A metagenomic insight into the Yangtze finless porpoise virome
 Zhigang Liu1,2*†
Zhigang Liu1,2*†  Xin Ding1† Muhammad Shahan Haider3 Farah Ali4 Han Yu1 Xin Chen1 Shuaishuai Tan1 Yuan Zu1 Wenlong Liu1 Bangzhi Ding1 Aifang Zheng1 Jinsong Zheng5 Zhengyi Qian6 Hassan Ashfaq7 Daoping Yu1,2
Xin Ding1† Muhammad Shahan Haider3 Farah Ali4 Han Yu1 Xin Chen1 Shuaishuai Tan1 Yuan Zu1 Wenlong Liu1 Bangzhi Ding1 Aifang Zheng1 Jinsong Zheng5 Zhengyi Qian6 Hassan Ashfaq7 Daoping Yu1,2  Kun Li8,9*
Kun Li8,9*- 1College of Life Science, Anqing Normal University, Anqing, China
- 2Research Center of Aquatic Organism Conservation and Water Ecosystem Restoration in Anhui Province, Anqing Normal University, Anqing, China
- 3Bahria University Medical and Dental College Karachi, Karachi, Pakistan
- 4Department of Theriogenology, Faculty of Veterinary and Animal Sciences, The Islamia University of Bahawalpur, Bahawalpur, Pakistan
- 5Institute of Hydrobiology, Chinese Academy of Sciences, Beijing, China
- 6Hubei Yangtze River Ecological Protection Foundation, Wuhan, China
- 7Institute of Continuing Education and Extension, University of Veterinary Animal Sciences, Lahore, Pakistan
- 8Institute of Traditional Chinese Veterinary Medicine, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
- 9MOE Joint International Research Laboratory of Animal Health and Food Safety, Nanjing Agricultural University, Nanjing, China
The Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis) inhabiting the Yantze River, China is critically endangered because of the influences of infectious disease, human activity, and water contamination. Viral diseases are one of the crucial factors that threatening the health of Yangtze finless porpoise. However, there are few studies which elaborate the viral diversity of Yangtze finless. Therefore, this study was performed to investigate the viral diversity of Yangtze finless by metagenomics. Results indicated that a total of 12,686,252 high-quality valid sequences were acquired and 2,172 virus reads were recognized. Additionally, we also obtained a total of 10,600 contigs. Phages was the most abundant virus in the samples and the ratio of DNA and RNA viruses were 69.75 and 30.25%, respectively. Arenaviridae, Ackermannviridae and Siphoviridae were the three most predominant families in all the samples. Moreover, the majority of viral genus were Mammarenavirus, Limestonevirus and Lambdavirus. The results of gene prediction indicated that these viruses play vital roles in biological process, cellular component, molecular function, and disease. To the best of our knowledge, this is the first report on the viral diversity of Yangtze finless porpoise, which filled the gaps in its viral information. Meanwhile, this study can also provide a theoretical basis for the establishment of the prevention and protection system for virus disease of Yangtze finless porpoise.
Introduction
Neophocaena asiaeorientalis ssp. asiaeorientalis also known as Yangtze finless porpoises (YFP) are fresh-water mammals that inhabit Yangtze River in People's Republic of China. They are critically endangered species with an estimated decreasing population of 500–1,800 (1). The survival of Yangtze finless porpoises is affected by multiple factors such as human activities, industrial development, mining, shipping, fishing, dams, water pollution and climate change. Efforts have been put into the research and conservation of this specie by protecting the areas, reserves and in situ conservation. Notably, the quantity of Yangtze finless porpoises is still gradually decreasing due to human activities over-fishing and water pollution of the Yangtze River. Water pollution is not only due to imbalance of the Yangtze River ecosystem but also caused the outbreak of viral and bacterial diseases, which posed a serious threat to the survival of Yangtze finless porpoises (2). Statistically, the population of Yangtze finless porpoises is <2,000 and it has been considered as the endangered species by the International Union for Conservation of Nature (IUCN) (3).
Viruses are one of the most abundant biological beings on the earth which are characterized by genomic flexibility, rapid transition, and smaller volume (4, 5). Viral habitat and diversity are important to formulate the prevention, control, and preparedness in case of emergency outbreaks (6). Moreover, this information is important to develop targeted drugs and vaccines against such diseases (6–8). The traditional virus identification/ detection methods include growth on cell culture and electron microscopy, serology, and molecular biology with each having its own benefits and limitations (9, 10). Recently, the high throughput sequencing-based techniques e.g., the metagenomics, has provided an important platform for the large-scale identification of known and novel viruses in a habitat (11, 12). A large amount of known and novel viruses has been identified from the pigs, ducks, turkey, pigeon, and bats by using viral metagenomics (13–17). However, little is known about the viral diversity in Yangtze finless porpoises. Therefore, the objective of this study was to investigate the viral diversity in Yangtze finless porpoise by metagenomics.
Materials and methods
Sample collection and processing
A total of 40 samples including heart, liver, spleen, lung, kidney, pancreas, lymph nodes and skin were collected from the dead Yangtze finless porpoises. The samples were collected from 2017 to 2019. Moreover, these samples were collected from 4 wild finless porpoises inhabiting the Yangtze River, aged 2–5, from the same site. The achieved samples were immediately stored in sterile plastic containers and transported to the laboratory and later stored at −80°C for further processing.
Nucleic acid extraction and library construction
First, the tissue samples were mixed and homogenized and then the debris of the tissues and cells were removed by low-speed centrifugation. Additionally, multiple virologic separation methods including filtration and ultracentrifugation were used for purifying and concentrating the virus-like particle (VLP) in the solution. The nucleic acid of the obtained VLP was extracted by the method of virus DNA (dsDNA, ssDNA) and RNA (ssRNA, dsRNA) co-extraction using viral DNA/RNA extraction kit (Beijing Quanshijin Biotechnology Co., LTD). Afterwards, the genomic libraries were constructed based on viral genome types. The DNA library of dsDNA genome was constructed directly, whereas ssDNA needs to be synthesized into double-stranded DNA before DNA library construction. Prior to the RNA library construction, the RNA genome needs to be converted into double-stranded cDNA through reverse transcription and two-strand synthesis. The total amount and purity of the DNA and RNA were evaluated, and the unqualified nucleic acids need to be re-extracted or undergo whole-genome amplification and purification. The qualified or amplified DNA samples were randomly shattered by an ultrasonic disruptor and the obtained short fragments of DNA were used for sequencing library construction. The final qualified libraries were used for high-throughput sequencing on the Illumina platform at Guangdong Meige Gene Technology Co., Ltd., China.
Bioinformatics analysis
After obtaining the metagenomic sequencing data, the quality of data was evaluated using Soapnuke software to ensure the credibility of subsequent analysis results. The paired reads with adapter and low quality were discarded. Moreover, duplicate reads generated by PCR amplification were also required to be removed. The finally obtained high-quality sequences were used for further data analysis. The clean reads were aligned to the specific host genome to remove the host sequence by using SOAPaligner (v2.0.5) and BWA software's (v0.7.17). Additionally, the high-quality sequences were required to compare with the virus database to obtain the virus composition information in the sample. Meanwhile, the NCBI taxonomy database was used to count the virus taxonomy annotation information. The obtained high-quality reads of each sample were assembled to achieve a longer contigs sequence by using Megahit, Trinity and IDBA software. The quantity, length, and N50 of assembled sequences were counted and the high-quality reads were aligned to the assembled sequence to evaluate the utilization of assembled reads. The reads were compared with the identified viral contigs and the RPKM value of each contig was also calculated. The RPKM value was calculated with the following formula: RPKM% = [Contig reads/Total mapped reads × Contig length] × 100%. The MetaGeneMark was used to predict the gene sequences of viral contigs and assess the number and length of the predicted genes. The predicted gene protein sequences were compared with UniProtKB/Swiss-Prot database to obtain functional annotation information.
Results
Sequences analyses
After the filtration of raw data, a total of 12,686,252 high-quality sequences were obtained (Table 1). The obtained high-quality sequences were compared with the ribosomal database (Silva.132) and the host (Neophocaena asiaeorientalis asiaeorientalis) database and the corresponding sequences which were <80% of the total length of reads were removed to avoid the influences of ribosome and host sequence on subsequent analysis (Table 2). Additionally, the high-quality sequences were compared with the virus database to quickly obtain the virus composition information in the sample and a total of 2,172 virus reads were found (Supplementary Table S1). The virus classification information was statistically analyzed based on the annotation information of the NCBI taxonomy database. Statistical analysis indicated that Phages was the most abundant virus, accounting for approximately 62.94% of the total number of viruses (Supplementary Table S2). Meanwhile, the proportion of DNA and RNA viruses were 69.75% and 30.25%, respectively (Supplementary Table S3). The top 11 preeminent families in the collected samples at the family level are shown in Figure 1A. At the family level, Arenaviridae, Ackermannviridae and Siphoviridae were the three most predominant families, whereas Inoviridae, Myoviridae, Podoviridae, Herpesviridae, Retroviridae, Togaviridae, Baculoviridae and Nairoviridae were represented with a lower abundance. At the level of genus, Mammarenavirus, Limestonevirus and Lambdavirus were observed as the predominant. Furthermore, other genera including unclassified_Inoviridae, unclassified_Myoviridae, unclassified_Podoviridae, Muromegalovirus, Gammaretrovirus, Alphavirus, Alphabaculovirus and Orthonairovirus were indicated with a lower abundance (Figure 1B).

Table 1. Statistical analysis of the sequence information.

Table 2. Statistical analysis of the sequence information after removing host sequence.
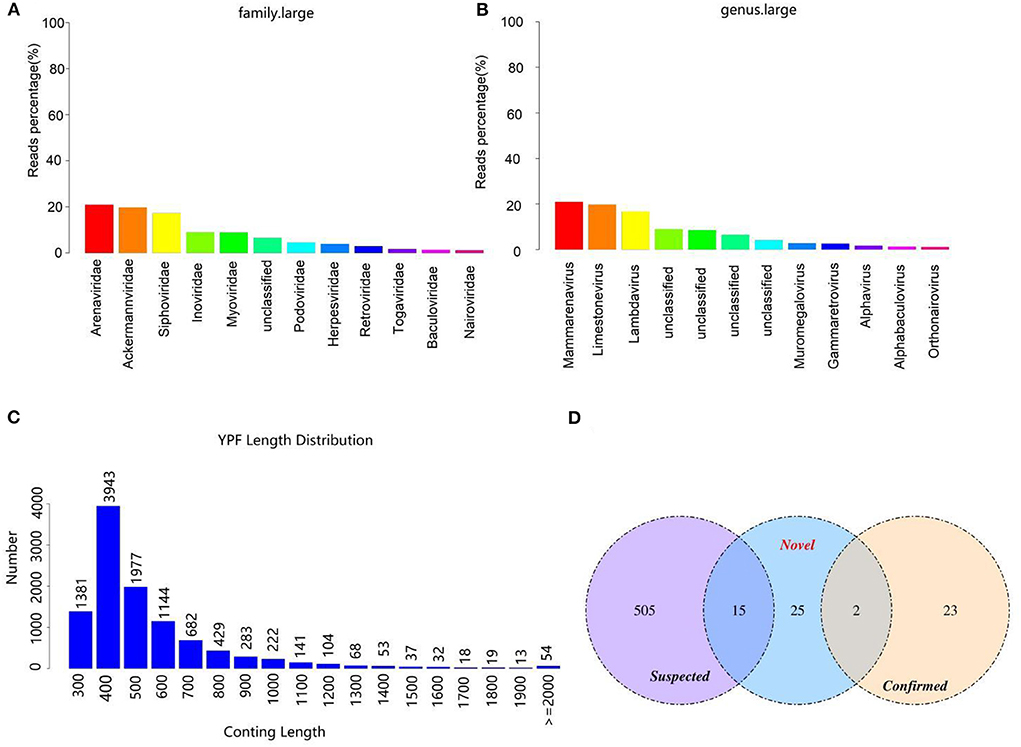
Figure 1. Viral composition and the length distribution of contigs. (A) The relative abundance of viral composition at the level of family. (B) The relative abundance of viral composition at the level of genus. (C) The length distribution of contigs. The abscissa represents the length of contigs and the ordinate indicates the number of contigs. (D) Comparison of the number of viruses contigs obtained by different methods.
Virus sequence identification
After removing the host sequence, 10600 contigs were totally obtained (Table 3) and the length distribution of contigs was displayed in Figure 1C. The obtained contigs were compared with the virus database. After comparison and screening, a total of 25 confirmed viral sequences and 520 suspected viral sequences were found (Table 4). Meanwhile, the percentage of DNA and RNA viral sequences in the total number of confirmed sequences were 56% and 44%, respectively (Supplementary Table S4). On the other hand, the DNA and RNA viral sequences accounted for 46.73% and 53.27% of the total suspected sequences, respectively (Supplementary Table S4). Moreover, the ratio of Phages in the confirmed and suspected viruses were 56.00% and 46.73%, respectively (Table 5). However, the identified sequence based on the comparison of the virus database existed some defects, because this method can only identify known viruses and the results may be false positives. Therefore, the variety of databases were increased to further identify viral sequences. Eventually, a total of 42 new viral sequences were identified (Figure 1D). Additionally, Myoviridae and Herpesviridae possessed the highest contig number at the family level (Figure 2A). The results of virus abundance statistics indicated that Nimaviridae and Whispovirus possessed the highest RPKM values at the family and genus levels, respectively (Figures 2B,C).

Table 3. The information of the obtained contigs.

Table 4. Statistical analysis of the viral sequence.

Table 5. Statistical analysis of viral types in the confirmed and suspected virus.
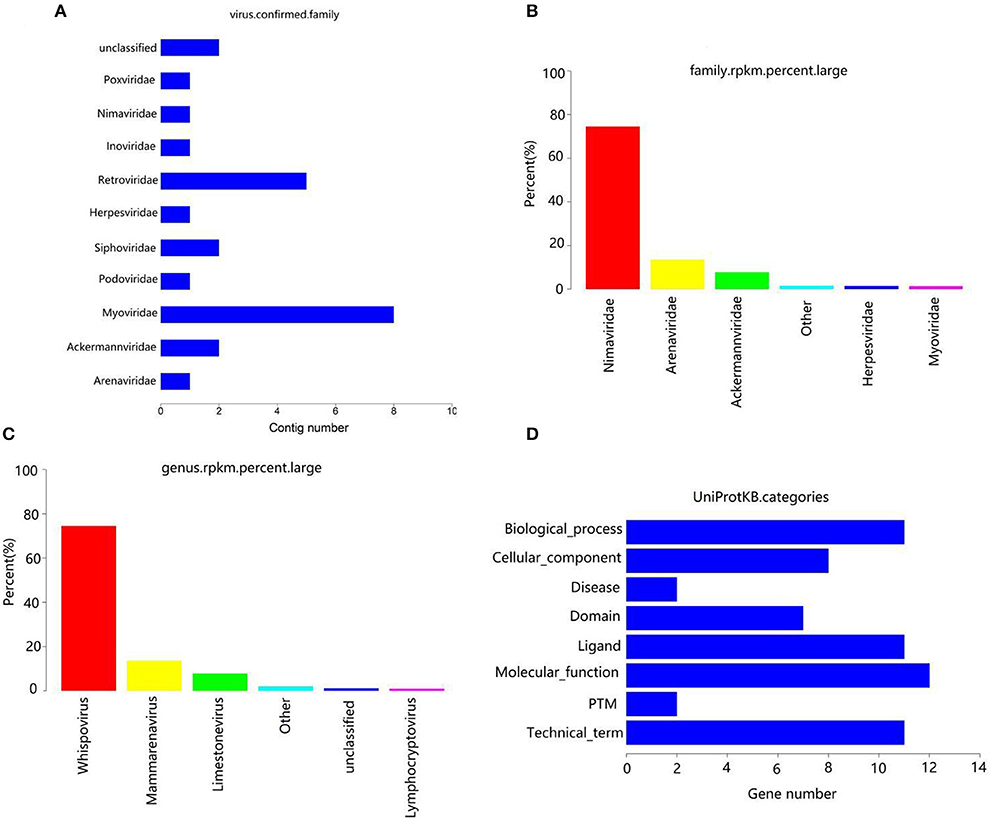
Figure 2. Annotation statistics of confirmed virus at the family level (A). Abscissa: quantity of contrags; ordinates: annotation information. (B) RPKM percentage statistics maps at family level. (C) RPKM percentage statistics maps at genus level. (D) Uniprotkb category statistics.
Gene prediction and functional analysis
The contigs of virus were genetically predicted using MetaGeneMark (v3.38) software and the sequences of the gene nucleic acid <150 bp was filtered. The results revealed that a total of 52 genes were predicted (Table 6). The protein sequences of genes were compared with the virus sequence of UniProtKB/Swiss-Prot database by using blastp (v2.9.0+) to obtain the functional information of the virus. The results indicated that these genes were closely related to biological process, cellular component, molecular function and disease (Figure 2D).

Table 6. Statistical analysis of gene prediction results.
Discussion
Viral diseases are one of the greatest health challenges in the contemporary world, posing a huge threat to animal and public health (18, 19). Multiple factors regarding the viral emergence e.g., the climate, population growth, changing diets, human activity and environmental pollution have been demonstrated (20). These factors affect the spread of viruses in humans and animals and may accelerate the emergence of viral epidemics. Consequently, an in-depth investigation of viral diversity is essential to have a base line for the prevention and development of drug strategies against these diseases. Traditional identification methods have certain restrictions mostly based on the lack of corresponding cell lines, antibodies, gene sequences and biosafety facilities (21–24). Metagenomics provide a modern alternative in identifying and discovering novel and existing microorganisms especially in the samples which was not possible to analyze previously (25). Despite, non-human and environmental samples would need further confirmation and databases (26, 27). Theoretically, metagenomics techniques can perform the genomic characterization and identification of most microorganisms contained in the samples with a generic lab procedure (28). With the popularization and application of metagenomics, the new viruses in mammals have been observed gradually (29, 30).
Bacteriophages are potentially the largest source of gene diversity and are the most abundant biological entities in the ecosphere as well as marine environment (25, 31, 32). Previous research has indicated that temperate phages remain in the host in a latent state, whereas pathogenic phages can infect the bacterium of host and exit the cell by lysis or budding (33). Our results revealed a high abundance of phages in the Yangtze finless porpoise, which was consistent with the previous findings in other animals (34). This may be due to phages which are normally present in higher abundance in marine waters (35). Herpes virus is a DNA virus with envelope and can be divided into four subfamilies (36). Previous studies revealed that herpes virus can infect multiple livestock, poultry and fish and damage their skin, mucosa and nervous tissue (37, 38). Notably, cyprinids are the most susceptible to the herpes virus among aquatic animals. Previous research demonstrated that cyprinids were one of the main foods for the Yangtze finless porpoise (39–43). Moreover, Pei et al. also found herpes virus in the liver of dead Yangtze finless porpoise, which was consistent with our results (44). Therefore, further research is needed to determine whether the Yangtze finless porpoise can infect the herpes virus through the food chain.
The Poxviridae is a type of DNA virus with the largest virions generally brick-shaped or elliptic (45). The Poxviridae composed of Orthpoxvirus, Parapoxvirus and Vaccinia (46). Previous research revealed that members of the family Poxviridae posed a serious threat to the health of pig, poultry, rabbit and ruminant and resulted in skin damage (47). Furthermore, Poxvirus can also infect marine mammals such as whales, dolphins, seals, causing ring-shaped, pinhole or tattoo-like skin injury (48).
Retroviruses are spherical RNA viruses with envelopes, including Tumorigenic virus, Lenti virus and Foamy virus (49). Although most members of the Retroviridae cannot directly result in diseases, they could induce cancer growth (50, 51). It was reported that Retroviruses mainly infect humans and can be transmitted through contaminated water (52). Currently, the water quality of the Yangtze River has seriously degraded due to the discharge of domestic sewage, industrial and agricultural waste (53, 54). However, whether Retrovirus could infect Yangtze finless porpoise through polluted water sources remains to be studied.
Early studies indicated that the members of the Arenaviridae can cause hemorrhage, meningoencephalitis, hemorrhagic fever and neurological diseases in animal and humans (55, 56). It is still unclear whether members of the family Arenaviridae can result in the diseases of Yangtze finless porpoise. Alphavirus consists of 27 viruses, all of which are transmitted by vector insects including mosquito, louse, bug and mite (57, 58). The most members of Alphavirus can naturally infect birds and rodents (59). Moreover, there have been reports of Alphavirus isolated from amphibians and reptiles (60, 61). Alphavirus can cause fever, rash, arthralgia and other symptoms in humans and animals and can even lead to death in severe cases (62, 63). Notably, Alphavirus was also found in the Yangtze finless porpoise through viral metagenome.
In summary, this study first revealed the viral diversity of the Yangtze finless porpoise by metagenomics. Results indicated that there were many viruses in the Yangtze finless porpoise, such as Arenaviridae, Ackermannviridae and Siphoviridae. Moreover, most of the viruses were reported for the first time in the Yangtze finless porpoise. However, the viral diversity can be influenced by multiple external and internal factors, such as individual biological difference, virus degradation. Considering the restriction of a small number of samples and experiment conditions, we cannot completely remove all the above-mentioned influencing factors. However, our research first attempted to reveal the viral diversity of the Yangtze finless porpoise and supplemented the gap in its virus information. Additionally, our study may provide a theoretical basis for further research on the pathogenic mechanism and prevention and treatment of viral diseases to understand the natural flora in these animals by the use of advanced techniques to know the basis and the possibility of diagnosis and therapy of infectious diseases in these animals.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was reviewed and approved by Ethics Committee of the Anqing Normal University.
Author contributions
ZL conceived and designed the experiment data curation. ZL and KL contributed sample collection and reagents preparation. MH, FA, ZL, XD, HY, XC, ST, YZ, WL, BD, AZ, JZ, ZQ, HA, and DY revised the manuscript. All the authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Hubei Yangtze River Ecological Protection Foundation-Dolphin-Vietnam Project, Anqing Normal University High-level Talent Introduction Program (2021), Aquatic Organism Protection and Water Ecological Restoration Project for External Development of Engineering Technology Research Center of Anhui Province Colleges and Universities (2021), and the key project of the Natural Science Foundation of Universities in Anhui Province (KJ2019A0551).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2022.922623/full#supplementary-material
References
1. Liu Z, Li A, Wang Y, Iqbal M, Zheng A, Zhao M, et al. Comparative analysis of microbial community structure between healthy and Aeromonas veronii-infected Yangtze finless porpoise. Microb Cell Fact. (2020) 19:123. doi: 10.1186/s12934-020-01383-4
2. Liu ZG, Zheng AF, Chen MM, Lian YX, Zhang XK, Zhang SZ, et al. Isolation and identification of pathogenic Aeromonas veronii from a dead Yangtze finless porpoise. Dis Aquat Organ. (2018) 132:13–22. doi: 10.3354/dao03288
3. Wang K, Wang D, Zhang X, Pfluger A, Barrett L. Range-wide Yangtze freshwater dolphin expedition: The last chance to see Baiji? Environ Sci Pollut Res Int. (2006) 13:418–24. doi: 10.1065/espr2006.10.350
4. Brussow H, Hendrix RW. Phage genomics: small is beautiful. Cell. (2002) 108:13–6. doi: 10.1016/S0092-8674(01)00637-7
5. Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, et al. The marine viromes of four oceanic regions. Plos Biol. (2006) 4:e368. doi: 10.1371/journal.pbio.0040368
6. He B, Li Z, Yang F, Zheng J, Feng Y, Guo H, et al. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS ONE. (2013) 8:e61950. doi: 10.1371/journal.pone.0061950
7. Yang L, Zhu W, Li X, Bo H, Zhang Y, Zou S, et al. Genesis and dissemination of highly pathogenic H5N6 avian influenza viruses. J Virol. (2017) 91:e02199. doi: 10.1128/JVI.02199-16
8. Guo Y, Liu H, Wang Q, Yi S, Niu J, Li D, et al. assessment of the protective efficacy of a feline calicivirus inactivated vaccine using in vivo FCV CH-JL2 infection. Pak Vet J. (in press). doi: 10.29261/pakvetj/2022.030
9. Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, et al. Uncovering earth's virome. Nature. (2016) 536:425–30. doi: 10.1038/nature19094
10. Zaheer I, Saleemi MK, Javed MT, Sajjad-ur-Rahman Abubakar M. Clinical investigation and molecular prevalence of Fowl adenoviruses of commercial poultry from division Faisalabad, Pakistan. Pak Vet J. (2022). doi: 10.29261/pakvetj/2022.056
11. Edridge A, Deijs M, van Zeggeren IE, Kinsella CM, Jebbink MF, Bakker M, et al. Viral Metagenomics on Cerebrospinal Fluid. Genes (Basel). (2019) 10:e50332. doi: 10.3390/genes10050332
12. Tang P, Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol. (2010) 5:177–89. doi: 10.2217/fmb.09.120
13. Day JM, Ballard LL, Duke MV, Scheffler BE, Zsak L. Metagenomic analysis of the turkey gut RNA virus community. Virol J. (2010) 7:313. doi: 10.1186/1743-422X-7-313
14. Shan T, Li L, Simmonds P, Wang C, Moeser A, Delwart E. The fecal virome of pigs on a high-density farm. J Virol. (2011) 85:11697–708. doi: 10.1128/JVI.05217-11
15. Chen GQ, Zhuang QY, Wang KC, Liu S, Shao JZ, Jiang WM, et al. Identification and survey of a novel avian coronavirus in ducks. PLoS ONE. (2013) 8:e72918. doi: 10.1371/journal.pone.0072918
16. Phan TG, Vo NP, Boros A, Pankovics P, Reuter G, Li OT, et al. The viruses of wild pigeon droppings. PLoS ONE. (2013) 8:e72787. doi: 10.1371/journal.pone.0072787
17. Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. (2010) 84:6955–65. doi: 10.1128/JVI.00501-10
18. Grubaugh ND, Ladner JT, Lemey P, Pybus OG, Rambaut A, Holmes EC, et al. Tracking virus outbreaks in the twenty-first century. Vat Microbiol. (2019) 4:10–19. doi: 10.1038/s41564-018-0296-2
19. Hafez H M. Avian adenoviruses infections with special attention to inclusion body hepatitis/hydropericardium syndrome and egg drop syndrome. Pak Vet J. (2021) 31:85–92. Available online at: http://www.pvj.com.pk/abstract/31_2/10-110.htm
20. Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. (2007) 447:279–83. doi: 10.1038/nature05775
21. Nooij S, Schmitz D, Vennema H, Kroneman A, Koopmans M. Overview of virus metagenomic classification methods and their biological applications. Front Microbiol. (2018) 9:749. doi: 10.3389/fmicb.2018.00749
22. Hayes S, Mahony J, Nauta A, van Sinderen D. Metagenomic approaches to assess bacteriophages in various environmental niches. Viruses. (2017) 9:e60127. doi: 10.3390/v9060127
23. Yozwiak NL, Skewes-Cox P, Stenglein MD, Balmaseda A, Harris E, DeRisi JL. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl Trop Dis. (2012) 6:e1485. doi: 10.1371/journal.pntd.0001485
24. Su W, Li Z, He Y, Yan X, Wang L, Peng D. Isolation and identification of a raccoon dog parvovirus and sequence analysis of its VP2 gene. Pak Vet J. (2021). doi: 10.29261/pakvetj/2022.027
25. Breitbart M, Salamon P, Andresen B, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA. (2002) 99:14250–5. doi: 10.1073/pnas.202488399
26. Culligan EP, Sleator RD, Marchesi JR, Hill C. Metagenomics and novel gene discovery: promise and potential for novel therapeutics. Virulence. (2014) 5:399–412. doi: 10.4161/viru.27208
27. Pallen MJ. Diagnostic metagenomics: potential applications to bacterial, viral and parasitic infections. Parasitology. (2014) 141:1856–62. doi: 10.1017/S0031182014000134
28. Wooley JC, Ye Y. Metagenomics: facts and artifacts, and computational challenges*. J Comput Sci Technol. (2009) 25:71–81. doi: 10.1007/s11390-010-9306-4
29. Shkoporov AN, Hill C. Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe. (2019) 25:195–209. doi: 10.1016/j.chom.2019.01.017
30. Fawaz M, Vijayakumar P, Mishra A, Gandhale PN, Dutta R, Kamble NM, et al. Duck gut viral metagenome analysis captures snapshot of viral diversity. Gut Pathog. (2016) 8:30. doi: 10.1186/s13099-016-0113-5
31. Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. (2004) 68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004
32. Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brussow H. Phage-host interaction: an ecological perspective. J Bacteriol. (2004) 186:3677–86. doi: 10.1128/JB.186.12.3677-3686.2004
33. Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol. (2008) 25:762–77. doi: 10.1093/molbev/msn023
34. Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, et al. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci USA. (2013) 110:12798–803. doi: 10.1073/pnas.1305956110
35. Bergh B, Rsheim K, Bratbak G, Heldal M. High abundance of viruses found in aquatic environments. Nature. (1989) 340:467–46 doi: 10.1038/340467a0
36. Lombardi L, Falanga A, Del GV, Palomba L, Galdiero M, Franci G, et al. A boost to the antiviral activity: Cholesterol tagged peptides derived from glycoprotein B of Herpes Simplex virus type I. Int J Biol Macromol. (2020) 162:882–93. doi: 10.1016/j.ijbiomac.2020.06.134
37. Di Lernia V, Mansouri Y. Epstein-Barr virus and skin manifestations in childhood. Int J Dermatol. (2013) 52:1177–84. doi: 10.1111/j.1365-4632.2012.05855.x
38. Lin GL, McGinley JP, Drysdale SB, Pollard AJ. Epidemiology and immune pathogenesis of viral sepsis. Front Immunol. (2018) 9:2147. doi: 10.3389/fimmu.2018.02147
39. Boutier M, Ronsmans M, Rakus K, Jazowiecka-Rakus J, Vancsok C, Morvan L, et al. Cyprinid herpesvirus 3: an archetype of fish alloherpesviruses. Adv Virus Res. (2015) 93:161–256. doi: 10.1016/bs.aivir.2015.03.001
40. Aziz S, Abdullah S, Abbas K, Zia MA. Effects of engineered zinc oxide nanoparticles on freshwater fish, Labeo rohita: Characterization of ZnO nanoparticles, acute toxicity and oxidative stress. Pak Vet J. (2020) 40:479–83. doi: 10.29261/pakvetj/2020.030
41. Ghaffar A, Hussain R, Noreen S, Abbas G, Chodhary IR, Khan A, et al. Dose and time-related pathological and genotoxic studies on thiamethoxam in fresh water fish (Labeo rohita) in Pakistan. Pak Vet J. (2020) 40(2):151–6. doi: 10.29261/pakvetj/2020.002
42. Aziz S, Abdullah S, Anwar H, Latif F, Mustfa W. Effect of engineered nickel oxide nanoparticles on antioxidant enzymes in fresh water fish, Labeo rohita. Pak Vet J. (2021) 41:424–8. doi: 10.29261/pakvetj/2021.044
43. Nabi G, Hao Y, Zeng X, Wang D. Assessment of yangtze finless porpoises (neophocaena asiaorientalis) through biochemical and hematological parameters. Zool Stud. (2017) 56:e31. doi: 10.6620/ZS.2017.56-31
44. Pei C, Lei XY, Yuan XP, Wang D, Zhao QZ, Zhang QY. Herpes-like virus infection in Yangtze finless porpoise (Neophocaena phocaenoides): pathology, ultrastructure and molecular analysis. J Wildl Dis. (2012) 48:235–7. doi: 10.7589/0090-3558-48.1.235
45. Tuazon KM, Ng E, Al RZ, Pandey P, Ruuls SR, Korner H, et al. TNF deficiency dysregulates inflammatory cytokine production, leading to lung pathology and death during respiratory poxvirus infection. Proc Natl Acad Sci USA. (2020) 117:15935–46. doi: 10.1073/pnas.2004615117
46. Fleming SB, Wise LM, Mercer AA. Molecular genetic analysis of orf virus: a poxvirus that has adapted to skin. Viruses. (2015) 7:1505–39. doi: 10.3390/v7031505
48. Barnett J, Dastjerdi A, Davison N, Deaville R, Everest D, Peake J, et al. Identification of novel cetacean poxviruses in cetaceans stranded in South West England. PLoS ONE. (2015) 10:e0124315. doi: 10.1371/journal.pone.0124315
49. Garcia-Montojo M, Doucet-O'Hare T, Henderson L, Nath A. Human endogenous retrovirus-K (HML-2): a comprehensive review. Crit Rev Microbiol. (2018) 44:715–38. doi: 10.1080/1040841X.2018.1501345
50. Premeaux TA, Javandel S, Hosaka K, Greene M, Therrien N, Allen IE, et al. Associations between plasma immunomodulatory and inflammatory mediators with VACS index scores among older HIV-infected adults on antiretroviral therapy. Front Immunol. (2020) 11:1321. doi: 10.3389/fimmu.2020.01321
51. Beghin JC, Ruelle J, Goubau P, Van der Linden D. Drug resistance in HIV-infected children living in rural South Africa: Implications of an antiretroviral therapy initiated during the first year of life. J Clin Virol. (2020) 129:104547. doi: 10.1016/j.jcv.2020.104547
52. Knight A, Haines J, Stals A, Li D, Uyttendaele M, Knight A, et al. A systematic review of human norovirus survival reveals a greater persistence of human norovirus RT-qPCR signals compared to those of cultivable surrogate viruses. Int J Food Microbiol. (2016) 216:40–9. doi: 10.1016/j.ijfoodmicro.2015.08.015
53. Zhang J, Liu L, Zhao Y, Li H, Lian Y, Zhang Z, et al. Development of a high-resolution emission inventory of agricultural machinery with a novel methodology: A case study for Yangtze River Delta region. Environ Pollut. (2020) 266:115075. doi: 10.1016/j.envpol.2020.115075
54. Sun J, Jin L, He T, Wei Z, Liu X, Zhu L, et al. Antibiotic resistance genes (ARGs) in agricultural soils from the Yangtze River Delta, China. Sci Total Environ. (2020) 740:140001. doi: 10.1016/j.scitotenv.2020.140001
55. Baral P, Pavadai E, Gerstman BS, Chapagain PP. In-silico identification of the vaccine candidate epitopes against the Lassa virus hemorrhagic fever. Sci Rep. (2020) 10:7667. doi: 10.1038/s41598-020-63640-1
56. Sudhakar H, Bhate J, Patra AK. Patent landscape of novel technologies for combating category-A Arenavirus infections. Expert Opin Ther Pat. (2020) 30:557–65. doi: 10.1080/13543776.2020.1755255
57. Filomatori CV, Merwaiss F, Bardossy ES, Alvarez DE. Impact of alphavirus 3'UTR plasticity on mosquito transmission. Semin Cell Dev Biol. (2020) 111:148–55. doi: 10.1016/j.semcdb.2020.07.006
58. Michie A, Ernst T, Chua IJ, Lindsay M, Neville PJ, Nicholson J, et al. Phylogenetic and timescale analysis of Barmah forest virus as inferred from genome sequence analysis. Viruses. (2020) 12:E70732. doi: 10.3390/v12070732
59. Nguyen W, Nakayama E, Yan K, Tang B, Le TT, Liu L, et al. Arthritogenic alphavirus vaccines: serogrouping versus cross-protection in mouse models. Vaccines (Basel). (2020) 8:e20209. doi: 10.3390/vaccines8020209
60. Aslam ML, Robledo D, Krasnov A, Moghadam HK, Hillestad B, Houston RD, et al. Quantitative trait loci and genes associated with salmonid alphavirus load in Atlantic salmon: implications for pancreas disease resistance and tolerance. Sci Rep. (2020) 10:10393. doi: 10.1038/s41598-020-67405-8
61. Jansen MD, Guarracino M, Carson M, Modahl I, Taksdal T, Sindre H, et al. Field evaluation of diagnostic test sensitivity and specificity for salmonid alphavirus (SAV) infection and pancreas disease (PD) in farmed atlantic salmon (Salmo salar L.) in Norway using Bayesian latent class analysis. Front Vet Sci. (2019) 6:419. doi: 10.3389/fvets.2019.00419
62. Torres-Ruesta A, Teo TH, Chan YH, Renia L, Ng L. Pathogenic Th1 responses in CHIKV-induced inflammation and their modulation upon Plasmodium parasites co-infection. Immunol Rev. (2020) 294:80–91. doi: 10.1111/imr.12825
Keywords: Yangtze finless porpoise, diversity, virus, metagenomics, Arenaviridae
Citation: Liu Z, Ding X, Haider MS, Ali F, Yu H, Chen X, Tan S, Zu Y, Liu W, Ding B, Zheng A, Zheng J, Qian Z, Ashfaq H, Yu D and Li K (2022) A metagenomic insight into the Yangtze finless porpoise virome. Front. Vet. Sci. 9:922623. doi: 10.3389/fvets.2022.922623
Received: 18 April 2022; Accepted: 15 August 2022;
Published: 02 September 2022.
Edited by:
Fazul Nabi, Lasbela University of Agriculture, Water and Marine Sciences, PakistanReviewed by:
Ahrar Khan, Shandong Vocational Animal Science and Veterinary College, ChinaXinyu Zhu, Duke University, United States
Samina Shabbir, Guangdong Academy of Science (CAS), China
Copyright © 2022 Liu, Ding, Haider, Ali, Yu, Chen, Tan, Zu, Liu, Ding, Zheng, Zheng, Qian, Ashfaq, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Li, lik2014@sina.com; Zhigang Liu, lzg12021@163.com
†These authors have contributed equally to this work