Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Collection of Muscle Biopsies
2.3. Measurement of Iron Isotopes in Muscle Biopsies by Inductively Coupled Plasma-Mass Spectrometry
2.4. Western Immunoblotting
2.5. Quantification of Mitochondrial DNA Deletion
2.6. Statistical Analysis
3. Results
3.1. Study Participants
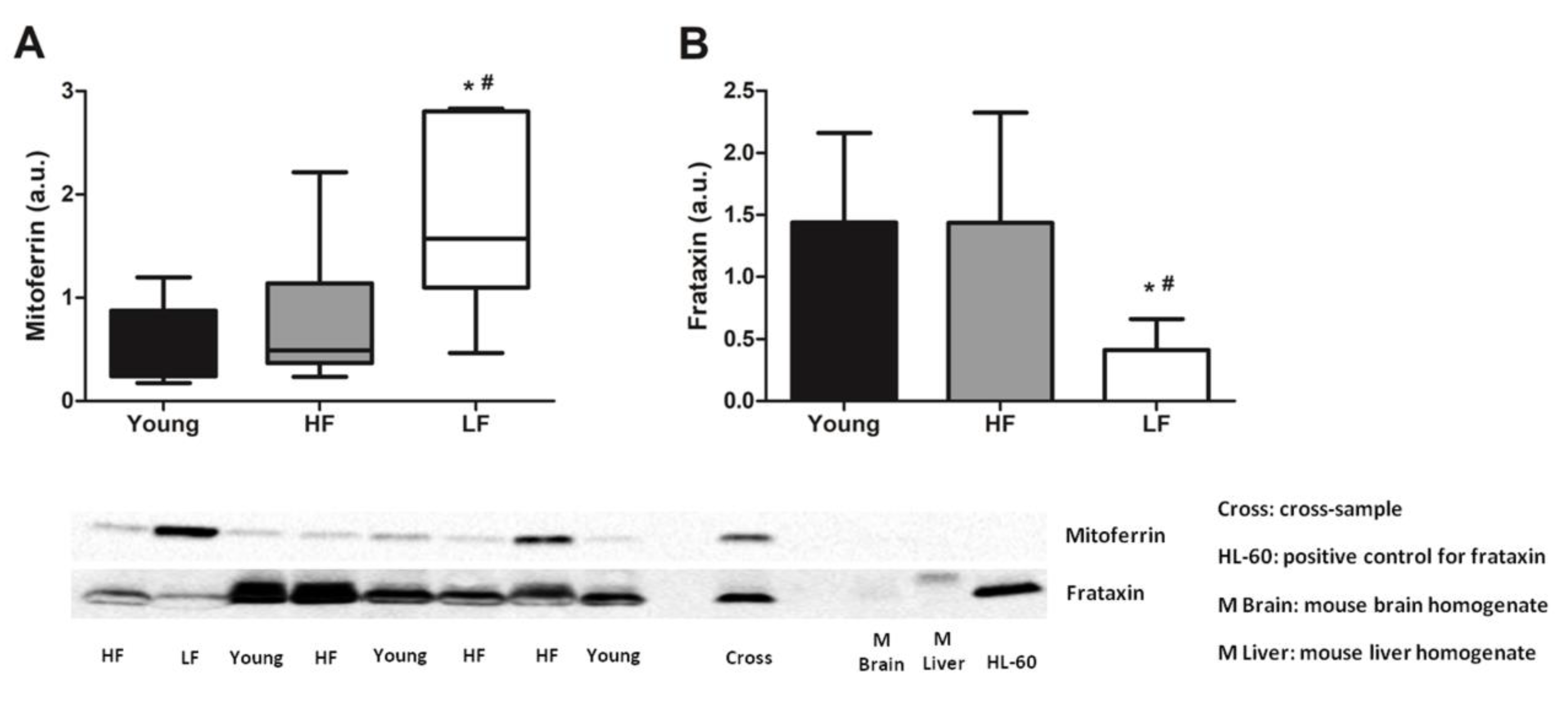
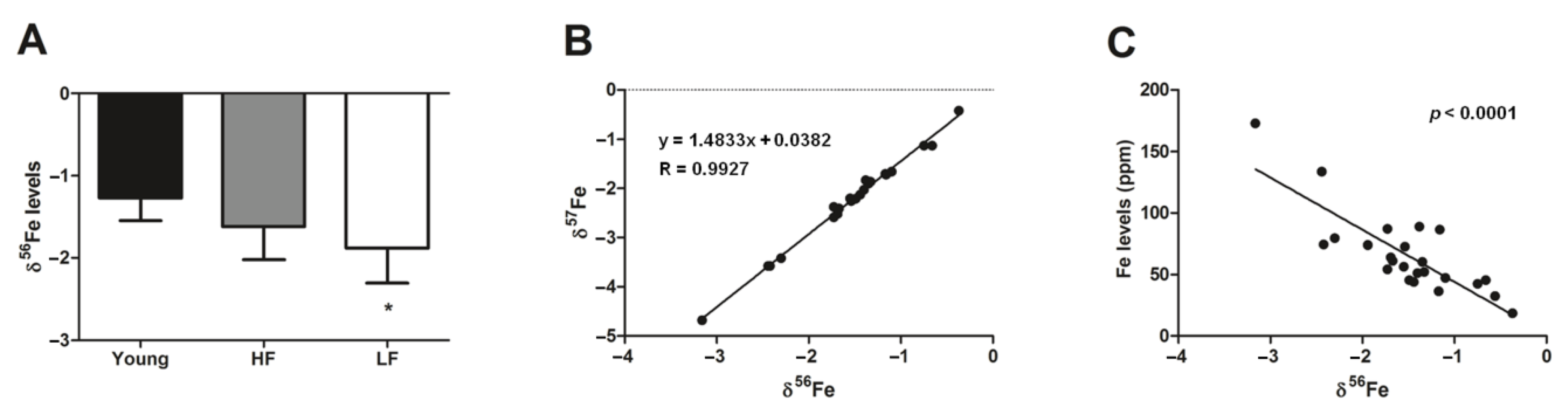
3.2. Measurement of Mitochondrial Iron Handling Proteins and Quantification of Iron Isotopes in Muscle Biopsies from Young and Old Participants
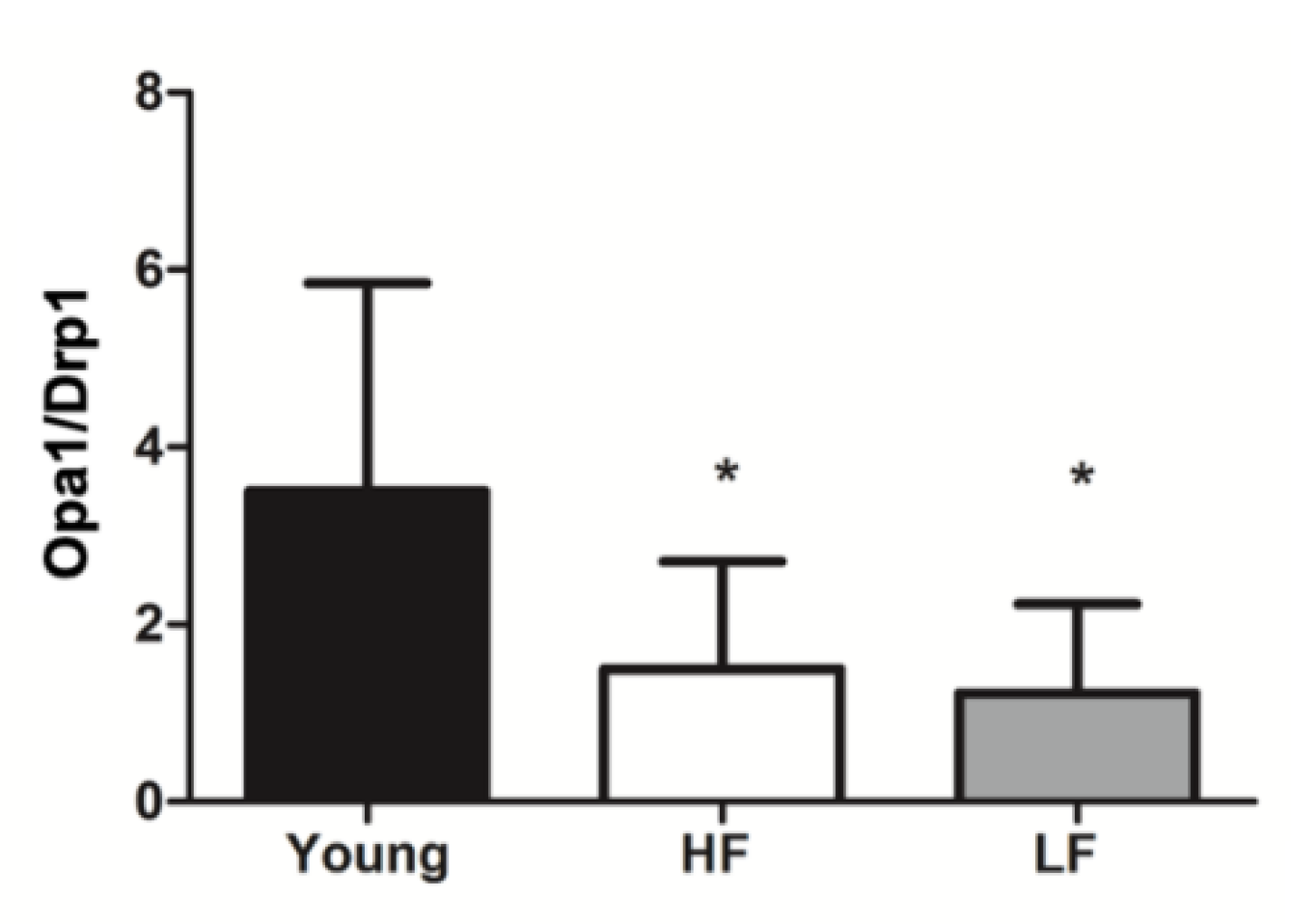
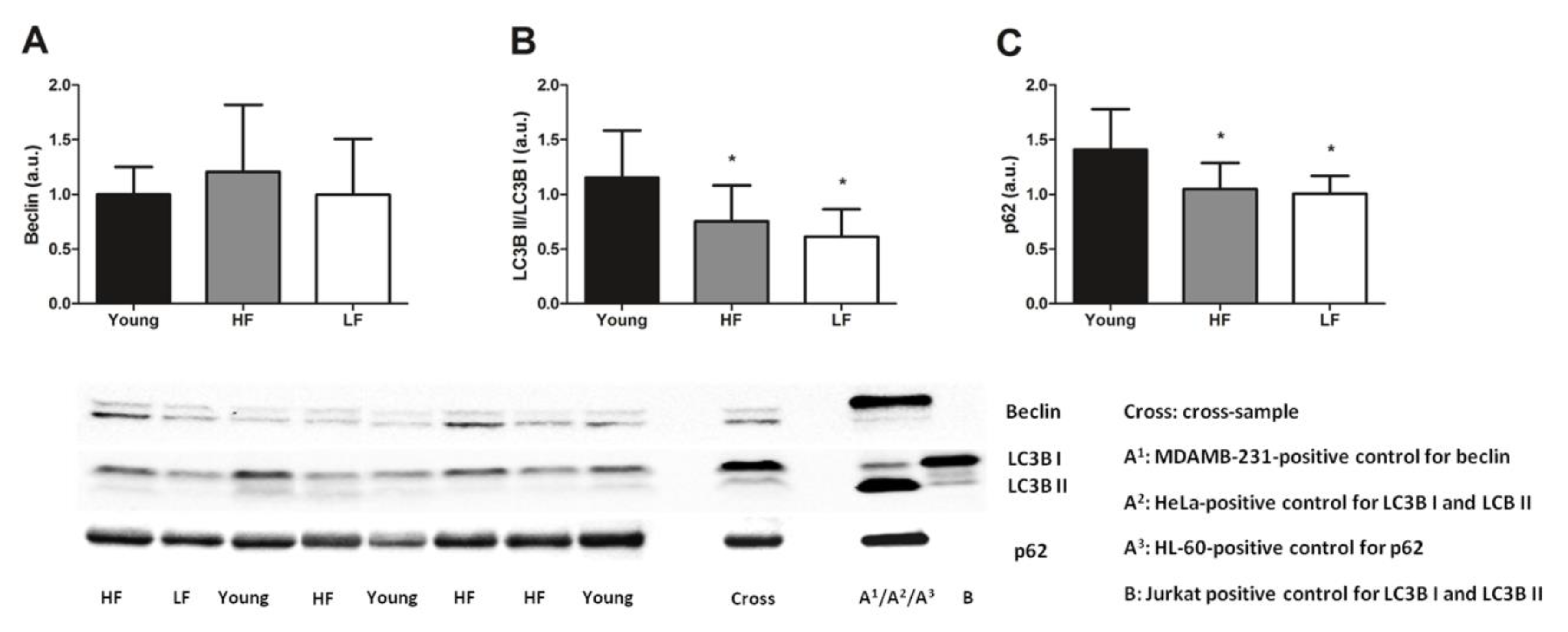
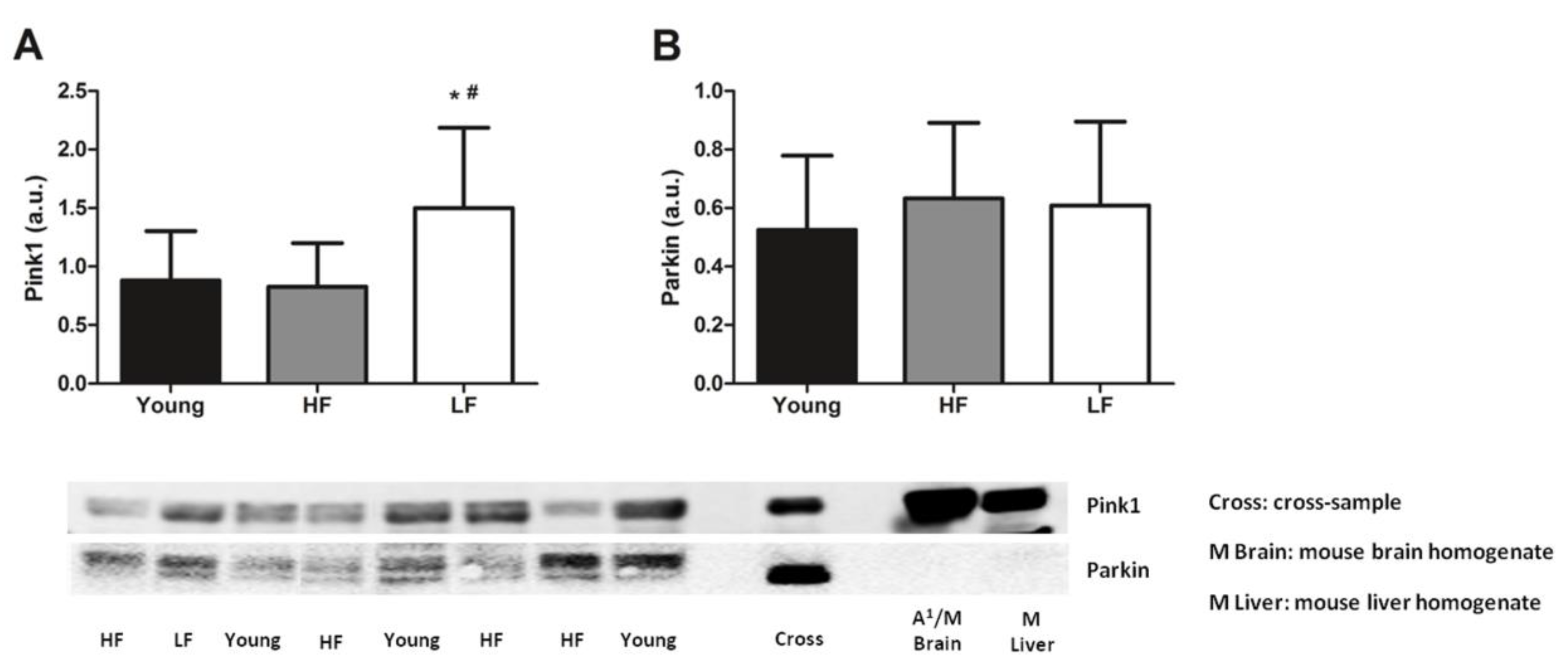
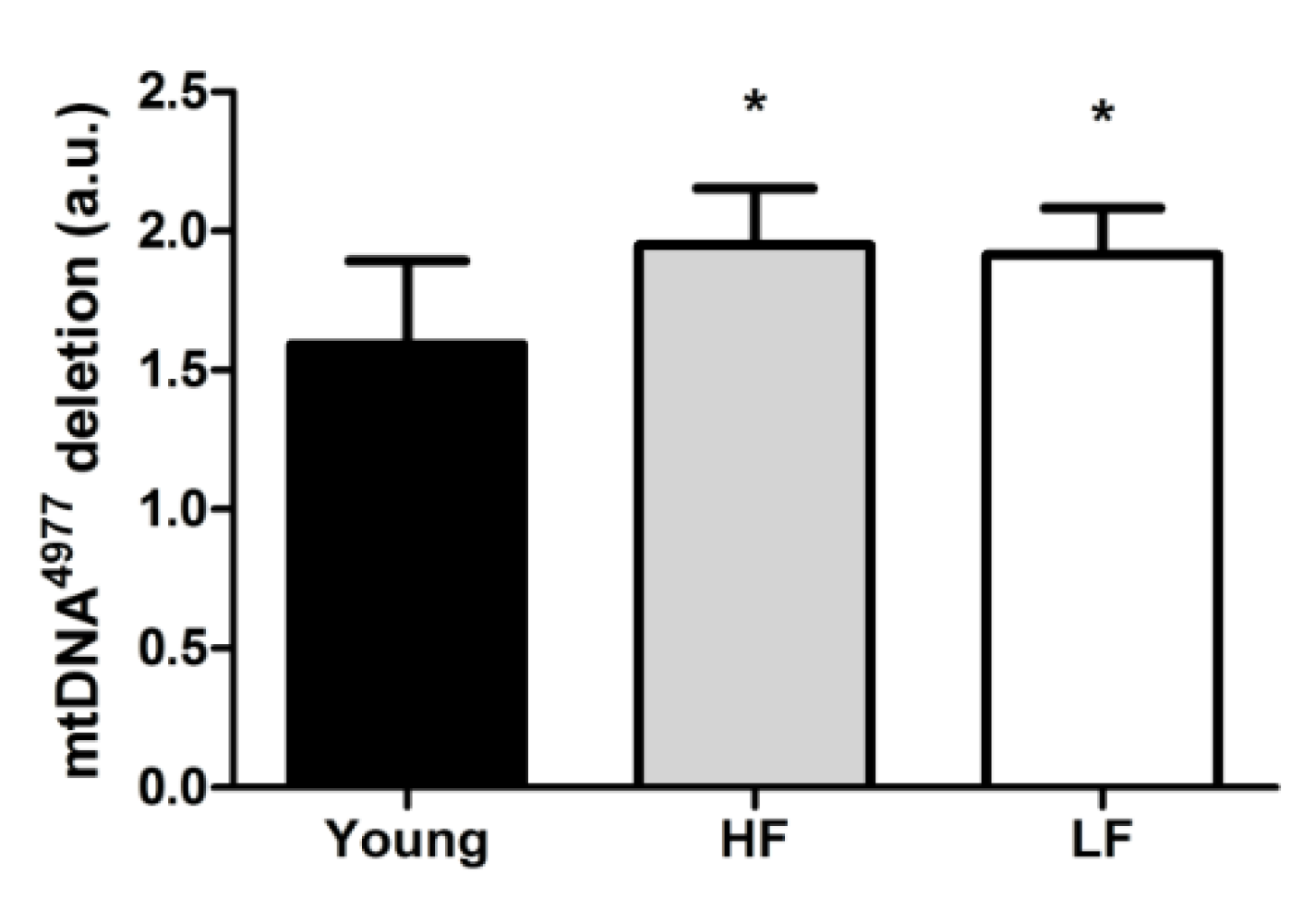
3.3. Protein Levels of Selected Markers of Mitochondrial Quality Control and Abundance of the mtDNA4977 Common Deletion
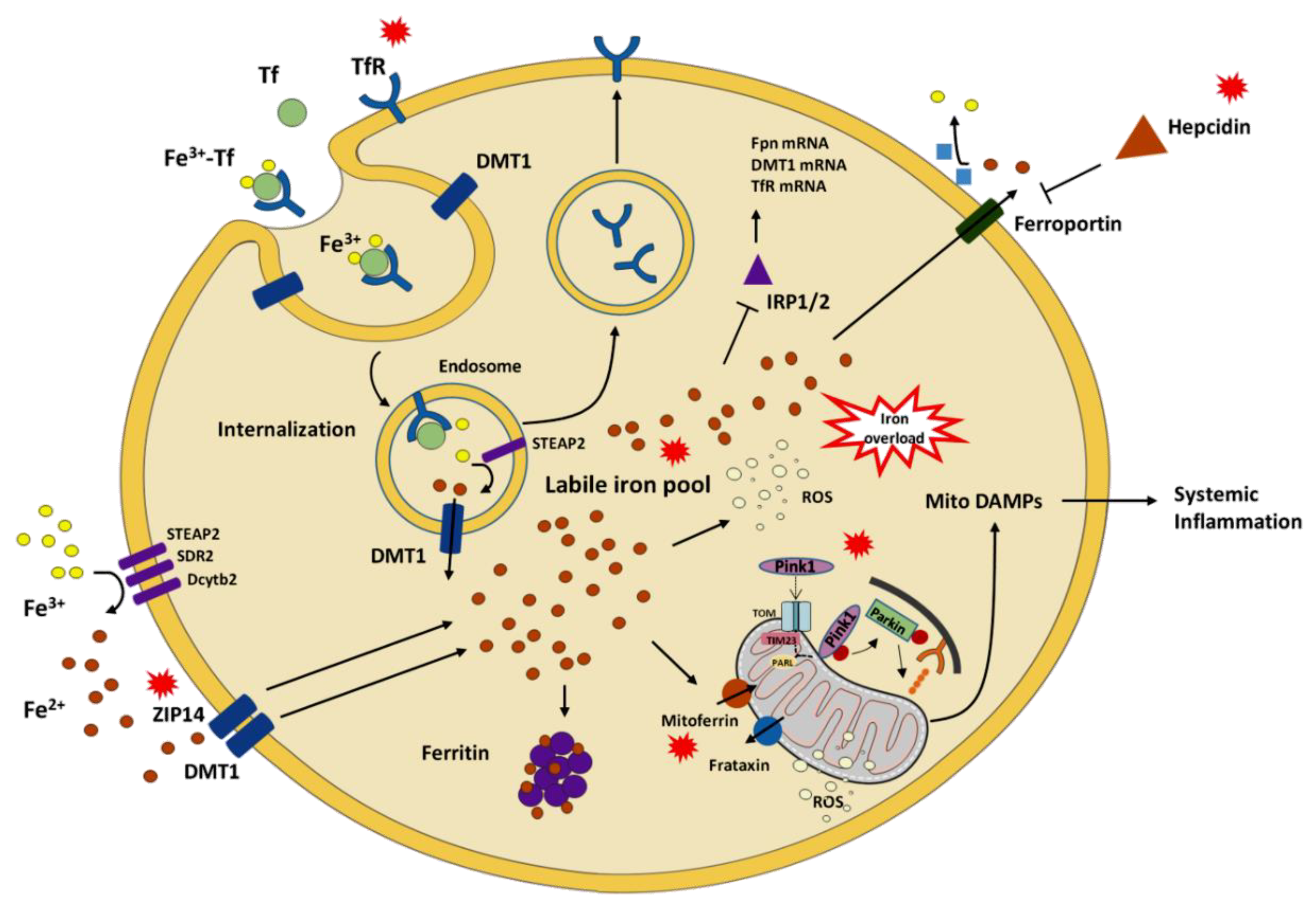
4. Discussion
5. Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aging Research: Protect Health and Prevent Disease|NIA. Available online: https://www.nia.nih.gov/about/aging-strategic-directions-research/goal-health-interventions#c3 (accessed on 14 October 2020).
- Chang, M.; Cohen-Mansfield, J.; Ferrucci, L.; Leveille, S.; Volpato, S.; De Rekeneire, N.; Guralnik, J.M. Incidence of loss of ability to walk 400 meters in a functionally limited older population. J. Am. Geriatr. Soc. 2004, 52, 2094–2098. [Google Scholar] [CrossRef]
- Guralnik, J.M.; Ferrucci, L.; Simonsick, E.M.; Salive, M.E.; Wallace, R.B. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N. Engl. J. Med. 1995, 332, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Pavasini, R.; Guralnik, J.; Brown, J.C.; di Bari, M.; Cesari, M.; Landi, F.; Vaes, B.; Legrand, D.; Verghese, J.; Wang, C.; et al. Short Physical Performance Battery and all-cause mortality: Systematic review and meta-analysis. BMC Med. 2016, 14, 215. [Google Scholar] [CrossRef] [Green Version]
- Fanning, J.; Rejeski, W.J.; Chen, S.H.; Guralnik, J.; Pahor, M.; Miller, M.E. Relationships Between Profiles of Physical Activity and Major Mobility Disability in the LIFE Study. J. Am. Geriatr. Soc. 2020, 68, 1476–1483. [Google Scholar] [CrossRef]
- Pahor, M.; Guralnik, J.M.; Anton, S.D.; Ambrosius, W.T.; Blair, S.N.; Church, T.S.; Espeland, M.A.; Fielding, R.A.; Gill, T.M.; Glynn, N.W.; et al. Impact and Lessons From the Lifestyle Interventions and Independence for Elders (LIFE) Clinical Trials of Physical Activity to Prevent Mobility Disability. J. Am. Geriatr. Soc. 2020, 68, 872–881. [Google Scholar] [CrossRef]
- Groessl, E.J.; Kaplan, R.M.; Rejeski, W.J.; Katula, J.A.; Glynn, N.W.; King, A.C.; Anton, S.D.; Walkup, M.; Lu, C.J.; Reid, K.; et al. Physical Activity and Performance Impact Long-term Quality of Life in Older Adults at Risk for Major Mobility Disability. Am. J. Prev. Med. 2019, 56, 141–146. [Google Scholar] [CrossRef]
- Coen, P.M.; Jubrias, S.A.; Distefano, G.; Amati, F.; Mackey, D.C.; Glynn, N.W.; Manini, T.M.; Wohlgemuth, S.E.; Leeuwenburgh, C.; Cummings, S.R.; et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 447–455. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.-M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.-D.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Marzetti, E.; Seo, A.Y.; Kim, J.-S.; Prolla, T.A.; Leeuwenburgh, C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech. Ageing Dev. 2010, 131, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Heckerman, D.; Traynor, B.J.; Picca, A.; Calvani, R.; Marzetti, E.; Hernandez, D.; Nalls, M.; Arepali, S.; Ferrucci, L.; Landi, F. Genetic variants associated with physical performance and anthropometry in old age: A genome-wide association study in the ilSIRENTE cohort. Sci. Rep. 2017, 7, 15879. [Google Scholar] [CrossRef]
- Timmers, P.R.H.J.; Wilson, J.F.; Joshi, P.K.; Deelen, J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nat. Commun. 2020, 11, 3570. [Google Scholar] [CrossRef]
- Zhang, A.S.; Enns, C.A. Iron homeostasis: Recently identified proteins provide insight into novel control mechanisms. J. Biol. Chem. 2009, 284, 711–715. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Rouault, T.A. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef] [Green Version]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef]
- Emerit, J.; Beaumont, C.; Trivin, F. Iron metabolism, free radicals, and oxidative injury. Biomed. Pharmacother. 2001, 55, 333–339. [Google Scholar] [CrossRef]
- Walczyk, T.; Von Blanckenburg, F. Deciphering the iron isotope message of the human body. Int. J. Mass Spectrom. 2005, 242, 117–134. [Google Scholar] [CrossRef]
- Albarède, F.; Telouk, P.; Lamboux, A.; Jaouen, K.; Balter, V. Isotopic evidence of unaccounted for Fe and Cu erythropoietic pathways. Metallomics 2011, 3, 926–933. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and disorders of iron metabolism. Annu. Rev. Med. 2011, 62, 347–360. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Taroni, F.; Cortopassi, G.A. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid. Redox Signal. 2006, 8, 506–516. [Google Scholar] [CrossRef]
- Boddaert, N.; Sang, K.H.L.Q.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Altun, M.; Edström, E.; Spooner, E.; Flores-Moralez, A.; Bergman, E.; Tollet-Egnell, P.; Norstedt, G.; Kessler, B.M.; Ulfhake, B. Iron load and redox stress in skeletal muscle of aged rats. Muscle Nerve 2007, 36, 223–233. [Google Scholar] [CrossRef]
- Hofer, T.; Marzetti, E.; Xu, J.; Seo, A.Y.; Gulec, S.; Knutson, M.D.; Leeuwenburgh, C.; Dupont-Versteegden, E.E. Increased iron content and RNA oxidative damage in skeletal muscle with aging and disuse atrophy. Exp. Gerontol. 2008, 43, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.H.; DeRuisseau, L.R.; Kavazis, A.N.; DeRuisseau, K.C. Plantaris muscle of aged rats demonstrates iron accumulation and altered expression of iron regulation proteins. Exp. Physiol. 2008, 93, 407–414. [Google Scholar] [CrossRef]
- Xu, J.; Knutson, M.D.; Carter, C.S.; Leeuwenburgh, C. Iron accumulation with age, oxidative stress and functional decline. PLoS ONE 2008, 3, e2865. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Hwang, J.C.Y.; Lees, H.A.; Wohlgemuth, S.E.; Knutson, M.D.; Judge, A.R.; Dupont-Versteegden, E.E.; Marzetti, E.; Leeuwenburgh, C. Long-term perturbation of muscle iron homeostasis following hindlimb suspension in old rats is associated with high levels of oxidative stress and impaired recovery from atrophy. Exp. Gerontol. 2012, 47, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Saini, S.K.; Calvani, R.; Landi, F.; Bernabei, R.; et al. Advanced Age Is Associated with Iron Dyshomeostasis and Mitochondrial DNA Damage in Human Skeletal Muscle. Cells 2019, 8, 1525. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.Y.; Xu, J.; Servais, S.; Hofer, T.; Marzetti, E.; Wohlgemuth, S.E.; Knutson, M.D.; Chung, H.Y.; Leeuwenburgh, C. Mitochondrial iron accumulation with age and functional consequences. Aging Cell 2008, 7, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Inter-Organelle Membrane Contact Sites and Mitochondrial Quality Control during Aging: A Geroscience View. Cells 2020, 9, 598. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Leeuwenburgh, C.; Coelho-Junior, H.J.; Bernabei, R.; Landi, F.; Marzetti, E. Targeting mitochondrial quality control for treating sarcopenia: Lessons from physical exercise. Expert Opin. Ther. Targets 2019, 23, 153–160. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Beli, R.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Bucci, C.; Guerra, F.; Marzetti, E. Older Adults with Physical Frailty and Sarcopenia Show Increased Levels of Circulating Small Extracellular Vesicles with a Specific Mitochondrial Signature. Cells 2020, 9, 973. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating Mitochondrial DNA at the Crossroads of Mitochondrial Dysfunction and Inflammation During Aging and Muscle Wasting Disorders. Rejuvenation Res. 2018, 21, 350–359. [Google Scholar] [CrossRef]
- Buford, T.W.; Lott, D.J.; Marzetti, E.; Wohlgemuth, S.E.; Vandenborne, K.; Pahor, M.; Leeuwenburgh, C.; Manini, T.M. Age-related differences in lower extremity tissue compartments and associations with physical function in older adults. Exp. Gerontol. 2012, 47, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Marzetti, E.; Lees, H.A.; Manini, T.M.; Buford, T.W.; Aranda, J.M.; Calvani, R.; Capuani, G.; Marsiske, M.; Lott, D.J.; Vandenborne, K.; et al. Skeletal muscle apoptotic signaling predicts thigh muscle volume and gait speed in community-dwelling older persons: An exploratory study. PLoS ONE 2012, 7, e32829. [Google Scholar] [CrossRef]
- Marzetti, E.; Landi, F.; Marini, F.; Cesari, M.; Buford, T.W.; Manini, T.M.; Onder, G.; Pahor, M.; Bernabei, R.; Leeuwenburgh, C.; et al. Patterns of circulating inflammatory biomarkers in older persons with varying levels of physical performance: A partial least squares-discriminant analysis approach. Front. Med. 2014, 1, 27. [Google Scholar] [CrossRef]
- Guralnik, J.M.; Simonsick, E.M.; Ferrucci, L.; Glynn, R.J.; Berkman, L.F.; Blazer, D.G.; Scherr, P.A.; Wallace, R.B. A short physical performance battery assessing lower extremity function: Association with self-reported disability and prediction of mortality and nursing home admission. J. Gerontol. 1994, 49, M85–M94. [Google Scholar] [CrossRef]
- Vasunilashorn, S.; Coppin, A.K.; Patel, K.V.; Lauretani, F.; Ferrucci, L.; Bandinelli, S.; Guralnik, J.M. Use of the Short Physical Performance Battery Score to predict loss of ability to walk 400 meters: Analysis from the InCHIANTI study. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 223–229. [Google Scholar] [CrossRef]
- Studenski, S.; Perera, S.; Patel, K.; Rosano, C.; Faulkner, K.; Inzitari, M.; Brach, J.; Chandler, J.; Cawthon, P.; Connor, E.B.; et al. Gait speed and survival in older adults. JAMA 2011, 305, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Pahor, M.; Guralnik, J.M.; Ambrosius, W.T.; Blair, S.; Bonds, D.E.; Church, T.S.; Espeland, M.A.; Fielding, R.A.; Gill, T.M.; Groessl, E.J.; et al. LIFE study investigators Effect of structured physical activity on prevention of major mobility disability in older adults: The LIFE study randomized clinical trial. JAMA 2014, 311, 2387–2396. [Google Scholar] [CrossRef]
- Marzetti, E.; Cesari, M.; Calvani, R.; Msihid, J.; Tosato, M.; Rodriguez-Mañas, L.; Lattanzio, F.; Cherubini, A.; Bejuit, R.; Di Bari, M.; et al. The “Sarcopenia and Physical fRailty IN older people: Multi-componenT Treatment strategies” (SPRINTT) randomized controlled trial: Case finding, screening and characteristics of eligible participants. Exp. Gerontol. 2018, 113, 48–57. [Google Scholar] [CrossRef]
- Maréchal, C.N.; Télouk, P.; Albarède, F. Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry. Chem. Geol. 1999, 156, 251–273. [Google Scholar] [CrossRef]
- Craddock, P.R.; Dauphas, N. Iron Isotopic Compositions of Geological Reference Materials and Chondrites. Geostand. Geoanal. Res. 2011, 35, 101–123. [Google Scholar] [CrossRef]
- Marzetti, E.; Carter, C.S.; Wohlgemuth, S.E.; Lees, H.A.; Giovannini, S.; Anderson, B.; Quinn, L.S.; Leeuwenburgh, C. Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech. Ageing Dev. 2009, 130, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, S.K.; Li, L.; Peek, C.B.; Kosmac, K.; Polonsky, T.S.; Tian, L.; Criqui, M.H.; Ferrucci, L.; Guralnik, J.M.; Kibbe, M.; et al. Associations of Poly (ADP-Ribose) Polymerase1 abundance in calf skeletal muscle with walking performance in peripheral artery disease. Exp. Gerontol. 2020, 140, 111048. [Google Scholar] [CrossRef]
- Picca, A.; Fracasso, F.; Pesce, V.; Cantatore, P.; Joseph, A.-M.; Leeuwenburgh, C.; Gadaleta, M.N.; Lezza, A.M.S. Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age 2013, 35, 1607–1620. [Google Scholar] [CrossRef] [Green Version]
- Nicassio, L.; Fracasso, F.; Sirago, G.; Musicco, C.; Picca, A.; Marzetti, E.; Calvani, R.; Cantatore, P.; Gadaleta, M.N.; Pesce, V. Dietary supplementation with acetyl-l-carnitine counteracts age-related alterations of mitochondrial biogenesis, dynamics and antioxidant defenses in brain of old rats. Exp. Gerontol. 2017, 98, 99–109. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef]
- Picca, A.; Sirago, G.; Pesce, V.; Lezza, A.M.S.; Calvani, R.; Bossola, M.; Villani, E.R.; Landi, F.; Leeuwenburgh, C.; Bernabei, R.; et al. Administration of enalapril started late in life attenuates hypertrophy and oxidative stress burden, increases mitochondrial mass, and modulates mitochondrial quality control signaling in the rat heart. Biomolecules 2018, 8, 177. [Google Scholar] [CrossRef] [Green Version]
- Chimienti, G.; Picca, A.; Fracasso, F.; Marzetti, E.; Calvani, R.; Leeuwenburgh, C.; Russo, F.; Lezza, A.M.S.; Pesce, V. Differences in liver TFAM binding to mtDNA and mtDNA damage between aged and extremely aged rats. Int. J. Mol. Sci. 2019, 20, 2601. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.K.; McDermott, M.M.; Picca, A.; Li, L.; Wohlgemuth, S.E.; Kosmac, K.; Peterson, C.A.; Tian, L.; Ferrucci, L.; Guralnik, J.M.; et al. Mitochondrial DNA damage in calf skeletal muscle and walking performance in people with peripheral artery disease. Free Radic. Biol. Med. 2020, 160, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Ravel-Chapuis, A.; Bélanger, G.; Côté, J.; Michel, R.N.; Jasmin, B.J. Misregulation of calcium-handling proteins promotes hyperactivation of calcineurin-NFAT signaling in skeletal muscle of DM1 mice. Hum. Mol. Genet. 2017, 26, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; López-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef]
- Summermatter, S.; Bouzan, A.; Pierrel, E.; Melly, S.; Stauffer, D.; Gutzwiller, S.; Nolin, E.; Dornelas, C.; Fryer, C.; Leighton-Davies, J.; et al. Blockade of Metallothioneins 1 and 2 Increases Skeletal Muscle Mass and Strength. Mol. Cell. Biol. 2017, 37, e00305-16. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron Regulatory Protein 1 Sustains Mitochondrial Iron Loading and Function in Frataxin Deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Lorenzi, M.; Menghi, A.; Galli, M.; Vitiello, R.; Randisi, F.; Bernabei, R.; Landi, F.; Marzetti, E. Mitochondrial dynamics signaling is shifted toward fusion in muscles of very old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp. Gerontol. 2017, 96, 63–67. [Google Scholar] [CrossRef]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Danel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.S.; Duce, J.A.; Devedjian, J.C.; et al. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J. Neural Transm. 2020, 127, 189–203. [Google Scholar] [CrossRef]
- Romano, A.D.; Paglia, A.; Bellanti, F.; Villani, R.; Sangineto, M.; Vendemiale, G.; Serviddio, G. Molecular aspects and treatment of iron deficiency in the elderly. Int. J. Mol. Sci. 2020, 21, 3821. [Google Scholar] [CrossRef]
- Marzetti, E.; Wohlgemuth, S.E.; Anton, S.D.; Bernabei, R.; Carter, C.S.; Leeuwenburgh, C. Cellular Mechanisms of Cardioprotection by Calorie Restriction: State of the Science and Future Perspectives. Clin. Geriatr. Med. 2009, 25, 715–732. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Wang, W.; Zhabyeyev, P.; Basu, R.; Mclean, B.; Fan, D.; Parajuli, N.; Desaulniers, J.; Patel, V.B.; Hajjar, R.J.; et al. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci. Rep. 2015, 5, 18132. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Desaulniers, J.; Dyck, J.R.B.; Kassiri, Z.; Oudit, G.Y. Resveratrol mediates therapeutic hepatic effects in acquired and genetic murine models of iron-overload. Liver Int. 2016, 36, 246–257. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer and Catalog Number | Type | Species | Dilution | Blocking | Detected Band MW (kDa) |
|---|---|---|---|---|---|---|
| Beclin | Cell Signaling Technology (Beverly, MA, USA) (#3738S) | Polyclonal | Rabbit | 1:2000 | 5% BSA TBS-t | 60 |
| Frataxin | Abcam (Cambridge, MA, USA) (ab110328) | Monoclonal | Mouse | 1:500 | 5% milk TBS-t | 23 |
| LC3B | Cell Signaling Technology (#2775S) | Polyclonal | Rabbit | 1:1000 | 5% BSA TBS-t | 14 (LC3B II) 16 (LC3B I) |
| Mitoferrin | Abcam (ab102959) | Polyclonal | Rabbit | 1:1000 | 5% milk TBS-t | 37 |
| SQSTM1/p62 | Cell Signaling Technology (#5114S) | Polyclonal | Rabbit | 1:2000 | 5% BSA TBS-t | 62 |
| Parkin | Abcam (ab77924) | Monoclonal | Mouse | 2 μg/mL | 2.5% milk TBS-t | 52 |
| Pink1 | Abcam (ab23707) | Polyclonal | Rabbit | 1:1000 | 5% milk TBS-t | 66 |
| Young (n = 11) | Old (n = 23) | |||
|---|---|---|---|---|
| Characteristic | HF (n = 16) | LF (n = 7) | p Value | |
| Age (years), median (IQR) | 22 (21–28) | 73 (71–81) * | 80.0 (80–83) * | <0.0001 a |
| Sex (women), n (%) | 5 (45.5) | 4 (25.0) | 5 (71.4) | 0.1076 b |
| BMI (kg/m2), mean ± SD | 24.9 ± 4.2 | 27.7 ± 3.6 | 27.8 ± 4.2 | 0.1604 c |
| Total iron levels (ppm), median (IQR) | 36.3 (32.9–58.8) | 76.8 (54.0–111.3) * | 82.9 (60.5–119.3) * | 0.0256 c |
| Number of diseases ¥, median (IQR) | 1.0 (0–2.0) | 1.5 (1.0–3.0) | 2 (1.0–4.0) | 0.1274 c |
| Number of medications #, median (IQR) | 1.5 (0–3.0) | 3.0 (1.0–8.0) | 3.0 (0.5–5.5) | 0.3112 c |
| SPPB summary score, median (IQR) | 11 (11–12) | 6 (6–7) | 0.0002 d | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Saini, S.K.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Wohlgemuth, S.E.; Xiao, R.; Calvani, R.; et al. Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults. Cells 2020, 9, 2579. https://doi.org/10.3390/cells9122579
Picca A, Saini SK, Mankowski RT, Kamenov G, Anton SD, Manini TM, Buford TW, Wohlgemuth SE, Xiao R, Calvani R, et al. Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults. Cells. 2020; 9(12):2579. https://doi.org/10.3390/cells9122579
Chicago/Turabian StylePicca, Anna, Sunil K. Saini, Robert T. Mankowski, George Kamenov, Stephen D. Anton, Todd M. Manini, Thomas W. Buford, Stephanie E. Wohlgemuth, Rui Xiao, Riccardo Calvani, and et al. 2020. "Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults" Cells 9, no. 12: 2579. https://doi.org/10.3390/cells9122579