JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future

 ,
,  , ,
, ,
Abstract
:1. Introduction
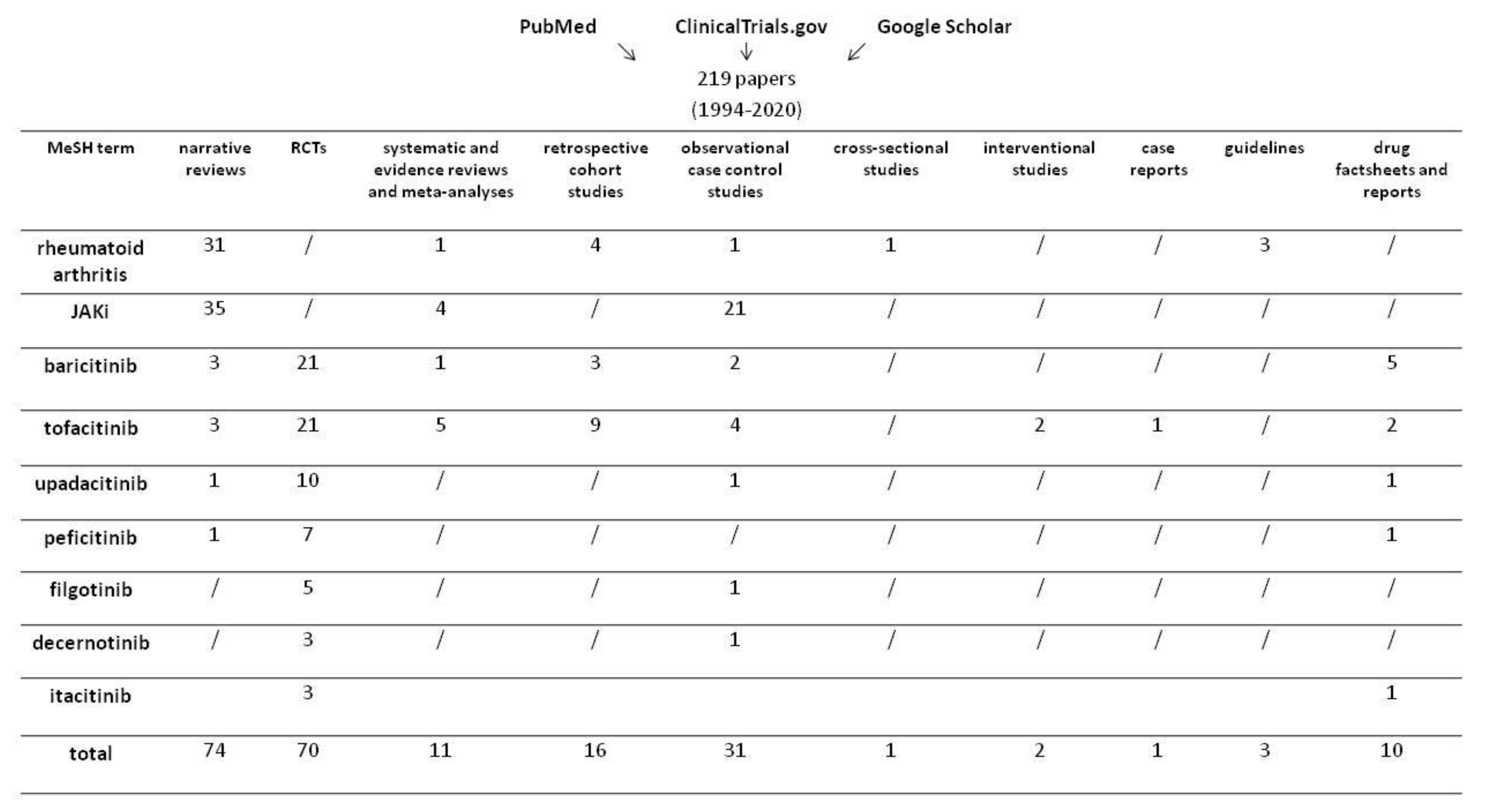
2. Methods
3. General Pharmacological Properties of JAKi
4. Baricitinib
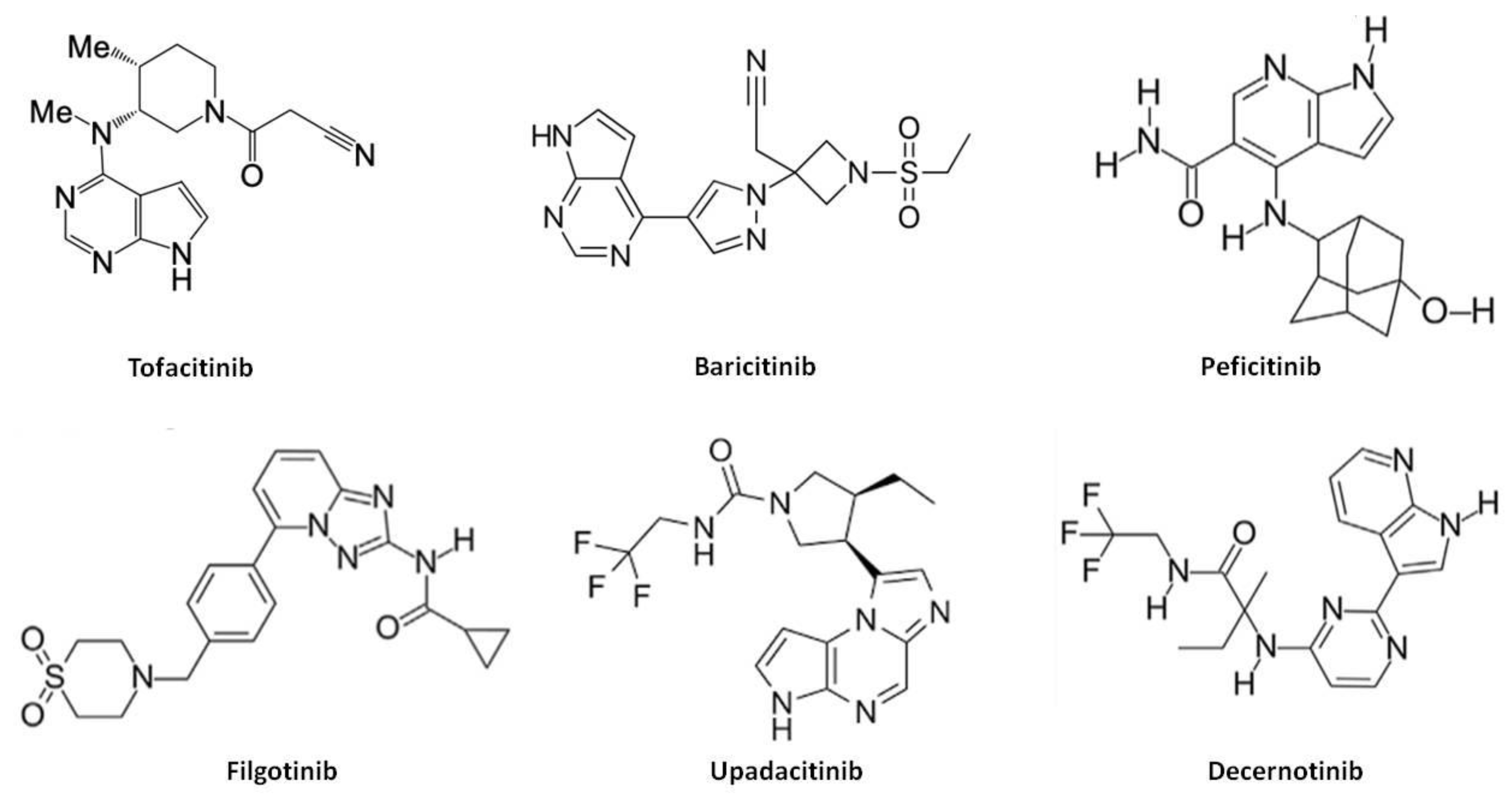
4.1. Chemical Structure, Pharmacokinetics, Pharmacodynamics and Mechanism of Action
4.2. Efficacy
4.3. Selected Populations
4.3.1. Pediatric Patients
4.3.2. Selected Ethnic Groups
4.4. Safety
4.5. Pharmacoeconomics
5. Tofacitinib
5.1. Chemical Structure, Pharmacokinetics, Pharmacodynamics and Mechanism of Action
5.2. Efficacy
5.3. Selected Populations
5.3.1. Pediatric Patients
5.3.2. Selected Ethnic Groups
5.4. Indirect Studies Comparing Tofacitinib Efficacy
5.5. Safety
5.6. Pharmacoeconomics
6. Second Generation JAKi
6.1. Upadacitinib
6.2. Peficitinib
6.3. Filgotinib
6.4. Decernotinib
6.5. Itacitinib
7. Concluding Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Areskoug-Josefsson, K.; Öberg, U. A literature review of the sexual health of women with rheumatoid arthritis. Musculoskelet. Care 2009, 7, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ahlmen, M.; Svensson, B.; Albertsson, K.; Forslind, K.; Hafström, I. Influence of gender on assessments of disease activity and function in early rheumatoid arthritis in relation to radiographic joint damage. Ann. Rheum. Dis. 2009, 69, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Grassi, W.; De Angelis, R.; Lamanna, G.; Cervini, C. The clinical features of rheumatoid arthritis. Eur. J. Radiol. 1998, 27, S18–S24. [Google Scholar] [CrossRef]
- Young, A.; Koduri, G. Extra-Articular manifestations and complications of rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2007, 21, 907–927. [Google Scholar] [CrossRef]
- Generali, E.; Cantarini, L.; Selmi, C. Ocular Involvement in Systemic Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2015, 49, 263–270. [Google Scholar] [CrossRef]
- Rugarli, C. Medicina Interna Sistematica. Sesta Edizione; Elsevier Srl: Milan, Italy, 2010; pp. 1710–1712. [Google Scholar]
- Scherer, H.U.; Häupl, T.; Burmester, G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.D.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002, 4, S265–S272. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; The RACI Consortium; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2013, 506, 376–381. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.A.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Han, B.; Fu, B.; Yarwood, A.; Thomson, W.; Symmons, D.P.M.; Worthington, J.; Young, A.; Hyrich, K.L.; et al. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality, and treatment response. JAMA Am. Med. Assoc. 2015, 313, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Malmström, V.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 2011, 23, 92–98. [Google Scholar] [CrossRef]
- Millar, K.; Lloyd, S.M.; McLean, J.S.; Batty, G.D.; Burns, H.; Cavanagh, J.; Deans, K.A.; Ford, I.; McConnachie, A.; McGinty, A.; et al. Personality, Socio-Economic Status and Inflammation: Cross-Sectional, Population-Based Study. PLoS ONE 2013, 8, e58256. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [Green Version]
- Ebringer, A.; Wilson, C. HLA molecules, bacteria and autoimmunity. J. Med. Microbiol. 2000, 49, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Evangelatos, G.; Fragoulis, G.E.; Koulouri, V.; Lambrou, G.I. MicroRNAs in rheumatoid arthritis: From pathogenesis to clinical impact. Autoimmun. Rev. 2019, 18, 102391. [Google Scholar] [CrossRef]
- Gaffen, S.L. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 365–370. [Google Scholar] [CrossRef]
- Silverman, G.J.; Carson, D.A. Roles of B cells in rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, S1–S6. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; Van Vollenhoven, R.F.; De Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.A.; Saag, K.G.; Bridges, S.L.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 68, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saag, K.G.; Koehnke, R.; Caldwell, J.R.; Brasington, R.; Burmeister, L.F.; Zimmerman, B.; Kohler, J.A.; Furst, D.E. Low dose long-term corticosteroid therapy in rheumatoid arthritis: An analysis of serious adverse events. Am. J. Med. 1994, 96, 115–123. [Google Scholar] [CrossRef]
- Donahue, K.E.; Gartlehner, G.; Jonas, D.E.; Lux, L.J.; Thieda, P.; Hansen, R.; Morgan, L.C.; Lohr, K.N. Systematic Review: Comparative Effectiveness and Harms of Disease-Modifying Medications for Rheumatoid Arthritis. Ann. Intern. Med. 2008, 148, 124. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 2013, 15, S2. [Google Scholar] [CrossRef] [Green Version]
- Curtis, J.R.; Singh, J.A. Use of biologics in rheumatoid arthritis: Current and emerging paradigms of care. Clin. Ther. 2011, 33, 679–707. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis. JAMA Am. Med. Assoc. 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Wang, D.; Li, Y.; Liu, Y.; Shi, G. The use of biologic therapies in the treatment of rheumatoid arthritis. Curr. Pharm. Biotechnol. 2014, 15, 542–548. [Google Scholar] [CrossRef]
- Yamaoka, K. Janus kinase inhibitors for rheumatoid arthritis. Curr. Opin. Chem. Biol. 2016, 32, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Pope, J.E.; Kruger, K.; Lippe, R.; Demasi, R.; Lula, S.; Kola, B. Efficacy of Monotherapy with Biologics and JAK Inhibitors for the Treatment of Rheumatoid Arthritis: A Systematic Review. Adv. Ther. 2018, 35, 1535–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.S.; Watowich, S.S. Jak-STAT Signaling Pathways. Encycl. Immunobiol. 2016, 3, 134–145. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef]
- Atzeni, F.; Talotta, R.; Nucera, V.; Marino, F.; Gerratana, E.; Sangari, D.; Masala, I.F.; Sarzi-Puttini, P. Adverse events, clinical considerations and management recommendations in rheumatoid arthritis patients treated with JAK inhibitors. Expert Rev. Clin. Immunol. 2018, 14, 945–956. [Google Scholar] [CrossRef]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. BioDrugs 2019, 33, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [Green Version]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 Drives Innate Immune Deficiency. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef]
- Rochman, Y.; Kashyap, M.; Robinson, G.W.; Sakamoto, K.; Gomez-Rodriguez, J.; Wagner, K.-U.; Leonard, W.J. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 19455–19460. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, J.-B.; Uyttenhove, C.; Lejeune, D.; Mui, A.; Groner, B.; Renauld, J.C. STAT5 activation is required for interleukin-9-dependent growth and transformation of lymphoid cells. Cancer Res. 2000, 60, 3971–3977. [Google Scholar] [PubMed]
- Kovanen, P.E.; Leonard, W.J. Cytokines and immunodeficiency diseases: Critical roles of the gammac-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 2004, 202, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; Van Dijk, T.; Raaijmakers, J.A.M.; Lammers, J.-W.J.; Koenderman, L.; De Groot, R.P. Activation of the STAT3/Acute Phase Response Factor Transcription Factor by Interleukin-5. J. Biol. Chem. 1995, 270, 25778–25784. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.H.; Patel, B.K.R.; Wang, L.-M.; Frankel, M.; Ellmore, N.; Flavell, R.A.; LaRochelle, W.J.; Pierce, J.H. Jak1 Expression Is Required for Mediating Interleukin-4-induced Tyrosine Phosphorylation of Insulin Receptor Substrate and Stat6 Signaling Molecules. J. Biol. Chem. 1997, 272, 6556–6560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, G.R.; Darnell, J.E. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.P.; Dickensheets, H.; Finbloom, D.S. The Interleukin-10 Signal Transduction Pathway and Regulation of Gene Expression in Mononuclear Phagocytes. J. Interferon Cytokine Res. 1999, 19, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Watford, W.T.; Hissong, B.D.; Bream, J.H.; Kanno, Y.; Muul, L.; O’Shea, J.J. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol. Rev. 2004, 202, 139–156. [Google Scholar] [CrossRef]
- Chiba, Y.; Goto, K.; Misawa, M. Interleukin-13-induced activation of signal transducer and activator of transcription 6 is mediated by an activation of Janus kinase 1 in cultured human bronchial smooth muscle cells. Pharmacol. Rep. 2012, 64, 454–458. [Google Scholar]
- Trivella, D.B.B.; Ferreira-Junior, J.R.; Dumoutier, L.; Renauld, J.-C.; Polikarpov, I. Structure and function of interleukin-22 and other members of the interleukin-10 family. Cell. Mol. Life Sci. 2010, 67, 2909–2935. [Google Scholar] [CrossRef]
- Andrés, R.M.; Hald, A.; Johansen, C.; Kragballe, K.; Iversen, L. Studies of Jak/STAT3 expression and signalling in psoriasis identifies STAT3-Ser727 phosphorylation as a modulator of transcriptional activity. Exp. Dermatol. 2013, 22, 323–328. [Google Scholar] [CrossRef]
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Baur, E.V.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-γ are not. J. Mol. Med. 2009, 87, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2015, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Shioya, M.; Nishida, A.; Bamba, S.; Tsujikawa, T.; Kim-Mitsuyama, S.; Fujiyama, Y. Expression of IL-24, an Activator of the JAK1/STAT3/SOCS3 Cascade, Is Enhanced in Inflammatory Bowel Disease. J. Immunol. 2009, 183, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Alunno, A.; Padjen, I.; Fanouriakis, A.; Boumpas, D.T. Pathogenic and Therapeutic Relevance of JAK/STAT Signaling in Systemic Lupus Erythematosus: Integration of Distinct Inflammatory Pathways and the Prospect of Their Inhibition with an Oral Agent. Cells 2019, 8, 898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Putheti, P.; Zhou, Q.; Liu, Q.; Gao, W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008, 19, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.; Kerr, I.; Stark, G. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Abdolvahab, M.H.; Mofrad, M.K.; Schellekens, H. Interferon Beta: From Molecular Level to Therapeutic Effects. Int. Rev. Cell Mol. Biol. 2016, 326, 343–372. [Google Scholar] [CrossRef]
- Morelli, M.; Scarponi, C.; Mercurio, L.; Facchiano, F.; Pallotta, S.; Madonna, S.; Girolomoni, G.; Albanesi, C. Selective Immunomodulation of Inflammatory Pathways in Keratinocytes by the Janus Kinase (JAK) Inhibitor Tofacitinib: Implications for the Employment of JAK-Targeting Drugs in Psoriasis. J. Immunol. Res. 2018, 2018, 7897263. [Google Scholar] [CrossRef]
- Biethahn, S.; Alves, F.; Wilde, S.; Hiddemann, W.; Spiekermann, K. Expression of granulocyte colony-stimulating factor- and granulocyte-macrophage colony-stimulating factor-associated signal transduction proteins of the JAK/STAT pathway in normal granulopoiesis and in blast cells of acute myelogenous leukemia. Exp. Hematol. 1999, 27, 885–894. [Google Scholar] [CrossRef]
- Al-Shami, A.; Mahanna, W.; Naccache, P.H. Granulocyte-Macrophage Colony-stimulating Factor-activated Signaling Pathways in Human Neutrophils. J. Biol. Chem. 1998, 273, 1058–1063. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Tahara, T.; Matsumoto, A.; Kato, T.; Miyazaki, H.; Ohashi, H. Functional analysis of the cytoplastic domain of the human Mpl receptor for tyrosine-phosphorylation of the signaling molecules, proliferation and differentiation. FEBS Lett. 1996, 395, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Smit, L.S.; Meyer, D.J.; Billestrup, N.; Norstedt, G.; Schwartz, J.; Carter-Su, C. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol. Endocrinol. 1996, 10, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Kuhrt, D.; Wojchowski, N.M. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015, 125, 3536–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Nagahisa, H.; Nagasawa, T.; Todokoro, K. Regulation of megakaryocytopoiesis by thrombopoietin and stromal cells. Leukemia 1997, 11, 435–438. [Google Scholar]
- Wada, N.; Hirako, S.; Takenoya, F.; Kageyama, H.; Okabe, M.; Shioda, S. Leptin and its receptors. J. Chem. Neuroanat. 2014, 61, 191–199. [Google Scholar] [CrossRef]
- Lehtonen, A.; Matikainen, S.; Miettinen, M.; Julkunen, I. Granulocyte-Macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J. Leukoc. Biol. 2002, 71, 511–519. [Google Scholar]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 2019, 146, dev167643. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Aittomäki, S.; Pesu, M. Therapeutic Targeting of the JAK/STAT Pathway. Basic Clin. Pharmacol. Toxicol. 2013, 114, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 258. [Google Scholar] [CrossRef] [Green Version]
- Li, W.X. Canonical and non-Canonical JAK–STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, P.B. Paradigm shifts in the cell biology of STAT signaling. Semin. Cell Dev. Biol. 2008, 19, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver-Morse, L.; Li, W.X. JAK-STAT in heterochromatin and genome stability. JAK-STAT 2013, 2, e26090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, E.H.S.; Miceli-Richard, C.; González-Gay, M.A.; Sinigaglia, L.; Schlichting, D.E.; Meszaros, G.; de la Torre, L.; Schulze-Koops, H. The effect of JAK1/JAK2 inhibition in rheumatoid arthritis: Efficacy and safety of baricitinib. Clin. Exp. Rheumatol. 2019, 37, 694–704. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Yarilina, A.; Xu, K.; Chen, J.; Ivashkiv, L.B. TNF activates calcium–nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc. Natl. Acad. Sci. USA 2011, 108, 1573–1578. [Google Scholar] [CrossRef] [Green Version]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-κB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013, 11, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Manukyan, I.; Galatioto, J.; Mascareno, E.; Bhaduri, S.; Siddiqui, M. Cross-Talk between calcineurin/NFAT and Jak/STAT signalling induces cardioprotective αB-crystallin gene expression in response to hypertrophic stimuli. J. Cell. Mol. Med. 2009, 14, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.-B.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S. The mechanism of action of tofacitinib—An oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 318–328. [Google Scholar]
- Westhovens, R. Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology 2019, 58, i27–i33. [Google Scholar] [CrossRef] [Green Version]
- Kremer, J.M.; Emery, P.; Camp, H.S.; Friedman, A.; Wang, L.; Othman, A.A.; Khan, N.; Pangan, A.L.; Jungerwirth, S.; Keystone, E.C. A Phase IIb Study of ABT-494, a Selective JAK-1 Inhibitor, in Patients with Rheumatoid Arthritis and an Inadequate Response to Anti-Tumor Necrosis Factor Therapy. Arthritis Rheumatol. 2016, 68, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Olumiant EPAR Product Information; EMA: London, UK, 2018.
- European Medicines Agency. Xeljanz EPAR Product Information; EMA: London, UK, 2018.
- Wollenhaupt, J.; Lee, E.B.; Curtis, J.R.; Silverfield, J.; Terry, K.; Soma, K.; Mojcik, C.; Demasi, R.; Strengholt, S.; Kwok, K.; et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: Final results of a global, open-label, long-term extension study. Arthritis Res. 2019, 21, 89. [Google Scholar] [CrossRef] [Green Version]
- Harigai, M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology 2019, 58, i34–i42. [Google Scholar] [CrossRef] [Green Version]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Frisdal, E.; Lesnik, P.; Olivier, M.; Robillard, P.; Chapman, M.J.; Huby, T.; Guerin, M.; Le Goff, W. Interleukin-6 Protects Human Macrophages from Cellular Cholesterol Accumulation and Attenuates the Proinflammatory Response. J. Biol. Chem. 2011, 286, 30926–30936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles-Schoeman, C.; Fleischmann, R.; Davignon, J.; Schwartz, H.; Turner, S.M.; Beysen, C.; Milad, M.; Hellerstein, M.K.; Luo, Z.; Kaplan, I.V.; et al. Potential Mechanisms Leading to the Abnormal Lipid Profile in Patients With Rheumatoid Arthritis Versus Healthy Volunteers and Reversal by Tofacitinib. Arthritis Rheumatol. 2015, 67, 616–625. [Google Scholar] [CrossRef]
- Jagpal, A.; Navarro-Millán, I. Cardiovascular co-morbidity in patients with rheumatoid arthritis: A narrative review of risk factors, cardiovascular risk assessment and treatment. BMC Rheumatol. 2018, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivera, P.A.; Lasa, J.S.; Bonovas, S.; Danese, S.; Peyrin-Biroulet, L. Safety of Janus Kinase Inhibitors in Patients With Inflammatory Bowel Diseases or Other Immune-mediated Diseases: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1554–1573.e12. [Google Scholar] [CrossRef]
- Jegatheeswaran, J.; Turk, M.; Pope, J.E. Comparison of Janus kinase inhibitors in the treatment of rheumatoid arthritis: A systemic literature review. Immunotherapy 2019, 11, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Mayence, A.; Eynde, J.J.V. Baricitinib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Janus Kinases. Pharmaceuticals 2019, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Olumiant, T.M. Product Monograph; Eli Lilly Canada Inc.: Toronto, ON, Canada, 2018; Revised on 9 April 2020. [Google Scholar]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, S.; Nakayamada, S.; Sakata, K.; Kitanaga, Y.; Ma, X.; Lee, S.; Ishii, A.; Yamagata, K.; Nakano, K.; Tanaka, Y. Janus Kinase Inhibitor Baricitinib Modulates Human Innate and Adaptive Immune System. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.; Takeuchi, T.; Schiff, M.; Schlichting, D.; Xie, L.; Issa, M.; Stoykov, I.; Lisse, J.; Martinez-Osuna, P.; Rooney, T.; et al. Efficacy and safety of long-term baricitinib with and without methotrexate for the treatment of rheumatoid arthritis: Experience with baricitinib monotherapy continuation or after switching from methotrexate monotherapy or baricitinib plus methotrexate. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.G.; Chen, X.; Lee, F.; Emm, T.; Scherle, P.A.; Lo, Y.; Punwani, N.; Williams, W.V.; Yeleswaram, S. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 1354–1361. [Google Scholar] [CrossRef]
- Zhang, X.; Chua, L.; Ernest, C.; Macias, W.; Rooney, T.; Tham, L.S. Dose/Exposure-Response Modeling to Support Dosing Recommendation for Phase III Development of Baricitinib in Patients with Rheumatoid Arthritis. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 804–813. [Google Scholar] [CrossRef]
- Olumiant. Monograph for Professionals-Drugs.com. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/207924Orig1s000ClinPharmR.pdf (accessed on 12 April 2017).
- Kawalec, P.; Śladowska, K.; Malinowska-Lipień, I.; Brzostek, T.; Kózka, M. New alternative in the treatment of rheumatoid arthritis: Clinical utility of baricitinib. Ther. Clin. Risk Manag. 2019, 15, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.; Schiff, M.; Van Der Heijde, D.; Ramos-Remus, C.; Spindler, A.; Stanislav, M.; Zerbini, C.A.F.; Gurbuz, S.; Dickson, C.; De Bono, S.; et al. Baricitinib, Methotrexate, or Combination in Patients With Rheumatoid Arthritis and No or Limited Prior Disease-Modifying Antirheumatic Drug Treatment. Arthritis Rheumatol. 2017, 69, 506–517. [Google Scholar] [CrossRef]
- Taylor, P.; Keystone, E.; van der Heijde, D.; Tanaka, Y.; Ishii, T.; Emoto, K.; Yang, L.; Arora, V.; Gaich, C.L.; Rooney, T.; et al. Baricitinib Versus Placebo or Adalimumab in Patients with Active Rheumatoid Arthritis (RA) and an Inadequate Response to Background Methotrexate Therapy: Results of a Phase 3 Study Peter. Arthritis Rheumatol. 2015, 67, 26–30. [Google Scholar]
- Dougados, M.; Van Der Heijde, D.; Chen, Y.-C.; Greenwald, M.; Drescher, E.; Liu, J.; Beattie, S.; De La Torre, I.; Rooney, T.; Schlichting, D.; et al. LB0001 Baricitinib, an Oral Janus Kinase (JAK)1/JAK2 Inhibitor, in Patients with Active Rheumatoid Arthritis (RA) and An Inadequate Response to CDMARD Therapy: Results of the Phase 3 RA-Build Study. Ann. Rheum. Dis. 2015, 74. [Google Scholar] [CrossRef]
- Genovese, M.; Kremer, J.; Zamani, O.; Ludivico, C.; Krogulec, M.; Xie, L.; Beattie, S.; Koch, A.; Cardillo, T.; Rooney, T.; et al. OP0029 Baricitinib, An Oral Janus Kinase (JAK)1/JAK2 Inhibitor, in Patients with Active Rheumatoid Arthritis (RA) and an Inadequate Response to TNF Inhibitors: Results of the Phase 3 RA-Beacon Study. Ann. Rheum. Dis. 2015, 74, 75–76. [Google Scholar] [CrossRef]
- European Medicines Agency. Olumiant (Baricitinib): EU Assessment Report; European Medicines Agency: London, UK, 2016.
- Van Der Heijde, D.; Dougados, M.; Chen, Y.-C.; Greenwald, M.; Drescher, E.; Klar, R.; Xie, L.; De La Torre, I.; Rooney, T.P.; Witt, S.L.; et al. Effects of baricitinib on radiographic progression of structural joint damage at 1 year in patients with rheumatoid arthritis and an inadequate response to conventional synthetic disease-modifying antirheumatic drugs. RMD Open 2018, 4, e000662. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijde, D.; Schiff, M.; Tanaka, Y.; Xie, L.; Meszaros, G.; Ishii, T.; Casillas, M.; Ortmann, R.A.; Emery, P. Low rates of radiographic progression of structural joint damage over 2 years of baricitinib treatment in patients with rheumatoid arthritis. RMD Open 2019, 5, e000898. [Google Scholar] [CrossRef] [Green Version]
- Keystone, E.C.; Taylor, P.C.; Tanaka, Y.; Gaich, C.; DeLozier, A.M.; Dudek, A.; Zamora, J.V.; Cobos, J.A.C.; Rooney, T.; De Bono, S.; et al. Patient-reported outcomes from a phase 3 study of baricitinib versus placebo or adalimumab in rheumatoid arthritis: Secondary analyses from the RA-BEAM study. Ann. Rheum. Dis. 2017, 76, 1853–1861. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Takeuchi, T.; Genovese, M.C.; Haraoui, B.; Li, Z.; Xie, L.; Klar, R.; Pinto-Correia, A.; Otawa, S.; Lopez-Romero, P.; De La Torre, I.; et al. Dose reduction of baricitinib in patients with rheumatoid arthritis achieving sustained disease control: Results of a prospective study. Ann. Rheum. Dis. 2018, 78, 171–178. [Google Scholar] [CrossRef]
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Kochi, Y.; Suzuki, A.; Yamada, R.; Yamamoto, K. Ethnogenetic heterogeneity of rheumatoid arthritis—Implications for pathogenesis. Nat. Rev. Rheumatol. 2010, 6, 290–295. [Google Scholar] [CrossRef]
- Alamanos, Y.; Drosos, A.A. Epidemiology of adult rheumatoid arthritis. Autoimmun. Rev. 2005, 4, 130–136. [Google Scholar] [CrossRef]
- Harrold, L.R.; Harrington, J.T.; Curtis, J.R.; Furst, D.E.; Bentley, M.J.; Shan, Y.; Reed, G.; Kremer, J.; Greenberg, J.D. Prescribing practices in a US cohort of rheumatoid arthritis patients before and after publication of the American College of Rheumatology treatment recommendations. Arthritis Rheumatol. 2012, 64, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Atsumi, T.; Amano, K.; Harigai, M.; Ishii, T.; Kawaguchi, O.; Rooney, T.P.; Akashi, N.; Takeuchi, T. Efficacy and safety of baricitinib in Japanese patients with rheumatoid arthritis: Subgroup analyses of four multinational phase 3 randomized trials. Mod. Rheumatol. 2017, 28, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Emoto, K.; Cai, Z.; Aoki, T.; Schlichting, D.; Rooney, T.; Macias, W. Efficacy and Safety of Baricitinib in Japanese Patients with Active Rheumatoid Arthritis Receiving Background Methotrexate Therapy: A 12-week, Double-blind, Randomized Placebo-controlled Study. J. Rheumatol. 2016, 43, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ishii, T.; Cai, Z.; Schlichting, D.; Rooney, T.; Macias, W. Efficacy and safety of baricitinib in Japanese patients with active rheumatoid arthritis: A 52-week, randomized, single-blind, extension study. Mod. Rheumatol. 2017, 28, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harigai, M.; Takeuchi, T.; Smolen, J.S.; Winthrop, K.L.; Nishikawa, A.; Rooney, T.P.; Saifan, C.G.; Issa, M.; Isaka, Y.; Akashi, N.; et al. Safety profile of baricitinib in Japanese patients with active rheumatoid arthritis with over 1.6 years median time in treatment: An integrated analysis of Phases 2 and 3 trials. Mod. Rheumatol. 2019, 30, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwald, M.K.; Fidelus-Gort, R.; Levy, R. A randomized dose-ranging, placebo-controlled study of INCB028050, a selective JAK1 and JAK2 inhibitor in subjects with active rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2010, 62 (Suppl. 10), 2172. [Google Scholar]
- Keystone, E.; Taylor, P.C.; Drescher, E.; Schlichting, D.E.; Beattie, S.D.; Berclaz, P.-Y.; Lee, C.H.; Fidelus-Gort, R.K.; Luchi, M.E.; Rooney, T.P.; et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann. Rheum. Dis. 2014, 74, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Keystone, E.; Van Der Heijde, D.; Weinblatt, M.E.; Morales, L.D.C.; Gonzaga, J.R.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Dougados, M.; Van Der Heijde, D.; Chen, Y.-C.; Greenwald, M.; Drescher, E.; Liu, J.; Beattie, S.; Witt, S.; De La Torre, I.; Gaich, C.; et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: Results from the RA-BUILD study. Ann. Rheum. Dis. 2016, 76, 88–95. [Google Scholar] [CrossRef]
- Genovese, M.C.; Kremer, J.M.; Zamani, O.; Ludivico, C.; Krogulec, M.; Xie, L.; Beattie, S.D.; Koch, A.E.; Cardillo, T.E.; Rooney, T.P.; et al. Baricitinib in Patients with Refractory Rheumatoid Arthritis. N. Engl. J. Med. 2016, 374, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Genovese, M.C.; Takeuchi, T.; Hyslop, D.L.; Macias, W.L.; Rooney, T.; Chen, L.; Dickson, C.L.; Camp, J.R.; Cardillo, T.E.; et al. Safety Profile of Baricitinib in Patients with Active Rheumatoid Arthritis with over 2 Years Median Time in Treatment. J. Rheumatol. 2018, 46, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Xiao, S.; Zhang, Z. Response to: ’Impact of Janus kinase inhibitors on the risk of cardiovascular events in patients with rheumatoid arthritis: Systematic review and meta-analysis of randomised controlled trials’ by Lee and Song. Ann. Rheum. Dis. 2019, 78, 1048–1054. [Google Scholar] [CrossRef]
- Schlueter, M.; Finn, E.; Díaz, S.; Dilla, T.; Inciarte-Mundo, J.; Fakhouri, W. Cost-effectiveness analysis of baricitinib versus adalimumab for the treatment of moderate-to-severe rheumatoid arthritis in Spain. Clin. Outcomes Res. 2019, 11, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Ravasio, R.; Antonelli, S.; Rogai, V.; Fakhouri, W.; Capron, J.P.; Losi, S. Mean cost per number needed to treat of baricitinib versus adalimumab in the treatment of rheumatoid arthritis in Italy. Glob. Reg. Health Technol. Assess. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wehler, E.; Boytsov, N.; Nicolay, C.; Herrera-Restrepo, O.; Kowal, S. A Budget Impact and Cost Per Additional Responder Analysis for Baricitinib for the Treatment of Moderate-to-Severe Rheumatoid Arthritis in Patients with an Inadequate Response to Tumor Necrosis Factor Inhibitors in the USA. PharmacoEconomics 2019, 38, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Bermejo, I.; Simpson, E.; Wong, R.; Scott, D.L.; Young, A.; Stevenson, M. Baricitinib for Previously Treated Moderate or Severe Rheumatoid Arthritis: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. PharmacoEconomics 2018, 36, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Product Monograph, Xeljanz (Tofacitinib); FDA: New York, NY, USA, 2018.
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Chan, G.L.; Krishnaswami, S.; Prakash, C. The Pharmacokinetics, Metabolism, and Clearance Mechanisms of Tofacitinib, a Janus Kinase Inhibitor, in Humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yuan, L.; Yang, J.; Lei, Y.; Zhang, H.; Xia, L.; Shen, H.; Lu, J. Changes in Serum Cytokines May Predict Therapeutic Efficacy of Tofacitinib in Rheumatoid Arthritis. Mediat. Inflamm. 2019, 2019, 5617431. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tomalin, L.; Lee, J.; Fitz, L.J.; Berstein, G.; Da Rosa, J.C.; Garcet, S.; Lowes, M.; Valdez, H.; Wolk, R.; et al. Reduction of Inflammatory and Cardiovascular Proteins in the Blood of Patients with Psoriasis: Differential Responses between Tofacitinib and Etanercept after 4 Weeks of Treatment. J. Investig. Dermatol. 2018, 138, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; McCormick, J.; Orr, C.; Connolly, M.; Fearon, U.; Veale, D.J. Tofacitinib regulates synovial angiogenesis in psoriatic arthritis through induction of negative feedback inhibitors. Arthritis Rheumatol. 2014, 66, S1222–S1223. [Google Scholar]
- Gao, W.; McGarry, T.; Orr, C.; McCormick, J.; Veale, D.J.; Fearon, U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann. Rheum. Dis. 2015, 75, 311–315. [Google Scholar] [CrossRef]
- Kubo, S.; Yamaoka, K.; Kondo, M.; Yamagata, K.; Zhao, J.; Iwata, S.; Tanaka, Y. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann. Rheum. Dis. 2013, 73, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.F.; Tanaka, Y.; Lamba, M.; Collinge, M.; Hendrikx, T.; Hirose, T.; Toyoizumi, S.; Hazra, A.; Krishnaswami, S. THU0178 Relationship Between NK Cell Count and Important Safety Events in Rheumatoid Arthritis Patients Treated with Tofacitinib. Ann. Rheum. Dis. 2015, 74, 258.3–259. [Google Scholar] [CrossRef]
- Weinhold, K.J.; Bukowski, J.F.; Brennan, T.V.; Noveck, R.J.; Staats, J.; Lin, L.; Stempora, L.; Hammond, C.; Wouters, A.; Mojcik, C.F.; et al. Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin. Immunol. 2018, 191, 10–20. [Google Scholar] [CrossRef]
- Rizzi, M.; Lorenzetti, R.; Fischer, K.; Staniek, J.; Janowska, I.; Troilo, A.; Strohmeier, V.; Erlacher, M.; Kunze, M.; Bannert, B.; et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J. Autoimmun. 2017, 77, 55–66. [Google Scholar] [CrossRef]
- Yoshida, H.; Hunter, C.A. The Immunobiology of Interleukin-27. Annu. Rev. Immunol. 2015, 33, 417–443. [Google Scholar] [CrossRef]
- Fasching, P.; Stradner, M.; Graninger, W.; Dejaco, C.F.J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules 2017, 22, 134. [Google Scholar] [CrossRef]
- Damsky, W.; King, B. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef]
- Fleischmann, R.; Kremer, J.M.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S. Placebo-Controlled Trial of Tofacitinib Monotherapy in Rheumatoid Arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.B.; Fleischmann, R.; Hall, S.; Wilkinson, B.; Bradley, J.D.; Gruben, D.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.V.; Zang, C.; et al. Tofacitinib versus Methotrexate in Rheumatoid Arthritis. N. Engl. J. Med. 2014, 370, 2377–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, J.; Li, Z.-G.; Hall, S.; Fleischmann, R.; Genovese, M.; Martín-Mola, E.; Isaacs, J.D.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2013, 159, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Van Der Heijde, D.; Tanaka, Y.; Fleischmann, R.; Keystone, E.C.; Kremer, J.M.; Zerbini, C.; Cardiel, M.H.; Cohen, S.B.; Nash, P.; Song, Y.-W.; et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheumatol. 2013, 65, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.F.; Fleischmann, R.; Cohen, S.; Lee, E.B.; Meijide, J.A.G.; Wagner, S.; Forejtová, Š.; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or Adalimumab versus Placebo in Rheumatoid Arthritis. N. Engl. J. Med. 2012, 367, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Burmester, G.R.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef]
- Yamanaka, H.; Tanaka, Y.; Takeuchi, T.; Sugiyama, N.; Yuasa, H.; Toyoizumi, S.; Morishima, Y.; Hirose, T.; Zwillich, S.H. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: An open-label, long-term extension study. Arthritis Res. 2016, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Emery, P.; Van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): A randomised, double-blind, controlled phase 4 trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.J.; Moots, R.J.; Luo, Z.; Demasi, R.; Soma, K.; Zhang, R.; Takiya, L.; et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017, 390, 457–468. [Google Scholar] [CrossRef]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.; Moots, R.; Luo, Z.; Tatulych, S.; Demasi, R.; Soma, K.; Zhang, R.; et al. Tofacitinib with and without Methotrexate Versus Adalimumab with Methotrexate for the Treatment of Rheumatoid Arthritis: Patient-Reported Outcomes from a Phase 3b/4 Randomized Trial. Arthritis Rheumatol. 2017, 69, 10. [Google Scholar]
- van der Heijde, D.; Wollenhaupt, J.; Cohen, S.B.; Strengholt, S.; Terry, K.; Kwok, K.; DeMasi, R.; Lazariciu, L.; Wang, L. Assessment of radiographic progression in patients with rheumatoid arthritis treated with tofacitinib: Data from an open-label long-term extension study over 3 years. Arthritis Rheumatol. 2017, 69, 10. [Google Scholar]
- Ruperto, N.; Brunner, H.I.; Zuber, Z.; Tzaribachev, N.; Kingsbury, D.J.; Foeldvari, I.; Horneff, G.; Smolewska, E.; Vehe, R.K.; Hazra, A.; et al. Pharmacokinetic and safety profile of tofacitinib in children with polyarticular course juvenile idiopathic arthritis: Results of a phase 1, open-label, multicenter study. Pediatr. Rheumatol. 2017, 15, 86. [Google Scholar] [CrossRef]
- Huang, Z.; Lee, P.Y.; Yao, X.; Zheng, S.; Li, T. Tofacitinib Treatment of Refractory Systemic Juvenile Idiopathic Arthritis. Pediatrics 2019, 143, e20182845. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Hossain, A.; Ghogomu, E.T.; Kotb, A.; Christensen, R.; Mudano, A.S.; Maxwell, L.J.; Shah, N.P.; Tugwell, P.; Wells, G.A. Biologics or tofacitinib for rheumatoid arthritis in incomplete responders to methotrexate or other traditional disease-modifying anti-rheumatic drugs: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Hossain, A.; Ghogomu, E.T.; Mudano, A.S.; Maxwell, L.J.; Buchbinder, R.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E.; Tugwell, P.; Wells, G.A. Biologics or tofacitinib for people with rheumatoid arthritis unsuccessfully treated with biologics: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2017, 2017, 818. [Google Scholar] [CrossRef]
- Vieira, M.-C.; Zwillich, S.H.; Jansen, J.P.; Smiechowski, B.; Spurden, D.; Wallenstein, G.V. Tofacitinib Versus Biologic Treatments in Patients With Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Tumor Necrosis Factor Inhibitors: Results From a Network Meta-analysis. Clin. Ther. 2016, 38, 2628–2641.e5. [Google Scholar] [CrossRef] [Green Version]
- Bergrath, E.; Gerber, R.A.; Gruben, D.; Lukic, T.; Makin, C.; Wallenstein, G.V. Tofacitinib versus Biologic Treatments in Moderate-to-Severe Rheumatoid Arthritis Patients Who Have Had an Inadequate Response to Nonbiologic DMARDs: Systematic Literature Review and Network Meta-Analysis. Int. J. Rheumatol. 2017, 2017. [Google Scholar] [CrossRef]
- Straub, R.H.; Tellenbach, C.; Herzog, L.; Scherer, A.; Moeller, B.; Ciurea, A.; Von Muehlenen, I.; Gabay, C.; Kyburz, D.; Brulhart, L.; et al. Comparative effectiveness of antitumour necrosis factor agents, biologics with an alternative mode of action and tofacitinib in an observational cohort of patients with rheumatoid arthritis in Switzerland. RMD Open 2020, 6, e001174. [Google Scholar] [CrossRef]
- Reed, G.W.; Gerber, R.A.; Shan, Y.; Takiya, L.; Dandreo, K.J.; Gruben, D.; Kremer, J.; Wallenstein, G. Real-World Comparative Effectiveness of Tofacitinib and Tumor Necrosis Factor Inhibitors as Monotherapy and Combination Therapy for Treatment of Rheumatoid Arthritis. Rheumatol. Ther. 2019, 6, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, R.; Kremer, J.; Tanaka, Y.; Gruben, D.; Kanik, K.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.; Wilkinson, B.; Zwillich, S.H.; et al. Efficacy and safety of tofacitinib in patients with active rheumatoid arthritis: Review of key Phase 2 studies. Int. J. Rheum. Dis. 2016, 19, 1216–1225. [Google Scholar] [CrossRef]
- Sands, B.; Taub, P.R.; Armuzzi, A.; Friedman, G.S.; Moscariello, M.; Lawendy, N.; Pedersen, R.D.; Chan, G.; Nduaka, C.I.; Quirk, D.; et al. Tofacitinib Treatment Is Associated With Modest and Reversible Increases in Serum Lipids in Patients With Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2020, 18, 123–132.e3. [Google Scholar] [CrossRef] [Green Version]
- Salgado, E.; Gomez-Reino, J.J. The JAK inhibitor tofacitinib for active rheumatoid arthritis: Results from Phase III trials. Int. J. Clin. Rheumatol. 2013, 8, 315–326. [Google Scholar] [CrossRef]
- Schulze-Koops, H.; Strand, V.; Nduaka, C.; Demasi, R.; Wallenstein, G.; Kwok, K.; Wang, L. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology 2016, 56, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Radominski, S.C.; Gómez-Reino, J.J.; Wang, L.; Krishnaswami, S.; Wood, S.P.; Soma, K.; Nduaka, C.I.; Kwok, K.; Valdez, H.; et al. Analysis of Infections and All-Cause Mortality in Phase II, Phase III, and Long-Term Extension Studies of Tofacitinib in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 2924–2937. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Tanaka, Y.; Mariette, X.; Curtis, J.R.; Lee, E.B.; Nash, P.; Winthrop, K.L.; Charles-Schoeman, C.; Thirunavukkarasu, K.; Demasi, R.; et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: Integrated analysis of data from the global clinical trials. Ann. Rheum. Dis. 2017, 76, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Curtis, J.R.; Lee, E.B.; Kaplan, I.V.; Kwok, K.; Geier, J.; Benda, B.; Soma, K.; Wang, L.; Riese, R. Tofacitinib, an oral Janus kinase inhibitor: Analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann. Rheum. Dis. 2015, 75, 831–841. [Google Scholar] [CrossRef]
- European Medicines Agency. Restrictions in Use of Xeljanz While EMA Reviews Risk of Blood Clots in Lungs; EMA: Amsterdam, The Netherlands, 2019.
- Lee, M.-Y.; Park, S.-K.; Park, S.-Y.; Byun, J.-H.; Lee, S.-M.; Ko, S.-K.; Lee, E.-K. Cost-effectiveness of Tofacitinib in the Treatment of Moderate to Severe Rheumatoid Arthritis in South Korea. Clin. Ther. 2015, 37, 1662–1676.e2. [Google Scholar] [CrossRef]
- Bellinvia, S.; Edwards, C.J. JAK Inhibitors in the Treatment Algorithm of Rheumatoid Arthritis: A Review. EMJ Rheumatol. 2018, 5, 59–65. [Google Scholar]
- Kulikov, A.Y.; Komarov, I.; Zinchuk, I. Pharmacoeconomic Analysis Tofacitinib Use in Rheumatoid Arthritis Treatment Scheme. Value Health 2014, 17, A381. [Google Scholar] [CrossRef] [Green Version]
- Claxton, L.; Taylor, M.; Soonasra, A.; Bourret, J.A.; Gerber, R.A. An Economic Evaluation of Tofacitinib Treatment in Rheumatoid Arthritis After Methotrexate or After 1 or 2 TNF Inhibitors from a U.S. Payer Perspective. J. Manag. Care Spéc. Pharm. 2018, 24, 1010–1017. [Google Scholar] [CrossRef]
- Claxton, L.; Jenks, M.; Wallenstein, G.; Moynagh, D.; Taylor, M.; Mendelsohn, A.M.; Bourret, J.A.; Singh, A.; Gerber, R.A. An Economic Evaluation of Tofacitinib Treatment in Rheumatoid Arthritis: Modeling the Cost of Treatment Strategies in the United States. J. Manag. Care Spéc. Pharm. 2016, 22, 1088–1102. [Google Scholar] [CrossRef]
- Uttley, L.; Bermejo, I.; Ren, S.; James, M.M.; Wong, R.; Scott, D.L.; Young, A.; Stevenson, M. Tofacitinib for Treating Rheumatoid Arthritis After the Failure of Disease-Modifying Anti-rheumatic Drugs: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. PharmacoEconomics 2018, 36, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Martinez-Sesmero, J.M.; Balsa, A.; Peral, C.; Montoro, M.; Valderrama, M.; Gómez, S.; De Andrés-Nogales, F.; Casado, M.A.; Oyagüez, I. Cost-effectiveness analysis of treatment sequences containing tofacitinib for the treatment of rheumatoid arthritis in Spain. Clin. Rheumatol. 2020, 1–12. [Google Scholar] [CrossRef]
- Parmentier, J.M.; Voss, J.; Graff, C.; Schwartz, A.; Argiriadi, M.; Friedman, M.; Camp, H.S.; Padley, R.J.; George, J.S.; Hyland, D.; et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbbVie Receives FDA Approval of RINVOQ™ (upadacitinib), an Oral JAK Inhibitor for the Treatment of Moderate to Severe Rheumatoid Arthritis. 2019. Available online: https://news.abbvie.com/news/press-releases/abbvie-receives-fda-approval-rinvoq-upadacitinib-an-oral-jak-inhibitor-for-treatment-moderate-to-severe-rheumatoid-arthritis.htm (accessed on 16 August 2019).
- Genovese, M.C.; Smolen, J.S.; Weinblatt, M.E.; Burmester, G.R.; Meerwein, S.; Camp, H.S.; Wang, L.; Othman, A.A.; Khan, N.; Pangan, A.L.; et al. Efficacy and Safety of ABT-494, a Selective JAK-1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2016, 68, 2857–2866. [Google Scholar] [CrossRef] [Green Version]
- Kameda, H.; Takeuchi, T.; Yamaoka, K.; Oribe, M.; Kawano, M.; Zhou, Y.; Othman, A.A.; Pangan, A.L.; Kitamura, S.; Meerwein, S.; et al. Efficacy and safety of upadacitinib in Japanese patients with rheumatoid arthritis (SELECT-SUNRISE): A placebo-controlled phase IIb/III study. Rheumatology 2020, keaa084. [Google Scholar] [CrossRef]
- Burmester, G.R.; Kremer, J.M.; Bosch, F.V.D.; Kivitz, A.; Bessette, L.; Li, Y.; Zhou, Y.; Othman, A.A.; Pangan, A.L.; Camp, H.S. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 2503–2512. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Combe, B.; Hall, S.; Rubbert-Roth, A.; Zhang, Y.; Zhou, Y.; Mohamed, M.-E.F.; Meerwein, S.; Pangan, A.L. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): A double-blind, randomised controlled phase 3 trial. Lancet 2018, 391, 2513–2524. [Google Scholar] [CrossRef]
- Smolen, J.S.; Pangan, A.L.; Emery, P.; Rigby, W.; Tanaka, Y.; Vargas, J.I.; Zhang, Y.; Damjanov, N.; Friedman, A.; Othman, A.A.; et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): A randomised, placebo-controlled, double-blind phase 3 study. Lancet 2019, 393, 2303–2311. [Google Scholar] [CrossRef]
- Euctr, S.K. A Study to Compare Upadacitinib (ABT-494) Monotherapy to Methotrexate Monotherapy in Subjects with Rheumatoid Arthritis (RA) Who Have Not Previously Taken Methotrexate (SELECT-EARLY). 2015. Available online: http://WwwWhoInt/Trialsearch/Trial2Aspx?TrialID=EUCTR2015-003334-27-SK (accessed on 31 March 2019).
- Fleischmann, R.; Pangan, A.L.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.; Li, Y.; Zhou, Y.; Othman, A.A.; et al. A phase 3, randomized, double-blind study comparing upadacitinib to placebo and to adalimumab, in patients with active rheumatoid arthritis with inadequate response to methotrexate. Arthritis Rheumatol. 2018, 70, 988–990. [Google Scholar]
- Fleischmann, R.; Genovese, M.C.; Enejosa, J.V.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.; Li, Y.; Song, I.-H. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann. Rheum. Dis. 2019, 78, 1454–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischmann, R.; Pangan, A.L.; Song, I.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y.; et al. Upadacitinib Versus Placebo or Adalimumab in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase III, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Coppola, S.; Feng, T.; Camp, H.O.A. Lack of clinically-relevant effect of upadacitinib on plasma exposures of rosuvastatin and atorvastatin. Clin. Pharmacol. Drug Dev. 2019, 8, S1. [Google Scholar]
- Astellas Pharma. Oral JAK Inhibitor Smyraf® Tablets Approved in Japan for the Treatment of rheUmatoid Arthritis (Including Prevention of Structural Joint Damage) in Patients Who Have an Inadequate Response to Conventional Therapies. 2019. Available online: https://www.astellas.com/en/news/14651 (accessed on 11 February 2020).
- Markham, A.; Keam, S.J. Peficitinib: First Global Approval. Drugs 2019, 79, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Parganas, E.; Wang, D.; Stravopodis, D.J.; Topham, D.J.; Marine, J.-C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; Van Deursen, J.M.; et al. Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, D.; Shibata, T.; Shibata, M.; Kaneko, Y.; Oda, K.; Nishimura, T.; Katashima, M.; Sekino, H.; Furihata, K.; Urae, A. Pharmacokinetics and Safety of a Single Oral Dose of Peficitinib (ASP015K) in Japanese Subjects with Normal and Impaired Renal Function. Clin. Drug Investig. 2019, 40, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.J.; Sawamoto, T.; Valluri, U.; Cho, K.; Lewand, M.; Swan, S.; Lasseter, K.; Matson, M.; Holman, J.; Keirns, J.; et al. Pharmacokinetics, Pharmacodynamics, and Safety of ASP015K (Peficitinib), a New Janus Kinase Inhibitor, in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2016, 5, 435–449. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Iwasaki, M.; Ishikura, H.; Saeki, S.; Kaneko, Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: A 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 2015, 75, 1057–1064. [Google Scholar] [CrossRef]
- Genovese, M.C.; Greenwald, M.; Codding, C.; Zubrzycka-Sienkiewicz, A.; Kivitz, A.J.; Wang, A.; Shay, K.; Wang, X.; Garg, J.P.; Cardiel, M.H. Peficitinib, a JAK Inhibitor, in Combination With Limited Conventional Synthetic Disease-Modifying Antirheumatic Drugs in the Treatment of Moderate-to-Severe Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 932–942. [Google Scholar] [CrossRef]
- Kivitz, A.J.; Gutierrez-Ureña, S.R.; Poiley, J.; Genovese, M.C.; Kristy, R.; Shay, K.; Wang, X.; Garg, J.P.; Zubrzycka-Sienkiewicz, A. Peficitinib, a JAK Inhibitor, in the Treatment of Moderate-to-Severe Rheumatoid Arthritis in Patients With an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2017, 69, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Takeuchi, T.; Tanaka, S.; Kawakami, A.; Iwasaki, M.; Song, Y.W.; Chen, Y.-H.; Wei, J.C.-C.; Lee, S.-H.; Rokuda, M.; et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: A randomised, double-blind, placebo-controlled phase III trial (RAJ3). Ann. Rheum. Dis. 2019, 78, 1320–1332. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Tanaka, Y.; Tanaka, S.; Kawakami, A.; Iwasaki, M.; Katayama, K.; Rokuda, M.; Izutsu, H.; Ushijima, S.; Kaneko, Y.; et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann. Rheum. Dis. 2019, 78, 1305–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menet, C.J.; Fletcher, S.R.; Van Lommen, G.; Geney, R.; Blanc, J.; Smits, K.; Jouannigot, N.; Deprez, P.; Van Der Aar, E.M.; Clement-Lacroix, P.; et al. Triazolopyridines as Selective JAK1 Inhibitors: From Hit Identification to GLPG0634. J. Med. Chem. 2014, 57, 9323–9342. [Google Scholar] [CrossRef]
- Namour, F.; Diderichsen, P.M.; Cox, E.; Vayssière, B.; Van Der Aa, A.; Tasset, C.; Klooster, G.V. Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clin. Pharmacokinet. 2015, 54, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, F.; Mazur, M.; Voloshyn, O.; Stanislavchuk, M.; Van Der Aa, A.; Namour, F.; Galien, R.; Meuleners, L.; van’t Klooster, G. Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of Filgotinib, a Selective JAK-1 Inhibitor, After Short-Term Treatment of Rheumatoid Arthritis: Results of Two Randomized Phase IIa Trials. Arthritis Rheumatol. 2017, 69, 1949–1959. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, A.; Kremer, J.; Ponce, L.; Cseuz, R.; Reshetko, O.V.; Stanislavchuk, M.; Greenwald, M.; Van Der Aa, A.; Vanhoutte, F.; Tasset, C.; et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: Results from a randomised, dose-finding study (DARWIN 2). Ann. Rheum. Dis. 2016, 76, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Westhovens, R.; Taylor, P.C.; Alten, R.; Pavlova, D.; Enríquez-Sosa, F.; Mazur, M.; Greenwald, M.; Van Der Aa, A.; Vanhoutte, F.; Tasset, C.; et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: Results from a randomised, dose-finding study (DARWIN 1). Ann. Rheum. Dis. 2016, 76, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.C.; Kalunian, K.; Gottenberg, J.-E.; Mozaffarian, N.; Bartok, B.; Matzkies, F.; Gao, J.; Guo, Y.; Tasset, C.; Sundy, J.S.; et al. Effect of Filgotinib vs Placebo on Clinical Response in Patients With Moderate to Severe Rheumatoid Arthritis Refractory to Disease-Modifying Antirheumatic Drug Therapy. JAMA Am. Med. Assoc. 2019, 322, 315–325. [Google Scholar] [CrossRef]
- Elwood, F.; Witter, D.J.; Piesvaux, J.; Kraybill, B.; Bays, N.; Alpert, C.; Goldenblatt, P.; Qu, Y.; Ivanovska, I.; Lee, H.-H.; et al. Evaluation of JAK3 Biology in Autoimmune Disease Using a Highly Selective, Irreversible JAK3 Inhibitor. J. Pharmacol. Exp. Ther. 2017, 361, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Farmer, L.J.; Ledeboer, M.W.; Hoock, T.; Arnost, M.J.; Bethiel, R.S.; Bennani, Y.L.; Black, J.J.; Brummel, C.L.; Chakilam, A.; Dorsch, W.A.; et al. Discovery of VX-509 (Decernotinib): A Potent and Selective Janus Kinase 3 Inhibitor for the Treatment of Autoimmune Diseases. J. Med. Chem. 2015, 58, 7195–7216. [Google Scholar] [CrossRef]
- Fleischmann, R.; Damjanov, N.S.; Kivitz, A.J.; Legedza, A.; Hoock, T.; Kinnman, N. A Randomized, Double-Blind, Placebo-Controlled, Twelve-Week, Dose-Ranging Study of Decernotinib, an Oral Selective JAK-3 Inhibitor, as Monotherapy in Patients With Active Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 334–343. [Google Scholar] [CrossRef]
- Genovese, M.C.; Van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an Oral Selective JAK-3 Inhibitor, in Combination With Methotrexate in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 68, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Yang, F.; Østergaard, M.; Kinnman, N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: Clinical and MRI findings. Ann. Rheum. Dis. 2016, 75, 1979–1983. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public Summary of Opinion on Orphan Designation Itacitinib for Treatment of Graft-Versus-Host Disease; EMA: London, UK, 2018.
- Genovese, M.C.; Jarosova, K.; Cieślak, D.; Alper, J.; Kivitz, A.; Hough, D.; Maes, P.; Pineda, L.; Chen, M.; Zaidi, F. Apremilast in patients with active rheumatoid arthritis: A phase II, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheumatol. 2015, 67, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, N.; Barbour, A.M.; Epstein, N.; Zhou, G.; Petusky, S.; Xun, Z.; Yuska, B.; Marbury, T.; Chen, X.; Yeleswaram, S.; et al. The Effect of Renal Impairment on the Pharmacokinetics and Safety of Itacitinib. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Darpo, B.; Xue, H.; Punwani, N.; He, K.; Barbour, A.M.; Epstein, N.; Landman, R.; Chen, X.; Yeleswaram, S. Evaluation of Clinical Cardiac Safety of Itacitinib, a JAK1 Inhibitor, in Healthy Participants. Clin. Pharmacol. Drug Dev. 2019. [Google Scholar] [CrossRef]
- Mok, C.C. The Jakinibs in systemic lupus erythematosus: Progress and prospects. Expert Opin. Investig. Drugs 2018, 28, 85–92. [Google Scholar] [CrossRef]
- Dodington, D.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Ligand | Type of Cytokine Receptor | Associated JAK Subtype | Associated STAT Subtype | Main Target Organ/Cell | Diseases Linked to an Altered Pathway | References |
|---|---|---|---|---|---|---|
| IL-2 | Type I | JAK1 JAK3 | STAT3 STAT5A/B | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease, ankylosing spondylitis | [38,39,40,41,42,43] |
| IL-3 | Type I | JAK2 | STAT3 STAT5A/B STAT6 | Th2 lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis | [38,39,44] |
| IL-4 | Type I | JAK1 JAK3 | STAT6 | naïve T lymphocytes, Th2 lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, psoriasis, ankylosing spondylitis, ulcerative colitis | [38,39,45] |
| IL-5 | Type I | JAK2 | STAT3 STAT5A/B STAT6 | Th2 lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis | [38,39,44] |
| IL-6 | Type I | JAK1 JAK2 TYK2 | STAT1 STAT3 | naïve T lymphocytes, human keratinocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis, psoriasis | [11,38,39,46,47] |
| IL-7 | Type I | JAK1 JAK3 | STAT3 STAT5A/B | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease, ankylosing spondylitis | [38,39,40,41,42,43] |
| IL-9 | Type I | JAK1 JAK3 | STAT3 STAT5A/B | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease, ankylosing spondylitis | [38,39,40,41,42,43] |
| IL-10 | Type II | JAK1 JAK2 TYK2 | STAT3 STAT5A/B | Treg lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus | [38,39,48] |
| IL-11 | Type I | JAK1 JAK2 TYK2 | STAT1 STAT3 | naïve T lymphocytes, immunoglobulin-producing B cells, hematopoietic stem cells, megakaryocyte progenitor cells | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis, anemia, leukopenia | [11,38,39,46,47] |
| IL-12 | Type I | JAK2 TYK2 | STAT4 | naïve T lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis | [11,38,39,49] |
| IL-13 | Type I | JAK1 JAK2 JAK3 TYK2 | STAT6 | Th2 lymphocytes, human bronchial smooth muscle cells | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, asthma | [38,39,50] |
| IL-15 | Type I | JAK1 JAK3 | STAT3 STAT5A/B | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease, ankylosing spondylitis | [38,39,40,41,42,43] |
| IL-19 | Type II | JAK1 JAK2 TYK2 | STAT3 | innate immune system | / | [38,39,51] |
| IL-20 | Type II | JAK1 JAK2 TYK2 | STAT3 | innate immune system, human keratinocytes | psoriasis | [38,39,52,53] |
| IL-21 | Type I | JAK1 JAK3 | STAT3 STAT5A/B | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease, ankylosing spondylitis | [38,39,40,41,42,43,54] |
| IL-22 | Type II | JAK1 JAK2 TYK2 | STAT1 STAT3 STAT5A/B | Th17 lymphocytes | alopecia, rheumatoid arthritis, systemic lupus erythematosus, Crohn’s disease, ankylosing spondylitis, psoriasis | [38,39,53] |
| IL-23 | Type I | TYK2 JAK2 | STAT3 STAT4 | naïve T lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis | [11,38,39,49,55] |
| IL-24 | Type II | JAK1 | STAT3 | immune cells, colonic epithelial cells | inflammatory bowel disease | [38,39,56] |
| IL-27 | Type I | JAK1 JAK2 TYK2 | STAT1 STAT2 STAT3 STAT4 STAT5A/B | Th1 lymphocytes, cytotoxic T cell, Treg lymphocytes | autoimmune disorders | [38,39,57] |
| IL-28 | Type II | JAK1 TYK2 | STAT1 STAT2 STAT3 STAT4 STAT5A/B | immune cells, human keratinocytes | / | [38,39,54] |
| IL-29 | Type II | JAK1 TYK2 | STAT1 STAT2 STAT3 STAT4 STAT5A/B | immune cells | / | [38,39,54] |
| IL-31 | Type I | JAK1 JAK2 | STAT1 STAT3 STAT5A/B | lung, skin, thymus, spleen, myelomonocytic cells | atopic dermatitis | [38,58] |
| IFN-α/β | Type II | JAK1 TYK2 | STAT1 STAT2 STAT4 STAT3 | naïve T lymphocytes, Th1 lymphocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis | [11,38,39,59,60] |
| IFN-γ | Type II | JAK1 TYK2 | STAT1 | naïve T lymphocytes, Th1 lymphocytes, human salivary glands, human keratinocytes | alopecia, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus, ulcerative colitis, Crohn’s disease, ankylosing spondylitis, psoriasis | [38,39,61] |
| GM-CSF and G-CSF | Type I | JAK2 | STAT3 STAT5A/B | cells belonging to the neutrophil lineage, from haemopoietic stem cells to mature neutrophils, antigen presenting cells | anemia, leukopenia, autoimmune disorders, acute myelogenous leukemia | [38,39,62,63] |
| GH | Type I | JAK2 | STAT3 STAT5A/B | all tissues | / | [38,39,50,64,65] |
| EPO | Type I | JAK2 | STAT5A/B | erythroid precursor cell at colony-forming units | anemia | [38,39,66] |
| Thrombopoietin | Type I | JAK2 | STAT1 STAT3 STAT5A/B | earliest erythroid progenitors | thrombocytopenia | [38,39,67] |
| Leptin | Type I | JAK2 | STAT3 STAT5A/B | brain, peripheral tissues | / | [38,39,68] |
| Study | RA-BEGIN MTX-Naïve (n = 588) | RA-BEAM MTX-IR (n = 1308) | RA-BUILD cDMARD-IR (n = 684) | RA-BEACON bDMARD-IR (n = 527) | RA-BEYOND OLE Study (n = 3073) |
|---|---|---|---|---|---|
| Inclusion criteria |
|
|
|
|
|
| Type of therapy | Monotherapy + combination therapy | Combination therapy | Combination therapy | Combination therapy | Monotherapy—patients who completed previous BARI RA studies |
| Background treatment | None/MTX | MTX | cDMARDs | cDMARDs | cDMARDs |
| Active comparator | MTX | ADA + MTX | |||
| Arms | (1) BARI 4 mg sid (2) BARI 4 mg sid + MTX (3) MTX 10 mg/week | (1) PBO (2) BARI 4 mg sid (3) ADA 40 mg/sc q2wk | (1) BARI 2 mg sid (2) BARI 4 mg sid (3) PBO | (1) BARI 2 mg sid (2) BARI 4 mg sid (3) PBO | (1) BARI 2 mg sid (2) BARI 4 mg sid |
| Duration (weeks) | 52 | 52 | 24 | 24 | Ongoing (completion estimated in 2024) |
| Primary endpoint | ACR20 (Week 24) | ACR20 (Week 12) | ACR20 (Week 12) | ACR20 (Week 12) | Long term Safety |
| Key secondary endpoint | Week 24: DAS28-CRP HAQ-DI mTSS SDAI remission | Week 12: DAS28-CRP HAQ-DI mTSS (Week 24) SDAI remission Morning Joint stiffness | Week 12: DAS28-CRP HAQ-DI SDAI remission Morning Joint stiffness | Week 12: DAS28-CRP HAQ-DI SDAI remission | Long term Efficacy |
| Main results (ACR20): | (Week 24) BARI 4 mg vs. MTX: 77% vs. 62% (p ≤ 0.01); BARI 4 mg vs. BARI 4 mg + MTX: 77% vs. 78% (Week 52) BARI 4 mg vs. MTX: 73% vs. 56% (p ≤ 0.05); BARI 4 mg vs. BARI 4 mg + MTX: 73% vs. 73% | (Week 12) BARI vs. PBO: 70% vs. 40% (p < 0.001); BARI vs. ADA: 70% vs. 61% (p = 0.014) (week 24) BARI vs. PBO: 74% vs. 37% (p < 0.001); BARI vs. ADA: 74% vs. 66% (p ≤ 0.05) | (Week 12) BARI 2 mg vs. PBO: 66% vs. 39% (p ≤ 0.001); BARI 4 mg vs. PBO: 62% vs. 39% (p ≤ 0.001) | (Week 12) BARI 2 mg vs. PBO: 49% vs. 27% (p < 0.001); BARI 4 mg vs. PBO: 55% vs. 27% (p < 0.001) (Week 24) BARI 2 mg vs. PBO: 45% vs. 27% (p ≤ 0.001); BARI 4 mg vs. PBO: 46% vs. 27% (p ≤ 0.001) | Currently recruiting |
| Number of Individual Cases by Reaction Groups (Updated on 28 February 2020) | |||
|---|---|---|---|
| Reaction Groups | VigiAccess | Eudravigilance | FAERS |
| Blood and lymphatic system disorders | 176 (2.0%) | 98 (3.5%) | 46 (2.2%) |
| Cardiac disorders | 117 (1.3%) | 57 (2.1%) | 54 (2.6%) |
| Congenital, familial and genetic disorders | 1 (0.0%) | 1(0.0%) | 2 (0.1%) |
| Ear and labyrinth disorders | 41 (0.5%) | 15 (0.5%) | 11 (0.5%) |
| Endocrine disorders | 7 (0.1%) | 1 (0.0%) | 1 (0.0%) |
| Eye disorders | 110 (1.3%) | 36 (1.3%) | 32 (1.6%) |
| Gastrointestinal disorders | 879 (10.0%) | 310 (11.2%) | 181(8.8%) |
| General disorders and administration site conditions | 1308 (14.9%) | 326 (11.8%) | 282(13.7%) |
| Hepatobiliary disorders | 66 (0.8%) | 42 (1.5%) | 46 (2.2%) |
| Immune system disorders | 88 (1.0%) | 15 (0.5%) | 26 (1.3%) |
| Infections and infestations | 2095 (23.9%) | 615 (22.2%) | 371 (18.0%) |
| Injury, poisoning and procedural complications | 348 (4.0%) | 77 (2.8%) | 91(4.4%) |
| Metabolism and nutrition disorders | 162 (1.8%) | 75 (2.7%) | 28 (1.4%) |
| Musculoskeletal and connective tissue disorders | 889 (10%) | 151 (5.4%) | 129 (6.3%) |
| Neoplasms benign, malignant and unspecified (including cyst and polyps) | 99 (1.1%) | 70 (2.5%) | 100 (4.9%) |
| Nervous system disorders | 566 (6.5%) | 175 (6.3%) | 158 (7.7%) |
| Pregnancy, puerperium and perinatal conditions | 4 (0.0%) | 4 (0.1%) | 1 (0.0%) |
| Product issues | 3 (0.0%) | 0 (0.0%) | 3 (0.1%) |
| Psychiatric disorders | 206 (2.3%) | 56 (2.0%) | 47 (2.3%) |
| Renal and urinary disorders | 134 (1.5%) | 40 (1.4%) | 37 (1.8%) |
| Reproductive system and breast disorders | 49 (0.6%) | 16 (0.6%) | 5 (0.2%) |
| Respiratory, thoracic and mediastinal disorders | 612 (7%) | 247 (8.9%) | 170 (8.3%) |
| Skin and subcutaneous tissue disorders | 504 (5.7%) | 199 (7.2%) | 89 (4.3%) |
| Surgical and medical procedures | 60 (0.7%) | 1 (0.0%) | 73 (3.5%) |
| Vascular disorders | 248 (2.8%) | 147 (5.3%) | 75 (3.6%) |
| Total | 8772 (100%) | 2774 (100%) | 2058 (100%) |
| Trial | ORAL Start MTX-naïve (n = 958) | ORAL Solo c/bDMARD-IR (n = 611) | ORAL Sync c/bDMARD-IR (n = 795) | ORAL Scan MTX-IR (n = 797) | ORAL Standard MTX-IR (n = 717) | ORAL Strategy MTX-IR (n = 1146) | ORAL Step TNF-IR (n = 399) |
|---|---|---|---|---|---|---|---|
| Participants | MTX-naïve patients with active RA | Active RA patients with inadequate response to ≥ 1 c/bDMARD receiving stable doses of antimalarial | Active RA patients with inadequate response to ≥ 1 c/bDMARD | Active RA patients receiving background MTX | Active RA patients receiving stable doses of MTX | Active RA patients receiving stable doses of MTX | Moderate to severe RA patients with inadequate response to anti-TNF drugs |
| Type of therapy | Monotherapy | Monotherapy | Combination therapy | Combination therapy | Combination therapy | Monotherapy | Combination therapy |
| Active Comparator | MTX | / | / | / | ADA | ADA + MTX non-inferiority | / |
| Background treatment | None | None | cDMARD | MTX | MTX | None | MTX |
| Arms | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) MTX | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) PBO advanced at 3 months to TOFA 5 mg bid or 10 mg bid | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) PBO advanced to TOFA 5 mg bid or 10 mg bid at 6 months (3 months for non-responders) | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) PBO advanced to TOFA 5 mg bid or 10 mg bid at 6 months (3 months for non-responders) | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) PBO advanced to TOFA 5 mg bid or 10 mg bid at 6 months (3 months for non-responders) (4) ADA | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) ADA + MTX | (1) TOFA 5 mg bid (2) TOFA 10 mg bid (3) PBO advanced to TOFA 5 mg bid or 10 mg bid at 3 months |
| Duration (months) | 24 | 6 | 12 | 24 | 12 | 12 | 6 |
| Features | X-Rays | X-Rays | |||||
| Coprimary endpoints | ∆mTSS ACR70 (month 6) | ACR20 HAQ-DI DAS28-ESR < 2.6 (month 3) | ACR20 DAS28-ESR < 2.6 (month 6) HAQ-DI (month 3) | ACR20 ∆mTSS DAS28-ESR < 2.6 (month 6) HAQ-DI (month 3) | ACR20 DAS28-(ESR) < 2.6 (month 6) HAQ-DI (month 3) | ACR50 (month 6) | ACR20 HAQ-DI DAS28-ESR < 2.6 (month 3) |
| Main results | (Month 6) ACR20 (% pts): 71.3 (p < 0.001) TOFA 5 mg; 76.1 (p ≤ 0.01) TOFA 10 mg; 50.5 MTX. ACR70 (% pts): 25.5 (p < 0.001) TOFA 5 mg; 37.7 (p ≤ 0.01) TOFA 10 mg; 12 MTX. HAQ-DI (change from BL): −0.8 (p < 0.001) TOFA 5 mg; −0.9 (p < 0.001) TOFA 10 mg; −0.6 MTX. DAS28-ESR < 2.6 (% pts): 14.6 (p ≤ 0.05) TOFA 5 mg; 21.8 (p ≤ 0.01) TOFA 10 mg; 7.6 MTX. ∆mTSS (from baseline): 0.2 (p < 0.001) TOFA 5 mg; <0.1 (p < 0.001) TOFA 10 mg; 0.8 MTX | (Month 3) ACR20 (% pts): 59.8 (p < 0.001) TOFA 5 mg; 65.7 (p < 0.001) TOFA 10 mg; 26.7 PBO. HAQ-DI (change from BL): −0.5 (p < 0.001) TOFA 5 mg; −0.57 (p < 0.001) TOFA 10 mg; −0.19 PBO. DAS28-ESR < 2.6 (% pts): 5.6 TOFA 5 mg; 8.7 TOFA 10 mg; 4.4 PBO | (Month 6) ACR20 (%pts): 52.7 (p < 0.001) TOFA 5 mg+ cDMARD; 56.6 (p < 0.001) TOFA 10 mg + cDMARD; 31.2 PBO + cDMARD. HAQ-DI (change from BL): −0.44 (p < 0.001) TOFA 5 mg + cDMARD; −0.53 (p < 0.001) TOFA 10 mg+ cDMARD; −0.21 PBO + cDMARD. DAS28-ESR < 2.6 (% pts): 8.5 (p ≤ 0.01) TOFA 5 mg + cDMARD; 12.5 (p < 0.001) TOFA 10 mg + cDMARD; 2.7 PBO + cDMARD | (Month 6) ACR20 (%pts): 51.5 (p < 0.001) TOFA 5 mg + MTX; 61.8 (p < 0.001) TOFA 10 mg + MTX; 25.3 PBO + MTX. DAS28-ESR < 2.6 (% pts): 7.2 TOFA 5 mg + MTX; 16.0 (p < 0.001) TOFA 10 mg + MTX; 1.6 PBO + MTX. HAQ-DI (change from BL): −0.40 TOFA 5 mg + MTX; −0.54 (p < 0.001) TOFA 10 mg + MTX; −0.15 PBO + MTX. ∆mTSS (from baseline): 0.12 TOFA 5 mg + MTX; 0.06 (p ≤ 0.05) TOFA 10 mg + MTX; 0.47 PBO + MTX | (Month 6) ACR20 (%pts): 51.5 (p < 0.001) TOFA 5 mg + MTX; 52.6 (p < 0.001) TOFA 10 mg + MTX; 47.2 (p < 0.001) ADA + MTX; 28.3 PBO+ MTX. HAQ-DI (change from BL): −0.55 (p < 0.001) TOFA 5 mg+ MTX; −0.61 (p < 0.001) TOFA 10 mg + MTX; −0.49 (p < 0.001) ADA + MTX; −0.24 PBO + MTX. DAS28-ESR < 2.6 (% pts): 7 (p ≤ 0.05) TOFA 5 mg + MTX; 12.5 (p < 0.001) TOFA 10 mg + MTX; 6.7 (p ≤ 0.05) ADA + MTX; 1.1 PBO + MTX | (Month 6) ACR20 (% pts): 65 TOFA 5 mg; 73.1 TOFA 5 mg + MTX; 71 ADA+ MTX. ACR50 (% pts): 38.3 TOFA 5 mg; 46 TOFA 5 mg + MTX; 44 ADA+ MTX. HAQ-DI (change from BL): −0.54 TOFA 5 mg; −0.59 TOFA 5 mg + MTX; −0.54 ADA + MTX. DAS28-ESR < 2.6 (% pts): 10.4 TOFA 5 mg; 12 TOFA 5 mg + MTX; 12.4 ADA + MTX. | (Month 3) ACR20 (% pts): 41.7 (p ≤ 0.01)) TOFA 5 mg + MTX; 48.1 (p < 0.001) TOFA 10 mg + MTX; 24.4 PBO. + MTX HAQ-DI (change from BL): −0.43 (p < 0.001) TOFA 5 mg + MTX; −0.46 (p < 0.001) TOFA 10 mg + MTX; −0.18 PBO + MTX. DAS28-ESR < 2.6 (% pts): 6.7 (p ≤ 0.05) TOFA 5 mg + MTX; 8.8 (p ≤ 0.05) TOFA 10 mg + MTX; 1.7 PBO + MTX |
| Number of Individual Cases by Reaction Groups (Updated on 28 February 2020) | |||
|---|---|---|---|
| Reaction Groups | VigiAccess | Eudravigilance | FAERS |
| Blood and lymphatic system disorders | 959 (0.8%) | 749 (1.3%) | 1276 (1.0%) |
| Cardiac disorders | 1302(1.1%) | 1240 (2.1%) | 1870 (1.4%) |
| Congenital, familial and genetic disorders | 51(0.0%) | 51 (0.1%) | 91 (0.1%) |
| Ear and labyrinth disorders | 792 (0.7%) | 442 (0.8%) | 985 (0.7%) |
| Endocrine disorders | 157 (0.1%) | 145 (0.2%) | 262 (0.2%) |
| Eye disorders | 1681 (1.4%) | 1173 (2.0%) | 2176 (1.6%) |
| Gastrointestinal disorders | 10,951 (9.4%) | 4894 (8.4%) | 12,200 (9.1%) |
| General disorders and administration site conditions | 29,273 (25.2%) | 10,533 (18.0%) | 31,152 (23.2%) |
| Hepatobiliary disorders | 595 (0.5%) | 537 (0.9%) | 939 (0.7%) |
| Immune system disorders | 1809 (1.6%) | 1052 (1.8%) | 2560 (1.9%) |
| Infections and infestations | 16,042 (13.8%) | 8940 (15.3%) | 16,908 (12.6%) |
| Injury, poisoning and procedural complications | 8189 (7.0%) | 5028 (8.6%) | 10,407 (7.8%) |
| Metabolism and nutrition disorders | 1385 (1.2%) | 980 (1.7%) | 1728 (1.3%) |
| Musculoskeletal and connective tissue disorders | 12,602 (10.8%) | 6497 (11.1%) | 15271 (11.4%) |
| Neoplasms benign, malignant and unspecified (including cyst and polyps) | 1587 (1.4%) | 1611 (2.8%) | 2358 (1.8%) |
| Nervous system disorders | 8910 (7.7%) | 4252 (7.3%) | 9929 (7.4%) |
| Pregnancy, puerperium and perinatal conditions | 29 (0.0%) | 33 (0.1%) | 59 (0.0%) |
| Product issues | 86 (0.1%) | 49 (0.1%) | 112 (0.1%) |
| Psychiatric disorders | 3026 (2.6%) | 1502 (2.6%) | 3567 (2.7%) |
| Renal and urinary disorders | 1508 (1.3%) | 1077 (1.8%) | 1869 (1.4%) |
| Reproductive system and breast disorders | 405 (0.3%) | 223 (0.4%) | 460 (0.3%) |
| Respiratory, thoracic and mediastinal disorders | 6594 (5.7%) | 3465 (5.9%) | 7725 (5.8%) |
| Skin and subcutaneous tissue disorders | 5706 (4.9%) | 2306 (3.9%) | 6684 (5.0%) |
| Surgical and medical procedures | 865 (0.7%) | 216 (0.4%) | 1289 (1.0%) |
| Vascular disorders | 1832 (1.6%) | 1516 (2.6%) | 2403 (1.8%) |
| Total | 116,336 (100%) | 58,511 (100%) | 134,280 (100%) |
| Drug | First generation JAKi | Second Generation JAKi | ||||
|---|---|---|---|---|---|---|
| active principle | Baricitinib | Tofacitinib | Upadacitinib | Peficitinib | Filgotinib | Decernotinib |
| brand name | Olumiant® | Xeljanz® | Rinvoq™ | Smyraf® | / | / |
| other name | INCB028050 LY3009104 | CP-690,550 | ABT-494 | ASP015K, JNJ-54781532 | GLPG0634/GS-6034 | VX-509 |
| target | JAK1 JAK2 | JAK1 JAK3 JAK2 TYK2 | JAK1 | JAK3 JAK1 | JAK1 | JAK3 |
| Dose | 2 mg sid | 5 mg bid | 15 mg sid | 150 mg sid or 100 mg depending on the patient’s condition | 100 mg or 200 mg sid | 50–150 mg bid |
| renal insufficiency | 1 mg once daily in patients with creatinine clearance between 30 and 60 mL/min Not recommended in patients with creatinine clearance < 30 mL/min | No dose adjustment in patients with mild (50–80 mL/min) or moderate (30–49 mL/min) renal impairment 5 mg once daily in patient with severe renal impairment (<30 mL/min) | No dose adjustment in patients with mild, moderate or severe renal impairment Not tested in subjects with end stage renal disease | No dose adjustment required in patients with renal impairment | / | / |
| liver failure | No dose adjustment in patients with mild or moderate hepatic impairment Not recommended in patients with severe hepatic impairment | No dose adjustment in patients with mild (Child Pugh A) hepatic impairment 5 mg once daily recommended in patient with moderate hepatic function (<Child Pugh B) Not recommended for use in patients with severe hepatic function (Child Pugh C) | No dose adjustment in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment Not recommended in patients with severe hepatic impairment (Child Pugh C) | 50 mg sid in patients with moderate liver dysfunction Contraindicated in patient with severe liver dysfunction | / | / |
| development stage for rheumatoid arthritis | Approved in ∼50 countries | Approved in ∼80 countries | Approved in US and Eu | Approved in Japan Under regulatory review in South Korea and Taiwan Under clinical development in China | Under regulatory review is US, EU and Japan | Development discontinued |
| approval | FDA approval: yes (2018) EMA approval: yes (2017) | FDA approval: yes (2012) EMA approval: yes (2017) | FDA approval: yes (2019) EMA approval: yes (2019) | FDA approval: no EMA approval: no Japan approval: yes (2019) | FDA approval: no EMA approval: no | FDA approval: no EMA approval: no |
| safety | Most frequent: throat and nose infections; herpes simplex virus infection; infections causing a sick stomach or diarrhea; urinary infections; pneumonia; thrombocytosis nausea; Uncommon: leukopenia; increase in serum creatine kinase; high serum levels of triglycerides; acne; weight gain | Most frequent: upper and lower airway infections; influenza; herpes zoster virus infection; urinary tract infections; abdominal pain; vomiting; diarrhea; nausea; gastritis; rash; headache; anemia; leukopenia; hyper-transaminasemia Uncommon: tuberculosis; diverticulitis; pyelonephritis; cellulitis; herpes simplex virus infection; viral gastroenteritis and other viral infections; blood creatinine increase; blood cholesterol increase; low density lipoprotein increase; weight increase Rare: sepsis; disseminated tuberculosis involving bones and other organs; other unusual infections; joint infection | Most frequent: upper respiratory tract infections (common cold, sinus infections); nausea; cough; fever Uncommon: serious infections; malignancies; thrombosis; gastrointestinal perforations; altered laboratory parameters; embryo-fetal toxicity Rare: major adverse cardiac events pulmonary embolism; venous thromboembolism | Most frequent: nasopharyngitis; herpes zoster virus infection; blood creatine kinase increase; lymphopenia Uncommon: pneumonia; pharyngitis; epipharyngitis; upper respiratory tract infections bronchitis; influenza; cystitis Rare: sepsis; gastrointestinal perforation | Most frequent: nasopharyngitis; nausea; bronchitis; headache; upper respiratory tract infection Uncommon: major adverse cardiac events pulmonary embolism; herpes zoster virus infection; deep vein thrombosis | Most frequent: nausea; headache; nasopharyngitis; diarrhea; upper respiratory tract infections; blood cholesterol increase; alanine aminotransferase increase Uncommon: serious infections |
| Licensed therapeutic indications | rheumatoid arthritis | rheumatoid arthritis, psoriatic arthritis, ulcerative colitis | rheumatoid arthritis | rheumatoid arthritis | Under regulatory review for rheumatoid arthritis | |
| PK | Tmax 1.5 h; t1/2 12.5 h | Tmax 0.5–1 h; t1/2 3.3 h | Tmax 2–4 h; t1/2 8-14 h | Tmax 1.1–2.1 h; t1/2 9.9–16.2 h (after multiple 30–200 mg twice daily dosages) | Tmax 0,5–3 h; t1/2 3.82–10.9 h (after single/multiple twice daily 25–450 mg dosages) Tmax 1–2; t1/2 (after single/multiple twice daily 30–300 mg dosages) | N.A. |
| IC50 | IC50JAK1 5.9 nM; IC50JAK2 5.7 nM; IC50JAK3 420 nM; IC50TYK2 60 nM | IC50JAK1 3.2 nM; IC50JAK2 4.1 nM; IC50JAK3 1.6 nM; IC50TYK2 34 nM | IC50JAK1 45 nM; IC50JAK2 109 nM; IC50JAK3 2.1 μM; IC50TYK2 4.7 µM | IC50JAK1 3.9 nM; IC50JAK2 5.0 nM; IC50JAK3 0.71 nM; IC50Tyk2 4.8 nM | IC50JAK1 10 nM; IC50JAK2 28 nM; IC50JAK3 810 nM; IC50Tyk2 110 nM | IC50JAK1 10 nM; IC50JAK2 10 nM; IC50JAK3 2.5 nM; IC50TYK2 10 nM |
| drug interactions | OAT3 inhibitors and CYP3A4 inhibitors (e.g., ketoconazole) and inducers (e.g., rifampicin) | CYP3A4 inhibitors (e.g., ketoconazole), medicinal products that result in both moderate inhibition of CYP3A4 as well as potent inhibition of CYP2C19 (e.g., fluconazole) and CYP inducers | CYP3A4 inhibitors (e.g., ketoconazole) and inducers (e.g., rifampicin) | Verapamil (P-glycoprotein inhibitor that increase AUC and Cmax of peficitinib by 27–39%) No clinically significant interaction with methotrexate and rosuvastatin | / | CYP3A4 inhibitors |
| / | / | / | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Dal Cin, L.; Brancati, S.; Scaglione, F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules 2020, 10, 1002. https://doi.org/10.3390/biom10071002
Angelini J, Talotta R, Roncato R, Fornasier G, Barbiero G, Dal Cin L, Brancati S, Scaglione F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules. 2020; 10(7):1002. https://doi.org/10.3390/biom10071002
Chicago/Turabian StyleAngelini, Jacopo, Rossella Talotta, Rossana Roncato, Giulia Fornasier, Giorgia Barbiero, Lisa Dal Cin, Serena Brancati, and Francesco Scaglione. 2020. "JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future" Biomolecules 10, no. 7: 1002. https://doi.org/10.3390/biom10071002