104.5 kcal mol−1 and 96.1 kcal mol−1, respectively [23]. To understand combustion behaviour and to identify their decomposition patterns, it is important to look at typical groups of potential biofuels. The growing interest in biofuels from biomass pyrolysis has motivated systematic investigations of different chemical families such as alcohols [7,25], aldehydes [26,27,28,29], acids [6], and oxygenated aromatics [30,31,32] to elucidate the effects of oxygenated groups on combustion chemistry. In this regard, the accurate evaluation of the fuel decomposition and oxidation reaction mechanisms of alcohols, acids, furans and aldehydes classes of oxygenated fuels is a valuable step toward the awareness of the reaction paths ruling the formation of relevant intermediates [20]. The shortlist of oxygenated species representative for the most relevant functional groups is reported in Figure 1, together with some related properties, which is provided in Table 1. More specifically, the lower heating value (LHV), the heat of vaporization (λ), and the autoignition temperature (AIT) were included as macroscopic properties. Additionally, the most relevant chemical groups involved in the H-abstraction reaction, referred to as abstracting agents from now on, were listed for each species, since the H-abstraction rules the activation step of biofuels in a wide range of conditions [33]. Other than the abstracting agent, the combustion behavior of flammable species can be expressed in terms of the overall reactivity under the given initial conditions. In this sense, an overview of the conditions used so far to collect either experimental (Exp) or numerical (Mod) data for the ignition delay time (IDT) and laminar burning velocity (Su) of these species is provided in Table 2, whereas a detailed analysis on the combustion mechanisms will be provided in the following sections.
3.1. Light Alcohols
The energy production via alcohols is primarily satisfied by using them as alternative fuels or additive in blends [7,25]. However, in the kinetic field, alcohols are commonly adopted as per the definition of a surrogate to mimic the combustion behaviour of more complex mixtures characterized by flexible compositions (e.g., biodiesels) [77]. Among them, the primary alcohols (such as methanol, ethanol, and butanol) are ideal for engine combustion [78]. These fuels have no negative temperature coefficient (NTC) behaviours and are all water-soluble [78]. Additionally, their moderate tendency to form soot and elevated octane rating make the light alcohols (i.e., ≤C5) good aspirants for lean to rich stratified combustion [79] and low-temperature combustion [7,80]. Moreover, in homogeneous charge compression ignition, methanol and ethanol have limited sensitivity to the equivalent ratio but high sensitivity to the temperature, while n-butanol has similar reactivity to equivalent ratios and temperatures like that of gasoline [81,82]. The average bond dissociation energies of alcohol fuels are around 105 kcal mol−1. Due to the good electron losing the ability of the hydroxyl functional group, the bond dissociation energies of the secondary C−H bond in the α-position largely decreases to ∼95 kcal mol−1 and that of β-position to ∼100 kcal mol−1 [7]. In addition, the location of the hydroxyl group (–OH) attached to the carbon atom in alcohol plays a crucial role in the physical-chemical properties. Further, this functional group acts as a radical chain terminating group following H-abstraction, which ends up hindering the cool flame reactivity [83,84]. The presence of the –OH functional group also helps them to suppress the NTC bearing of other fuels [78].
3.1.1. Methanol
The high H/C ratio, the lack of C–C bonds, and the high latent heat of methanol help to reduce the peak temperature and, ultimately, result in low NOx emissions. Besides, the low molecular weight and high oxygen content of methanol result in a high combustion speed and high-octane number, thereby providing an elevated thermal efficiency [47]. Bowman [43] conducted both experimental and numerical studies of methanol–air mixture behind reflected shockwaves over the temperature range of 1545–2180 K and pressures of 1.5–4.2 atm. The times required to obtain the maximum concentrations of CO and O-atom were taken as ignition delay times. Fieweger et al. [85] reported the self-ignition features of various fuel species, including stoichiometric methanol/air mixtures at pressures of 13 and 40 bar and a temperature range of 800–1200 K. The point at which the CH band emission and maximum change in the rate of pressure occurred was defined as the ignition delay time. Moreover, the high-temperature ignition delay time of C1–C4 primary alcohols under pressures of 2, 10, and 12 atm were studied by Noorani et al. [45], and CH emissions were considered as a measure of the ignition delay time. Methanol oxidation under a rapid compression machine (RCM) was hardly reported in the literature, and the most commonly used data was reported by Kumar and Sung [44] and was performed at an equivalence ratio of 0.25–1.00, a pressure of 7–30 bar, and temperature range of 850–1100 K. The maximum rate of the pressure increase was used to define the ignition delay time. Cathonnet et al. [86] performed pyrolysis experiments of methanol using a static reactor at a pressure of 0.3–0.5 atm and a temperature range of 875–975 K. Additionally, methanol oxidation in a stirred reactor at nearly atmospheric pressure and a temperature range of 650–700 K was studied by Aniolek and Wilk [87]. Recently, a comprehensive study on the ignition phenomena of a stoichiometric methanol/oxygen/argon mixture was reported at the pressure range of 12–24 bar and temperature range of 840–1000 K under a rapid compression machine (RCM) by Wang et al. [47]. On the other hand, a flame speciation study was reported by Akrich et al. [34]. From the study, several species such as CH3OH, O2, H2O, H2, CO, and CO2 were measured as a function of the distance from the burner, and H, OH, and HO2 were found to be responsible species for H-abstraction during methanol oxidation. A laminar flame speed study of methanol at atmospheric pressure and a temperature range of 298–368 K using a counter-flow twin flame method was reported [88]. Furthermore, Liao et al. [89] studied the laminar burning velocity of methanol–air mixtures at 358 K using the spherical combustion bomb technique. This study emphasized the decrease in Markstein lengths with an increasing equivalence ratio. Veloo et al. [8] experimentally reported the laminar flame speed of methanol using the counter-flow configuration technique at atmospheric pressure and an unburned mixture temperature of 343 K. From the study, CH2O, HCO, and H were reported to be the dominating radical species. Additionally, the laminar burning velocity of methanol–air mixtures at atmospheric pressure and a temperature ranging from 298 to 358 K was reported using the heat flux method [90].
Having robust experimental data, the need for a detailed kinetic model has been growing, and Mech 15.34 is the first kinetic mechanism model available to predict the experimental data during engine-relevant conditions [91]. Westbrook and Dryer [92] developed the first comprehensive detailed methanol kinetic model that accounted for both high and intermediate temperatures. Besides, many important rate constants for the thermal decomposition of methanol and hydroxymethyl (CH2OH) and the abstract reaction rate constants for H and OH were estimated. However, the lack of elementary rate constants, methoxy radical (CH3O) formation, and reaction path information hindered this work. Later, Norton and Dryer updated the kinetic model using more reliable rate constants and a coherent set of thermochemical parameters [93]. Similarly, a model that was applied to both a premixed laminar flame speed [94,95] and autoignition in a spark-ignition engine [95] was developed, and a good agreement with the experimental data was observed. Besides, HO2 + H→ products and the decomposition of hydroxymethyl (CH2OH) were identified as important steps for the determination of the flame speed. The OH abstraction reaction was reported to be a predominant fuel consumption route in the methanol mechanism. Decades later, Aranda et al. [96] developed a detailed kinetic model for methanol oxidation and validated it with experimental data reported at high pressure (20–100 bar) and intermediate temperatures (600–900 K). As well, the rate constants of important reactions (Equations (1)–(4)) were obtained by ab initio calculations. Similar oxidation pathways with those of high temperature and low-pressure reactions were revealed, while model predictions at a high pressure for onset reactions were particularly sensitive to H-abstraction by a hydroperoxyl radical, as shown below (5).
3.1.2. Ethanol
Due to its high demand in the gasoline engine, ethanol combustion has been widely studied using different experimental systems (e.g., shock tubes [97,98,99], rapid compression machines [50,100,101], plate burner [102,103], counter-flow twin flames [104,105], and constant volume chambers [106,107]. In addition, the oxidation of ethanol under several conditions has been experimentally investigated in flow reactors as well [39]. Barraza-Botet et al. [50] reported that the H-abstraction by HO2 (Equation (6)) significantly affected the overall reactivity of ethanol in ethanol oxidation.
The oxidation of ethanol under shock tubes was investigated by Mathieu et al. [52] by measuring the ignition delay times and water time history profiles of the species at a temperature of 944–1580 K; a pressure range of 1.3–53 atm; and equivalence ratios of 0.5, 1, and 2. From the study, it has been revealed that most of the models used were not accurately reproduced the experimental data at temperature < 1300 K. Similarly, recent studies performed by Laich et al. [51] have shown that the CO time–histories and ignition delay times behind reflected shockwaves under elevated pressures are fairly predicted by the existing mechanisms at elevated temperatures, whereas significant deviations were observed at low temperatures. Bimolecular methyl radical and hydroperoxide radical reaction (Equation (7)) and H-abstraction reaction at the α position of ethanol (Equation (6)) were identified as the reactions significantly affecting the low-temperature chemistry.
CH3 + HO2 → CH3O + OH
Xu et al. [58] conducted an experimental study on premixed laminar combustion with laser-induced spark ignition (LISI) and electric spark ignition (SI) at a low initial temperature and atmospheric pressure. Similar conditions were investigated by using the counterflow flame. Veloo et al. [8]. Katoch et al. [59] performed an experimental study of laminar burning velocities of ethanol–air mixtures at different initial temperatures. Moreover, laminar burning velocities of ethanol–water–air mixtures studied using the heat flux method under adiabatic conditions has been reported [56]. From the study, C2H4, CH3CHO, CH2O, CH4, and CH3, were revealed as major intermediate species. Besides, three H-abstraction sites (at CH3, CH2, or OH) were observed where the abstraction from CH3 led to C2H4 and OH production, from where the CH2 site led to the production of CH3CHO and H, and finally, abstraction from the OH group led to the formation of CH3CH2O. With the extensive experimental data present in the literature, the demand for the relevant detailed kinetic model for the prediction of the combustion parameters is growing and attracting attention. Given this, Dunphy and Simmie [108] developed a kinetic mechanism for ethanol comprising 30 species and 97 reactions. The authors used the detailed mechanism previously reported for methanol, assembled with additional reactions that accounted for ethanol combustion, obtaining satisfactorily predictions for the shock tube experimental data at high temperatures and pressure of 2–3.4 bar. Marinov [109] developed a kinetic model for ethanol oxidation by assembling the sub-mechanisms reported in the literature for methane, hydrogen, ethane, ethylene, and propane oxidation. The model was validated using numerous experimental data of ignition delay times, laminar flame speeds, and species concentrations in the temperature range of 1000–1700 K and the pressure of 1–4.5 atm and was in excellent agreement with the experimental data. Decades before, ethanol kinetic mechanisms were developed basing mostly on a shock tube ignition delay analysis [108,110,111] and validated with limited experimental conditions. Using the mechanism developed by Marinov as a base, Li et al. [112,113] updated the kinetic model. A new mechanism model called AramcoMech1.3 was developed [114] for the combustion of C1–C2 hydrocarbons (methane, ethane, ethylene, acetylene, and acetaldehyde) and oxygenated species such as methanol and ethanol. Mittal et al. [101] validated the model with experimental ignition delay time data and confirmed the higher accuracy of the model than other reported kinetic models. A detailed kinetic mechanism developed by the University of San Diego is comparatively small, yet detailed; however, it has been widely used in research works [115]. Alternatively, reduced mechanisms were produced [56,116] based on the one developed by LLNL, resulting in a significant reduction in computational time with a poor impact on the accuracy of the estimations. Recently, a mechanism on ethanol pyrolysis at high pressures was published by Hashemi et al. [117], whereas a new detailed mechanism was produced by Zyada and Samimi-Abianeh [118] through automated tools, i.e., a reaction mechanism generator (RMG). Several kinetic mechanisms have been reported in the literature [36] at different operating conditions for ethanol. Through the improvement in computational methods, numerically generated mechanisms are becoming more powerful. However, it is still hard to make a comprehensive mechanism suitable for all operating ranges based on the information available in the current literature [109,116]. Roy and Askari [119] developed a new ethanol detailed kinetic mechanism called PCRL-Mech1 (with 67 species and 1016 reactions) at the engine-relevant conditions based on RMG. The important reactions were selected by a sensitivity and path flux analysis, and the rate parameters of these reactions were adjusted during the development of the new mechanism. The model showed an excellent agreement with experimental results of the laminar burning velocity (obtained at temperature (300–600 K), pressure (1–10 atm); equivalence ratio (0.6–1.4); and ignition delay time (at a temperature within the range 820–1450 K, pressures included in 3.3–80 atm, and equivalence ratio ranging from 0.3–2). Besides, H, OH, O2, and HO2 were reported to be the radicals responsible for H-abstraction during ethanol oxidation. The experimental ignition delay time and laminar burning velocity data available for the ethanol/air mixtures reported in the current literature are shown below in Figure 5. As well, a reduced reaction pathway is presented in Figure 6.
3.1.3. Butanol
Due to its high octane number, high energy density, hydrophobicity, and compatibility with existing internal combustion engines, the use of butanol as a fuel source has attracted the attention of many researchers [120]. In light of this, Feng et al. [121] and Gu et al. [122] studied the laminar burning velocity of butanol–air mixtures at various temperatures, pressures, and equivalence ratios. The latter article concluded that functional groups and branching are the main factors affecting the laminar burning velocity of butanol/air mixtures, and the molecular structure has the least effect on flame instability. The laminar flame speed of n-butanol was experimentally measured at atmospheric pressure in the counterflow configuration by Veloo et al. [8] and Veloo and Egolfopoulos [123]. The results showed that the propagation of n-butanol/air flame is slightly faster than that of sec-butanol/air flame and iso-butanol/air flame, and the propagation speed of tert-butanol/air flame is significantly slower than that of the other three isomers. Wu and Law [124] studied the laminar flame speed and flame chemistry of butanol isomers at a pressure range of 1–5 bar.
From the computational study, the designed kinetic model was found to accurately predict the laminar burning velocity of n-butanol and sec-butanol, whereas the mechanism overestimated and underestimated this parameter for iso-butanol and tert-butanol, respectively. On the other hand, Moss et al. [125] studied the ignition delay times for all four isomers of butanol under reflected shockwaves and showed hydrogen abstraction by OH as the most responsible radical for alcohol consumption. The reaction mechanisms of all isomers of butanol were developed and validated with various experimental data: tert-butanol pyrolysis in a shock tube [126], butanol and tert-butanol oxidation products in the flow reactor [127], n-butanol oxidation in the jet stirring reactor [36], and iso-butanol counterflow non-premixed flames [128]. Moreover, Stranic et al. [60] measured the ignition delay times of butanol isomers containing 4% O2 diluted in argon by using shock tube machines under a wide range of reaction conditions. To evaluate the effect of n-butanol/heptane mixtures, Zhang et al. [129] studied the experimental and numerical study of n-butanol/heptane auto-ignition behind reflected shock tubes. Recently, Pelucchi et al. [80] performed an experimental and modelling study on the combustion of C3–C6 linear alcohols at temperature ranges of 550–1100 K; the pressure of 10; 30 bar; and equivalence ratio of 0.5, 1.0, and 2.0. Good agreement between the experimental and modelling results and no NTC behaviour was observed for ethanol and propanol at both pressures and for n-butanol at P = 10 bar. Furthermore, as indicated in the study conducted by Dagaut et al. [36] on the detailed combustion chemistry of n-butanol, it has been observed that, for the comparable experimental conditions, the partition between different reaction channels mostly depends on the equivalence ratio (flame stoichiometry). Based on that, the authors recommended a similarly detailed combustion analysis for the other typical biofuel families and intermediate species. From this, one can understand that to accurately predict the combustion of a particular fuel, providing global parameters such as octane number or ignition delay is not enough, since combustion is a complex process where different free radicals and reactive intermediates play a key role. Besides, it has been reported that the combustion process sensitively depends on the molecular properties of the corresponding fuel [130]. Many signs of progress have also been made in the development of kinetic models describing the chemical kinetics of butanol oxidation. Dagaut et al. [36] studied the chemical kinetic modelling of n-butanol oxidation at a pressure of 10 atm and wide ranges of equivalence ratios in a jet stirred reactor. CO, CO2, H2, H2O, C1–C4 hydrocarbons, and C1–C4 oxygenated compounds were the main decomposition products. The proposed kinetic mechanism indicated H-atom abstraction from α, β, and γ carbon atoms as the dominant decomposition pathway for n-butanol oxidation. Similarly, Sarathy et al. [67] performed the kinetic modelling of n-butanol combustion at atmospheric pressure and equivalence ratios of 0.5, 1, and 2 in a jet stirred reactor. Considering the laminar flame speeds and species concentrations, the authors modelled the oxidation mechanism using an improved detailed chemical kinetic mechanism containing 118 species and 878 reactions. H-atom abstraction and β-scission were shown as the key reaction pathways of the combustion process. Grana et al. [131] conducted kinetic modelling for the combustion of butanol isomers (n-C4H9OH, sec-C4H9OH, iso-C4H9OH, and tert-C4H9OH) using a hierarchal approach and validated it with the burning velocity experimental data. The flame structures and overall combustion characteristics of the four butanol isomers were found to be similar. Moreover, to better understand the combustion chemistry of both linear and branched-chain alcohols, Sarathy et al. [132] performed a comprehensive chemical kinetic model study on the combustion of four butanol isomers. A model accounting for the high- and low-temperature chemistry of linear and branched alcohol has been proposed. The reaction of 1-hydroxybutyl radical with O2 to formaldehyde/ketone and water was reported to be the key step under low pressure. The model implicated H-atom abstraction reactions as a prevalent reaction in the oxidation of butanol for premixed flames under low pressure, while β-scission reactions were revealed to be an important factor for higher temperature reactions (above 800 K). Experimental data available in the current literature on the overall reactivity of n-butanol in the oxidative environment has been reported in terms of IDT and Su under several initial conditions (Figure 7), whereas simplified reaction pathways were produced for all the isomers (Figure 8 and Figure 9).
3.2. Carboxylic Acids
Oxygenated fuels with carboxylic acid functionality, especially acetic acid, are the dominant fractions in the tar released from biomass pyrolysis [133,134,135], and an accurate description of biofuel combustion must take into account the formation of these relevant intermediates. Most importantly, on top of their use as fuel surrogate components [134], oxygenated species can be intended as intermediates formed through the decomposition of hydrocarbon. Hence, they are essential in the hierarchical nature of kinetic models [7].
3.2.1. Acetic Acid
The experimental research of acid combustion poses a huge challenge to the combustion community due to issues related to adsorption [136], corrosion [14], and dimerization [137]. Indeed, only a few experimental studies in the literature are available, as reviewed in recent works [14,15]. Many researchers have measured and reported organic acid emissions from spark-ignition engines [138] and rapid compression engines [139]. The studies indicated that, out of the total hydrocarbon emissions from the combustion engines, organic acid emissions measured in spark-ignition engines are 4–27%, with acetic acid being the most important. Numerical and experimental studies of acetic acid combustion in laminar premixed flames were reported by Leplat and Vandooren [70]. Apart from its combustion chemistry, the study also reported ketene as an intermediate product. Mackie and Doolan [68] studied the thermal decomposition kinetics of acetic acid and its products in a single pulse shock tube within the temperature range of 1300–1950 K. As part of this, decomposition kinetics having 21 species and 46 reactions were modelled and simulated using experimental data. From the decomposition kinetics, decarboxylation and dehydration were confirmed to be the two key decomposition reactions producing methane and carbon dioxide, on the one hand, through (Equation (8)) and ketene and water, on the other, through (Equation (9)), respectively. Ketene further decomposed to a methyl radical and CO2, followed by a further reaction of the methyl radical with CH to form C2H4 and CO. Besides, methyl radicals were revealed to play an important role in determining the main products.
CH3COOH → CH4 + CO2
CH3COOH → CH2CO + H2O
Similarly, Gg. Wagner and Zabel [69] studied the further decomposition kinetics of ketene (CH2CO) behind reflected shocks at low pressure and reported the degradation rate constant-coefficient K = 3.6 × 1015 exp (–248 kJ mol−1 K−1) cm3 mol−1 s−1. In the same way, the gas-phase reactivity analysis of acetic acid, rate constant estimation, and kinetic simulation were studied by Cavallotti et al. [37]. The 1D master equation was also integrated on the potential energy surface (PES) to determine the rate coefficient of acetic acid degradation under a wide range of temperatures (700–2100 K) and pressures (0.1–100 atm). The simulation showed a gradual decrease in the reaction rate at a temperature above 1200 K and a pressure of smaller than 10 atm. Besides, H-abstraction by H, OH, OOH, O2, and CH3 was reported to be the responsible radicals in the decomposition of acetic acid [37]. Lately, Zhang et al. [13] studied the laminar flame propagation and kinetic modelling of acetic acid at a low initial temperature and atmospheric pressure. The authors indicated the pathway related to ketene consumption (Equation (10)) as the main in the propagation of acetic acid flames.
CH2CO + H → CH3 + CO
3.2.2. Crotonic Acid
Crotonic acid is the major intermediate product from bioplastic (e.g., polyhydroxy-butyrate, PHB) degradation, as reported by many researchers demonstrating the conversion of PHB to 3-hydroxybutyric acid (3HBA) and crotonic acid. However, the degradation kinetics and reaction mechanisms of these monomers have been largely overlooked. As a result, reliable combustion parameter data (such as ignition delay time, laminar burning velocity, or species profiles) are lacking. For instance, Li and Strathmann [16] studied the hydrothermal degradation and kinetic mechanism of PHB conversion to 3HBA and crotonic acid and, further, the decomposition of crotonic acid to carbon dioxide and propylene. From the developed kinetic network model, it was found that crotonic acid is mainly generated by hydration to 3HBA, followed by a synergistic dehydration–decarboxylation route to propylene and CO2. Additionally, the rate constants for each reaction were determined [16]. Nevertheless, the ignition delay and laminar flame speed of crotonic acid have not been reported so far.
3.3. Light Aldehydes
Acetaldehyde is a key intermediate in the oxidation of hydrocarbons and alcohols, especially ethanol, which is increasingly being used as a fuel for automobiles. However, it is one of the most abundant toxic oxidative emissions from the combustion of biofuels [140,141], and its atmospheric reaction generates several secondary pollutants [142,143]. Thus, the pyrolysis mechanism study of this intermediate at various reaction conditions can help to understand the overall combustion mechanism of hydrocarbons and alcohol-based fuels [71]. In this regard, several authors reported on the degradation kinetics and combustion chemistry of acetaldehyde. For instance, Sivaramakrishnan et al. [144] conducted a study on the theoretical calculations of acetaldehyde (C2H4O) and ethoxide (C2H5O) potential energy surfaces (PES) and updated the kinetic model of acetaldehyde pyrolysis. The study revealed C–C bond fission with a minor contribution from the roaming mechanism to form CH4 and CO as the main decomposition pathway of acetaldehyde during high-temperature processing. The model developed by the author incorporates a master equation for the analysis of H + CH2CHOH as a primary reaction mechanism for the removal of CH2CHOH. The governing H-abstraction route at the aldehydic site was found to form a carbonyl radical (Rn–CO), which quickly further decomposes to an alkyl radical (Rn) and CO. Based on that, there is a general implication that the low-temperature oxidation of the generic Cn aldehyde degraded to Cn−1 alkyl radicals [26].
To better understand the combustion parameters, the ignition delay times of acetaldehyde behind shock tube waves under ranges of reaction conditions were reported by Mével et al. [72]. additionally, a sensitivity analysis, energy release, and rate of production were conducted, indicating four important elementary reactions (Equations (11)–(14)) taking place during acetaldehyde pyrolysis and oxidation:
CH3CHO → CH3 + HCO
CH3CHO + CH3 → CH3CO + CH4
CH3CHO + H → CH3CO + H2
CH3CHO + CH3 → CH2HCO + CH4
In the end, due to the huge differences observed during the research, the authors recommended the need for new experimental and detailed numerical studies. Tao et al. [145] reported nearly 40 species in laminar and premixed flames of acetaldehyde. Christensen et al. [146] studied the laminar burning velocities at atmospheric pressure and different initial temperatures. Similarly, Christensen and Konnov [147] reported the laminar burning velocity of diacetyl and the updated sub-mechanism model of acetaldehyde and CH3CO in their model. Halstead et al. [148] studied the kinetic development of acetaldehyde in the perspective of the cool flame feature and suggested models containing 14 steps. From the study, acetyl was found to play a significant role in the chain-branching process through CH3CO → CH3CO3 → CH3CO3H → CH3CO2 + OH. The theoretical work reported by Felton et al. [149] and the detailed kinetic model developed by Cavanagh et al. [150] supported the result of Halstead et al. [148].
Nevertheless, Gibson et al. [151] came up with another cool flame phenomena of acetaldehyde to be processed by CH3OOH (CH3 → CH3OO → CH3OOH → CH3O + OH). On the other hand, the study conducted by Kaiser et al. [152] revealed the radical decomposition reaction (Equation (15)) and O2 addition to acetyl (Equation (16)) as the main determining step of the chain-branching process.
CH3CO → CH3 + CO
CH3CO + O2 → CH3CO3
Recently, researchers [27,153] have developed a kinetic model for the low-temperature oxidation of acetaldehyde, as well as C3 and C4 aldehydes. Zhang et al. [40] studied the oxidation of acetaldehyde under a wide range of conditions and revealed CH3OO, CH3OOH, and HOOCOCHO as the main oxidation products. Besides, H-abstracting agents were found to be processed by H, OH, HO2, CH3, O2, CH3COOO, CH3OO, and CH3O. At the lean condition, OH was found to be the most important H-abstracting agent. It was concluded that CH3COOOH and CH3OOH are the main decomposition pathways of acetaldehyde oxidation via the chain-branching reaction, and the reactions related to methyl oxidation were reported to be very sensitive to CH3OO and CH3OOH under the studied conditions [40]. Bentz et al. [154] studied the shock tube thermal decomposition of CH3CHO and CH3CHO + H at a temperature within 1250–1650 K and a pressure range of 1–5 bar. Combining their results and the low-temperature data from other studies, the authors reported the acetaldehyde rate constant expression as K = 6.6 × 10−18 exp (−800 K/T) cm3 s−1 for the temperature range of 300–2000 K. Moreover, Hidaka et al. [155] studied the pyrolysis of acetaldehyde oxidation behind reflected shockwave tubes using single-pulse methods. The study considered different fuel concentrations (2.0% CH3CHO, 4.0% CH3CHO, and 5.0% CH3CHO) diluted with Ar under the temperature range of 1000–1700 K and pressure of 1.2 and 3.0 atm. The (Equations (17)–(19)) reactions were mentioned to be the most important initiation reactions and (Equations (20) and (21)) as the most crucial reactions responsible for acetaldehyde pyrolysis.
CH3CHO → CH3 + CHO
CH3CHO → CH4 + CO
CH3CHO→ CH2CO + H2
CH3CHO + H → CH2CHO + H2
CH3CHO + CH3 → CH2CHO + CH4
Similarly, Ernst et al. [156] conducted acetaldehyde pyrolysis behind reflected shockwaves under a temperature range of 1350–1650 K. The results revealed the decomposition as a first-order reaction with a rate constant expression of K = 1.2 × 1016 exp (−81.74 kcal/RT) s−1. The experimental ignition delay time and laminar burning velocity data of acetaldehyde oxidation reported in the current literature are shown below in Figure 12. Furthermore, Figure 13 reports a simplified schematization of the oxidation pathway of acetaldehyde.
3.4. Heterocyclic Organic Compounds
Due to their high energy density (~30 MJ L−1), better resistance to undesired ignition, high research octane number, better engine efficiency, and lower emissions, furans and their derivatives are a focus of the current research expertise [42]. Furan, a promising biofuel candidate catalytically produced from second-generation biofuels, has attracted the attention of many fuel researchers. Oxygenated fuels like furan were reported to have significantly lower HC, NOx, PM, and CO emissions than their corresponding conventional fuels without compromising their performances [157]. Despite these benefits, some of them have a low energy density, water miscibility, and lower vapour pressure. In addition to their higher octane number and knock-resisting tendency, in recent times, furan-based fuels are progressively emerging because of their high energy density and non-miscibility in water compared to alcohols such as ethanol [74]. The thermal decomposition reaction of furan has been extensively studied because furan and its derivatives play an important role in understanding the combustion of coal and the biomass [158], and also, it is an interesting model compound that helps us to understand the vibrational relaxation and unimolecular decomposition of molecules [159]. The auto-ignition behaviour of furan and other biofuels was studied under a rapid compression machine, and the results revealed 2-methylfuran (2-MF) and ethanol to have an analogous knock inhibition capacity [160]. Wang et al. [161] and Wu et al. [162] compared the combustion and emissions of 2-MF with ethanol, gasoline, and 2-dimethylfuran (2-DMF) in a single-cylinder spray engine and found a higher efficiency, excellent combustion stability, antiknock ability, and lower aldehyde emissions for 2-MF than gasoline. For a better understanding, the laminar flame speed of furan-based fuel combustion has been studied at elevated temperatures and in the presence of an air mixture [163]. The same author reported the flame structure of the DMF/Ar/O2 mixture premixed at low pressure [162], showing that furan and 2-MF are stable intermediates in DMF flames. Tian et al. [76] studied the species distribution measurements for premixed furan/oxygen/argon flames at low pressure. Moreover, the decomposition chemistry of furan has been studied in shock tubes [164] and flow reactors [165]. Similarly, Cullis and Norris [166] investigated the decomposition of furan at 1173–1323 K and atmospheric pressure. Methane (CH4), acetylene (C2H2), ethylene (C2H4), and benzene (C6H6) were found to be the main products observed during the degradation. Besides, the decomposition of furan over the temperature range of 1050–1460 K and a pressure range of 2.6–3.6 atm was studied by Lifshitz et al. [167]. The two main furan decomposition pathways were reported to be CO + pC3H4 and C2H2 + CH2CO. Grela et al. [165] conducted a low-pressure (10−3 Torr) pyrolysis of furan over the temperature range of 1050–1270 K and revealed the occurrence of C3H4 (allene, aC3H4/propyne, and pC3H4) and CO. As well, the author proposed the high-pressure Arrhenius expression for furan pyrolysis to be K∞ = 1015.6 exp (−73.5 kcal mol−1/RT) s−1. In the same way, recently, the degradation of furan over the temperature range of 960–1085 K and pressure of 1 Torr was reported [168]. The study revealed CO and C3H4 as the main products of the degradation and rate coefficient expression of K = 1012.9 exp (−65.7 kcal mol−1/RT) s−1. From the experimental investigation of the premixed furan/oxygen/argon flames, the H-abstractions were confirmed to be mainly by H, OH, and CH3 radicals [76]. The authors also developed a kinetic mechanism that can predict their experimental work and obtained good agreement between the two results. Later on, Wei et al. [74] studied the ignition delay times of furan behind reflected shockwaves over the temperature range of 1320–1880 K and pressure of 1.2–10.4 atm. In addition to these experimental studies, a theoretical study of furan decomposition kinetics was conducted through quantum chemical methods [169,170]. Besides, furan also proved to be an important component of tobacco smoke [171] and was selected as a model to burn fuel, which can reduce NO formations [172]. The study modified the chemical kinetic mechanism developed by Tian et al. [76] to justify their results and showed that the model produced a reasonable agreement with the experiment. The authors concluded that the most important fuel consumption path under these conditions was triggered by unimolecular decomposition. Lately, Liu et al. [9] studied the combustion chemistry and flame structure of furan group biofuels (furan, methyl-furan, and dimethyl-furan) using molecular-beam mass spectrometry and gas chromatography at low pressures (20 and 40 mbar) and two equivalence ratios (1 and 1.7). Coming to the computational part, despite the abundant experimental investigations of furan decomposition, the theoretical calculations concerning these degradation reactions were hardly reported in the literature. For instance, Liu et al. [173] and Tian et al. [76] revealed the thermal degradation of furan with a density functional B3LYP for geometries and QCISD(T) for energies. However, the study did not mention information about the decomposition rate constants of furan. Additionally, Sendt et al. [169] came up with parameters of numerous crucial reactions associated with furan calculated at the CASSCF, CASPT2, and G2-(MP2) levels. The author accordingly presented a kinetic mechanism and validated it with measurements by Organ and Mackie [164]. They concluded that the formation of cyclic intermediates caused by the 1,2-H transfer and the formation of decomposition products (CO + pC3H4) are the main ways of furan pyrolysis reactions. Although there is an abundancy of information on the thermal degradation of this species, very poor understandings have been reported on its combustion, so far. Thus, the detailed combustion chemistry of furan at engine-relevant conditions is lacking. Figure 14 reports the experimental data available in the current literature for the ignition delay time of the furan/air mixtures. The mechanism determining the oxidation of furan is reported, in a simplified version, in Figure 15.
4. Future Challenges
This review showed that more experimental data on the key combustion characteristics are needed to verify the model performance. As an example, methanol may show random preignition during compression–ignition, causing the ignition delay time to be much shorter than expected. The source of this preignition is still unknown. Moreover, the relationship between methyl chemistry and acetaldehyde chemistry due to the weak C–C bond in the acetyl group (CH3CO) that eventually leads to the decomposition of the acetyl group to CH3 and CO is limited in the literature. From the species profile of CH4 and HCO, the amount of CH4 produced in ethanol/air flames is higher than that of the methanol/air flames. This is because the amount of CH3CHO produced in ethanol is higher than that of methanol, while a higher production of CH2O was observed from the methanol/air flames that favoured the formation of HCO under fuel-rich conditions. However, no clear information is available on these species under lean conditions. The kinetic study of furan, which is one of the primary structures of coal, is hardly reported. Since furan and its derivatives are unsaturated species, the accurate prediction of the branching ratios of pyrolysis and combustion products relies on the detailed analysis of the addition reactions of the H and OH radicals. It should be emphasized that almost no information is available in the literature for these types of reactions. Besides, the low-temperature oxidation mechanism of furan and its derivatives is still unknown. Moreover, assessing the possible consequent harmful emissions during the combustion of oxygenated fuels, especially alcohols and furans, needs further research efforts. Not to mention, the laminar burning velocity data of furan combustion is very limited in the literature. Due to technical difficulties, the number of experimental studies characterizing the combustion of acid-based fuels has been considerably limited in the literature, ultimately hindering the numerical study of the species. It has also been observed that additional experimental data on the high-temperature pyrolysis and oxidation of high molecular weight acid-based fuels are needed for the in-depth understanding of the kinetic effects of carboxylic functional groups. More specifically, to the best of our knowledge, no ignition delay time and laminar flame speed data have been reported so far on crotonic acid combustion. Similarly, the ignition delay time data for acetic acid combustion is not reported. In the end, for the accurate prediction of chemically sensitive low-temperature combustion systems, detailed knowledge on the fuel-specific reaction kinetics is very crucial and hardly reported in the literature.
5. Conclusions
The goal of this review was to analyze the recent research trends on the combustion and modelling of common intermediates derived from the decomposition of biofuels. To this aim, several experimental and numerical studies on the ignition behaviours and reactivity of oxygenated species such as alcohols, aldehydes, furans, and acids were reviewed to make them commercially beneficial. The important intermediate radicals and the main reactions affecting the combustion parameters were discussed. As part of this, data on the basic combustion parameters (laminar burning velocity, ignition delay times, and pyrolysis species profiles) under a wide range of reaction conditions were extensively investigated. The detailed modelling efforts were also considered. Generally, the distinctive chemical structures of these fuels concerning their intermediate and product species matter in their kinetic modelling and, thus, in-depth knowledge of how these intermediates are formed/consumed are criteria for a better understanding of bio-derived fuel combustion and emissions.
Author Contributions
F.M.W.: methodology, software, formal analysis, and writing—original draft preparation; G.P.: methodology, formal analysis, data curation, visualization, and writing—review and editing; and E.S.: supervision, conceptualization, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Solomon, S.; Qin, D.; Manning, M.; Chen, Z.; Marquis, M.; Averyt, K.B.; Tignor, M.; Miller, H.L. Climate Change 2007—The Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the IPCC; Cambridge University Press: Cambridge, MA, USA, 2007; Volume 4. [Google Scholar]
- Yasunaga, K.; Mikajiri, T.; Sarathy, S.M.; Koike, T.; Gillespie, F.; Nagy, T.; Simmie, J.M.; Curran, H.J. A shock tube and chemical kinetic modeling study of the pyrolysis and oxidation of butanols. Combust. Flame 2012, 159, 2009–2027. [Google Scholar] [CrossRef]
- Agarwal, A.K. Biofuels (alcohols and biodiesel) applications as fuels for internal combustion engines. Prog. Energy Combust. Sci. 2007, 33, 233–271. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Pelucchi, M.; Bissoli, M.; Rizzo, C.; Zhang, Y.; Somers, K.; Frassoldati, A.; Curran, H.; Faravelli, T. A Kinetic Modelling Study of Alcohols Operating Regimes in a HCCI Engine. SAE Int. J. Engines 2017, 10, 2354–2370. [Google Scholar] [CrossRef] [Green Version]
- Namysl, S.; Pelucchi, M.; Herbinet, O.; Frassoldati, A.; Faravelli, T.; Battin-Leclerc, F. A first evaluation of butanoic and pentanoic acid oxidation kinetics. Chem. Eng. J. 2019, 373, 973–984. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Battin-Leclerc, F.; et al. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part I: Reaction Classes, Rate Rules, Model Lumping, and Validation. Energy Fuels 2020, 34, 14688–14707. [Google Scholar] [CrossRef]
- Veloo, P.S.; Wang, Y.L.; Egolfopoulos, F.; Westbrook, C.K. A comparative experimental and computational study of methanol, ethanol, and n-butanol flames. Combust. Flame 2010, 157, 1989–2004. [Google Scholar] [CrossRef]
- Liu, D.; Togbé, C.; Tran, L.-S.; Felsmann, D.; Oßwald, P.; Nau, P.; Koppmann, J.; Lackner, A.; Glaude, P.-A.; Sirjean, B.; et al. Combustion chemistry and flame structure of furan group biofuels using molecular-beam mass spectrometry and gas chromatography—Part I: Furan. Combust. Flame 2014, 161, 748–765. [Google Scholar] [CrossRef] [Green Version]
- Ubando, A.T.; Africa, A.D.M.; Maniquiz-Redillas, M.C.; Culaba, A.B.; Chen, W.-H. Reduction of particulate matter and volatile organic compounds in biorefineries: A state-of-the-art review. J. Hazard. Mater. 2021, 403, 123955. [Google Scholar] [CrossRef]
- Nugroho, Y.K.; Zhu, L. Platforms planning and process optimization for biofuels supply chain. Renew. Energy 2019, 140, 563–579. [Google Scholar] [CrossRef]
- Huheey, J.; Cottrell, T. The Strengths of Chemical Bonds; Butterworths: London, UK, 1958. [Google Scholar]
- Zhang, X.; Mei, B.; Ma, S.; Zhang, Y.; Cao, C.; Li, W.; Ye, L.; Li, Y. Characterizing the fuel-specific combustion chemistry of acetic acid and propanoic acid: Laminar flame propagation and kinetic modeling studies. Proc. Combust. Inst. 2020, 38, 449–457. [Google Scholar] [CrossRef]
- Christensen, M.; Konnov, A.A. Laminar burning velocity of acetic acid + air flames. Combust. Flame 2016, 170, 12–29. [Google Scholar] [CrossRef]
- Marshall, P.; Glarborg, P. Ab initio and kinetic modeling studies of formic acid oxidation. Proc. Combust. Inst. 2015, 35, 153–160. [Google Scholar] [CrossRef]
- Li, Y.; Strathmann, T.J. Kinetics and mechanism for hydrothermal conversion of polyhydroxybutyrate (PHB) for wastewater valorization. Green Chem. 2019, 21, 5586–5597. [Google Scholar] [CrossRef]
- Fisher, E.; Pitz, W.; Curran, H.; Westbrook, C. Detailed chemical kinetic mechanisms for combustion of oxygenated fuels. Proc. Combust. Inst. 2000, 28, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Dafnomilis, I.; Hoefnagels, R.; Pratama, Y.W.; Schott, D.L.; Lodewijks, G.; Junginger, M. Review of solid and liquid biofuel demand and supply in Northwest Europe towards 2030—A comparison of national and regional projections. Renew. Sustain. Energy Rev. 2017, 78, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.; Saville, B.; MacLean, H. Ethanol Use in the United States: Status, Threats and the Potential Future. In Global Bioethanol; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 34–62. [Google Scholar]
- Kohse-Höinghaus, K.; Osswald, P.; Cool, T.A.; Kasper, T.; Hansen, N.; Qi, F.; Westbrook, C.K.; Westmoreland, P.R. Biofuel Combustion Chemistry: From Ethanol to Biodiesel. Angew. Chem. Int. Ed. 2010, 49, 3572–3597. [Google Scholar] [CrossRef]
- Saggese, C.; Frassoldati, A.; Cuoci, A.; Faravelli, T.; Ranzi, E. A wide range kinetic modeling study of pyrolysis and oxidation of benzene. Combust. Flame 2013, 160, 1168–1190. [Google Scholar] [CrossRef]
- Cossu, R.; Liu, J.; Pivato, A.; Ragazzi, M. Biomass to biofuels: Challenges and opportunities. Renew. Energy 2020, 158, 1–2. [Google Scholar] [CrossRef]
- Ruscic, B. Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [Google Scholar] [CrossRef] [Green Version]
- Pelucchi, M.; Cavallotti, C.; Ranzi, E.; Frassoldati, A.; Faravelli, T. Relative Reactivity of Oxygenated Fuels: Alcohols, Aldehydes, Ketones, and Methyl Esters. Energy Fuels 2016, 30, 8665–8679. [Google Scholar] [CrossRef] [Green Version]
- Nau, P.; Seipel, A.; Lucassen, A.; Brockhinke, A.; Kohse-Höinghaus, K. Intermediate species detection in a morpholine flame: Contributions to fuel-bound nitrogen conversion from a model biofuel. Exp. Fluids 2010, 49, 761–773. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Frassoldati, A.; Herbinet, O.; Battin-Leclerc, F.; Faravelli, T. An experimental and kinetic modelling study of n-C4C6 aldehydes oxidation in a jet-stirred reactor. Proc. Combust. Inst. 2019, 37, 389–397. [Google Scholar] [CrossRef]
- Pelucchi, M.; Ranzi, E.; Frassoldati, A.; Faravelli, T. Alkyl radicals rule the low temperature oxidation of long chain aldehydes. Proc. Combust. Inst. 2017, 36, 393–401. [Google Scholar] [CrossRef]
- Pelucchi, M.; Somers, K.P.; Yasunaga, K.; Burke, U.; Frassoldati, A.; Ranzi, E.; Curran, H.J.; Faravelli, T. An experimental and kinetic modeling study of the pyrolysis and oxidation of n-C3C5 aldehydes in shock tubes. Combust. Flame 2015, 162, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Veloo, P.; Dagaut, P.; Togbe, C.; Dayma, G.; Sarathy, S.; Westbrook, C.; Egolfopoulos, F. Jet-stirred reactor and flame studies of propanal oxidation. Proc. Combust. Inst. 2013, 34, 599–606. [Google Scholar] [CrossRef]
- Pelucchi, M.; Cavallotti, C.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Ranzi, E. Detailed kinetics of substituted phenolic species in pyrolysis bio-oils. React. Chem. Eng. 2019, 4, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Namysl, S.; Pelucchi, M.; Maffei, L.P.; Herbinet, O.; Stagni, A.; Faravelli, T.; Battin-Leclerc, F. Experimental and modeling study of benzaldehyde oxidation. Combust. Flame 2020, 211, 124–132. [Google Scholar] [CrossRef]
- Maffei, L.P.; Pelucchi, M.; Faravelli, T.; Cavallotti, C. Theoretical study of sensitive reactions in phenol decomposition. React. Chem. Eng. 2020, 5, 452–472. [Google Scholar] [CrossRef]
- Coniglio, L.; Bennadji, H.; Glaude, P.-A.; Herbinet, O.; Billaud, F. Combustion chemical kinetics of biodiesel and related compounds (methyl and ethyl esters): Experiments and modeling—Advances and future refinements. Prog. Energy Combust. Sci. 2013, 39, 340–382. [Google Scholar] [CrossRef] [Green Version]
- Akrich, R.; Vovelle, C.; Delbourgo, R. Flame profiles and combustion mechanisms of methanol-air flames under reduced pressure. Combust. Flame 1978, 32, 171–179. [Google Scholar] [CrossRef]
- Chen, C.-C.; Liaw, H.-J.; Shu, C.-M.; Hsieh, Y.-C. Autoignition Temperature Data for Methanol, Ethanol, Propanol, 2-Butanol, 1-Butanol, and 2-Methyl-2,4-pentanediol. J. Chem. Eng. Data 2010, 55, 5059–5064. [Google Scholar] [CrossRef]
- Dagaut, P.; Sarathy, S.M.; Thomson, M. A chemical kinetic study of n-butanol oxidation at elevated pressure in a jet stirred reactor. Proc. Combust. Inst. 2009, 32, 229–237. [Google Scholar] [CrossRef]
- Cavallotti, C.; Pelucchi, M.; Frassoldati, A. Analysis of acetic acid gas phase reactivity: Rate constant estimation and kinetic sim-ulations. Proc. Combust. Inst. 2019, 37, 539–546. [Google Scholar] [CrossRef]
- Wilhoit, R.C.; Chao, J.; Hall, K.R. Thermodynamic Properties of Key Organic Oxygen Compounds in the Carbon Range C1 to C4. Part 1. Properties of Condensed Phases. J. Phys. Chem. Ref. Data 1985, 14, 1–175. [Google Scholar] [CrossRef]
- Li, J.; Kazakov, A.; Chaos, M.; Dryer, F.L. Chemical kinetics of ethanol oxidation. In Proceedings of the 5th US Combustion Meeting, San Diego, CA, USA, 25–28 March 2007. [Google Scholar]
- Zhang, X.; Ye, L.; Li, Y.; Zhang, Y.; Cao, C.; Yang, J.; Zhongyue, Z.; Zhen, H.; Fei, Q. Acetaldehyde oxidation at low and intermediate temperatures: An experimental and kinetic modeling investigation. Combust. Flame 2018, 191, 431–441. [Google Scholar] [CrossRef]
- Christensen, E.; Fioroni, G.M.; Kim, S.; Fouts, L.; Gjersing, E.; Paton, R.; McCormick, R. Experimental and theoretical study of oxidative stability of alkylated furans used as gasoline blend components. Fuel 2018, 212, 576–585. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Akih-Kumgeh, B. Recent Trends in the Production, Combustion and Modeling of Furan-Based Fuels. Energies 2018, 11, 512. [Google Scholar] [CrossRef] [Green Version]
- Bowman, C.T. A shock-tube investigation of the high-temperature oxidation of methanol. Combust. Flame 1975, 25, 343–354. [Google Scholar] [CrossRef]
- Kumar, K.; Sung, C.-J. Autoignition of methanol: Experiments and computations. Int. J. Chem. Kinet. 2011, 43, 175–184. [Google Scholar] [CrossRef]
- Noorani, K.E.; Akih-Kumgeh, B.; Bergthorson, J. Comparative High Temperature Shock Tube Ignition of C1−C4 Primary Alcohols. Energy Fuels 2010, 24, 5834–5843. [Google Scholar] [CrossRef]
- Pinzón, L.; Mathieu, O.; Mulvihill, C.; Schoegl, I.; Petersen, E. Ignition delay time and H2O measurements during methanol oxidation behind reflected shock waves. Combust. Flame 2019, 203, 143–156. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, Y.; Liu, W.; Wang, Z. Investigation of methanol ignition phenomena using a rapid compression machine. Combust. Flame 2020, 211, 147–157. [Google Scholar] [CrossRef]
- Gülder, Ö.L. Laminar burning velocities of methanol, ethanol and isooctane-air mixtures. Symp. Int. Combust. 1982, 19, 275–281. [Google Scholar] [CrossRef]
- Xiao, P.; Lee, C.-F.; Wu, H.; Liu, F. Effects of hydrogen addition on the laminar methanol-air flame under different initial temperatures. Renew. Energy 2020, 154, 209–222. [Google Scholar] [CrossRef]
- Botet, C.B.; Wagnon, S.; Wooldridge, M.S. Combustion Chemistry of Ethanol: Ignition and Speciation Studies in a Rapid Compression Facility. J. Phys. Chem. A 2016, 120, 7408–7418. [Google Scholar] [CrossRef]
- Laich, A.R.; Ninnemann, E.; Neupane, S.; Rahman, R.; Barak, S.; Pitz, W.J.; Vasu, S.S. High-pressure shock tube study of ethanol oxidation: Ignition delay time and CO time-history measurements. Combust. Flame 2020, 212, 486–499. [Google Scholar] [CrossRef]
- Mathieu, O.; Pinzón, L.T.; Atherley, T.M.; Mulvihill, C.R.; Schoel, I.; Petersen, E.L. Experimental study of ethanol oxidation behind reflected shock waves: Ignition delay time and H2O laser-absorption measurements. Combust. Flame 2019, 208, 313–326. [Google Scholar] [CrossRef]
- Wadkar, C.; Chinnathambi, P.; Toulson, E. Analysis of rapid compression machine facility effects on the auto-ignition of ethanol. Fuel 2020, 264, 116546. [Google Scholar] [CrossRef]
- Leplat, N.; Dagaut, P.; Togbé, C.; Vandooren, J. Numerical and experimental study of ethanol combustion and oxidation in laminar premixed flames and in jet-stirred reactor. Combust. Flame 2011, 158, 705–725. [Google Scholar] [CrossRef]
- Konnov, A.; Meuwissen, R.; de Goey, L. The temperature dependence of the laminar burning velocity of ethanol flames. Proc. Combust. Inst. 2011, 33, 1011–1019. [Google Scholar] [CrossRef]
- Van Treek, L.; Lavadera, M.L.; Seidel, L.; Mauss, F.; Konnov, A.A. Experimental and modelling study of laminar burning velocity of aqueous ethanol. Fuel 2019, 257, 116069. [Google Scholar] [CrossRef]
- Xu, C.; Wang, H.; Zhou, K.; Li, X.; Zhou, W.; Liu, W.; Wang, C. Laminar Burning Velocity of Premixed Ethanol–Air Mixtures with Laser-Induced Spark Ignition Using the Constant-Volume Method. Energy Fuels 2019, 33, 7749–7758. [Google Scholar] [CrossRef]
- Xu, C.; Hu, Y.; Li, X.; Zhou, X.; Zhong, A. Comparative experimental study of ethanol-air premixed laminar combustion characteristics by laser induced spark and electric spark ignition. Korean J. Chem. Eng. 2017, 34, 574–579. [Google Scholar] [CrossRef]
- Katoch, A.; Millán-Merino, A.; Kumar, S. Measurement of laminar burning velocity of ethanol-air mixtures at elevated temperatures. Fuel 2018, 231, 37–44. [Google Scholar] [CrossRef]
- Stranic, I.; Chase, D.P.; Harmon, J.T.; Yang, S.; Davidson, D.F.; Hanson, R.K. Shock tube measurements of ignition delay times for the butanol isomers. Combust. Flame 2012, 159, 516–527. [Google Scholar] [CrossRef]
- Hantouche, M.; Sarathy, S.M.; Knio, O.M. Global sensitivity analysis of n-butanol ignition delay times to thermodynamics class and rate rule parameters. Combust. Flame 2020, 222, 355–369. [Google Scholar] [CrossRef]
- Gu, X.; Huang, Z.; Wu, S.; Li, Q. Laminar burning velocities and flame instabilities of butanol isomers–air mixtures. Combust. Flame 2010, 157, 2318–2325. [Google Scholar] [CrossRef]
- Katoch, A.; Alfazazi, A.; Sarathy, S.M.; Kumar, S. Experimental and numerical investigations on the laminar burning velocity of n-butanol + air mixtures at elevated temperatures. Fuel 2019, 249, 36–44. [Google Scholar] [CrossRef]
- Talukder, N.; Lee, K.Y. Laminar flame speeds for n-butanol/air mixtures at elevated pressures and temperatures: An experimental and numerical study. J. Mech. Sci. Technol. 2018, 32, 1827–1834. [Google Scholar] [CrossRef]
- Broustail, G.; Seers, P.; Halter, F.; Moréac, G.; Mounaim-Rousselle, C. Experimental determination of laminar burning velocity for butanol and ethanol iso-octane blends. Fuel 2011, 90, 1–6. [Google Scholar] [CrossRef]
- Broustail, G.; Halter, F.; Seers, P.; Moréac, G.; Mounaïm-Rousselle, C. Experimental determination of laminar burning velocity for butanol/iso-octane and ethanol/iso-octane blends for different initial pressures. Fuel 2013, 106, 310–317. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Thomson, M.; Togbé, C.; Dagaut, P.; Halter, F.; Mounaim-Rousselle, C. An experimental and kinetic modeling study of n-butanol combustion. Combust. Flame 2009, 156, 852–864. [Google Scholar] [CrossRef]
- Mackie, J.; Doolan, K.R. High-temperature kinetics of thermal decomposition of acetic acid and its products. Int. J. Chem. Kinet. 1984, 16, 525–541. [Google Scholar] [CrossRef]
- Wagner, H.G.; Zabel, F. Zum thermischen Zerfall von Keten in der Gasphase. Ber. Bunsenges. Phys. Chem. 1971, 75, 114–118. [Google Scholar] [CrossRef]
- Leplat, N.; Vandooren, J. Numerical and experimental study of the combustion of acetic acid in laminar premixed flames. Combust. Flame 2012, 159, 493–499. [Google Scholar] [CrossRef]
- Yasunaga, K.; Kubo, S.; Hoshikawa, H.; Kamesawa, T.; Hidaka, Y. Shock-tube and modeling study of acetaldehyde pyrolysis and oxidation. Int. J. Chem. Kinet. 2008, 40, 73–102. [Google Scholar] [CrossRef]
- Mével, R.; Chatelain, K.; Catoire, L.; Green, W.H.; Shepherd, J.E. Chemical Kinetics of Acetaldehyde Pyrolysis and Oxidation. In Proceedings of the 9th US National Combustion Meeting, Cincinnati, OH, USA, 17–20 May 2015. [Google Scholar]
- Tao, T.; Kang, S.; Sun, W.; Wang, J.; Liao, H.; Moshammer, K.; Hansen, N.; Law, C.K.; Yang, B. A further experimental and modeling study of acetaldehyde combustion kinetics. Combust. Flame 2018, 196, 337–350. [Google Scholar] [CrossRef]
- Wei, L.; Tang, C.; Man, X.; Jiang, X.; Huang, Z. High-Temperature Ignition Delay Times and Kinetic Study of Furan. Energy Fuels 2012, 26, 2075–2081. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, N.; Yang, M.; Liu, Y.; Tang, C.; Huang, Z. Ignition delay time measurement and kinetic modeling of furan, and comparative studies of 2,3-dihydrofuran and tetrahydrofuran at low to intermediate temperatures by using a rapid compression machine. Combust. Flame 2020, 213, 226–236. [Google Scholar] [CrossRef]
- Tian, Z.; Yuan, T.; Fournet, R.; Glaude, P.-A.; Sirjean, B.; Battin-Leclerc, F.; Zhang, K.; Qi, F. An experimental and kinetic investigation of premixed furan/oxygen/argon flames. Combust. Flame 2011, 158, 756–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.; Liu, H.; Zheng, Z. Chemical Kinetics Study on Combustion of Ethanol/biodiesel/n-heptane. Renew. Energy 2020, 148, 150–167. [Google Scholar] [CrossRef]
- Gainey, B.; Lawler, B. The role of alcohol biofuels in advanced combustion: An analysis. Fuel 2021, 283, 118915. [Google Scholar] [CrossRef]
- Kwon, H.; Lapointe, S.; Zhang, K.; Wagnon, S.W.; Pitz, W.J.; Zhu, J.; McEnally, C.S.; Pfefferle, L.D.; Xuan, Y. Sooting tendencies of 20 bio-derived fuels for advanced spark-ignition engines. Fuel 2020, 276, 118059. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Faravelli, T. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part II: Speciation Measurements in a Jet-Stirred Reactor, Ignition Delay Time Measurements in a Rapid Compression Machine, Model Validation, and Kinetic Analysis. Energy Fuels 2020, 34, 14708–14725. [Google Scholar] [CrossRef]
- Mack, J.H.; Schuler, D.; Butt, R.H.; Dibble, R. Experimental investigation of butanol isomer combustion in Homogeneous Charge Compression Ignition (HCCI) engines. Appl. Energy 2016, 165, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.; Tuner, M.; Verhelst, S. Influence of Injection Strategies on Engine Efficiency for a Methanol PPC Engine. SAE Tech. Pap. Ser. 2019, 2, 653–671. [Google Scholar] [CrossRef]
- Togbé, C.; Halter, F.; Foucher, F.; Mounaim-Rousselle, C.; Dagaut, P. Experimental and detailed kinetic modeling study of 1-pentanol oxidation in a JSR and combustion in a bomb. Proc. Combust. Inst. 2011, 33, 367–374. [Google Scholar] [CrossRef]
- Togbé, C.; Dagaut, P.; Mzé-Ahmed, A.; Diévart, P.; Halter, F.; Foucher, F. Experimental and Detailed Kinetic Modeling Study of 1-Hexanol Oxidation in a Pressurized Jet-Stirred Reactor and a Combustion Bomb. Energy Fuels 2010, 24, 5859–5875. [Google Scholar] [CrossRef]
- Fieweger, K.; Blumenthal, R.; Adomeit, G. Self-ignition of S.I. engine model fuels: A shock tube investigation at high pressure. Combust. Flame 1997, 109, 599–619. [Google Scholar] [CrossRef]
- Cathonnet, M.; Boettner, J.C.; James, H. Étude expérimentale et simulation de la pyrolyse du méthanol. J. Chim. Phys. Phys. Chim. Biol. 1979, 76, 183–189. [Google Scholar] [CrossRef]
- Aniolek, K.W.; Wilk, R.D. Preflame Oxidation Characteristics of Methanol. Energy Fuels 1995, 9, 395–405. [Google Scholar] [CrossRef]
- Egolfopoulos, F.N.; Du, D.; Law, C. A comprehensive study of methanol kinetics in freely-propagating and burner-stabilized flames, flow and static reactors, and shock tubes. Combust. Sci. Technol. 1992, 83, 33–75. [Google Scholar] [CrossRef]
- Liao, S.; Jiang, D.; Huang, Z.; Shen, W.; Yuan, C.; Cheng, Q. Laminar burning velocities for mixtures of methanol and air at elevated temperatures. Energy Convers. Manag. 2007, 48, 857–863. [Google Scholar] [CrossRef]
- Vancoillie, J.; Christensen, M.; Nilsson, E.J.K.; Verhelst, S.; Konnov, A.A. Temperature Dependence of the Laminar Burning Velocity of Methanol Flames. Energy Fuels 2012, 26, 1557–1564. [Google Scholar] [CrossRef] [Green Version]
- Burke, U.; Metcalfe, W.K.; Burke, S.M.; Heufer, K.A.; Dagaut, P.; Curran, H.J. A detailed chemical kinetic modeling, ignition delay time and jet-stirred reactor study of methanol oxidation. Combust. Flame 2016, 165, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, C.K.; Dryer, F.L. Comprehensive Mechanism for Methanol Oxidation. Combust. Sci. Technol. 1979, 20, 125–140. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. Some New Observations on Methanol Oxidation Chemistry. Combust. Sci. Technol. 1989, 63, 107–129. [Google Scholar] [CrossRef]
- Grotheer, H.; Kelm, S. Elementary Reactions in the Methanol Oxidation System. Implications on the Modeling of Laminar Burning Velocities. In Proceedings of the III International Seminar on Flame Structure, Alma-Ata, Kazakhstan, 18–22 September 1989. [Google Scholar]
- Grotheer, H.-H.; Kelm, S.; Driver, H.S.T.; Hutcheon, R.J.; Lockett, R.D.; Robertson, G.N. Elementary Reactions in the Methanol Oxidation System. Part I: Establishment of the Mechanism and Modelling of Laminar Burning Velocities. Ber. Bunsenges. Phys. Chem. 1992, 96, 1360–1376. [Google Scholar] [CrossRef]
- Aranda, V.; Christensen, J.M.; Alzueta, M.U.; Glarborg, P.; Gersen, S.; Gao, Y.; Marshall, P. Experimental and Kinetic Modeling Study of Methanol Ignition and Oxidation at High Pressure. Int. J. Chem. Kinet. 2013, 45, 283–294. [Google Scholar] [CrossRef]
- Cancino, L.R.; Oliveira, A.A.M.; Friki, M.; Schulz, C. Thermal Oxidation of Ethanol: Experimental and Numerical Analysis of Ignition Chemistry of Ethanol-Air Mixtures in Shock Heated Gases. In Proceedings of the Twenty Seventh International Symposium in Shock Waves, St. Petersburg, Russia, 19–24 July 2009. [Google Scholar]
- Cancino, L.; Fikri, M.; Oliveira, A.; Schulz, C. Ignition delay times of ethanol-containing multi-component gasoline surrogates: Shock-tube experiments and detailed modeling. Fuel 2011, 90, 1238–1244. [Google Scholar] [CrossRef]
- Aghsaee, M.; Nativel, D.; Bozkurt, M.; Fikri, M.; Chaumeix, N.; Schulz, C. Experimental study of the kinetics of ethanol pyrolysis and oxidation behind reflected shock waves and in laminar flames. Proc. Combust. Inst. 2015, 35, 393–400. [Google Scholar] [CrossRef]
- Lee, C.; Vranckx, S.; Heufer, K.A.; Khomik, S.V.; Uygun, Y.; Olivier, H.; Fernandez, R.X. On the Chemical Kinetics of Ethanol Oxidation: Shock Tube, Rapid Compression Machine and Detailed Modeling Study. Z. Phys. Chem. 2012, 226, 1–28. [Google Scholar] [CrossRef]
- Mittal, G.; Burke, S.M.; Davies, V.A.; Parajuli, B.; Metcalfe, W.K.; Curran, H.J. Autoignition of ethanol in a rapid compression machine. Combust. Flame 2014, 161, 1164–1171. [Google Scholar] [CrossRef] [Green Version]
- Van Lipzig, J.; Nilsson, E.; de Goey, L.; Konnov, A. Laminar burning velocities of n-heptane, iso-octane, ethanol and their binary and tertiary mixtures. Fuel 2011, 90, 2773–2781. [Google Scholar] [CrossRef]
- Dirrenberger, P.; Glaude, P.; Bounaceur, R.; Le Gall, H.; da Cruz, A.P.; Konnov, A.; Battin-Leclerc, F. Laminar burning velocity of gasolines with addition of ethanol. Fuel 2014, 115, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Vagelopoulos, C.; Egolfopoulos, F.; Law, C. Further considerations on the determination of laminar flame speeds with the counterflow twin-flame technique. Symp. Int. Combust. 1994, 25, 1341–1347. [Google Scholar] [CrossRef]
- Fiala, T.; Sattelmayer, T. Nonpremixed Counterflow Flames: Scaling Rules for Batch Simulations. J. Combust. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, H.; Zheng, Z.; Liu, J.; Reitz, R.D.; Yao, M. Development of a combined reduced primary reference fuel-alcohols (methanol/ethanol/propanols/butanols/n-pentanol) mechanism for engine applications. Energy 2016, 114, 542–558. [Google Scholar] [CrossRef]
- Hinton, N.; Stone, R.; Cracknell, R.; Olm, C. Aqueous ethanol laminar burning velocity measurements using constant volume bomb methods. Fuel 2018, 214, 127–134. [Google Scholar] [CrossRef]
- Dunphy, M.P.; Simmie, J.M. High-temperature oxidation of ethanol. Part 1. Ignition delays in shock waves. J. Chem. Soc. Faraday Trans. 1991, 87, 1691–1696. [Google Scholar] [CrossRef]
- Marinov, N.M. A detailed chemical kinetic model for high temperature ethanol oxidation. Int. J. Chem. Kinet. 1999, 31, 183–220. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. An experimental and modeling study of ethanol oxidation kinetics in an atmospheric pressure flow reactor. Int. J. Chem. Kinet. 1992, 24, 319–344. [Google Scholar] [CrossRef]
- Natarajan, K.; Bhaskaran, K. An Experimental and Analytical Investigation of High Temperature Ignition of Ethanol; Indian Institute of Tech Madras Department of Mechanical Engineering: Tamil Nadu, India, 1981. [Google Scholar]
- Li, J.; Kazakov, A.; Dryer, F.L. Ethanol pyrolysis experiments in a variable pressure flow reactor. Int. J. Chem. Kinet. 2001, 33, 859–867. [Google Scholar] [CrossRef]
- Li, J.; Kazakov, A.; Dryer, F.L. Experimental and Numerical Studies of Ethanol Decomposition Reactions. J. Phys. Chem. A 2004, 108, 7671–7680. [Google Scholar] [CrossRef]
- Metcalfe, W.K.; Burke, S.M.; Ahmed, S.S.; Curran, H.J. A Hierarchical and Comparative Kinetic Modeling Study of C1−C2 Hydrocarbon and Oxygenated Fuels. Int. J. Chem. Kinet. 2013, 45, 638–675. [Google Scholar] [CrossRef]
- Lutz, A.E.; Kee, R.J.; Miller, J.A. SENKIN: A FORTRAN Program for Predicting Homogeneous Gas Phase Chemical Kinetics with Sensitivity Analysis; Sandia National Labs.: Livermore, CA, USA, 1988. [Google Scholar]
- Millán-Merino, A.; Fernández-Tarrazo, E.; Sánchez-Sanz, M.; Williams, F.A. A multipurpose reduced mechanism for ethanol combustion. Combust. Flame 2018, 193, 112–122. [Google Scholar] [CrossRef]
- Hashemi, H.; Christensen, J.M.; Gersen, S.; Levinsky, H.; Klippenstein, S.; Glarborg, P. High-pressure oxidation of methane. Combust. Flame 2016, 172, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Zyada, A.; Samimi-Abianeh, O. Ethanol Kinetic Model Development and Validation at Wide Ranges of Mixture Temperatures, Pressures, and Equivalence Ratios. Energy Fuels 2019, 33, 7791–7804. [Google Scholar] [CrossRef]
- Roy, S.; Askari, O. A New Detailed Ethanol Kinetic Mechanism at Engine-Relevant Conditions. Energy Fuels 2020, 34, 3691–3708. [Google Scholar] [CrossRef]
- Geng, Z.; Xu, L.; Li, H.; Wang, J.; Huang, Z.; Lu, X. Shock tube measurements and modeling study on the ignition delay times of n-butanol/dimethyl ether mixtures. Energy Fuels 2014, 28, 4206–4215. [Google Scholar] [CrossRef]
- Feng, P.; Gu, X.; Huang, Z. Measurement of laminar burning velocities of iso-butanol-air mixtures. Chin. Sci. Bull. 2010, 55, 2046–2056. [Google Scholar] [CrossRef]
- Gu, X.; Li, Q.; Huang, Z.; Zhang, N. Measurement of laminar flame speeds and flame stability analysis of tert-butanol–air mixtures at elevated pressures. Energy Convers. Manag. 2011, 52, 3137–3146. [Google Scholar] [CrossRef]
- Veloo, P.S.; Egolfopoulos, F. Flame propagation of butanol isomers/air mixtures. Proc. Combust. Inst. 2011, 33, 987–993. [Google Scholar] [CrossRef]
- Wu, F.; Law, C.K. Laminar Flame Speed, Markstein Length and Flame Chemistry of the Butanol Isomers From 1 atm to 5 atm; Western States Section/Combustion Institute: Park City, UT, USA, 2013. [Google Scholar]
- Moss, J.T.; Berkowitz, A.M.; Oehlschlaeger, M.A.; Biet, J.; Warth, V.; Glaude, P.-A.; Battin-Leclerc, F. An Experimental and Kinetic Modeling Study of the Oxidation of the Four Isomers of Butanol. J. Phys. Chem. A 2008, 112, 10843–10855. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, T.K.; Lna, M.C.; Lin, C.-Y.; Sanders, W.A. Thermal Decomposition of t-Butyl Alcohol in Shock Waves. Combust. Sci. Technol. 1990, 71, 219–232. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. The flow reactor oxidation of C1−C4 alcohols and MTBE. Symp. Int. Combust. 1991, 23, 179–185. [Google Scholar] [CrossRef]
- Liu, W.; Kelley, A.P.; Law, C.K. Non-premixed ignition, laminar flame propagation, and mechanism reduction of n-butanol, iso-butanol, and methyl butanoate. Proc. Combust. Inst. 2011, 33, 995–1002. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, S.; Zhang, Y.; Tang, C.; Jiang, X.; Hu, E.; Huang, Z. Experimental and modeling study of the auto-ignition of n-heptane/n-butanol mixtures. Combust. Flame 2013, 160, 31–39. [Google Scholar] [CrossRef]
- Kohse-Höinghaus, K. Combustion in the future: The importance of chemistry. Proc. Combust. Inst. 2020, 38, 1–56. [Google Scholar] [CrossRef]
- Grana, R.; Frassoldati, A.; Faravelli, T.; Niemann, U.; Ranzi, E.; Seiser, R.; Cattolica, R.; Seshadri, K. An experimental and kinetic modeling study of combustion of isomers of butanol. Combust. Flame 2010, 157, 2137–2154. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Vranckx, S.; Yasunaga, K.; Mehl, M.; Oßwald, P.; Metcalfe, W.K.; Westbrook, C.K.; Pitz, W.J.; Kohse-Höinghaus, K.; Fernandes, R.X.; et al. A comprehensive chemical kinetic combustion model for the four butanol isomers. Combust. Flame 2012, 159, 2028–2055. [Google Scholar] [CrossRef]
- Oasmaa, A.; Solantausta, Y.; Arpiainen, V.; Kuoppala, E.; Sipilä, K. Fast Pyrolysis Bio-Oils from Wood and Agricultural Residues. Energy Fuels 2010, 24, 1380–1388. [Google Scholar] [CrossRef]
- Bertero, M.; de la Puente, G.; Sedran, U. Fuels from bio-oils: Bio-oil production from different residual sources, characterization and thermal conditioning. Fuel 2012, 95, 263–271. [Google Scholar] [CrossRef]
- Onarheim, K.; Solantausta, Y.; Lehto, J. Process Simulation Development of Fast Pyrolysis of Wood Using Aspen Plus. Energy Fuels 2014, 29, 205–217. [Google Scholar] [CrossRef]
- Doolan, K.R.; Mackie, J.; Reid, C.R. High temperature kinetics of the thermal decomposition of the lower alkanoic acids. Int. J. Chem. Kinet. 1986, 18, 575–596. [Google Scholar] [CrossRef]
- Elwardany, A.; Nasir, E.F.; Es-Sebbar, E.; Farooq, A. Unimolecular decomposition of formic and acetic acids: A shock tube/laser absorption study. Proc. Combust. Inst. 2015, 35, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Zervas, E.; Montagne, X.; Lahaye, J. Influence of Fuel Composition on the Emission of Oxygenated Pollutants (Organic Acids, Alcohols and Carbonyl Compounds) from a SI Engine. Tech. Chron. Sci. J. 2004, 1, 49–58. [Google Scholar]
- Zervas, E.; Montagne, X.; Lahaye, J. Emission of Alcohols and Carbonyl Compounds from a Spark Ignition Engine. Influence of Fuel and Air/Fuel Equivalence Ratio. Environ. Sci. Technol. 2002, 36, 2414–2421. [Google Scholar] [CrossRef]
- Fontaras, G.; Karavalakis, G.; Kousoulidou, M.; Ntziachristos, L.; Bakeas, E.; Stournas, S.; Samaras, Z. Effects of low concentration biodiesel blends application on modern passenger cars. Part 2: Impact on carbonyl compound emissions. Environ. Pollut. 2010, 158, 2496–2503. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Oßwald, P.; Hansen, N.; Kohse-Höinghaus, K. Alcohol combustion chemistry. Prog. Energy Combust. Sci. 2014, 44, 40–102. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Effects of Ethanol (E85) versus Gasoline Vehicles on Cancer and Mortality in the United States. Environ. Sci. Technol. 2007, 41, 4150–4157. [Google Scholar] [CrossRef]
- Gaffney, J.S.; Marley, N.A. The impacts of combustion emissions on air quality and climate—From coal to biofuels and beyond. Atmos. Environ. 2009, 43, 23–36. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Michael, J.V.; Harding, L.B.; Klippenstein, S. Resolving Some Paradoxes in the Thermal Decomposition Mechanism of Acetaldehyde. J. Phys. Chem. A 2015, 119, 7724–7733. [Google Scholar] [CrossRef]
- Tao, T.; Sun, W.; Yang, B.; Hansen, N.; Moshammer, K.; Law, C.K. Investigation of the chemical structures of laminar premixed flames fueled by acetaldehyde. Proc. Combust. Inst. 2017, 36, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.; Abebe, M.T.; Nilsson, E.J.; Konnov, A.A. Kinetics of premixed acetaldehyde + air flames. Proc. Combust. Inst. 2015, 35, 499–506. [Google Scholar] [CrossRef]
- Christensen, M.; Konnov, A.A. Laminar burning velocity of diacetyl + air flames. Further assessment of combustion chemistry of ketene. Combust. Flame 2017, 178, 97–110. [Google Scholar] [CrossRef]
- Halstead, M.P.; Prothero, A.; Quinn, C.P. A mathematical model of the cool-flame oxidation of acetaldehyde. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1971, 322, 377–403. [Google Scholar] [CrossRef]
- Felton, P.; Gray, B.; Shank, N. Low temperature oxidation in a stirred flow reactor—II. Acetaldehyde (theory). Combust. Flame 1976, 27, 363–376. [Google Scholar] [CrossRef]
- Cavanagh, J.; Cox, R.; Olson, G. Computer modeling of cool flames and ignition of acetaldehyde. Combust. Flame 1990, 82, 15–39. [Google Scholar] [CrossRef]
- Gibson, C.; Gray, P.; Griffiths, J.; Hasko, S. Spontaneous ignition of hydrocarbon and related fuels: A fundamental study of thermokinetic interactions. Symp. Int. Combust. 1985, 20, 101–109. [Google Scholar] [CrossRef]
- Kaiser, E.W.; Westbrook, C.K.; Pitz, W.J. Acetaldehyde oxidation in the negative temperature coefficient regime: Experimental and modeling results. Int. J. Chem. Kinet. 1986, 18, 655–688. [Google Scholar] [CrossRef]
- Pelucchi, M. Kinetic modeling of the low temperature cool flames of acetaldehyde in a well stirred reactor. In Proceedings of the Meeting of the Italian Section of the Combustion Institute, Lecce, Italy, 20–23 September 2015. [Google Scholar]
- Bentz, T.; Striebel, F.; Olzmann, M. Shock-Tube Study of the Thermal Decomposition of CH 3CHO and CH 3CHO + H Reaction. J. Phys. Chem. A 2008, 112, 6120–6124. [Google Scholar] [CrossRef]
- Hidaka, Y.; Kubo, S.; Hoshikawa, T.; Wakamatsu, H. Shock-tube study of acetaldehyde pyrolysis. In Shock Waves; Springer: Berlin/Heidelberg, Germany, 2005; pp. 603–608. [Google Scholar]
- Ernst, J.; Spindler, K.; Wagner, H.G. Untersuchungen zum thermischen Zerfall von Acetaldehyd und Aceton. Ber. Bunsenges. Phys. Chem. 1976, 80, 645–650. [Google Scholar] [CrossRef]
- Wei, L.; Tang, C.; Man, X.; Huang, Z. Shock-Tube Experiments and Kinetic Modeling of 2-Methylfuran Ignition at Elevated Pressure. Energy Fuels 2013, 27, 7809–7816. [Google Scholar] [CrossRef]
- Graedel, T.E.; Hawkins, D.T.; Claxton, L.D. Atmospheric Chemical Compounds: Sources, Occurrence and Bioassay; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Fulle, D.; Dib, A.A.; Kiefer, J.H.; Zhang, Q.; Yao, A.J.; Kern, R.D. Pyrolysis of Furan at Low Pressures: Vibrational Relaxation, Unimolecular Dissociation, and Incubation Times. J. Phys. Chem. A 1998, 102, 7480–7486. [Google Scholar] [CrossRef]
- Ohtomo, M.; Nishikawa, K.; Suzuoki, T.; Miyagawa, H.; Koike, M. Auto-ignition Characteristics of Biofuel Blends for SI Engines. SAE Tech. Pap. Ser. 2011. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Daniel, R.; Ghafourian, A.; Herreros, J.; Shuai, S.; Ma, X. Combustion characteristics and emissions of 2-methylfuran compared to 2,5-dimethylfuran, gasoline and ethanol in a DISI engine. Fuel 2013, 103, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Huang, Z.; Yuan, T.; Zhang, K.; Wei, L. Identification of combustion intermediates in a low-pressure premixed laminar 2,5-dimethylfuran/oxygen/argon flame with tunable synchrotron photoionization. Combust. Flame 2009, 156, 1365–1376. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Z.; Wang, X.; Jin, C.; Tang, C.; Wei, L.; Law, C.K. Laminar burning velocities and flame instabilities of 2,5-dimethylfuran–air mixtures at elevated pressures. Combust. Flame 2011, 158, 539–546. [Google Scholar] [CrossRef]
- Organ, P.P.; Mackie, J. Kinetics of pyrolysis of furan. J. Chem. Soc. Faraday Trans. 1991, 87, 815–823. [Google Scholar] [CrossRef]
- Grela, M.A.; Amorebieta, V.T.; Colussi, A.J. Very low pressure pyrolysis of furan, 2-methylfuran and 2,5-dimethylfuran. The stability of the furan ring. J. Phys. Chem. 1985, 89, 38–41. [Google Scholar] [CrossRef]
- Cullis, C.; Norris, A. The pyrolysis of organic compounds under conditions of carbon formation. Carbon 1972, 10, 525–537. [Google Scholar] [CrossRef]
- Lifshitz, A.; Bidani, M.; Bidani, S. Thermal reactions of cyclic ethers at high temperatures. III. Pyrolysis of furan behind reflected shocks. J. Phys. Chem. 1986, 90, 5373–5377. [Google Scholar] [CrossRef]
- Bruinsma, O.S.; Tromp, P.J.; Nolting, H.J.D.S.; Moulijn, J.A. Gas phase pyrolysis of coal-related aromatic compounds in a coiled tube flow reactor. Fuel 1988, 67, 334–340. [Google Scholar] [CrossRef]
- Sendt, K.; Bacskay, G.B.; Mackie, J. Pyrolysis of Furan: Ab Initio Quantum Chemical and Kinetic Modeling Studies. J. Phys. Chem. A 2000, 104, 1861–1875. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Zuo, T. The pyrolysis mechanism of furan revisited. Chem. Phys. Lett. 2000, 325, 457–464. [Google Scholar] [CrossRef]
- Holzer, G.; Oró, J.; Bertsch, W. Gas chromatographic-mass spectrometric evaluation of exhaled tobacco smoke. J. Chromatogr. A 1976, 126, 771–785. [Google Scholar] [CrossRef]
- Guarneri, F.; Ikeda, E.; Mackie, J. A Study of Furan as a Model Oxygenated Reburn Fuel for Nitric Oxide Reduction. Energy Fuels 2001, 15, 743–750. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Zhai, L. Theoretical investigation of unimolecular decomposition channels of furan4. J. Comput. Chem. 1998, 19, 240–249. [Google Scholar] [CrossRef]
Figure 1.
Global research trends of oxygenated biofuels for the last ten years (a), for the chemical classes (b), and countries (c).
Figure 1.
Global research trends of oxygenated biofuels for the last ten years (a), for the chemical classes (b), and countries (c).

Figure 2.
Simplified pathway representative of alternatives for biomass transformation toward biofuels, adapted from Cossu et al. [22].
Figure 2.
Simplified pathway representative of alternatives for biomass transformation toward biofuels, adapted from Cossu et al. [22].

Figure 3.
Ignition delay time (a) and laminar burning velocity (b) of the methanol/air mixture under different conditions.
Figure 3.
Ignition delay time (a) and laminar burning velocity (b) of the methanol/air mixture under different conditions.

Figure 4.
A simplified reaction pathway for methanol oxidation.

Figure 5.
Ignition delay time (a) and laminar burning velocity (b) of the ethanol/air mixture under different conditions.
Figure 5.
Ignition delay time (a) and laminar burning velocity (b) of the ethanol/air mixture under different conditions.

Figure 6.
A simplified reaction pathway for ethanol oxidation.

Figure 7.
Ignition delay time (a) and laminar burning velocity (b) of the n-butanol/air mixture under different conditions.
Figure 7.
Ignition delay time (a) and laminar burning velocity (b) of the n-butanol/air mixture under different conditions.

Figure 8.
A simplified reaction pathway for 1-butanol (a) and 2-butanol (b) oxidation.

Figure 9.
A simplified reaction pathway for iso-butanol (a) and tert-butanol (b) oxidation.

Figure 10.
Laminar burning velocity data for the acetic acid/air mixture at atmospheric pressure [14].
Figure 10.
Laminar burning velocity data for the acetic acid/air mixture at atmospheric pressure [14].

Figure 11.
A simplified reaction pathway for acetic acid oxidation.

Figure 12.
Ignition delay time (a) and laminar burning velocity (b) of the acetaldehyde/air mixture under different conditions. Note that laminar burning velocity measurements refer to the data reported by Christensen and Konnov [14] exclusively.
Figure 12.
Ignition delay time (a) and laminar burning velocity (b) of the acetaldehyde/air mixture under different conditions. Note that laminar burning velocity measurements refer to the data reported by Christensen and Konnov [14] exclusively.

Figure 13.
A simplified reaction pathway for acetaldehyde oxidation.

Figure 14.
Ignition delay time data for the furan/air mixture under different conditions.

Figure 15.
A simplified reaction pathway for furan oxidation.


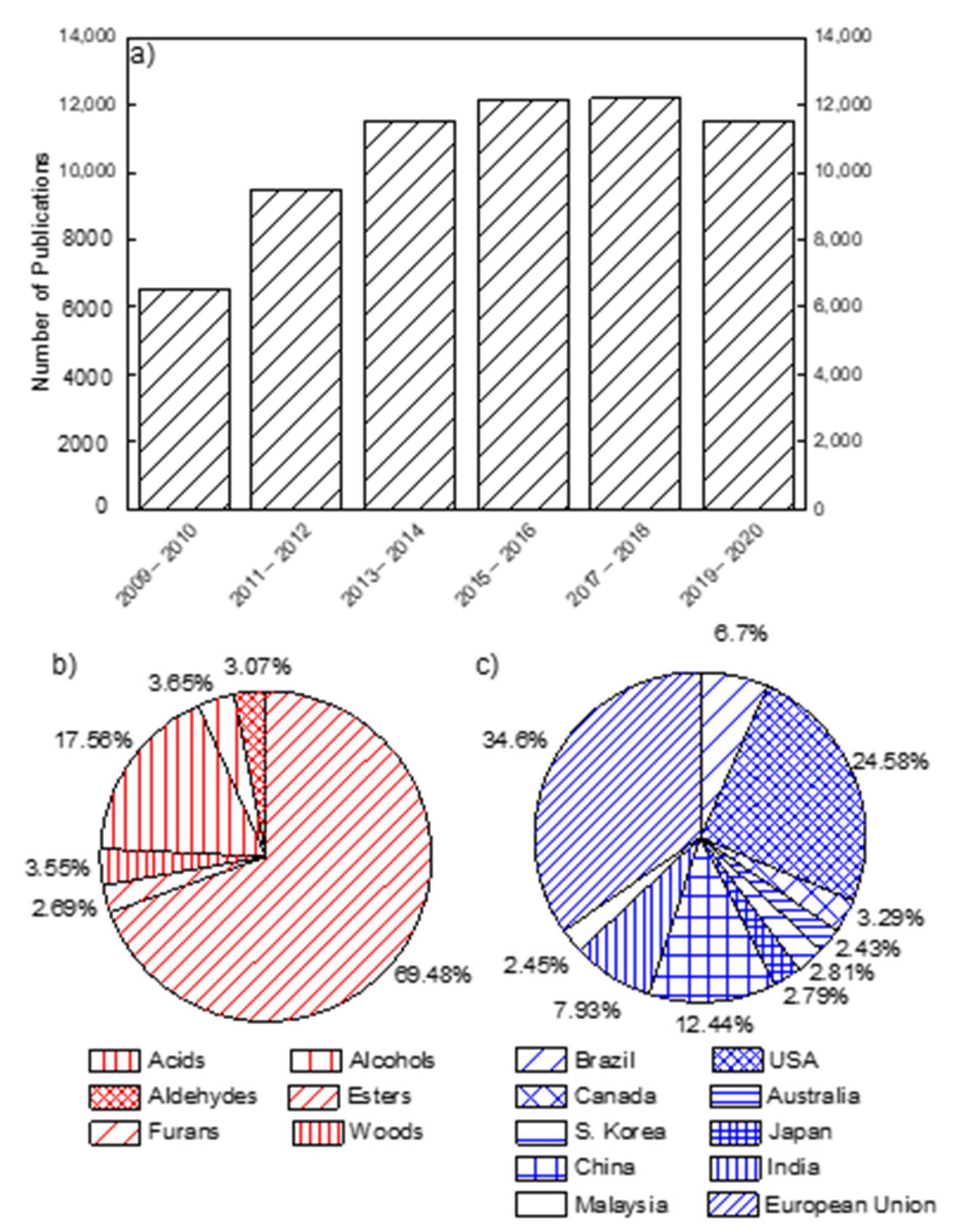{kind=link}
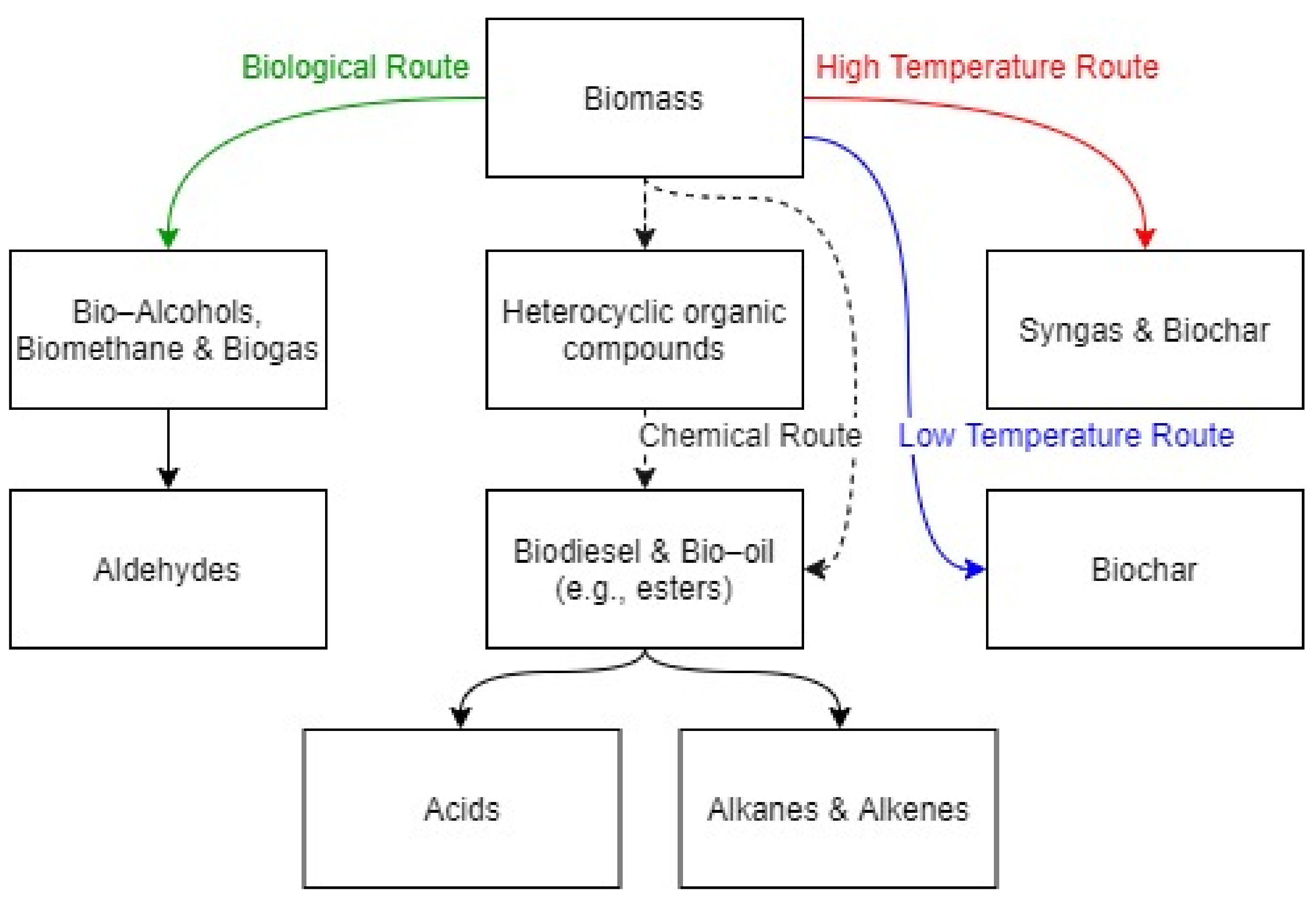{kind=link}
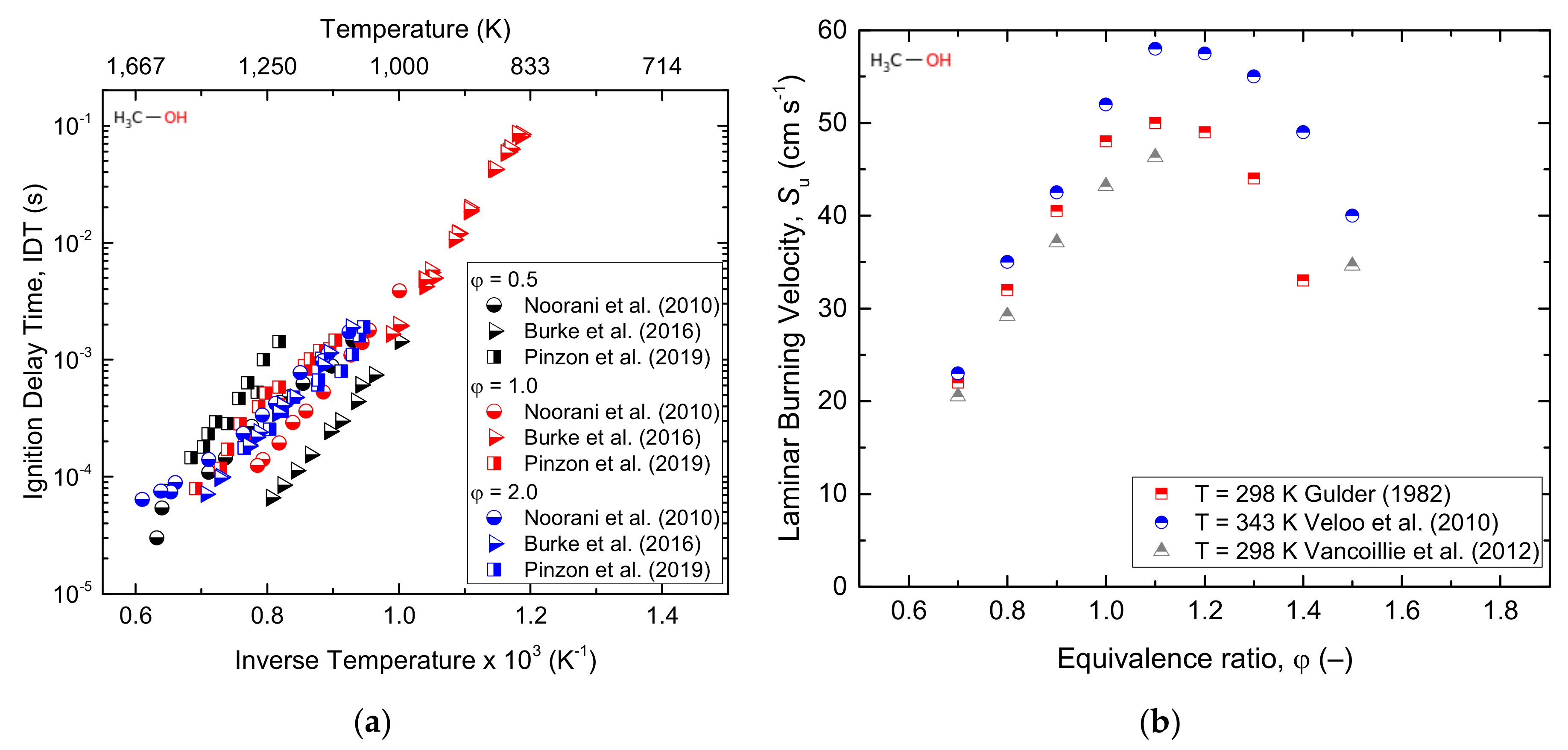{kind=link}
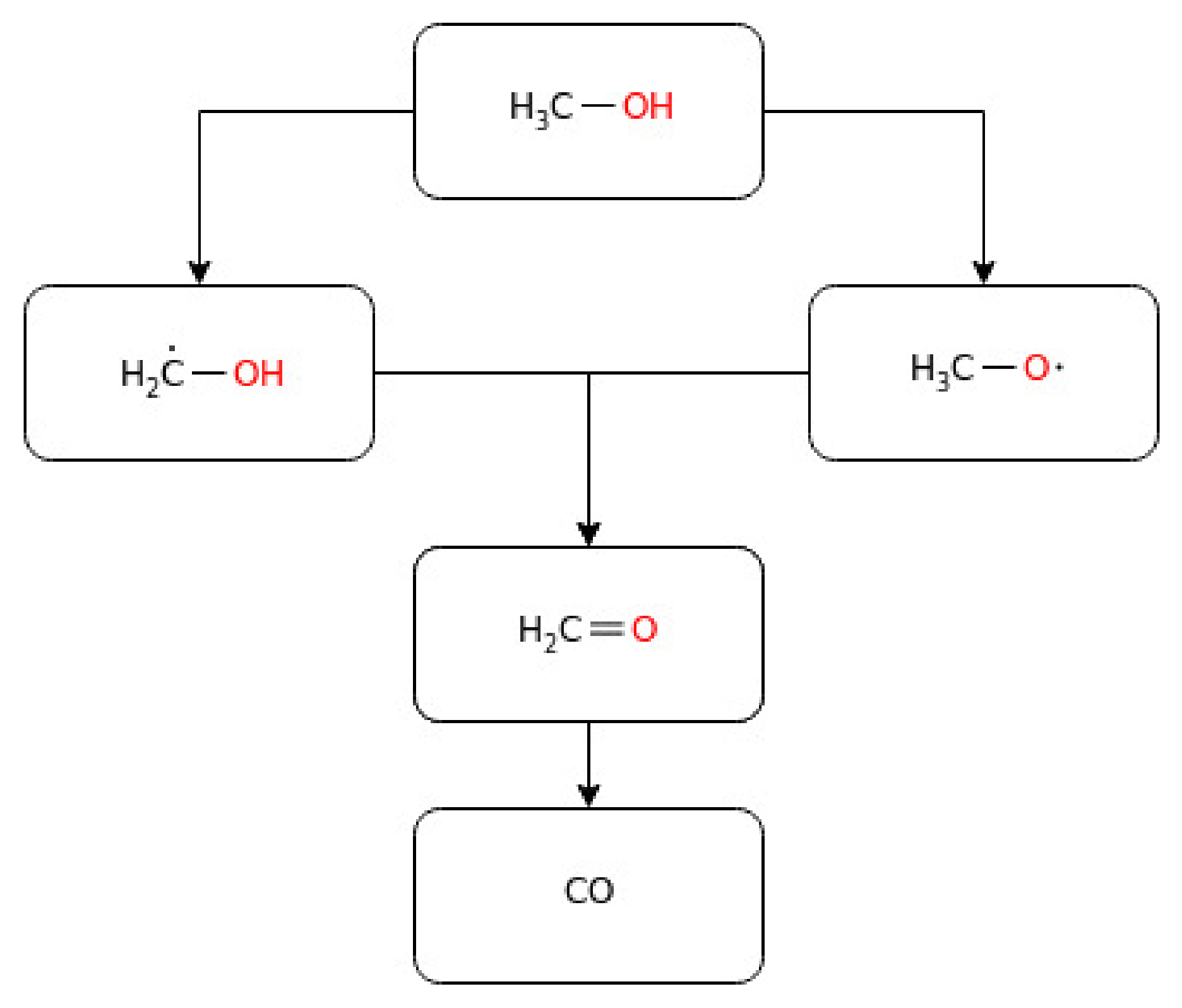{kind=link}
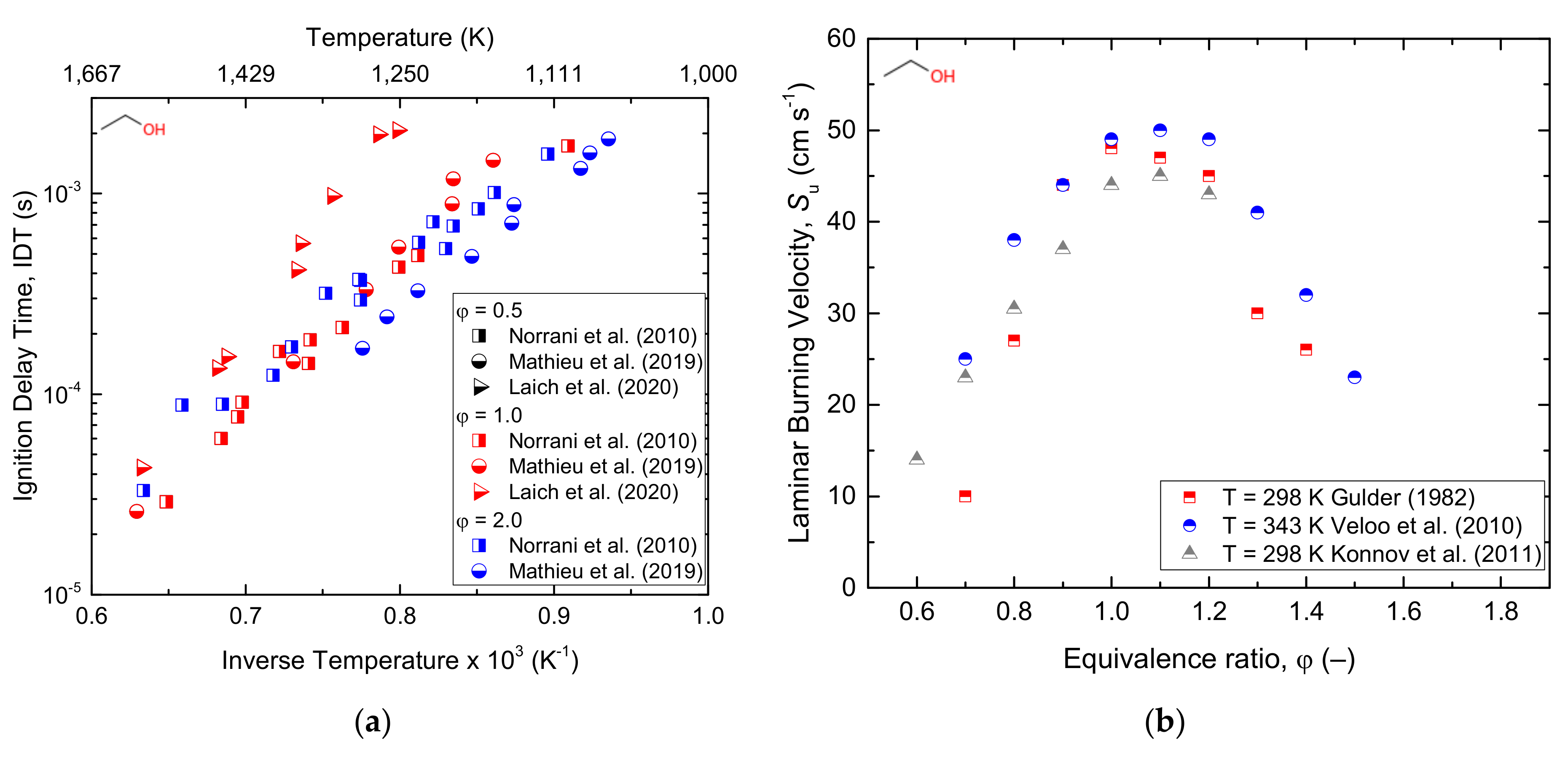{kind=link}
{kind=link}
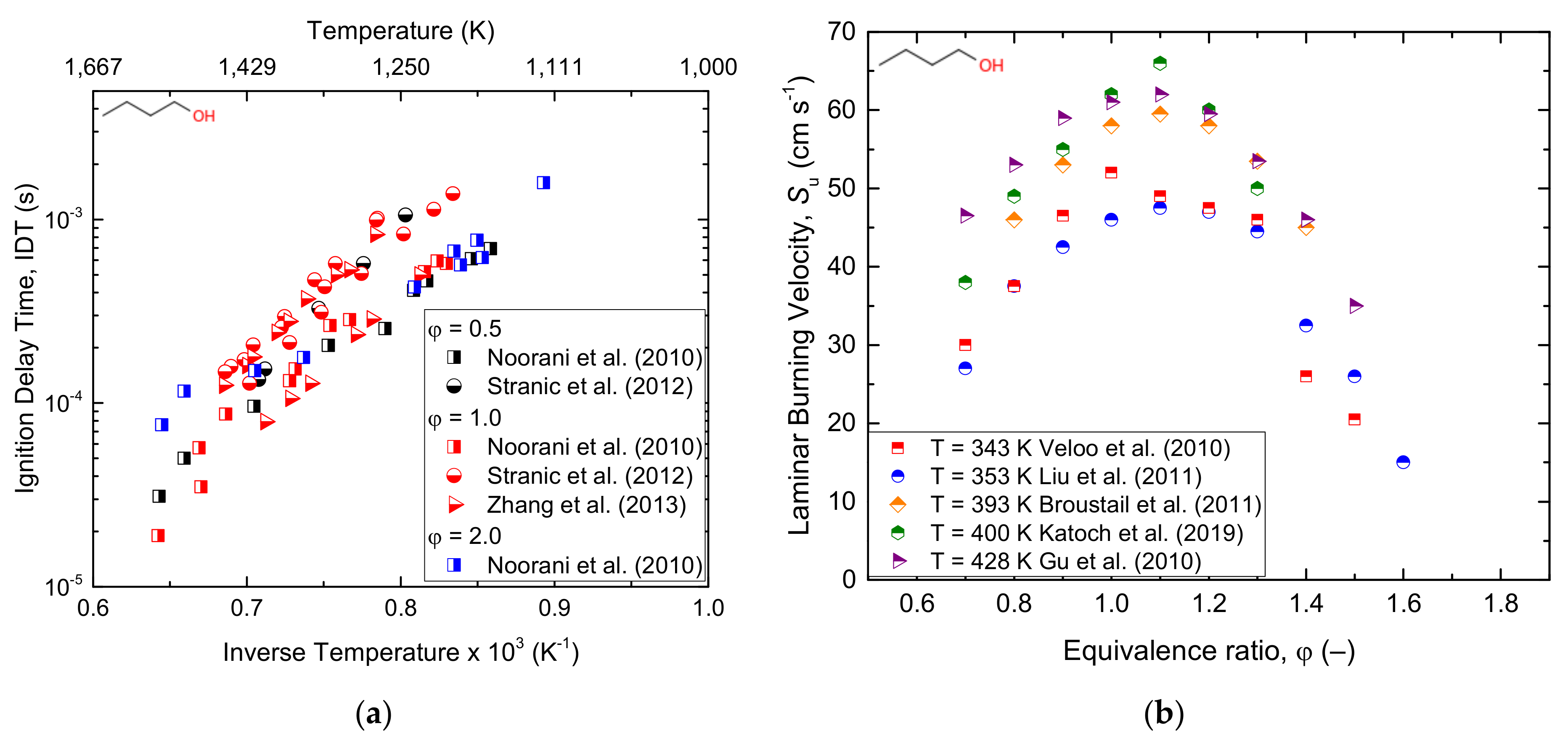{kind=link}
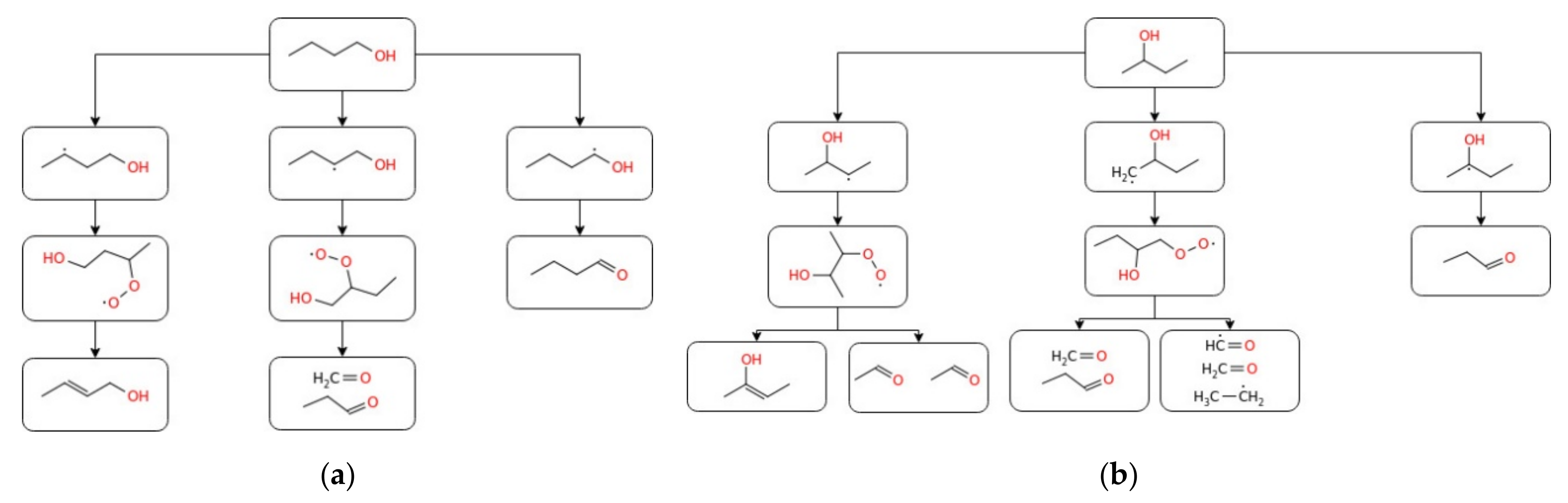{kind=link}
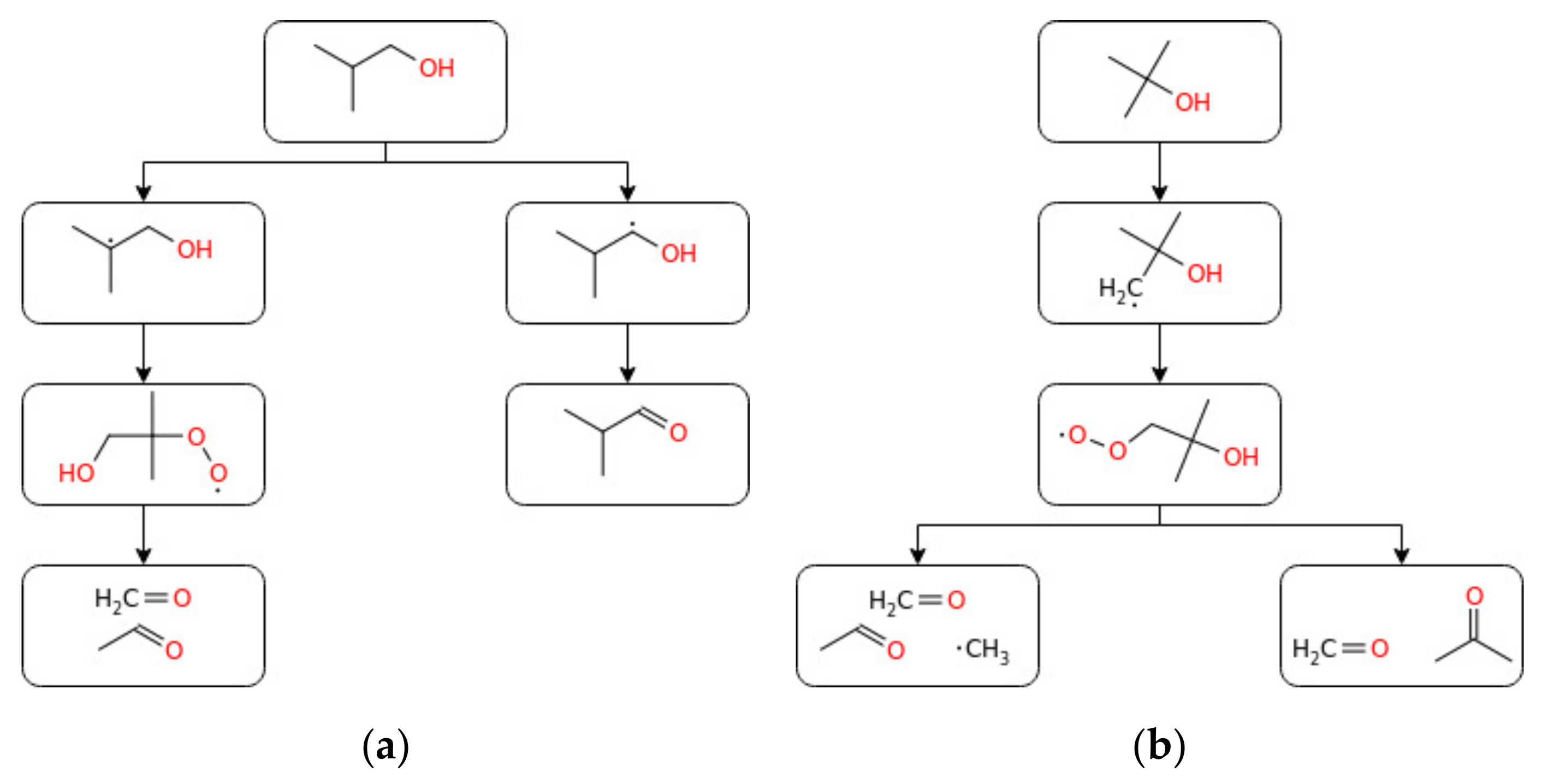{kind=link}
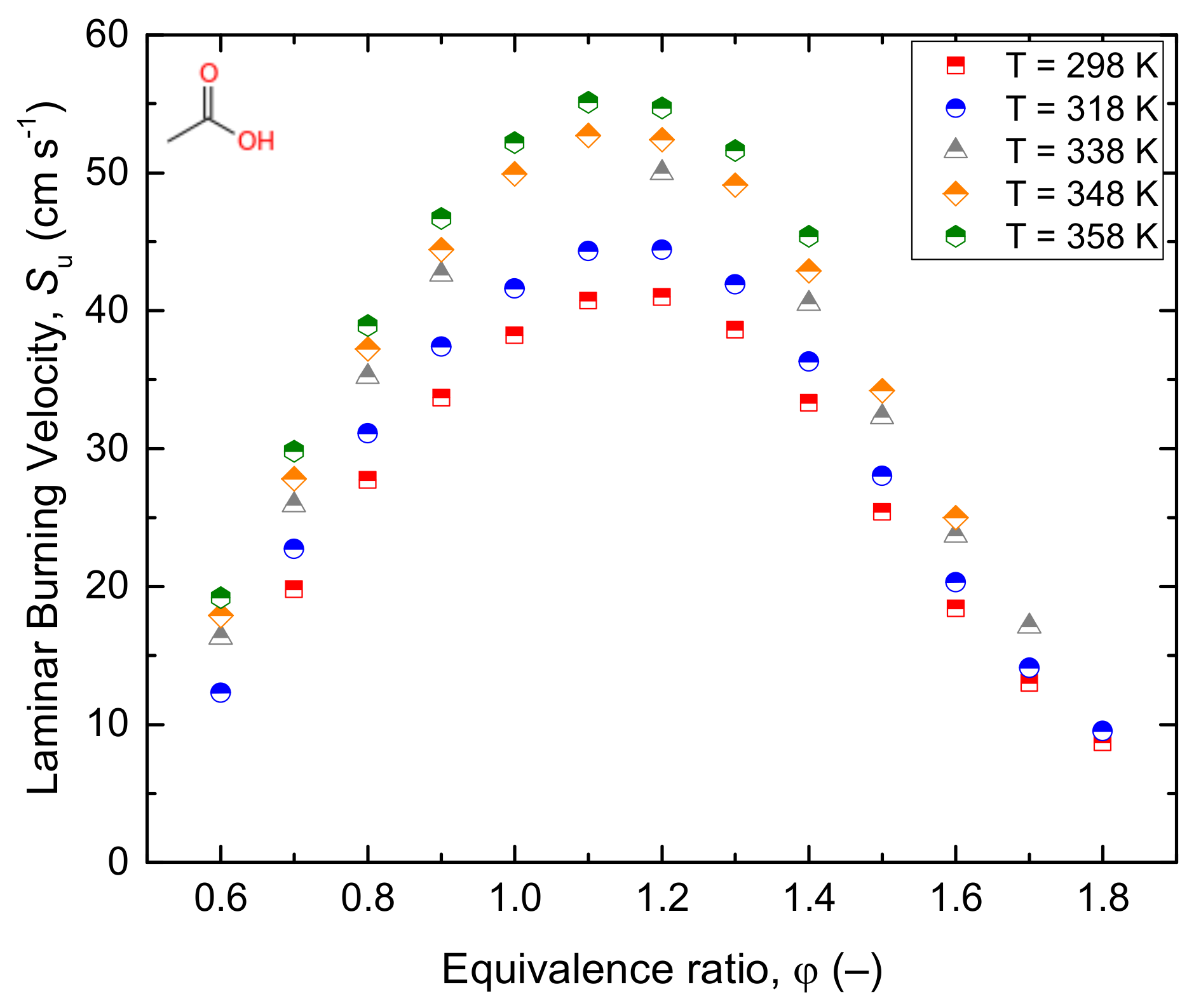{kind=link}
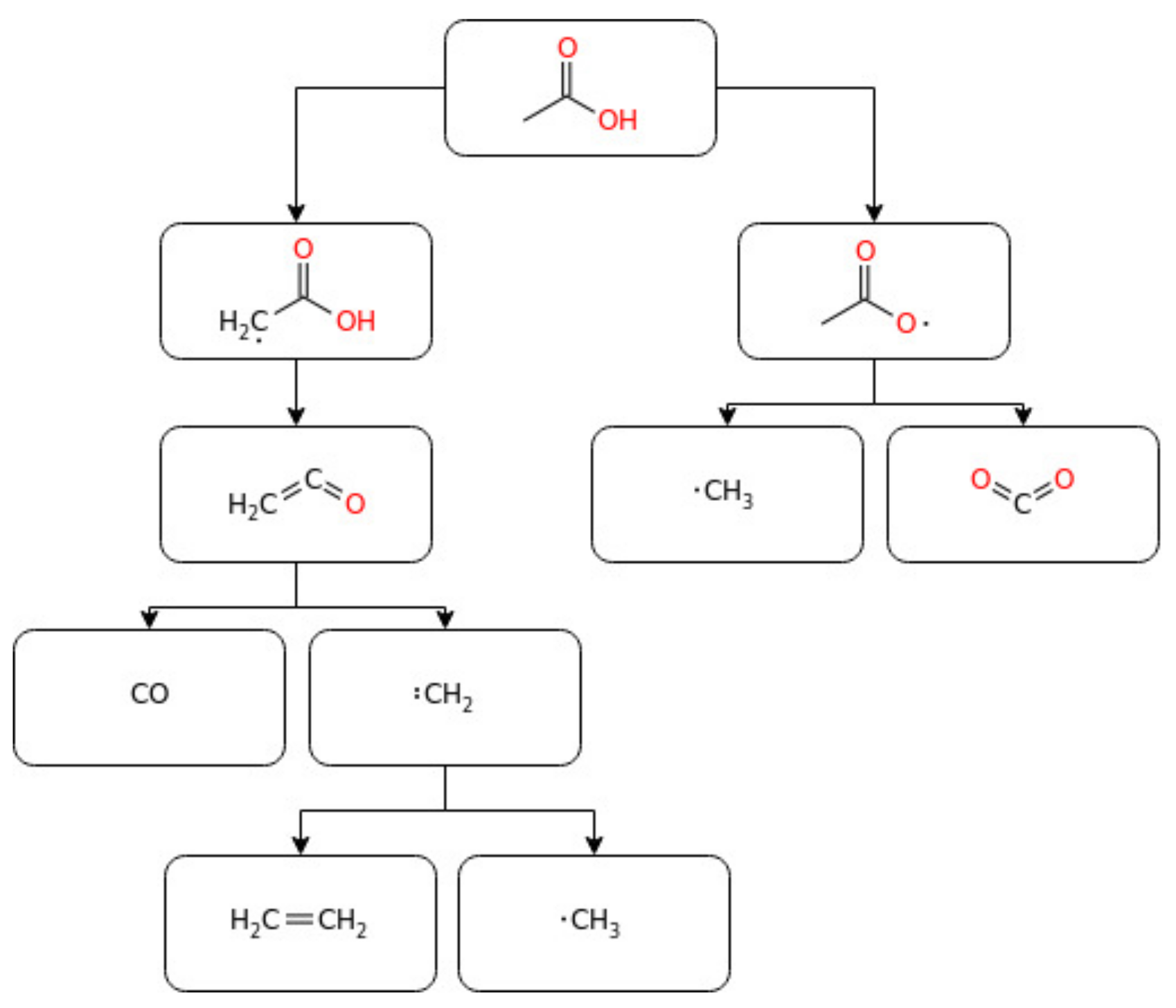{kind=link}
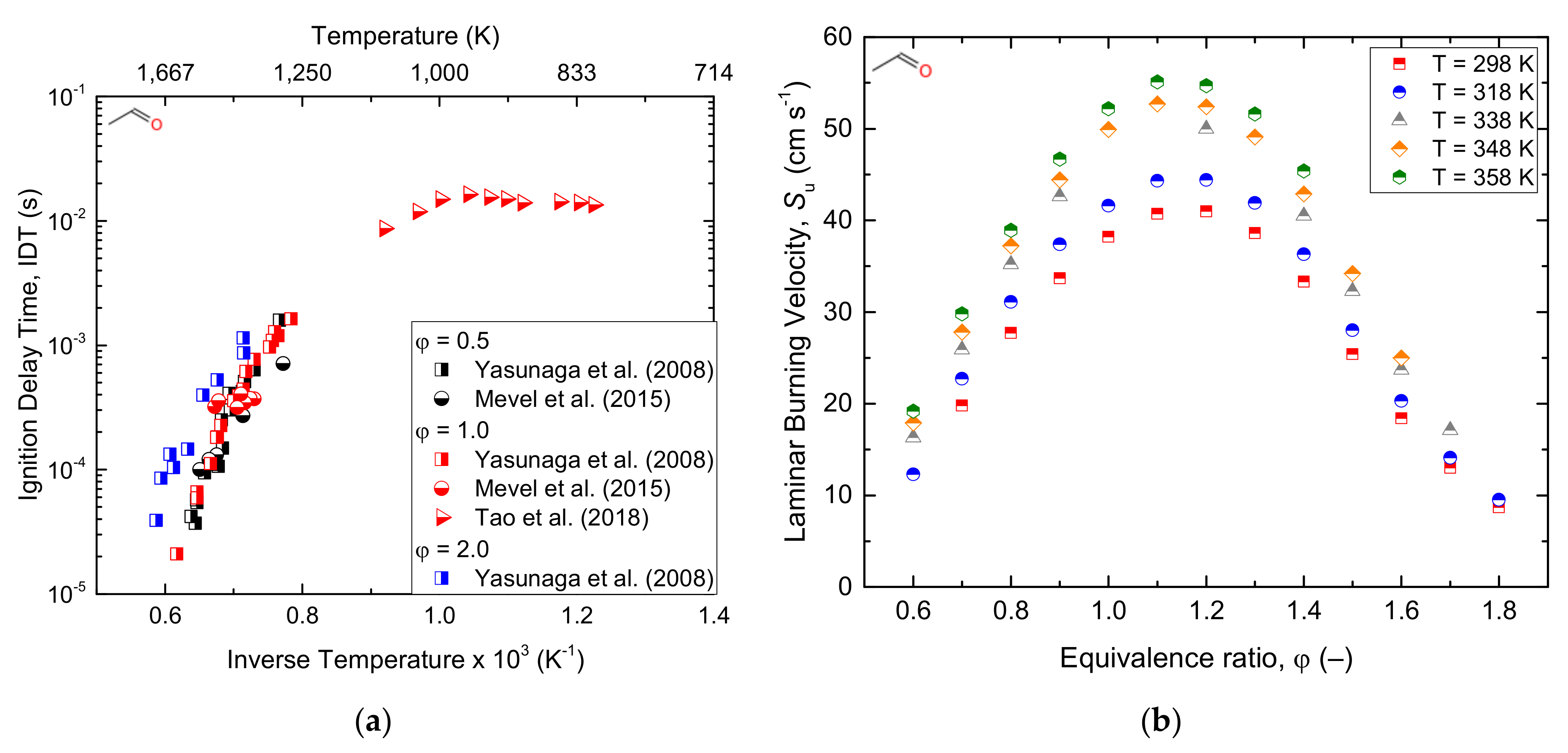{kind=link}
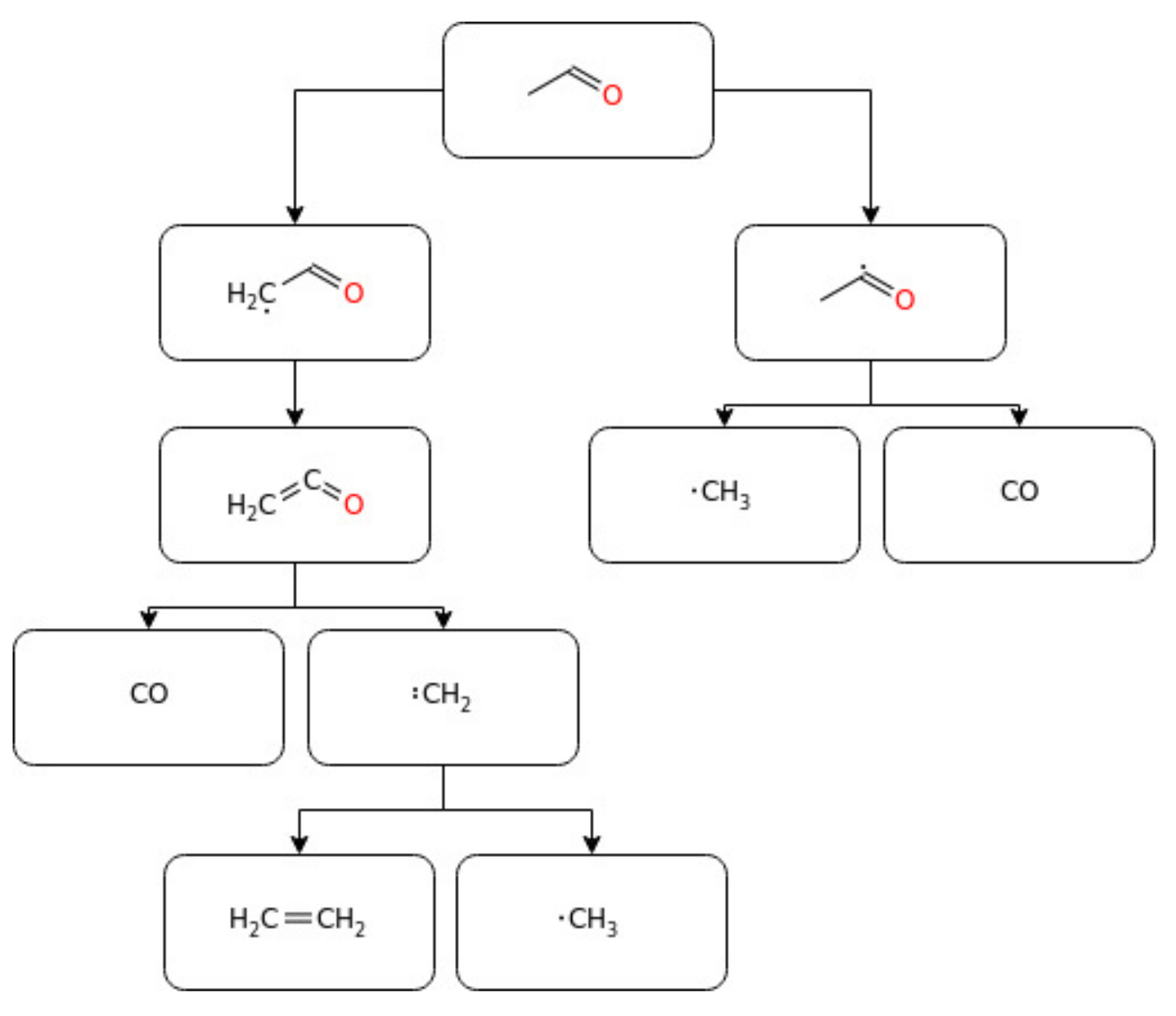{kind=link}
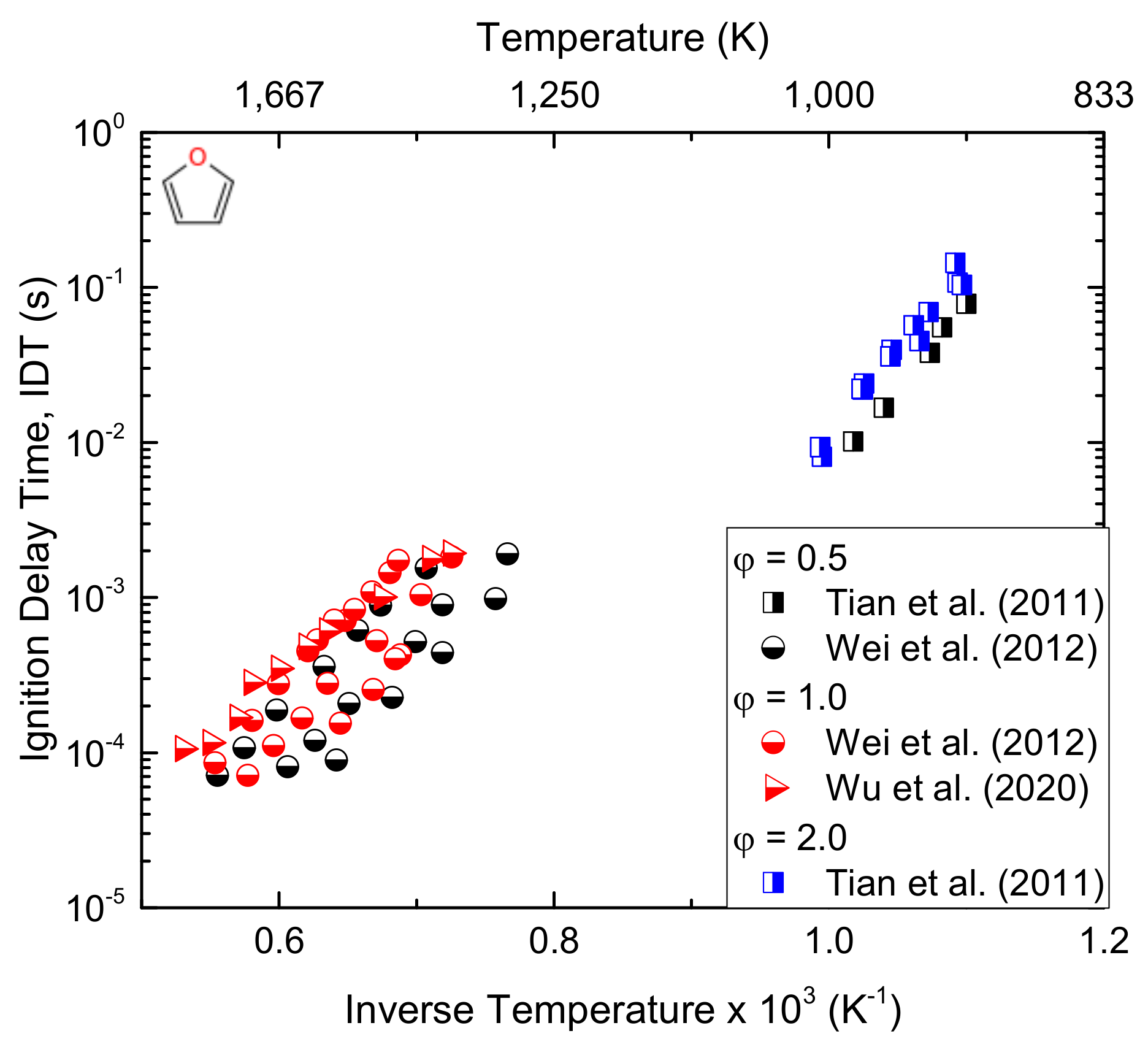{kind=link}
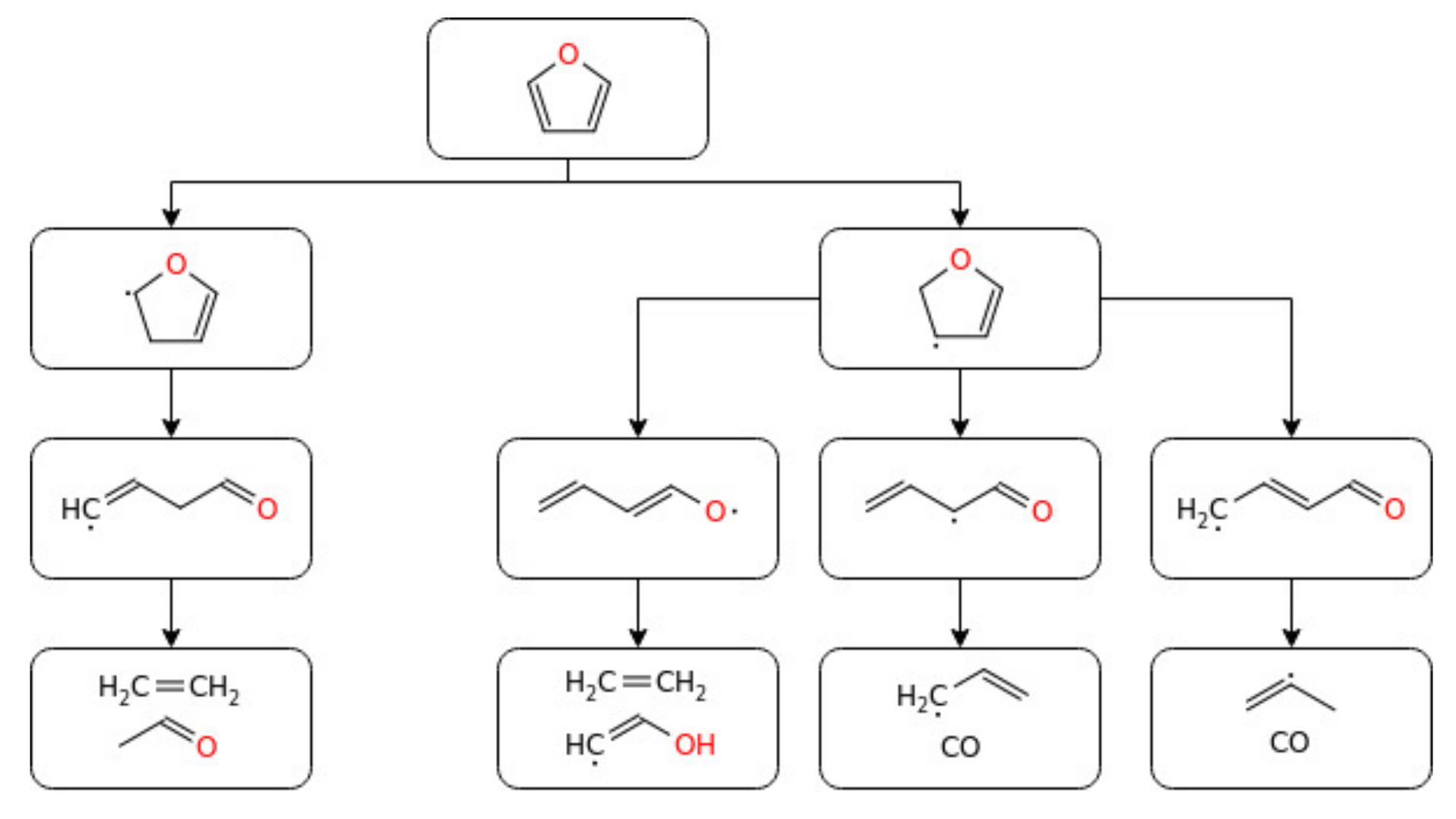{kind=link}
Table 1.
Physicochemical properties of some promising oxygenated fuels: LHV = lower heating value; λ = heat of vaporization; AIT = autoignition temperature.
Table 1.
Physicochemical properties of some promising oxygenated fuels: LHV = lower heating value; λ = heat of vaporization; AIT = autoignition temperature.
| Fuel | Structure | LHV (MJ L−1) | λ (kJ kg−1) | AIT (°C) | Abstracting Agents | Ref. |
|---|---|---|---|---|---|---|
| Methanol |  | 15.8 | 1103 | 465 | H, OH, HO2 | [34,35] |
| Ethanol |  | 21.4 | 904 | 420 | H, OH, O2, HO2 | [20,35] |
| Butanol |  | 26.9 | 582 | 343 | CH3, OH, HO2 | [35,36] |
| Acetic acid |  | 10.1 | 700 | 485 | H, OH, H O2, O2, CH3 | [30,37,38,39] |
| Crotonic acid |  | No data | 649 | 396 | No data | [38] |
| Acetaldehyde |  | 18.8 | 569 | 155 | H, OH, HO2, CH3, O2 | [40] |
| Furan |  | 23.2 | 406 | 390 | H, OH, CH3 | [41,42] |
Table 2.
Experimental and numerical study data of selected biofuel combustion. φ = equivalent ratio; IDT = ignition delay times; Su = laminar burning velocity.
Table 2.
Experimental and numerical study data of selected biofuel combustion. φ = equivalent ratio; IDT = ignition delay times; Su = laminar burning velocity.
| Parameter | Approach | T (K) | P (atm) | φ (–) | Ref. |
|---|---|---|---|---|---|
| Methanol | |||||
| IDT | Exp/Mod | 1545–2180 | 1.5–4.2 | 1 | [43] |
| IDT | Exp/Mo | 850–1100 | 7–30 | 0.25–1 | [44] |
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 940–1540 | 1–14.9 | 0.5, 1, 2 | [46] |
| IDT | Exp | 840–1000 | 12–24 | 1.0 | [47] |
| Su | Exp | 300 | 1 | 0.7–1.4 | [48] |
| Su | Exp/Mod | 340–450 | 0.02 | 0.8, 1, 1.2 | [49] |
| Ethanol | |||||
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 880–1150 | 3–10 | 1 | [50] |
| IDT | Exp/Mod | 960–1580 | 17.8–23.9 | 0.5, 1 | [51] |
| IDT | Exp/Mod | 944–1589 | 1.3–53 | 0.5, 1, 2 | [52] |
| IDT | Exp/Mod | 800–875 | 20 | 1.0 | [53] |
| Su | Exp/Mod | 300–453 | 1 | 0.8–1.4 | [54] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.55 | [55] |
| Su | Exp | 358 | 1 | 0.7–1.4 | [56] |
| Su | Exp | 358 | 1–3 | 0.7–1.4 | [57] |
| Su | Exp/Mod | 358 | 1 | 0.6–1.6 | [58] |
| Su | Exp/Mod | 350–620 | 1 | 0.7–1.3 | [59] |
| Su | Exp | 300 | 1 | 0.7–1.4 | [48] |
| Butanol | |||||
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 1050–1600 | 1.5–43 | 0.5, 1 | [60] |
| IDT | Exp/Mod | 1000–1800 | 1–4 | 0.5, 1 | [2] |
| IDT | Exp/Mod | 700–1000 | 10–80 | 1.0 | [61] |
| Su | Exp/Mod | 428 | 1, 2.5, 7.5 | 0.7–1.5 | [62] |
| Su | Exp/Mod | 350–600 | 1 | 0.7–1.3 | [63] |
| Su | Exp | 400 | 5 | 0.7–1.5 | [64] |
| Su | Exp/Mod | 393 | 1 | 0.8–1.4 | [65] |
| Su | Exp/Mod | 423 | 1 | 0.7–1.4 | [66] |
| Su | Exp/Mod | 350 | 1 | 0.8–1.2 | [67] |
| Acetic acid | |||||
| IDT | Exp/Mod | 1300–1950 | 0.3–0.7 | - | [68] |
| IDT | Exp/Mod | 700–2100 | 0.1–100 | No data | [37] |
| IDT | Exp | 1300–2000 | No data | No data | [69] |
| Su | Exp/Mod | 423 | 1 | 0.7–1.4 | [13] |
| Su | Exp/Mod | 673–973 | 0.05 | 0.77–1.05 | [70] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.8 | [14] |
| Crotonic acid | |||||
| IDT, Su | No data | No data | No data | No data | |
| Acetaldehyde | |||||
| IDT | Exp/Mod | 1000–1700 | 1.2–2.8 | 1 | [71] |
| IDT | Exp/Mod | 1295–1580 | 3–3.98 | 0.5, 1, 1.5 | [72] |
| IDT | Exp/Mod | 1000–1100 | 10 | 1 | [73] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.8 | [14] |
| Su | Exp/Mod | 460–900 | 0.7–0.71 | 0.5–4 | [40] |
| Furan | |||||
| IDT | Exp/Mod | 1320–1880 | 1.2–10.4 | 0.5, 1, 2 | [74] |
| IDT | Exp/Mod | 850–1050 | 18–33 | 0.5, 1, 2 | [75] |
| Su | Exp/Mod | 300–400 | 0.046 | 1.4, 1.8, 2.2 | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mosisa Wako, F.; Pio, G.; Salzano, E. Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel. Energies 2021, 14, 3562. https://doi.org/10.3390/en14123562
AMA Style
Mosisa Wako F, Pio G, Salzano E. Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel. Energies. 2021; 14(12):3562. https://doi.org/10.3390/en14123562
Chicago/Turabian StyleMosisa Wako, Fekadu, Gianmaria Pio, and Ernesto Salzano. 2021. "Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel" Energies 14, no. 12: 3562. https://doi.org/10.3390/en14123562
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.