Preparation and Characterization of Electrospun Food Biopackaging Films of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Derived From Fruit Pulp Biowaste
 Beatriz Melendez-Rodriguez1 Jinneth L. Castro-Mayorga1 Maria A. M. Reis2 Chris Sammon3
Beatriz Melendez-Rodriguez1 Jinneth L. Castro-Mayorga1 Maria A. M. Reis2 Chris Sammon3  Luis Cabedo4 Sergio Torres-Giner1
Luis Cabedo4 Sergio Torres-Giner1  Jose M. Lagaron1*
Jose M. Lagaron1*- 1Novel Materials and Nanotechnology Group, Institute of Agrochemistry and Food Technology (IATA), Spanish Council for Scientific Research (CSIC), Paterna, Spain
- 2UCIBIO-REQUIMTE, Chemistry Department, Faculty of Sciences and Technology, Universidade NOVA de Lisboa, Caparica, Portugal
- 3Materials and Engineering Research Institute, Sheffield Hallam University, Sheffield, United Kingdom
- 4Polymers and Advanced Materials Group, Universitat Jaume I, Castellón, Spain
In the present study, circular economy based and potentially low-cost poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) was produced by mixed microbial cultures derived from fruit pulp, an industrial by-product of the juice industry. Three different chemical routes, namely non-extraction, extraction with sodium hypochlorite (NaClO), and extraction with chloroform, in combination with filtering and centrifugation, were explored to purify the biopolymer and find the most optimal solution for its processing via electrospinning. The resultant ultrathin fiber mats of the different extracted PHBV materials were thermally post-processed at different temperatures in order to obtain continuous films adequate for food packaging applications. The resultant films were characterized in terms of morphology, crystallinity as well as thermal, mechanical, and barrier properties. The results showed that extraction with both chloroform and NaClO with a post-treatment of filtering and centrifugation of the PHBV-containing biomass were necessary refining steps to allow its processing by electrospinning. In particular, the PHBV extracted with chloroform presented the highest degree of purity, resulting in more transparent films with lower wettability and higher flexibility. The here-formulated electrospun films made of biomass derived from biowaste exhibit great potential as interlayers or coatings for food biopackaging applications.
Introduction
The growing concern for plastic waste disposal of petroleum-based materials has intensified the study and development of bio-based and biodegradable polymers, particularly those synthesized from agro-industrial residues (Babu et al., 2013). Polyhydroxyalkanoates (PHAs) comprise a family of biodegradable biopolyesters synthesized by hundred species of Gram-positive (G+) and Gram-negative (G−) bacteria (Rehm, 2003). PHAs provide a particularly good alternative to fossil-derived polymers, showing the highest potential to replace polyolefins in packaging applications (Fabra et al., 2014; Mutlu et al., 2018). The most widely studied PHA is poly(3-hydroxybutyrate) (PHB), which possesses similar thermal and mechanical properties than polystyrene (PS) and isotactic polypropylene (iPP) (Savenkova et al., 2000). PHB homopolyester has, however, excessive brittleness and a narrow processing temperature window, which limit its use for packaging applications (Reis et al., 2008). Instead, its copolymer with 3-hydroxyvalerate (HV), i.e., poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) has a much lower crystallinity, decreased stiffness, and lower melting temperature (Chen and Wang, 2002; Martínez-Sanz et al., 2016).
The production of PHAs based on open mixed cultures is a sustainable alternative to reduce costs (Dias et al., 2006; Serafim et al., 2008). However, large investments are needed in the processes of fermentation, isolation, purification, etc. Nowadays, main research efforts currently focus on reducing the costs of the fermentation and downstream processes and make the PHAs' industrial production more competitive (Jacquel et al., 2007). In this sense, the production of PHA by mixed crops from renewable resources is both economically and environmentally attractive. In particular, the use of food waste as the raw material for the PHA production is a more economical and sustainable industrial form within the frame of the Circular Economy (Colombo et al., 2017). For example, fermented cheese whey (CW), whose potential due to high contents in proteins and sugars is mostly not fully valorized at present, can be used as the feeding solution for PHA production (Colombo et al., 2016; Martínez-Abad et al., 2016; Domingos et al., 2018). Nevertheless, the extraction methods should be optimized in order to properly extract the biopolymer from the cell walls, especially in mixed cultures, where is more complex due to these are more resistant to cell hydrolysis than pure cultures (Samorı̀ et al., 2015). Moreover, the chemical digestion methods adopted to disrupt the cell wall and release PHAs can affect the purity and molecular weight (MW) of the obtained polymer (Kunasundari and Sudesh, 2011) while the large amounts of chemicals used can also increase the recovery cost and make the process environmentally unfriendly. To address these issues, improvements in the methods of purification and extraction are being currently explored.
Electrospinning is a physical process used for the formation of continuous polymer fibers with ultrathin diameters, generally in the sub-micrometer range, through the action of an external high-voltage electric field. At a critical high voltage, typically 5–35 kV, the polymer solution droplets distort and form the so-called Taylor's cone. This erupts from the solution to form a charged polymer jet whose size and morphology are affected by the solution properties, mainly viscosity, surface tension, and conductivity of the polymer solution, and also by the process conditions, namely applied voltage, flow-rate, and tip-to-collector distance (Doshi and Reneker, 1995; Torres-Giner et al., 2008). In the field of packaging, the electrospun fiber structure represents a good option from a point of view of mechanical reinforcement (Torres-Giner, 2011), but it can also enhance the barrier performance of biopolymer matrices (Busolo et al., 2009; Torres-Giner et al., 2014). The use of electrospinning in active food packaging by the incorporation into the fibers of both nanofillers or active substances can add an extra value to the final product, changing the packed food condition to extend the shelf life and improve safety and/or sensory properties (Torres-Giner et al., 2016b).
Furthermore, the application of a thermal post-treatment below the biopolymer's melting temperature (Tm), habitually referred as to annealing, offers the possibility to produce continuous films of more application interest in packaging. This technology is particularly very promising to valorize forms of processing renewable polymers in the form of interlayers or coatings with enhanced flexibility and transparency while potentially retaining the barrier performance to gases and vapors of conventional films prepared by compression molding or extrusion (Fabra et al., 2016; Echegoyen et al., 2017). In comparison with films obtained by compression molding, electrospun PHB films have showed better optical properties, similar barrier performance, and higher elongation at break and toughness (Cherpinski et al., 2017). In addition, electrospun PHA films have been recently applied as coatings on fiber-based packaging layers to improve their moisture resistance (Cherpinski et al., 2018). Other recent studies have been focused on the incorporation of antimicrobial ingredients in the electrospun PHA fibers, which can be thereafter integrated as active layers in packaging structures. For example, electrospun mats made of PHBV derived from CW and containing zinc oxide (ZnO) were thermally post-processed as coatings on PHBV films to develop antimicrobial nanocomposites (Castro-Mayorga et al., 2017). Similarly, in another study, copper oxide (CuO) nanoparticles were loaded into PHBV electrospun coatings to form antimicrobial coatings with increased capacity (Castro Mayorga et al., 2018).
The objective of this study is to assess different routes of extraction for PHBV-containing biomass derived from an industrial biowaste of fruit pulps for its optimal processing by electrospinning. Moreover, the obtained electrospun PHBV mats were subjected to a post-processing annealing treatment to prepare and characterize continuous films of relevance in food packaging applications.
Experimental
Materials
PHBV copolyester was produced at pilot-plant scale at Universidade NOVA (Lisboa, Portugal) using mixed microbial cultures fed with fermented fruits. These were obtained from fruit pulps supplied by SumolCompal S.A. (Portugal) as an industrial residue of the juice industry, which were taken from the stored barrels that did not comply with the criteria to be processed.
Commercial PHBV was ENMAT™ Y1000P, produced by Tianan Biologic Materials (Ningbo, China) and delivered in the form of pellets. According to the manufacturer, the molar fraction of HV in the commercial copolymer is 3%.
2,2,2-trifluoroethanol (TFE), ≥99% purity, and D-limonene, with 98% purity, were obtained from Sigma Aldrich S.A. (Madrid, Spain). Sodium hypochlorite, 10–15% active chloride, was supplied by Scharlab, S.L. (Barcelona, Spain) while chloroform, stabilized with ethanol and 99.8% purity, was purchased from Panreac S.A. (Barcelona, Spain). Sodium bicarbonate (NaHCO3) was supplied by JMGS (Odivelas, Portugal).
Production of Unpurified PHBV Powder
PHBV was produced using mixed microbial cultures applying a three-stage process: (1) acidogenic fermentation of the agro-food waste to produce a mixture of fermentation products, i.e., the precursors for PHA biosynthesis; (2) culture selection, where microorganisms were selected based on their PHA storage ability through alternating periods of feast and famine; (3) PHA production, in which the selected microorganisms were fed with the fermentation products mixture resultant from the acidogenic step in order to accumulate PHA up to the culture's maximum capacity.
The experimental setup consisted of three pilot-scale reactors. The fruit pulp waste was first fermented in a 60-L up-flow anaerobic sludge blanket reactor (UASB) inoculated with anaerobic granules from a full-scale anaerobic digester. The acidogenic reactor operated using an organic loading rate (OLR) of 29 ± 2 g-COD·L−1·d−1, a hydraulic retention time (HRT) of 1 day, a pH value of 4.7 ± 0.1 (controlled by adding NaHCO3 in the feed), and a temperature of 30 ± 1°C. The obtained stream of fermentation products had an average composition of 5 ± 1% lactate, 17 ± 1% acetate, 7 ± 1% propionate, 9 ± 1% ethanol, 57 ± 2% butyrate, and 5 ± 1% valerate (% COD basis) and was further used to fed the two subsequent steps of the process. A 100-L aerobic sequencing batch reactor (SBR) inoculated with activated sludge from the Muleta Wastewater Treatment Plant, in Portugal, was used for the enrichment of the PHA-producing culture. The SBR operated at a feast and famine regime in 12 h cycles, comprising four periods: feeding (12 min), aeration (11 h), settling (45 min), and withdrawal (15 min). The OLR was maintained at 350 C-mM·d−1 and the HRT and SRT were 1 and 4 days, respectively. In the feeding phase, the stream obtained in the acidogenic reactor was fed as the carbon source together with a mineral solution (Serafim et al., 2004) to adjust the reactor's OLR. Nitrogen and phosphorus concentrations were adjusted, keeping the C/N/P molar ratio at 100/7/1. PHA production was performed in a 50-L aerobic fed-batch reactor inoculated with 25 L of biomass purged from the SBR at the end of famine phase (ca. 10.5 h from cycle started). The reactor was fed with the fermented fruit pulp waste in a pulse-wise mode controlled by the dissolved oxygen (DO) response. When DO increased, a new pulse of carbon source was provided until no DO response was observed, indicating that the culture reached its maximum capacity.
A PHBV cell content of ca. 70% (wt.-%) was attained at the end of the accumulation assay. The molar fraction of HV in the copolymer was ca. 20 mol.-%, as determined by gas chromatography with flame ionization detector (GC-FID) according to the method described by Lanham et al. (2013). Samples were calibrated through standard curves with a solution made of a commercial copolymer of PHBV with 12% HV molar content and heptadecane as internal standard, both supplied by Sigma Aldrich Química S.A. (Sintra, Portugal).
Finally, lyophilization of the PHA-containing medium was performed neutralizing the liquid material. Then, it was centrifuged at 4,000 rpm during 15 min and the resultant pellet was washed three times with distilled water. The obtained material was stored at −80°C for at least 3 h and freeze-dried for a week to produce the unpurified PHBV powder.
Processing Routes
The unpurified lyophilized PHBV powder was processed using three different routes. Figure 1 gathers a schematic description of the here-employed procedures. Table 1 summarizes the different PHBV solution samples used for electrospinning with their resultant yields determined using Equation (1):
Where Wi is the initial dry weight (100 g) and Wf is the final weight obtained after drying the resultant solution samples at 110°C for 24 h.
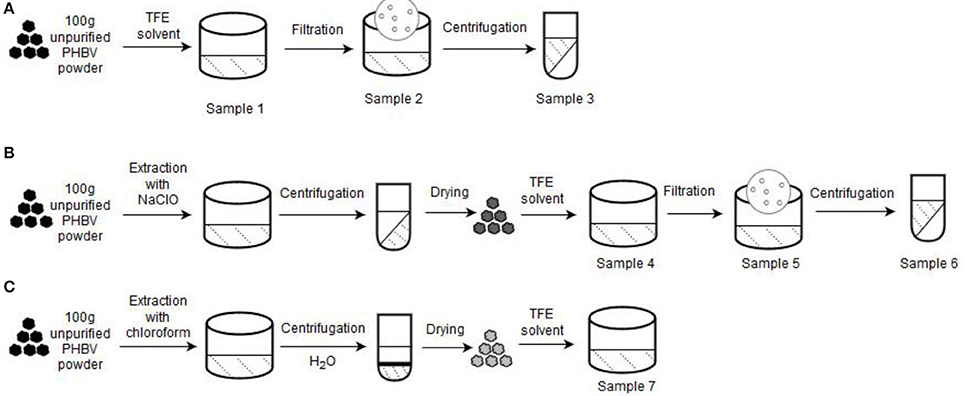
Figure 1. Processing routes for the preparation of the poly(3-hydroxybutyrate- co-3-hydroxyvalerate) (PHBV) solutions used for electrospinning: (A) Non-extraction; (B) Extraction with sodium hypochlorite (NaClO); (C) Extraction with chloroform. A starting sample of 100 g of unpurified PHBV powder was used in each process.

Table 1. Description and yield of the different poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) solutions used for electrospinning based on extraction routes with sodium hypochlorite (NaClO) and chloroform.
Non-Extraction Procedure
About 100 g of unpurified PHBV powder was dissolved in TFE at 8 wt.-% under magnetic stirring for 120 h at room temperature (sample 1). Then, part of this PHBV solution was filtered using Funnels Buchner Kartell disks, 2.5-mm mesh, from Scharlab S.L. (Barcelona, Spain) (sample 2). Finally, part of the filtered PHBV solution was additionally centrifugated at 13,000 rpm for 5 min (sample 3) in an Avanti J-26S XP Centrifuge (Beckman Coulter).
Extraction Procedure With NaCLO
Second route was based on the NaClO extraction method (Villano et al., 2014; Samorı̀ et al., 2015). Briefly, 10 wt.-% unpurified PHBV powder was mixed with pre-diluted NaClO (5% Cl2). The mixture was stirred for 3 h at room temperature to degrade the non-PHA cellular material. The resultant PHBV solution was then centrifugated for 15 min at 6,500 rpm and washed four times with distilled water. Afterwards, it was dried in 2 steps, first at 40°C for 2 days, and then at 70°C for 5 h. The resultant powder was dissolved in TFE at 8 wt.-% under magnetic stirring for 48 h at 50°C (sample 4). After this, the resultant PHBV solution was filtered (sample 5) and also centrifuged at 13,000 rpm for 5 min (sample 6).
Extraction Procedure With Chloroform
Third route followed a chloroform-based extraction method (Fiorese et al., 2009). For this, the unpurified PHBV powder was dissolved in chloroform at 5 wt.-%. The mixture was then stirred for 24 h at 50°C to degrade the non-PHA cellular material. Later on, the solution was transferred to centrifugation tubes in which distilled water was added at 50 wt.-%. After shaking the tubes manually, these were centrifuged for 5 min at 4,000 rpm. Afterwards, the PHBV suspension was recovered from the bottom of the tubes with a pipette and transferred to beakers, leaving them in the extractor hood until the solvent was completely evaporated. The resultant powder was dissolved in TFE at 2 wt.-% under magnetic stirring for 24 h at 50°C (sample 7).
Characterization of PHBV Solutions
Prior to electrospinning, all the prepared PHBV solutions were characterized in terms of their viscosity, surface tension, and conductivity. The apparent viscosity (ηa) was determined at 100 s−1 using a rotational viscosity meter Visco BasicPlus L from Fungilab S.A. (San Feliu de Llobregat, Spain) equipped with a low-viscosity adapter (LCP). The surface tension was measured following the Wilhemy plate method using an EasyDyne K20 tensiometer from Krüss GmbH (Hamburg, Germany). The conductivity was evaluated using a conductivity meter XS Con6 from Lab-box (Barcelona, Spain). All measurements were carried out at room temperature in triplicate.
Electrospinning Process
All PHBV solutions were processed by electrospinning using a high-throughput dual polarization Fluidnatek® LE-500 pilot-plant device manufactured by Bioinicia S.L. (Valencia, Spain). The equipment was operated in the lab configuration mode by means of a motorized single needle injector, scanning horizontally toward a roll-to-roll collector. Table 2 includes the different tested PHBV solutions in TFE with their final processing conditions, which were optimized for each solution. All experiments were carried out at room conditions, i.e., 25°C and 40% RH. The collected fiber mats were dried in a desiccator at 0% RH for 15 days to completely remove the remaining solvent.
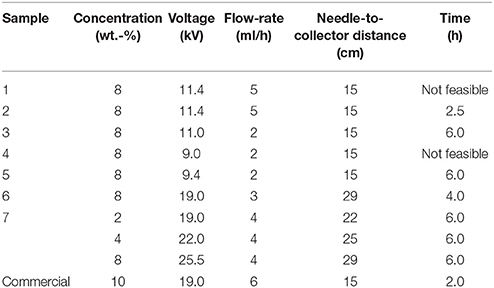
Table 2. Selected optimal conditions during electrospinning for the different poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) solutions.
Film Preparation
The resultant electrospun PHBV mats were subjected to annealing process in a 4122-model press from Carver, Inc. (Wabash, IN, USA). This was analyzed in the temperature range from 80 to 145°C, for 10 s, without pressure. The post-processed samples had an average thickness of ~60 μm.
Characterization
Scanning Electron Microscopy
The morphologies of the electrospun PHBV fibers and resultant films were observed by scanning electron microscopy (SEM) using an S-4800 device from Hitachi (Tokyo, Japan). The samples were fixed to beveled holders using conductive double-sided adhesive tape and sputtered with a mixture of gold-palladium under vacuum prior to observation. An accelerating voltage of 10 kV was used and the estimation of the dimensions was performed by means of the Aperture software from Apple (Cupertino, CA, USA) using a minimum of 20 SEM micrographs in their original magnification.
Contact Angle Measurements
Measurements of contact angle were performed in a Video-Based Contact Angle Meter model Theta Lite TL 101 from Biolin Scientific (Espoo, Finland). Data were obtained by analyzing the shape of a distilled water drop of 5 μL, placed over the sample surface, taken after 10 s of the droplet-surface contact. Measurements were carried out at room conditions. Image analyses were carried out by the OneAttension software. At least, three replicates were made for each sample.
FTIR Spectroscopy
Fourier transform infrared (FTIR) spectra were collected coupling the attenuated total reflection (ATR) accessory Golden Gate of Specac, Ltd (Orpington, UK) to the Tensor 37 FTIR equipment (Bruker, Germany). Single spectra were collected in the wavelength range from 4,000 to 600 cm−1 by averaging 20 scans at a resolution of 4 cm−1.
Variable temperature FTIR was performed on a Nicolet Nexus FTIR instrument from Thermo Fisher Scientific Inc (Wilmington, DE, USA) coupled to a variable temperature single reflection diamond ATR sampling accessory of Specac Ltd. (Orpington, UK). Spectra were collected by averaging 64 scans at 4 cm−1 resolution using the blank ATR crystal at the same temperature as the background. Spectra were collected at 10°C intervals from 30 to 100°C and thereafter at 5°C intervals up to 180°C. To ensure the validity of the selected temperature, spectra were not collected until the digital reading on the temperature controller remained constant.
WAXD
Wide angle X-ray diffraction (WAXD) was performed using a Bruker AXS D4 ENDEAVOR diffractometer (Billerica, MA, USA). The samples were scanned, at room temperature, in the reflection mode using incident Cu K-alpha radiation (k = 1.54 Å), while the generator was set up at 40 kV and 40 mA. The data were collected over the range of scattering angles (2θ) comprised in the 2–40° range.
Thermal Analysis
Thermal transitions were studied by differential scanning calorimetry (DSC) on a DSC-7 analyzer from PerkinElmer, Inc. (Waltham, MA, USA), equipped with a cooling accessory Intracooler 2 also from PerkinElmer, Inc. A three-step program, under nitrogen atmosphere with a flow-rate of 20 mL/min, was applied: A first heating step from −30 to 180°C, followed by a cooling step to −30°C, and completed by a second heating to 200°C. The heating and cooling rates were set as 10°C/min and the typical sample weight was ~3 mg while an empty aluminum pan was used as reference. Calibration was performed using an indium sample. All tests were carried out, at least, in duplicate. The glass transition temperature (Tg), cold crystallization temperature (Tcc), enthalpy of cold crystallization (ΔHcc), melting temperature (Tm), and enthalpy of melting (ΔHm) were obtained from the heating scans, while the crystallization temperature from the melt (Tc) and enthalpy of crystallization (ΔHc) were determined from the cooling scan.
Thermogravimetric analysis (TGA) was performed in a TG-STDA model TGA/STDA851e/LF/1600 thermobalance from Mettler-Toledo, LLC (Columbus, OH, USA). The samples, with a weight of about 15 mg, were heated from 50 to 900°C, at a heating rate of 10°C/min under a nitrogen flow-rate of 50 mL/min.
Mechanical Tests
Tensile tests of the PHBV films were performed according to ASTM standard method D638 using an Instron 4400 universal testing machine from Instron (Norwood, MA, USA) equipped with a 1-kN load cell. The tests were performed, at room conditions, with 115 × 16 mm2 stamped dumb-bell shaped specimens using a cross-head speed of 10 mm/min. Samples were conditioned to the test conditions for 24 h prior to tensile assay. A minimum of six specimens were measured for each sample.
Permeability Tests
The water vapor permeability (WVP) of the film samples was determined using the gravimetric method ASTM E96-95 in triplicate. For this, 5 mL of distilled water was placed inside a Payne permeability cup (diameter of 3.5 cm) from Elcometer Sprl (Hermallesous-Argenteau, Belgium). The film was not in direct contact with water but exposed to 100% RH on one side and secured with silicon rings. They were placed within a desiccator, sealed with dried silica gel, at 0% RH cabinet at 25°C. The control samples were cups with aluminum films to estimate solvent loss through the sealing. The cups were weighted periodically using an analytical balance (±0.0001 g). WVP was calculated from the regression analysis of weight loss data vs. time, and the weight loss was calculated as the total loss minus the loss through the sealing. The permeability was obtained by multiplying the permeance by the film thickness.
Similar as described above for WVP, limonene permeability (LP) was measured placing inside the Payne permeability cups 5 mL of D-limonene. The cups containing the films were placed at controlled room conditions of 25°C and 40% RH. The limonene vapor permeation rates were estimated from the steady-state permeation slopes and the weight loss was calculated as the total cell loss minus the loss through the sealing. LP were calculated taking into account the average film thickness in each case. The samples were tested in triplicate.
The oxygen permeability coefficient was derived from the oxygen transmission rate (OTR) measurements that were recorded at 60% RH and 25°C, in duplicate, using an Oxygen Permeation Analyzer M8001 from Systech Illinois (Thame, UK). The humidity equilibrated samples were purged with nitrogen, before exposure to an oxygen flow of 10 mL/min. The exposure area during the test was 5 cm2 for each sample. In order to obtain the oxygen permeability (OP), film thickness and gas partial pressure were considered.
Statistical Analysis
Statistical differences across solution and material properties were evaluated with a two-tailed unpaired t-test using GraphPad Prism 7.04 of GraphPad Software Company (La Jolla, CA, USA). Mean values and standard deviations were also calculated. Significant differences were used at the 99% confidence level (p < 0.01).
Results and Discussion
PHBV Powders
Figure 2 shows the appearance of the obtained powders of the various PHBV samples. As it can be seen in Figure 2.1, the unpurified PHBV powder presented a light brown color, indicating the presence of biomass impurities derived from the pulp fruits and the fermentation process. The PHBV powder processed without any purification method but filtered, shown in Figure 2.2, presented a similar color. However, when this sample was filtered and centrifugated, the brown color became more intense, as it can be observed in Figure 2.3. Nevertheless, it is possible that the powder developed a dark color during the drying process. In any case, this suggests that most of the impurities still remained in the unpurified PHBV samples. After treatment with NaClO, the powder became clearly whiter, as shown in Figure 2.4. This color change can be related to both the extraction of impurities of the pulp fruits and the intrinsic whitening effect of bleach. However, the PHBV sample again developed the brown color for both the sample filtered (Figure 2.5) and the sample filtered and centrifugated (Figure 2.6). This would confirm that the color development was favored during drying by most likely Maillard browning reactions. In the case of the PHBV powder extracted with chloroform, as seen in Figure 2.7, it presented a yellowish color.

Figure 2. Pictures of the different poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) powders obtained after drying from the extraction routes of the different solutions from sample 1 to sample 7.
The visual aspect of here-obtained PHBV powders can be, therefore, related to the resultant content of biomass impurities derived from the pulp fruits and the fermentation process. As it can be seen in previous Table 1, the samples reached different yield values at each process, indicating that the removal of non-PHA material was achieved to different extends. In addition, one can observe that both the filtration and centrifugation processes further reduced the impurities content, producing a reduction of the yield values. For instance, the yield values of the NaClO- and chloroform-extracted powders were 56 and 40%, respectively. However, since it was necessary for the PHBV powder processed with NaClO to be filtered and centrifugated, the final yields generated with this route ranged in values of 48–28%.
Electrospun PHBV Fibers
All the here-prepared PHBV solutions were intended to be processed by electrospinning. However, due to their high impurities, samples 1 and 4 clogged the injector of the electrospinning device and hence could not be processed, being ruled out of the study. Samples 2, 3, 5, 6, and 7 were successfully processed by electrospinning and their optimal processing conditions are gathered in the cited Table 2.
Figure 3 shows the resultant electrospun mats obtained from the different PHBV solutions. In all cases, the electrospinning process generated a mat composed of non-woven fibers with different sizes. In particular, the mean diameters of the electrospun fibers obtained from the solution samples made of unpurified PHBV were 0.71 ± 0.12 μm (Figure 3A), after filtration (sample 2), and 0.75 ± 0.14 μm (Figure 3B), after filtration and centrifugation (sample 3). It is also worthy to mention that both samples, however, presented certain difficulties during electrospinning due to their high content of particles in suspension. For the solutions of PHBV extracted with NaClO, the fiber sizes significantly increased from 0.65 ± 0.24 μm (Figure 3C), after filtration (sample 5), to 1.91 ± 0.40 μm (Figure 3D), after filtration and centrifugation (sample 6). In the case of sample 7, i.e., the solution made of PHBV extracted with chloroform, the mean fiber diameter was 1.32 ± 1.12 μm (Figure 3E). The morphology of the electrospun fibers obtained from a solution made of commercial PHBV, processed in similar conditions, was also included for comparison purposes. These fibers presented a mean diameter of 1.21 ± 0.27 μm (Figure 3F).

Figure 3. Scanning electron microscopy (SEM) images of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibers of: (A) Sample 2, (B) Sample 3, (C) Sample 5, (D) Sample 6, (E) Sample 7, and (F) Commercial sample.
Therefore, the application of the extraction procedures increased the fiber size, which were further increased after centrifugation in the case of the NaClO route. This change in morphology suggests that the purity of the biopolymer improved, which in turn increased its content in the TFE solutions. The increase in fiber size with polymer content can be ascribed to an increase in the viscoelastic properties of the material processed by electrospinning (Torres-Giner et al., 2008). It is also worthy to mention that in the case of samples 2 and 5, i.e., samples that were not centrifugated, these solutions generated some beaded morphologies and/or big agglomerates. This fact can be related to the presence of remaining impurities in the PHBV solutions. Interestingly, these beaded-like structures were not observed in the case of sample 7, which was not centrifugated, suggesting that the extraction route with chloroform was more efficient than that with NaClO.
Due to the difficulties faced during electrospinning and the presence of beads in their fiber mats, the study was further continued only with samples 6 and 7. In order to correlate the obtained morphologies with the characteristics of each PHBV, the solution properties were characterized in terms of their viscosity, surface tension, and conductivity. In the case of sample 7, different PHBV concentrations were also tested in order to find out the best processing conditions for the materials. Table 3 displays the values of viscosity, surface tension, and conductivity of samples 6 and 7 as well as the properties of the solution made of commercial PHBV. From this table, it can be observed that the solution made of PHBV extracted with chloroform, at a concentration of 8 wt.-%, presented a significantly higher viscosity, i.e., ~28300 cP, than the PHBV solution processed with NaClO at the same concentration, i.e., ~6300 cP. Indeed, sample 7 could not be electrospun at 8 or even 4 wt.-%, but it was processed at 2 wt.-%. This PHBV solution provided a viscosity value of ~300 cP. The viscosity of the commercial benchmark PHBV solution was ~690 cP, i.e., between the viscosity range of both extracted PHBV solutions. In this sense, it has been reported that relatively high viscosities are desirable to obtain uniform electrospun fibers from biopolymers, however excessively high viscosities can also yield low processability due to instabilities during electrospinning (Sreekumar et al., 2017). In the case of surface tension, all PHBV solutions presented similar values, i.e., in the 20–27 mN/m range. All PHBV solutions also presented similar values of conductivity, varying from 1 to 4 μS/cm, with the exception of sample 6. For the latter PHBV solution, conductivity was considerably higher, i.e., 162.5 μS/cm, which is a factor that limits its processability by electrospinning. The observed higher conductivity may be related to the presence of ions coming from the bleach treatment. Although solution properties can anticipate processability issues during electrospinning, habitually it is difficult to elucidate the effect of a single property without considering the impact of the other ones (Torres-Giner et al., 2017).
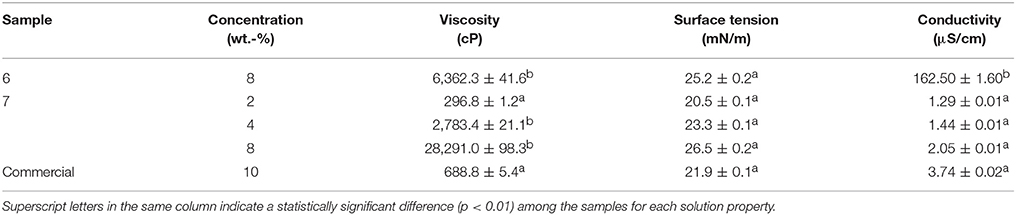
Table 3. Solution properties of the poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) samples.
Electrospun PHBV Films
The electrospun mats obtained from solution samples 6 and 7 were subjected to a thermal post-treatment in order to produce continuous films of more application interest in, for instance, packaging. In order to ascertain the film-forming process, the cryofracture surfaces of the electrospun mats after post-treatment at different temperatures were analyzed. Figure 4 shows the SEM images, both taken at low, i.e. 1,000x, and high magnifications, i.e., 5,000x, of the cross-sections of the electrospun PHBV mats. As one can observe, up to 110°C, the electrospun PHBV material preserved their original fiber-based morphology though the mat porosity was gradually reduced with increasing temperature. At a temperature of 125°C, a compact packing rearrangement of the electrospun fibers was produced. This phenomenon successfully resulted in the formation of a continuous film with a very low porosity. However, at the highest tested temperature, i.e., 145°C, the film integrity was lost and some large voids were formed probably due to partial material melting and/or degradation. Similar film cross-sections were recently observed by Cherpinski et al. (2017) for electrospun commercial PHB fibers post-processed at 160°C, a temperature below the homopolyester melting point. It was reported that the morphology of the starting electrospun non-woven fibers mat was lost due to the ultrathin fiber coalescence during the annealing step.

Figure 4. Scanning electron microscopy (SEM) images taken at 1,000x (Left) and 5,000x (Right) on the cross-sections of the electrospun poly(3-hydroxybutyrate-co-3-valerate) (PHBV) mats processed at different temperatures: Room temperature (25°C), 80, 110, 125, and 145°C. Scale markers of 50 and 10 μm.
Based on this observation, a post-processing temperature of 125°C was selected for annealing the electrospun mats obtained from samples 6 and 7. Figure 5 shows the visual aspect of both films to ascertain their contact transparency. Simple naked eye examination of this figure indicates that both PHBV materials generated films with similar surfaces but the contact transparency was higher in the film obtained from sample 7, i.e., in the film made of PHBV material purified with chloroform. This suggests that the latter film presented lower crystallinity and/or higher purity than the film produced with PHBV material extracted with NaClO.

Figure 5. Visual aspect of the electrospun poly(3-hydroxybutyrate-co-3-valerate) (PHBV) films obtained from: (A) Sample 6 and (B) Sample 7.
Characterization of the Electrospun PHBV Materials
The PHBV films obtained from samples 6 and 7 were characterized in terms of their hydrophobicity and thermal, mechanical, and barrier performance. These properties were analyzed in order to ascertain their potential use in packaging applications.
Contact Angle
The water contact angle of the PHBV films obtained from samples 6 and 7 were determined. Both electrospun films presented similar values, i.e., 78.2° ± 1.6 and 85.6° ± 0.3 for sample 6 and 7, respectively. Therefore, both electrospun films were highly hydrophobic suggesting that polar biomass residues were efficiently removed. The higher contact value observed for sample 7, i.e., the film obtained from the electrospun fibers treated with chloroform, further supports the fact that this film presented higher purity. Electrospun films made of commercial PHB and PHBV were recently applied to coat nanopapers, resulting in contact angle values in the 75-85° range (Cherpinski et al., 2018).
Thermal Properties
Table 4 displays the thermal transitions, for the three-step program, carried out by DSC for the electrospun fibers and annealed films of the two different extracted PHBV materials. The DSC curves of all thermal steps are gathered for each sample in Figure 6. During the first heating, one can observe that cold crystallization was not unambiguously observed in the fibers or the films for both PHBV materials. Additionally, melting was produced in a single peak in the 136–144°C range. Higher values of Tm were observed in the film samples in comparison to the fibers, suggesting that the films developed crystalline structures with thicker lamellae thickness or less defective crystals. In relation to the cooling step, it can be observed that both the fibers and film obtained from sample 6 crystallized from the melt in a single step, showing a clear Tc value at ~65.5 and 71°C, respectively. For sample 7, however, both PHBV materials, i.e., fibers and film, cold crystallized to a significant extent during the second heating step, presenting values of Tcc at ~52 and 65.5°C, respectively. Sample 7 did not exhibit a clear crystallization peak during the cooling step. These observations suggest that sample 6 was more easy to crystallize during cooling than sample 7, probably because of the presence of more nucleating impurities in the former sample.
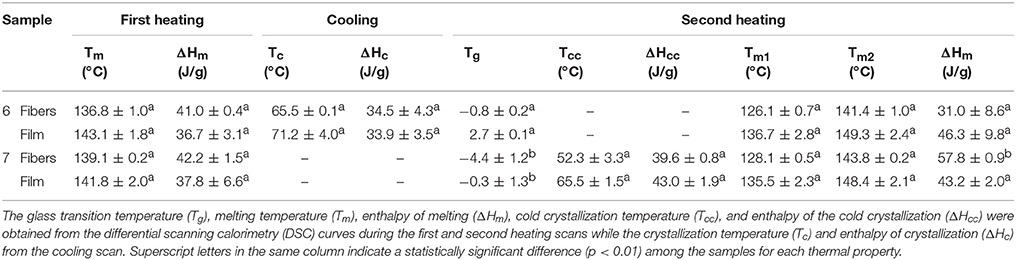
Table 4. Thermal properties of the poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibers and films.
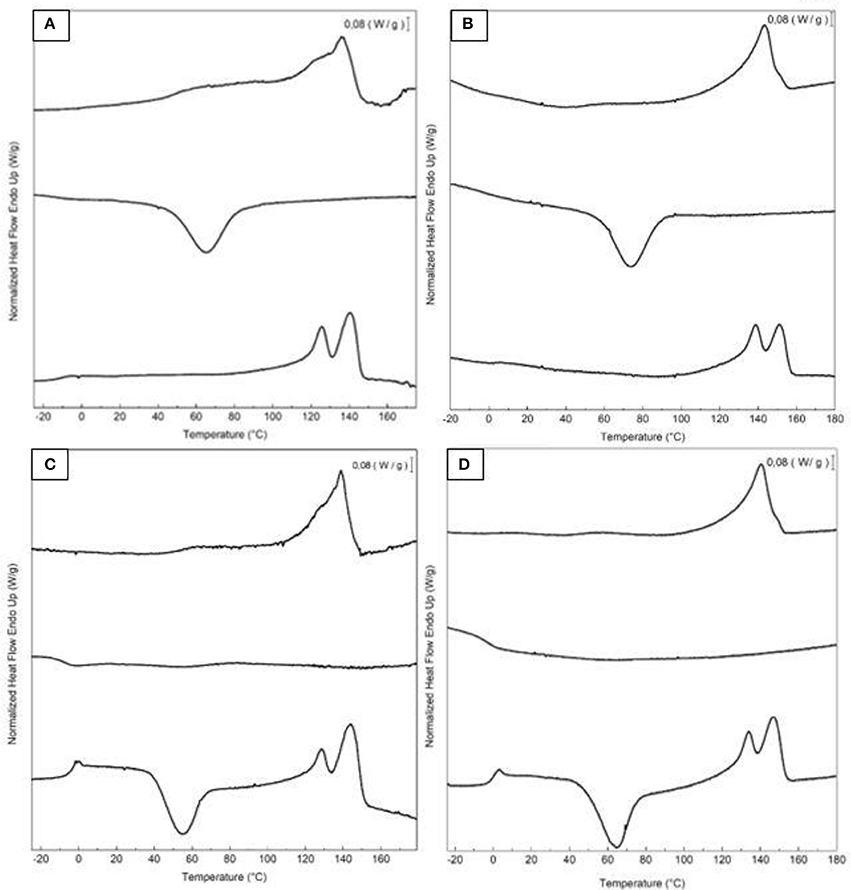
Figure 6. First heating, cooling, and second heating curves, shown from top to bottom, of the electrospun poly(3-hydroxybutyrate-co-3-valerate) (PHBV): (A) Fibers obtained from sample 6; (B) Film obtained from sample 6; (C) Fibers obtained from sample 7; (D) Film obtained from sample 7.
During the second heating, the glass transition of the PHBV was clearly observed, which occurred in the temperature range ranging from −5 to 3°C for all samples. In the case of sample 7, a slight endothermic peak during the glass transition was also noticed. This can be ascribed to a stress relaxation process of the amorphous phase during the heating run. Briefly, the amorphous polymer chains may attain a lower-free energy state during cooling. As a result of this frozen segmental mobility, the sample presents smaller free volume, enthalpy, and potential energy (Cowie et al., 1998). Therefore, when these materials are reheated, more energy is required to surpass the glass transition, resulting in a small endothermic peak associated with the change in heat capacity triggered by the Tg (Hutchinson et al., 1999). In addition, all samples showed a bimodal endothermic peak. A first melting temperature was observed in the range of 126–137°C, followed by a more intense second melting temperature in the 141–150°C range. Similar observations were previously found by Zhang et al. (2014) in a PHA copolyester, indicating that multiple melting peaks are linked to crystal reorganization upon melting by which imperfect crystals order into spherulites with thicker lamellar thicknesses and then melt at higher temperatures. In addition, the melting peaks at low temperatures could be also ascribed to the crystalline phase of the HV-rich fractions due to the relatively high HV content, i.e., 20 mol.-%. Additionally, similar to the first heating, higher Tm values were observed in the films that in the fibers. For instance, in the PHBV materials obtained from sample 7, the Tm values were in range of 128.1–143.8°C, for the fibers, whereas the values ranged in 135.5–148.4°C, for the film.
In comparison with other previous studies, the here-studied PHBV films exhibited a lower melting profile than other PHBV films prepared by solving casting, i.e., Tm values at 145–157°C for HV 12 mol.-% (Sanchez-Garcia et al., 2008). Similarly, PHBV materials with HVs of 3 and 18 mol.-% prepared by melt mixing also showed higher Tm values, i.e., 169.6 and 173.2°C, and Tc values, i.e., 107.1°C (Castro Mayorga et al., 2018). In relation to other electrospun films, (Cherpinski et al., 2018) reported two melting points at 162.2 and 177.3°C for PHB films while Tc was 110.4°C. The lower values in the present study can be related to the higher HV content of the here-processed PHBV materials.
Table 5 shows the thermal stability obtained by TGA of the electrospun fibers and films obtained from samples 6 and 7. One can observe that both processes generated materials with a similar thermal stability though sample 6, i.e., the PHBV materials extracted with NaClO, presented slightly better thermal values. In particular, the onset degradation temperature (T5%) of both materials obtained from sample 6 occurred at 268°C while the thermal decomposition of sample 7 started at 263 and 267°C, for its fibers and film, respectively. In addition, the thermal degradation temperature (Tdeg) was also higher in sample 6, i.e., 294°C, with a mass loss of only around 61%. In the case of sample 7, Tdeg values were at 285 and 290°C, for its fibers and film, respectively, presenting a mass loss of ~75%. Finally, the amount of residual mass was lower in the chloroform-extracted PHBV materials, in the 3–3.5% range, than the NaClO-extracted ones, i.e., 6–6.5%.
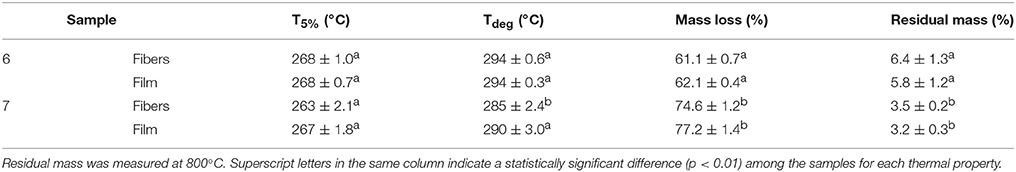
Table 5. The onset degradation temperature, defined as the temperature at 5% weight loss (T5%), degradation temperature (Tdeg), and residual mass of the poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibers and films.
Previous electrospun PHBV films with a HV content of 3 mol.-% showed a T5% value of 269°C and a Tdeg value of 290.8°C with a residual mass of 5.1% (Castro-Mayorga et al., 2017), which are relatively close to the ones obtained here. Interestingly, previously prepared PHBV films with a HV content of 18 mol.-% presented lower thermal stability, i.e., 249.8°C (Castro-Mayorga et al., 2016), which is an indication that the here-obtained PHBV films could have less impurities since it is known that the thermal degradation mechanism in PHA is easily accelerated by the presence of fermentation residues (Hablot et al., 2008).
Crystallinity
WAXD experiments were conducted on both films and fiber mats obtained from samples 6 and 7 to assess their crystallinity since DSC, being a dynamic method, does not provide an accurate description of crystallinity for this biopolymer as discussed earlier (Cherpinski et al., 2017; Castro Mayorga et al., 2018). The diffractograms, included in Figure 7, presented two main peaks located at 13.5° and 16.9° (2θ), followed by three other minor reflections at ca. 22°, 25°, and 27° (2θ). These peaks can be found in the four plots, thus revealing a similar semicrystalline nature for all the samples. According to the literature, these peaks correspond to the (020), (110), (111), (130), and (040) lattice planes of the orthorhombic unit cell of PHB (Mottina et al., 2016). This is in agreement with previous literature reporting that the crystalline structure of PHBV with HV contents below 37% is that of the PHB homopolymer (Kunioka et al., 1989).
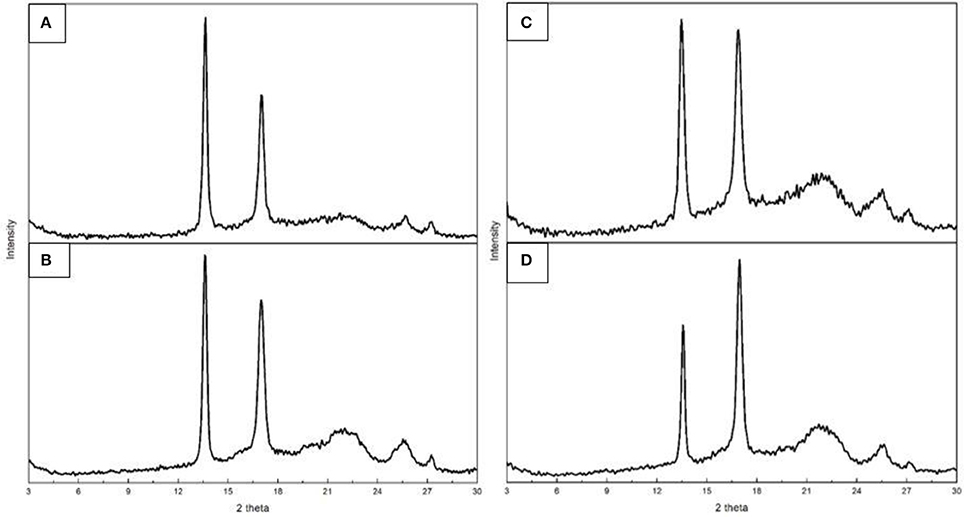
Figure 7. Wide angle X-ray diffraction (WAXD) patterns of the electrospun poly(3-hydroxybutyrate-co-3-valerate) (PHBV): (A) Fibers obtained from sample 6; (B) Film obtained from sample 6; (C) Fibers obtained from sample 7; (D) Film obtained from sample 7.
In regard to the crystallinity determined by WAXD, one can observe in Table 6 that the materials obtained from sample 6 were seen to exhibit higher crystallinity than those obtained from sample 7 while the films tended to show slightly higher crystallinity than the fibers, albeit the differences were very small. DSC seemed thus unable to discriminate among the samples, since all materials exhibited similar enthalpies of melting (see previous Table 4). As a result, and based on our previous study (Cherpinski et al., 2017), ATR-FTIR spectra as a function of temperature were recorded for the various materials with the aim of assessing their molecular order.
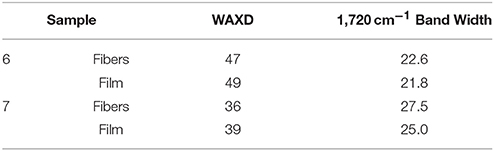
Table 6. WAXD crystallinity and ATR-FTIR 1720 cm−1 band width at half height.
Figure 8 gathers, as an example, two different view plots of the FTIR spectra taken across temperature of the electrospun fibers obtained from sample 7. Additionally, Figure 9 shows the evolution with temperature of the ratio of absorbance of the bands 1230/1453 and the 1,720 cm−1 band width at half height for the electrospun fibers obtained from samples 6, 7 and from the commercial PHBV solution. The latter two spectral features have been previously connected with the molecular order (crystallinity) in the polymer (Cherpinski et al., 2017). Thus, higher band ratios and lower 1,720 cm−1 band widths were correlated with higher crystallinity in the biopolymer (Cherpinski et al., 2017). The strongest peak, seen at ca. 1,720 cm−1, has been assigned to the stretching vibration of the carbonyl group (C = O) in PHA copolyesters, corresponding to the intramolecular bonding of their crystalline state, while the complex and multiple peaks in the region from 1,000 to 880 cm−1 have been related to the stretching bands of the carbon–carbon single bond (C–C) (Torres-Giner et al., 2016a). Finally, the ester-related band was observed at ~1,080 cm−1 and the band at ~1,020 cm−1 is known to arise from C–O and C–O–C stretching vibrations of ester groups in biopolyesters (Torres-Giner et al., 2011). In Figure 8 one can easily see that sample 7 underwent an increase in the intensity of the 1,720 cm−1 carbonyl band, and also of other peaks, with increasing temperature up to a maximum at around 120°C and then a decrease in band intensity, concomitant band broadening, and shift toward higher wavenumbers suggesting a decrease in molecular order and eventually melting. Figure 9 more clearly depicts for the three materials the evolution of the cited two molecular order spectroscopic indexes used. From this figure, it can be easily appreciated that for the fibers at room temperature the fibers obtained from the commercial PHBV solution and sample 6 were more crystalline than those obtained from sample 7, in good agreement with the WAXD results. With increasing temperature, samples 6 and 7 underwent an increase in molecular order with maximum around 120°C, in agreement with melting and recrystallization events suggested previously to explain crystallization, cold crystallization, and multiple melting endotherms during the DSC experiments.
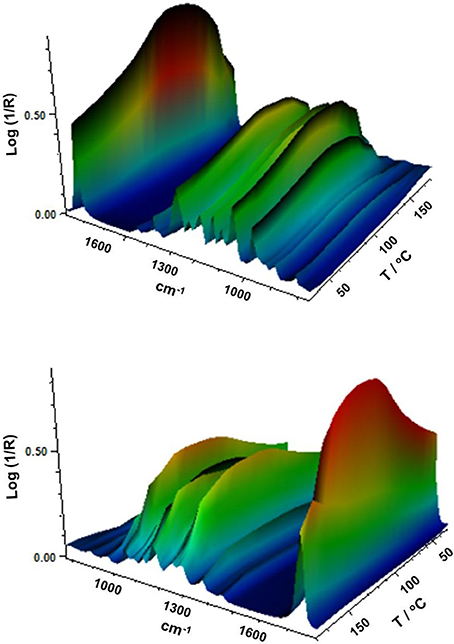
Figure 8. Fourier transform infrared (FTIR) spectra taken across temperature of the electrospun poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibers obtained from sample 7.
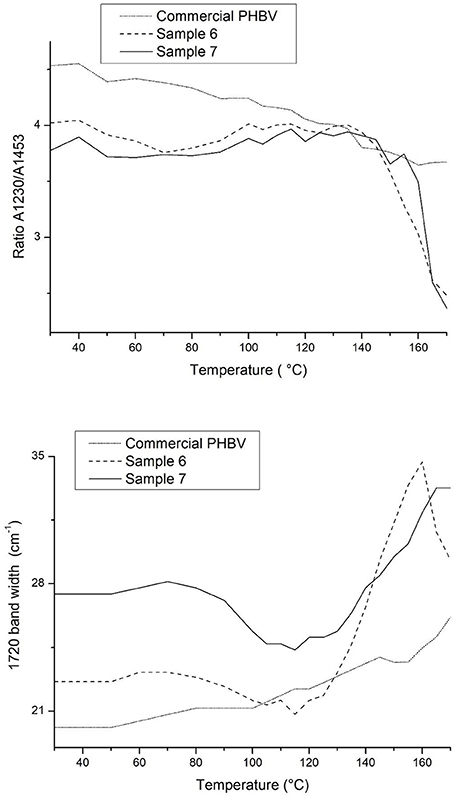
Figure 9. Evolution with temperature of the ratio of absorbance of the bands 1230/1453 and the 1720 cm−1 band width at half height for the electrospun poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibers obtained from samples 6, 7 and from the commercial PHBV solution.
Finally, Table 6 also gathers the 1,720 cm−1 band width for the fibers and films obtained from samples 6 and 7. This value was actually in good agreement with the WAXD results, again supporting that this simple and rapid method may be alternative to WAXD for rapid assessment of relative crystallinity among different samples, as opposed to the dynamic more commonly method often used based on DSC.
Mechanical Properties
Table 7 displays the values of the elastic modulus (E), tensile strength at break (σb), elongation at break (εb), and toughness (T) of the electrospun PHBV films calculated from their strain-stress curves. In general, both films presented characteristics of a brittle material associated to PHBV, showing low εb and T values, below 3% and 0.5 mJ/m3, respectively. However, in regard to the mechanical strength properties, the film samples presented a dissimilar performance. For the electrospun film obtained from sample 7, it can be observed that the specimens presented a relative low modulus, i.e., 434 MPa, while this was around three times higher for that obtained from sample 6. It is hypothesized that the presence of more impurities and higher crystallinity could be behind the higher rigidity associated to the film produced with sample 6.
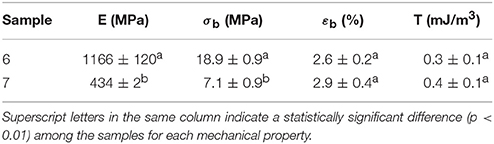
Table 7. Mechanical properties in terms of tensile modulus (E), tensile strength at break (σb), elongation at break (εb), and toughness (T) of the electrospun poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) films obtained from samples 6 and 7.
In comparison to PHB films prepared by conventional compression molding, it has been recently observed that equivalent annealed electrospun films exhibited higher elongation at break and toughness while similar mechanical strength (Cherpinski et al., 2017). Therefore, a more balanced mechanical performance can be achieved in biopolymer films prepared by electrospinning followed by optimal post-processing. The higher flexibility and toughness observed for the film produced with sample 7 makes it potentially more interesting for uses in flexible packaging applications.
Barrier Properties
Table 8 gathers the WVP, LP, and OP values of the electrospun PHBV films obtained from samples 6 and 7. From this table, it can be observed that the electrospun film obtained with the PHBV treated with chloroform, i.e., sample 7, presented the lowest WVP value, of 5.25 × 10−15 kg·m·m−2·Pa−1·s−1. This value was about 6 times lower than that observed for the PHBV film obtained from sample 6, i.e., 3.29 x 10−14 kg·m·m−2·Pa−1·s−1. This suggests that, in spite of the film prepared with NaClO being more crystalline, the film sample prepared with chloroform presented lower wettability, free volume, and presumably also lower porosity, which can be related to the presence of impurities. The here-reported values are close to this of the benchmark petroleum derived polyethylene terephthalate (PET) films, i.e., 2.30 × 10−15 kg·m·m−2·Pa−1·s−1 (Lagarón, 2011), widely employed for medium barrier food packaging applications. Additionally, the here-prepared electrospun PHBV films showed lower WVP values than these of PHBV films prepared by solvent casting, i.e., 1.27 × 10−14 kg·m·m−2·Pa−1·s−1 (Sanchez-Garcia et al., 2008), while these were higher but in the same range than these of compression-molded PHB films, i.e., 1.7 × 10−15 kg·m·m−2·Pa−1·s−1 (Sanchez-Garcia et al., 2007).
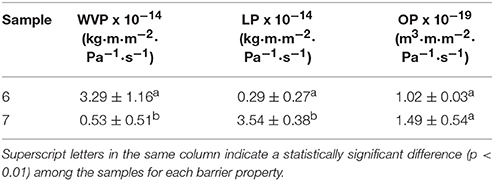
Table 8. Values of water vapor permeability (WVP), D-limonene permeability (LP), and oxygen permeability (OP) of the electrospun poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) films obtained from samples 6 and 7.
In relation to LP, a strong plasticizer for PHA polymers, the highest barrier performance was observed for the PHBV film prepared from sample 6. Thus, the LP value for the electrospun film obtained with PHBV treated with NaClO was 2.97 × 10−15 kg·m·m−2·Pa−1·s−1, which is similar to that observed for a compression-molded PHB film, i.e., 8.8 × 10−15 kg·m·m−2·Pa−1·s−1 (Sanchez-Garcia et al., 2007). LP reached a value of 3.54 × 10−14 kg·m·m−2·Pa−1·s−1 for the film prepared with sample 7. This can be related to the higher purity of this film, resulting in higher uptake and hence permeation for this sample. This value was lower, i.e., higher barrier, than those previously reported by Sanchez-Garcia et al. (2008) for PHBV films with 12 mol.-% HV prepared by solvent casting, i.e., 1.99 × 10−13 kg·m·m−2·Pa−1·s−1, and for PET films obtained by compression molding, i.e., 1.17 × 10−13 kg·m·m−2·Pa−1·s−1.
Finally, both electrospun PHBV films presented a good barrier performance to oxygen. In particular, the OP values were 1.02 × 10−19 m3·m·m−2·Pa−1·s−1, for the film obtained from sample 6, and 1.49 × 10−19 m3·m·m−2·Pa−1·s−1, for the one obtained from sample 7. These values are slightly lower than the OP value reported by Cherpinski et al. (2018), i.e., 1.20 × 10−18 m3·m·m−2·Pa−1·s−1, for an electrospun PHB film with a thickness of 142 μm. In this sense, it should be taken into account that OP is mainly a diffusivity-driven property since oxygen is a non-condensable small gas molecule and, hence, it is more sensitive to the material free volume, defects, porosity, morphological differences, crystallinity, etc. This suggests that the here-obtained electrospun films presented a relatively good uniformity and low porosity. Resultant OP values are also slightly lower than these reported for conventional 100-μm PHB films prepared by Sanchez-Garcia et al. (2007) using compression molding, i.e., 2.24 × 10−19 m3·m·m−2·Pa−1·s−1. In addition, in a more practical context for food packaging applications, the OP values of these electrospun PHBV films are in the range of PET films, i.e., 1.35 × 10−19 m3·m·m−2·Pa−1·s−1 and two order of magnitude lower than those of low-density polyethylene (LDPE) films, i.e., 2.15 × 10−17 m3·m·m−2·Pa−1·s−1 and only 1.5–2 higher than those of high-barrier ethylene–vinyl alcohol copolymer (EVOH) films, i.e., 7.7 × 10−20 m3·m·m−2·Pa−1·s−1 (Lagarón, 2011).
Conclusions
Circular economy and potentially low-cost PHBV-containing biomass, produced in a pilot plant scale from mixed microbial cultures fed with fruit pulp biowaste, was optimally subjected to two extraction process, namely NaClO and chloroform, and subsequently electrospun and post-processed by annealing to form continuous films with high transparency. The resultant PHBV films were seen to have differences in wettability, crystallinity, thermal stability, and mechanical and barrier properties depending on the route of purification applied. In view of the overall results, the biomass purified using chloroform resulted in lower crystallinity materials with higher toughness and barrier to moisture. Hence, this route seemed to be more favorable for the development of electrospun PHBV films as coatings or interlayers. The barrier data indicated that these new materials could potentially substitute their petroleum-based counterpart PET to applied in medium barrier food packaging applications.
Author Contributions
BM-R and ST-G purified the biomass, prepared the films, carry out most of the characterization and drafted the manuscript. JC-M and MR synthesized and provided the biomass. CS and LC conducted the crystallinity analysis. Conceptualization was devised by JL. Supervision ST-G and JL. Project Administration JL. MR and LC fermented and characterize the biomass. CS carried out the FTIR experiments as a function of temperature. LC carried out the mechanical measurements. JL designed the work, supervised the execution and interpretation of all experiments and carried out the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Unidad Asociada IATA-UJI Plastics Technology and the Spanish Ministry of Economy and Competitiveness (MINECO) project AGL2015-63855-C2-1-R and the H2020 EU projects YPACK (reference number 773872) and RES URBIS (reference number 730349) for funding. BM-R and ST-G would also like to thank the MINECO's FPI (BES-2016-077972) and Juan de la Cierva (IJCI-2016-29675) programs, respectively.
References
Babu, R. P., O'connor, K., and Seeram, R. (2013). Current progress on bio-based polymers and their future trends. Prog. Biomater. 2:8. doi: 10.1186/2194-0517-2-8
Busolo, M. A., Torres-Giner, S., and Lagaron, J. M. (2009). “Enhancing the gas barrier properties of polylactic acid by means of electrospun ultrathin zein fibers,” in Annual Technical Conference - ANTEC, Conference Proceedings (Chicago, IL), 2763–2767.
Castro-Mayorga, J. L., Fabra, M. J., and Lagaron, J. M. (2016). Stabilized nanosilver based antimicrobial poly(3-hydroxybutyrate-co-3-hydroxyvalerate) nanocomposites of interest in active food packaging. Innov. Food Sci. Emerg. Technol. 33, 524–533. doi: 10.1016/j.ifset.2015.10.019
Castro-Mayorga, J. L., Fabra, M. J., Pourrahimi, A. M., Olsson, R. T., and Lagaron, J. M. (2017). The impact of zinc oxide particle morphology as an antimicrobial and when incorporated in poly(3-hydroxybutyrate-co-3-hydroxyvalerate) films for food packaging and food contact surfaces applications. Food Bioproducts Process. 101, 32–44. doi: 10.1016/j.fbp.2016.10.007
Castro Mayorga, J. L., Fabra Rovira, M. J., Cabedo Mas, L., Sánchez Moragas, G., and Lagarón Cabello, J. M. (2018). Antimicrobial nanocomposites and electrospun coatings based on poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and copper oxide nanoparticles for active packaging and coating applications. J. Appl. Polym. Sci. 135:45673. doi: 10.1002/app.45673
Chen, L. J., and Wang, M. (2002). Production and evaluation of biodegradable composites based on PHB–PHV copolymer. Biomaterials 23, 2631–2639. doi: 10.1016/S0142-9612(01)00394-5
Cherpinski, A., Torres-Giner, S., Cabedo, L., and Lagaron, J. M. (2017). Post-processing optimization of electrospun submicron poly(3-hydroxybutyrate) fibers to obtain continuous films of interest in food packaging applications. Food Addit. Contamin. A Chem. Anal. Control Exposure Risk Assess. 34, 1817–1830. doi: 10.1080/19440049.2017.1355115
Cherpinski, A., Torres-Giner, S., Vartiainen, J., Peresin, M. S., Lahtinen, P., and Lagaron, J. M. (2018). Improving the water resistance of nanocellulose-based films with polyhydroxyalkanoates processed by the electrospinning coating technique. Cellulose 25, 1291–1307. doi: 10.1007/s10570-018-1648-z
Colombo, B., Favini, F., Scaglia, B., Sciarria, T. P., D'imporzano, G., Pognani, M., et al. (2017). Enhanced polyhydroxyalkanoate (PHA) production from the organic fraction of municipal solid waste by using mixed microbial culture. Biotechnol. Biofuels 10:201. doi: 10.1186/s13068-017-0888-8
Colombo, B., Pepè Sciarria, T., Reis, M., Scaglia, B., and Adani, F. (2016). Polyhydroxyalkanoates (PHAs) production from fermented cheese whey by using a mixed microbial culture. Bioresour. Technol. 218, 692–699. doi: 10.1016/j.biortech.2016.07.024
Cowie, J. M. G., Harris, S., and Mcewen, I. J. (1998). Physical aging in Poly(vinyl acetate). 2. Relative rates of volume and enthalpy relaxation. Macromolecules 31, 2611–2615.
Dias, J. M. L., Lemos, P. C., Serafim, L. S., Oliveira, C., Eiroa, M., Albuquerque, M. G. E., et al. (2006). Recent advances in polyhydroxyalkanoate production by mixed aerobic cultures: from the substrate to the final product. Macromol. Biosci. 6, 885–906. doi: 10.1002/mabi.200600112
Domingos, J. M. B., Puccio, S., Martinez, G. A., Amaral, N., Reis, M. A. M., Bandini, S., et al. (2018). Cheese whey integrated valorisation: Production, concentration and exploitation of carboxylic acids for the production of polyhydroxyalkanoates by a fed-batch culture. Chem. Eng. J. 336, 47–53. doi: 10.1016/j.cej.2017.11.024
Doshi, J., and Reneker, D. H. (1995). Electrospinning process and applications of electrospun fibers. J. Electrostat. 35, 151–160.
Echegoyen, Y., Fabra, M. J., Castro-Mayorga, J. L., Cherpinski, A., and Lagaron, J. M. (2017). High throughput electro-hydrodynamic processing in food encapsulation and food packaging applications: viewpoint. Trends Food Sci. Technol. 60, 71–79. doi: 10.1016/j.tifs.2016.10.019
Fabra, M. J., López-Rubio, A., and Lagaron, J. M. (2016). Use of the electrohydrodynamic process to develop active/bioactive bilayer films for food packaging applications. Food Hydrocoll. 55, 11–18. doi: 10.1016/j.foodhyd.2015.10.026
Fabra, M. J., Sánchez, G., Lopez-Rubio, A., and Lagaron, J. M. (2014). Microbiological and ageing performance of polyhydroxyalkanoate based multilayer structures of interest in food packaging. LWT Food Sci. Technol. 59, 760–767. doi: 10.1016/j.lwt.2014.07.021
Fiorese, M. L., Freitas, F., Pais, J., Ramos, A. M., De Aragão, G. M. F., and Reis, M. (2009). Recovery of polyhydroxybutyrate (PHB) from Cupriavidus necator biomass by solvent extraction with 1,2-propylene carbonate. Eng. Life Sci. 9, 454–461. doi: 10.1002/elsc.200900034
Hablot, E., Bordes, P., Pollet, E., and Avérous, L. (2008). Thermal and thermo-mechanical degradation of poly(3-hydroxybutyrate)-based multiphase systems. Polym. Degrad. Stab. 93, 413–421. doi: 10.1016/j.polymdegradstab.2007.11.018
Hutchinson, J., Smith, S., Horne, B., and, M., and Gourlay, G. (1999). Physical aging of polycarbonate: enthalpy relaxation, creep response, and yielding behavior. Macromolecules 32, 5046–5061.
Jacquel, N., Lo, C. W., Wu, H. S., Wei, Y. H., and Wang, S. S. (2007). Solubility of polyhydroxyalkanoates by experiment and thermodynamic correlations. AIChE J. 53, 2704–2714. doi: 10.1002/aic.11274
Kunasundari, B., and Sudesh, K. (2011). Isolation and recovery of microbial polyhydroxyalkanoates. Express Polym. Lett. 5, 620–634. doi: 10.3144/expresspolymlett.2011.60
Kunioka, M., Tamaki, A., and Doi, Y. (1989). Crystalline and thermal properties of bacterial copolyesters: poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate). Macromolecules 22, 694–697.
Lagarón, J. M. (2011). “Multifunctional and nanoreinforced polymers for food packaging,” in Multifunctional and Nanoreinforced Polymers for Food Packaging (Cambridge: Woodhead Publishing Limited), 1–28.
Lanham, A. B., Ricardo, A. R., Albuquerque, M. G. E., Pardelha, F., Carvalheira, M., Coma, M., et al. (2013). Determination of the extraction kinetics for the quantification of polyhydroxyalkanoate monomers in mixed microbial systems. Process Biochem. 48, 1626–1634. doi: 10.1016/j.procbio.2013.07.023
Martínez-Abad, A., Cabedo, L., Oliveira, C. S. S., Hilliou, L., Reis, M., and Lagarón, J. M. (2016). Characterization of polyhydroxyalkanoate blends incorporating unpurified biosustainably produced poly(3-hydroxybutyrate-co-3-hydroxyvalerate). J. Appl. Polym. Sci. 133. doi: 10.1002/app.42633
Martínez-Sanz, M., Lopez-Rubio, A., Villano, M., Oliveira, C. S. S., Majone, M., Reis, M., et al. (2016). Production of bacterial nanobiocomposites of polyhydroxyalkanoates derived from waste and bacterial nanocellulose by the electrospinning enabling melt compounding method. J. Appl. Polym. Sci. 133:42633. doi: 10.1002/app.42486
Mottina, A. C., Ayres, E., Orefice, R. L., and Câmara, J. J. D. (2016). What changes in poly(3-hydroxybutyrate) (PHB) when processed as electrospun nanofibers or thermo-compression molded film? Mater. Res. 19, 57–66. doi: 10.1590/1980-5373-MR-2015-0280
Mutlu, G., Calamak, S., Ulubayram, K., and Guven, E. (2018). Curcumin-loaded electrospun PHBV nanofibers as potential wound dressing material. J. Drug Deliv. Sci. Technol. 43, 185–193. doi: 10.1016/j.jddst.2017.09.017
Rehm, B. H. A. (2003). Polyester synthases: natural catalysts for plastics. Biochem. J. 376, 15–33. doi: 10.1042/bj20031254
Reis, K. C., Pereira, J., Smith, A. C., Carvalho, C. W. P., Wellner, N., and Yakimets, I. (2008). Characterization of polyhydroxybutyrate-hydroxyvalerate (PHB-HV)/maize starch blend films. J. Food Eng. 89, 361–369. doi: 10.1016/j.jfoodeng.2008.04.022
Samorì, C., Abbondanzi, F., Galletti, P., Giorgini, L., Mazzocchetti, L., Torri, C., et al. (2015). Extraction of polyhydroxyalkanoates from mixed microbial cultures: Impact on polymer quality and recovery. Bioresour. Technol. 189, 195–202. doi: 10.1016/j.biortech.2015.03.062
Sanchez-Garcia, M. D., Gimenez, E., and Lagaron, J. M. (2007). Novel PET nanocomposites of interest in food packaging applications and comparative barrier performance with biopolyester nanocomposites. J. Plastic Film Sheet. 23, 133–148. doi: 10.1177/8756087907083590
Sanchez-Garcia, M. D., Gimenez, E., and Lagaron, J. M. (2008). Morphology and barrier properties of solvent cast composites of thermoplastic biopolymers and purified cellulose fibers. Carbohydr. Polym. 71, 235–244. doi: 10.1016/j.carbpol.2007.05.041
Savenkova, L., Gercberga, Z., Nikolaeva, V., Dzene, A., Bibers, I., and Kalnin, M. (2000). Mechanical properties and biodegradation characteristics of PHB-based films. Process Biochem. 35, 573–579. doi: 10.1016/S0032-9592(99)00107-7
Serafim, L. S., Lemos, P. C., Albuquerque, M. G. E., and Reis, M. (2008). Strategies for PHA production by mixed cultures and renewable waste materials. Appl. Microbiol. Biotechnol. 81, 615–628. doi: 10.1007/s00253-008-1757-y
Serafim, L. S., Lemos, P. C., Oliveira, R., and Reis, M. (2004). Optimization of polyhydroxybutyrate production by mixed cultures submitted to aerobic dynamic feeding conditions. Biotechnol. Bioeng. 87, 145–160. doi: 10.1002/bit.20085
Sreekumar, S., Lemke, P., Moerschbacher, B. M., Torres-Giner, S., and Lagaron, J. M. (2017). Preparation and optimization of submicron chitosan capsules by water-based electrospraying for food and bioactive packaging applications. Food Addit. Contamin. A Chem. Anal. Control Exposure Risk Assess. 34, 1795–1806. doi: 10.1080/19440049.2017.1347284
Torres-Giner, S. (2011). “Electrospun nanofibers for food packaging applications,” in Multifunctional and Nanoreinforced Polymers for Food Packaging, ed J. M. Lagaron (Cambridge: Woodhead Publishing Limited), 108–125.
Torres-Giner, S., Gimenez, E., and Lagaron, J. M. (2008). Characterization of the morphology and thermal properties of Zein Prolamine nanostructures obtained by electrospinning. Food Hydrocoll. 22, 601–614. doi: 10.1016/j.foodhyd.2007.02.005
Torres-Giner, S., Gimeno-Alcañiz, J. V., Ocio, M. J., and Lagaron, J. M. (2011). Optimization of electrospun polylactide-based ultrathin fibers for osteoconductive bone scaffolds. J. Appl. Polym. Sci. 122, 914–925. doi: 10.1002/app.34208
Torres-Giner, S., Martinez-Abad, A., and Lagaron, J. M. (2014). Zein-based ultrathin fibers containing ceramic nanofillers obtained by electrospinning. II. Mechanical properties, gas barrier, and sustained release capacity of biocide thymol in multilayer polylactide films. J. Appl. Polym. Sci. 131, 9270–9276. doi: 10.1002/app.40768
Torres-Giner, S., Montanes, N., Boronat, T., Quiles-Carrillo, L., and Balart, R. (2016a). Melt grafting of sepiolite nanoclay onto poly(3-hydroxybutyrate-co-4-hydroxybutyrate) by reactive extrusion with multi-functional epoxy-based styrene-acrylic oligomer. Eur. Polym. J. 84, 693–707. doi: 10.1016/j.eurpolymj.2016.09.057
Torres-Giner, S., Pérez-Masiá, R., and Lagaron, J. M. (2016b). A review on electrospun polymer nanostructures as advanced bioactive platforms. Polym. Eng. Sci. 56, 500–527. doi: 10.1002/pen.24274
Torres-Giner, S., Wilkanowicz, S., Melendez-Rodriguez, B., and Lagaron, J. M. (2017). Nanoencapsulation of aloe vera in synthetic and naturally occurring polymers by electrohydrodynamic processing of interest in food technology and bioactive packaging. J. Agric. Food Chem. 65, 4439–4448. doi: 10.1021/acs.jafc.7b01393
Villano, M., Valentino, F., Barbetta, A., Martino, L., Scandola, M., and Majone, M. (2014). Polyhydroxyalkanoates production with mixed microbial cultures: from culture selection to polymer recovery in a high-rate continuous process. N. Biotechnol. 31, 289–296. doi: 10.1016/j.nbt.2013.08.001
Keywords: PHBV, electrospinning, packaging, biowaste, circular economy
Citation: Melendez-Rodriguez B, Castro-Mayorga JL, Reis MAM, Sammon C, Cabedo L, Torres-Giner S and Lagaron JM (2018) Preparation and Characterization of Electrospun Food Biopackaging Films of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Derived From Fruit Pulp Biowaste. Front. Sustain. Food Syst. 2:38. doi: 10.3389/fsufs.2018.00038
Received: 02 March 2018; Accepted: 26 June 2018;
Published: 17 July 2018.
Edited by:
Miguel Cerqueira, Laboratório Ibérico Internacional de Nanotecnologia (INL), PortugalReviewed by:
Miriam Dupas Hubinger, Universidade Estadual de Campinas, BrazilGuadalupe Virginia Nevárez-Moorillón, Autonomous University of Chihuahua, Mexico
Copyright © 2018 Melendez-Rodriguez, Castro-Mayorga, Reis, Sammon, Cabedo, Torres-Giner and Lagaron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose M. Lagaron, lagaron@iata.csic.es