 Yongtan Li
Yongtan Li Jun Zhang
Jun Zhang Shijie Wang
Shijie Wang Yiwen Zhang
Yiwen Zhang Minsheng Yang
Minsheng Yang- 1Forest Department, Forestry College, Hebei Agricultural University, Baoding, China
- 2Hebei Key Laboratory for Tree Genetic Resources and Forest Protection, Baoding, China
Pyrus hopeiensis is a valuable but endangered wild resource in the genus Pyrus. It has been listed as one of the 120 wild species with tiny population in China. The specie has been little studied. A preliminary study of propagation modes in P. hopeiensis was performed through seed propagation, hybridization, self-crossing trials, bud grafting, branch grafting, and investigations of natural growth. The results showed that the population size of P. hopeiensis was very small, the distribution range was limited, and the habitat was extremely degraded. In the wild population, natural hybridization and root tiller production were the major modes of propagation. Whole genome re-sequencing of the 23 wild and cultivated accessions from Pyrus species collected was performed using an Illumina HiSeq sequencing platform. The sequencing depth range was 26.56x−44.85x and the average sequencing depth was 32x. Phylogenetic tree and principal component analyses (PCA) based on SNPs showed that the wild Pyrus species, such as PWH06, PWH07, PWH09, PWH10, PWH13, and PWH17, were closely related to both P. hopeiensis HB-1 and P. hopeiensis HB-2. Using these results in combination with morphological characteristics, it speculated that P. hopeiensis populations may form a natural hybrid group with frequent gene exchanges between and within groups. A selective elimination analysis on the P. hopeiensis population were performed using Fst and π radio and a total of 381 overlapping genes including SAUR72, IAA20, HSFA2, and RKP genes were obtained. These genes were analyzed by gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) function enrichment. And four KEGG pathways, including lysine degradation, sphingolipid metabolism, other glycan degradation, and betaine biosynthesis were significantly enriched in the P. hopeiensis population. Our study provided information on genetic variation, evolutionary relationships, and gene enrichment in P. hopeiensis population. These data will help reveal the evolutionary history and origin of P. hopeiensis and provide guidelines for subsequent research on the locations of functional genes.
Introduction
Pyrus hopeiensis (2n = 34), belonging to Pyrus, is a rare wild resource of the genus that is unique to China. It is distributed in the provinces of Hebei and Shandong. Because of its limited distribution and population declines, P. hopeiensis has been classified as “the wild plants with tiny population” (Li et al., 2018) that urgently requires protection. It is an arbor. The leaves are ovate, broadly ovate to almost round, with long or short acuminate apices. The white flowers are clustered in umbrella-shaped racemes. Fruits are brown and generally have four ventricles, rarely five. The fruit has multiple lenticels and stone cells. The flowering period is during April, and the fruiting period is from September to October. Pu et al. (1985) also regarded P. hopeiensis as a separate species based on karyotype. Jiang et al. (1992) divided the genus Pyrus into 14 species, including P. hopeiensis, which was recognized by morphological characteristics. Based on the external morphology of the species, Yu (1984) suggested that it may have originated as a natural hybrid between Pyrus ussuriensis and Pyrus phaeocarpa and it is secondary species in the evolutionary process. However, the evolutionary origins of the species require further study.
Pears have significant phenotypic variation. Further studies of the genetic diversity of pear, and the identification of interesting gene-controlling traits will be key elements in future pear genetic breeding programs. However, pear is self-incompatible, and has a complex genetic background, a highly heterozygous genome, and a long breeding cycle, which make it difficult for breeders to directly determine phenotype-genotype relationships (Wang et al., 2016; Zhang et al., 2018). Moreover, it is easy to hybridize between Pyrus species, and the differences of biological and morphological characters between species or varieties are not obvious, which greatly increases the difficulty of phylogenetic evolution and classification of pears. The whole genome resequencing is to resequence the genomes of different individuals or populations of species with existing reference genomes in order to detected a large number of genetic variation information including SNP, Indel, SV, CNV etc. With the continuing publication of the pear genome (Wu et al., 2013; Linsmith et al., 2019; Ou et al., 2019; Dong et al., 2020) and the rapid development of high-throughput sequencing technologies, it provided a foundation for a genome-wide variation analysis of germplasm resources and the functional gene mining, phylogenetic analysis, population genetic structure, genetic diversity, analyses and selective elimination (Zhou et al., 2020), which is of great significance for the study of molecular breeding and population evolution of species. Li et al. (2020) used whole-genome resequencing to analyze the evolution and domestication characteristics of 69 lotus accessions founding that the flower lotus was biphyletic and genetically heterogeneous, whereas the rhizome and seed lotus were monophyletic and genetically homogeneous. Besides they identified a total of 1,346 selected regions in different lotus. Further analysis showed that genes in these regions were related to the important domestication traits of lotus such as insect resistance, seed weight, size and nutritional quality, freezing, and heat stress resistance.
To date, there are few reports on P. hopeiensis. The number, distribution, origins, and evolution of wild P. hopeiensis trees remain unclear. Information on the unique adaptability and genetic responses of P. hopeiensis to environmental pressure is lacking, and little is known of adaptation and genetic control of survival in natural habitats. In this study, we conducted a preliminary investigation about the quantity, distribution, natural environment, and reproductive mechanisms of P. hopeiensis and performed the first whole genome re-sequencing of 23 Pyrus accessions and carried out a detailed study of genomic mutation sites. And the SNPs obtained were used for population structure analysis of P. hopeiensis and other pears. Through a selective elimination analysis, the selection area of wild P. hopeiensis under natural conditions were accurately screened. At the same time, Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) function enrichment analysis of the screened genes were also performed. Our analyses will improve understanding of the selection pressures on P. hopeiensis during the evolutionary process and will provide a basis for the effective use of P. hopeiensis resources.
Materials and Methods
Sample Collection and Genomic DNA Extraction
In 2019, a survey of P. hopeiensis populations and other wild resources in Xingshuyuan and on Changyushan in Changli, Hebei Province were conducted. And a total of 20 wild Pyrus accessions and three locally cultivated Pyrus accessions were collected (Supplementary Table 1). The samples included Pyrus hopeiensis HB-1 (n = 3), Pyrus hopeiensis HB-2 (n = 2), Pyrus bretschneideri 'Guali' (n = 3), Pyrus betulifolia (n = 2), Pyrus ussuriensis Maxin. cv. Jingbaili (n = 1), Pyruscommunis L. cv. Early Red Comice (n = 1), and other wild pears (n = 11). The leaf and fruit morphologies of part of the wild plants collected were shown in Figure 1. The total DNA from fresh young leaves of the 23 Pyrus accessions were extracted using plant DNA extraction kit (TIANGEN, Beijing). And the extracts were sent to Novogene Co., Ltd. (Beijing) for whole genome re-sequencing. Agarose gel electrophoresis was used to analyze the RNA quality, degradation and contamination. The NanoDrop spectrophotometer (Thermofisher, Shanghai) was used to determine the purity of the DNA, and Qubit fluorometry (Thermofisher) was used to accurately quantify the DNA concentrations. Finally, the optical density (OD) values of DNA ranged between 1.8 and 2.0 and the content above 1.5 ug were used to build the library.

Figure 1. Images of leaves and fruit of the nine wild pears (PWH01, PWH04, PWH06, PWH07, PWH09, PWH10, PWH12, PWH18, PWH20) included in this study.
Pollination Tests
Cross-pollination and self-pollination (Supplementary Table 2) trials were performed at the end of March and in early April 2017 and 2018. The anthers were collected 1 day before flowering and the pollen were collected for pollination after dispersal at indoor. For self-pollination tests, two or three lateral flowers were selected for bagging before flowering and the other flowers were picked. Moreover, the artificial pollination was also performed. In the cross-pollination trials, the androecia of the flowers were removed 1 day before pollination and the flowers were bagged after pollination.
Library Construction and Whole Genome Re-sequencing
The qualified DNA samples were randomly broken into fragments of 350 bp in length using a Covaris (Guangzhou) ultra-sonic shearer. A Truseq Library Construction Kit (Illumina, Shanghai) was used to build the library. The entire library was prepared in the following steps: end repair, polyA tail addition, sequencing connector addition, purification, PCR amplification. Then the constructed library was sequenced with an Illumina HiSeq sequencing system. To obtain clean data, the low quality paired reads were removed based on the following criteria: reads with adapters, paired reads with N content exceeding 10% of the length of the read and low-quality (Q ≤ 5) paired reads contained in single-end sequencing reads that exceeded 50% of the length of the read.
Variant Detection and Annotation
The effective sequencing data were compared with the reference genome of Pyrus communis Bartlett DH Genome v2.0 (https://www.rosaceae.org/species/pyrus/pyrus_communis/genome_v2.0) using BWA (Li and Durbin, 2009) software (parameter: mem -t 4 -k 32 -M). SAMtools (Li et al., 2009) and Picard (http://picard.sourceforge.net) software were used to remove duplicates.
SAMtools software was used to detect the SNPs and insertion-deletion mutations (InDels) in 23 Pyrus accessions and ANNOVAR software was used for annotation (Wang et al., 2010). To reduce the detection error rate, SNPs with quality values (MQ) <20 and SNPs with read support numbers <4 were filtered out. The snpEff software (https://pcingola.github.io/SnpEff/) was used to annotate the filtered SNPs based on the Pyrus communis Bartlett DH Genome v2.0 genome. And the SNPs were classified according to their impact. Meanwhile, the insertion and deletion of small fragments <50 bp in length were detected.
Breakdancer (Chen et al., 2009) and CNVnator software (Abyzov et al., 2011) were used to detect structure variants (SVs) and copy number variants (CNVs), respectively. Breakdancer software was used to detect insertion (INS), deletion (DEL), inversion (inversion, INV), intra-chromosomal translocation (ITX), and inter-chromosomal translocation (CTX). The SVs with support numbers of paired-end (PE) reads <2 were filtered out. CNVnator was used to detect different read coverage depths on the genome to determine potential deletion and duplication. Then ANNOVAR was used to perform functional annotations of the all gene mutations detected. Finally, the circos (Krzywinski et al., 2009) were used to visualize the structural variation in P. hopeiensis.
Population Structure Analysis
A neighbor-joining tree was constructed using TreeBeST 1.9.2 software (Jin et al., 2012) with 1000 bootstrap replicates based on SNPs detected in whole genome resequencing analysis. Principal component analysis (PCA) was performed using gcta64 1.92.2 software (Yang et al., 2011). Population clustering analysis were calculated by ADMIXTURE 1.3.0 software (Alexander et al., 2009). The number of clusters was varied from K = 2 to K = 9 and the lowest cross-validation error was used to determine the optimal K value in order to determine the ideal population structure.
Selective Elimination Analysis
In order to detect the selected regions of P. hopeiensis, and to determine the identities of the selected genes to explain the adaptation mechanism in population evolution and domestication at the molecular level, PopGenome software (Pfeifer et al., 2014) was used to perform a sliding window according to physical length (100 kb as the window and 10 kb as the step size) based on the filtered high-quality SNPs to analyzed the π ratio (Lin et al., 2014) and the genetic differentiation index (Fst) (Hudson et al., 1992) between the two populations. At the same time, the significant regions were screened by the top 5% to identified the genes in these selected regions based on the Fst and π ratio analyses. And the Fst and π ratio values were combined to obtain the candidate genes in the selected region and to complete the construction of the Venn diagram. All relevant charts were plotted by R scripts (Team, 2013).
Variant Functional Annotation and Enrichment Analysis
GO and KEGG pathway enrichment analyses were conducted on the selected area of the genome. The numbers of genes enriched in different GO entries were counted and the GO entries significantly enriched in the candidate region were determined. Besides, the KEGG enrichment analysis was also performed to identify the significantly enriched pathways. The enriched pathways and genes were selected stringently using an adjusted probability (P < 0.05).
Results and Analysis
Distribution and Habitats of Pyrus hopeiensis
At present, the P. hopeiensis were found only in Changli, Hebei Province and Laoshan, Shandong Province. Between 2016 and 2019, multiple surveys of the numbers, habitats and distribution of the species in Changli were conducted. The surveys showed that P. hopeiensis had disappeared in many places; the trees were found only in Xingshuyuan and Changyushan village (Figure 2A). Based on phenotype and previous SSR analysis, it initially believed that two main types of P. hopeiensisr existed: P. hopeiensis HB-1 and P. hopeiensis HB-2. The P. hopeiensis HB-1 population occurred mainly in Xingshuyuan. More than 100 P. hopeiensis specimens were found, most of which were shrubs generated by root sprouting. And only 10 tree form specimens remained. The maximum age of the individuals was ca. 50 years. In the early stage of our work, some individuals with SSR molecular markers were tested and no differences were found among these specimens. The P. hopeiensis HB-2 populations found only five strains were distributed on Changyushan. After verification using SSR molecular markers, no differences were found among the individuals. The P. hopeiensis populations lived under very harsh conditions (Figure 2B). They grew mostly on cliffs and had bare roots. As the fruits of P. hopeiensis are small and inedible, local people mostly used the trees as rootstocks or felled them to provide new areas for development. So few P. hopeiensis individuals survived.
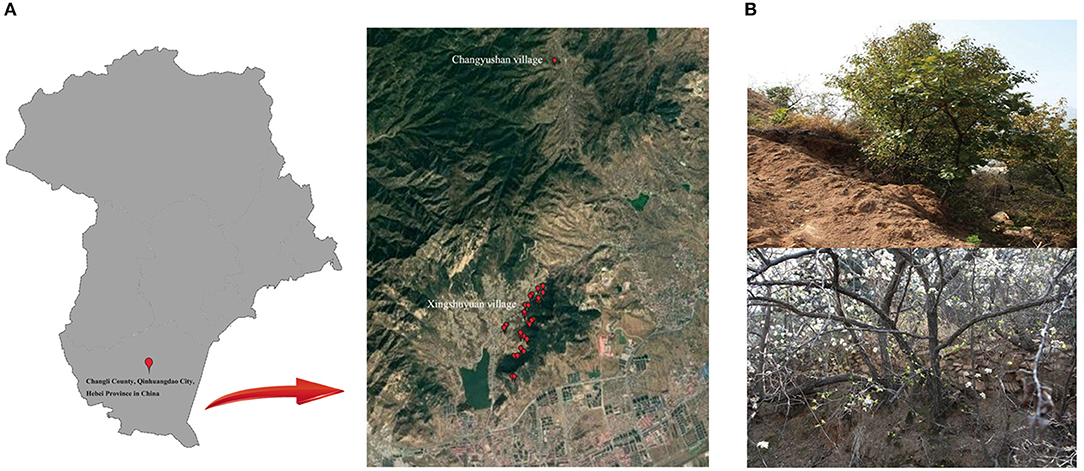
Figure 2. (A) Distribution of part of the Pyrus hopeiensis; (B) Habitat of Pyrus hopeiensis.
Preliminary Study of the Reproduction Mode in Pyrus hopeiensis
Using P. hopeiensis individuals (No. 1 and No. 5) as the female parents, and the five cultivated pear strains (Huangguan, Yali, Weixian red pear, Xuehua, and Dunzi pear) as the male parents, the hybridization experiments were performed and a total of 1,190 hybrid seeds were obtained. When P. hopeiensis were used as the male parent and Xianghong and Yali as the female parents, these crosses produced a total of 281 hybrid seeds (Supplementary Table 2). The plant height and ground diameter of the offspring plants from each cross (now planted in the Changli Institute for Pomology) were measured. The average height of progeny plants from hybrid combination 7 (No. 5 P. hopeiensis HB-1 × Yali) was 106.63 cm, the highest among crosses (Figure 3A). The progeny plants of hybrid combination 4 (No. 1 P. hopeiensis HB-1 × Xue hua) had the largest average ground diameter (8.9 cm) (Figure 3B). The plant height and ground diameter varied among different offspring. The results showed that the progeny of different cross combinations were quite different. The progeny of Yali and P. hopeiensis had greater average plant heights and ground diameters than the progeny of Xianghong and P. hopeiensis. In total, 1,839 natural hybrid seeds collected of P. hopeiensis. After sand storage, accelerated budding and sowing, 466 progeny plants of P. hopeiensis were obtained in the following year. These individuals were planted at the Changli Institute for Pomology, where they were growing well. The plant heights ranged between 69.11 and 99.37 cm (Figure 3A), and the ground diameter ranged between 6.34 and 8.23 cm (Figure 3B).
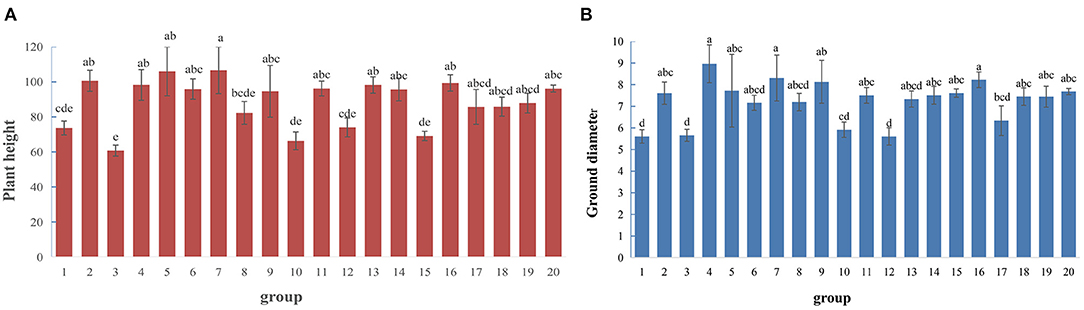
Figure 3. Plant height (A) and ground diameter (B) of the hybrid combination.
The bagging for self-crossing of P. hopeiensis in Xingshuyuan obtained no fruits. Thus, it provisionally categorized P. hopeiensis as self-incompatible. The different P. hopeiensis plants were cross pollinated and the same individuals were self-pollinated (Supplementary Table 2). Again, no fruits were produced indicating that P. hopeiensis was self-incompatible.
The stems of sterile seedlings of P. hopeiensis were used as source materials for culture. A medium was optimized to establish tissue cultures and a rapid propagation system for P. hopeiensis and 1,000 rooted tissue culture seedlings were obtained. In addition, 80 P. hopeiensis seedlings obtained by bud grafting grew well. The one-year-old branches of P. hopeiensis as scions, and 1- or 2-year-old seedlings as rootstocks were collected for grafting and propagation. A total of 2,200 P. hopeiensis seedlings were obtained and 1,900 of these were provided to diverse ex-situ conservation bases.
The investigation of the natural propagation mode of P. hopeiensis showed that individuals at some sites were clustered. We excavated the roots and found that the specimens were derived from root tillers, and no seedlings were found. An earlier SSR molecular marker detection investigation showed that P. hopeiensis trees occurring in Xingshuyuan belonged to a single genotype and had low genetic diversity. Thus, it came to the provisional conclusion that P. hopeiensis reproduced mainly by root tillers under natural conditions.
In summary, it postulated that P. hopeiensis could be bred through hybridization, tissue culture, bud grafting, branch grafting and propagation by root tillers. Moreover, it was self incompatible but propagated through root tillers in its natural environment.
Variation in the Pyrus Genome
The sequencing produced 399.361G raw data from 23 Pyrus accessions, and 388.774G clean data after filtering. The sequencing quality was high (Q20 ≥ 96.35%, Q30 ≥ 90.36%) (Supplementary Table 3). Paired-end sequencing reads were mapped to the reference genome (P. communis Bartlett DH Genome v2.0). The reference genome size was 498,265,991 bp and the GC content (%) was 37.47%.
The comparison ratio of all samples were between 92.75 and 98.15%, the coverage depth was between 26.56x and 44.85x, and the 1X coverage (with at least one base coverage) was >85.95% (Supplementary Table 4). The comparison result was normal and suitable for subsequent mutation detection and related analyses.
The genome-wide variations in 23 Pyrus accessions included 107,133,072 single nucleotide polymorphisms (SNPs), 14,186,626 insertions or deletions (InDels), 290,443 copy number variations (CNVs), and 1,317,828 structural variations (SVs). The numbers of SNPs detected ranged from 2,980,930 (P. communi L. cv. Early Red Comic) to 5,211,026 (PWH07). Most mutation sites were located in non-coding regions (Table 1), such as intron and intergenic regions. Among them, 58.71% of SNPs were located in intergenic regions, 14.48% of SNPs were located in intron regions, and 9.98% were located in coding regions (Figure 4A). The SNP in coding regions included 107,289–242,631 synonymous substitutions and 124,051–253,183 non-synonymous substitutions (Figure 4B), giving non-synonymous-to synonymous substitution ratios (dN/dS) in the range 1.030–1.156 showing that the numbers of non-synonymous mutations exceeded the numbers of synonymous mutations. The transition/transversion (ts/tv) ratio values were in the range 1.743–1.939 and the heterozygosity rate of different Pyrus accessions varied greatly, ranging from 0.19 to 0.53%, with an average of 0.44%. Based on the type of change and its predicted effect, 1.53% of the SNPs were predicted to have high impacts (e.g., stop codon gaining, frameshift), 1.38% moderate impacts (e.g., non-synonymous change, non-disruptive frameshift), and 0.15% low impacts (e.g., synonymous coding/start/stop, start gained).
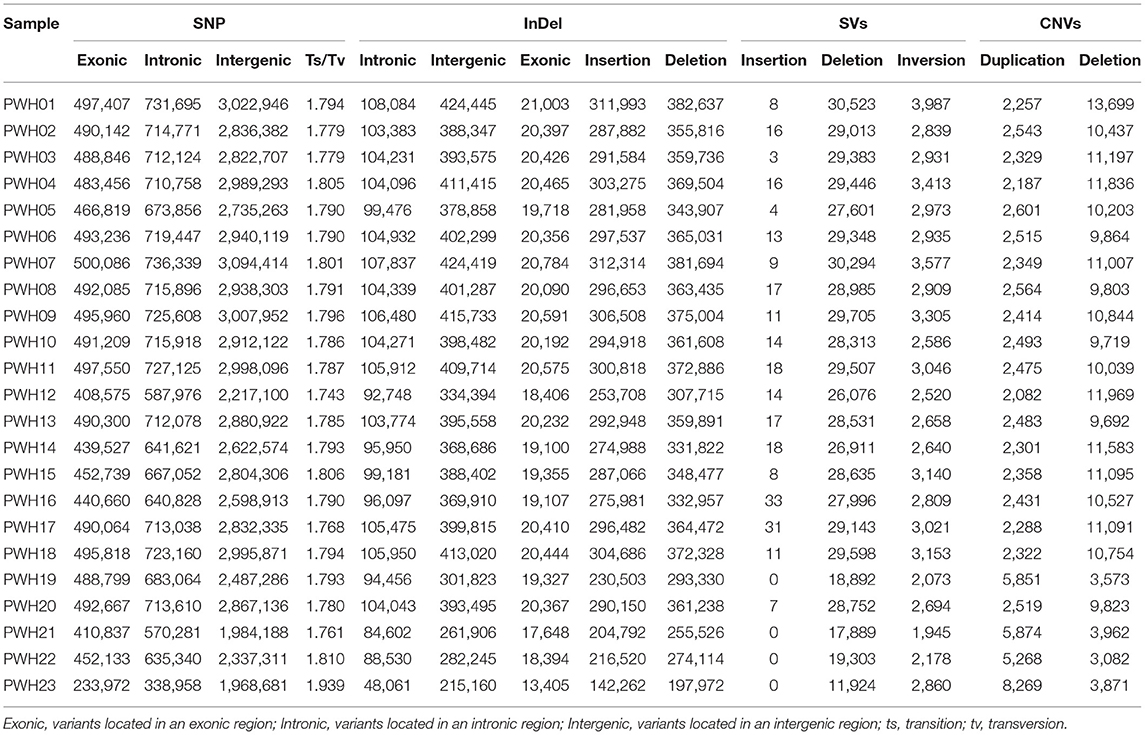
Table 1. Genome-wide variations identified in 23 pyrus accessions.
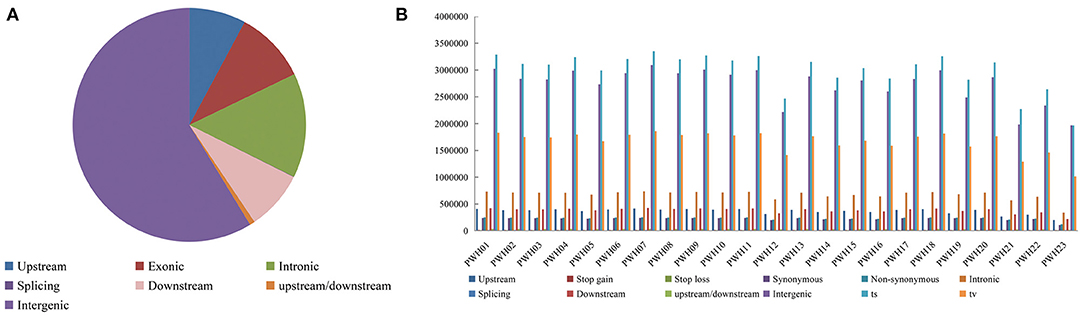
Figure 4. (A) Distribution of SNPs in genomic regions. (B) Number of different SNPs in genomic regions per species.
Regarding InDels, the number of InDels mutations detected ranged from 340,234 (P. communis L. cv. Early Red Comic) to 694,630 (PWH01). And the total number of InDel variants amounted to 14,186,626, of which 6,355,526 were insertions and 7,831,100 deletions showing that the numbers of deletions exceeded the insertions. Besides, the Indel heterozygous rates of different Pyrus accessions differed, ranging from 0.16 to 0.47‰ (average 0.39‰). Among those with potential functional consequences, 3.18% were located in gene exons and 0.077% were located in splice site regions. What is more, 60.43% (8,572,988) were distributed in the intergenic region, 16.01% (2,271,908) were distributed in the intron region, and 3.18% (18,046) were distributed in the coding region among all the detected Indels. And the vast majority of Indels were located upstream or downstream from genes and intergenic regions (Figure 5A). Moreover, the lengths of insertions and deletions were in the range 1–50 nucleotides (Figure 5B) (Supplementary Tables 5, 6).
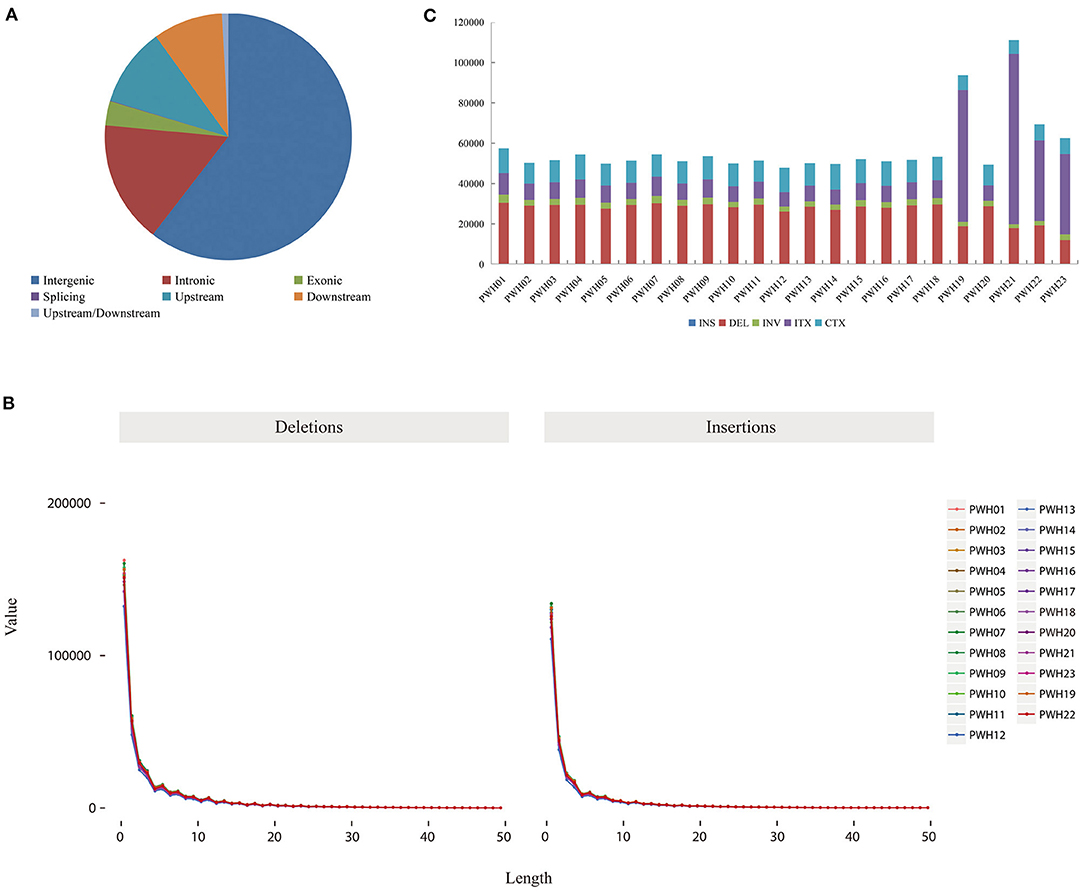
Figure 5. (A) Distribution of SNPs in genomic regions. (B) The distribution of small insertions and deletions in genomic regions per species. (C) The number of different SVs in genomic regions per species.
The numbers of SVs among the 23 Pyrus accessions ranged between 47,827 (PWH12) and 111,172 (PWH21) (Figure 5C). The deletions, insertions, inversions, ITX, and CTX accounted for 46.73% (615,768), 0.02% (268), 5.02% (6,6192), 29.55% (389,468), and 18.68% (246,132) of the detected SVs, respectively. And the CNVs ranged from 8,350 (P. ussuriensis Maxin. cv. Jingbaili) to 15,956 (PWH01), with an average of 12,628. Besides, the number of duplications ranged from 2,082 (PWH12) to 8,269 (P. communis L. cv. Early Red Comice) and the number of deletions ranged from 3,082 (P. ussuriensis Maxin. cv. Jingbaili) to 1,3699 (PWH01). Among these Pyrus species, 34,867 CNV and 198,137 SV were detected in P. hopeiensis HB-1, and 24,856 CNV and 100,806 SV were detected in P. hopeiensis HB-2, respectively (Figure 6).
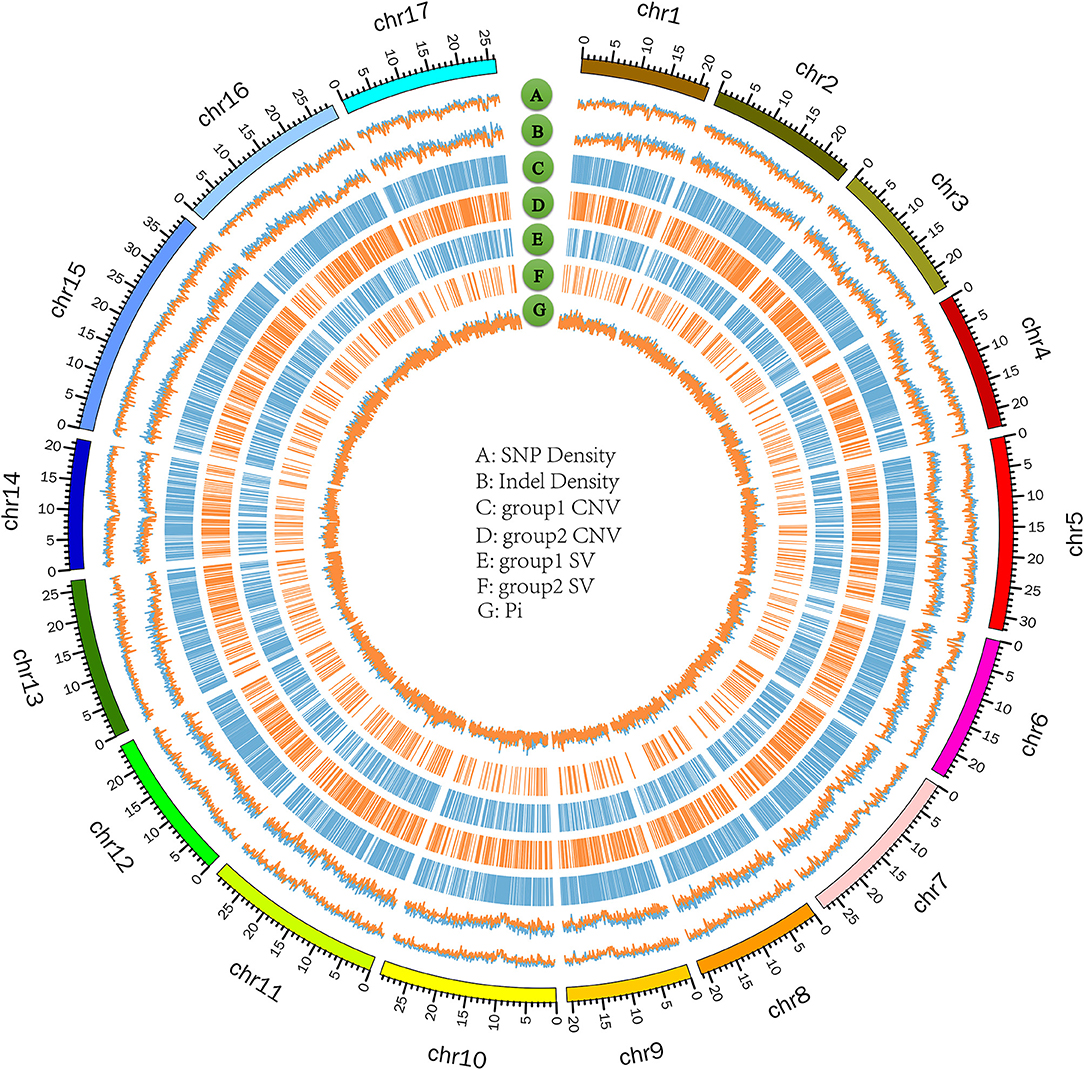
Figure 6. Circos plot on 17 chromosomes of P. hopeiensis. (A) SNP density; blue refers to group 1, orange to group 2. (B) Indel density; blue refers to group 1, orange to group 2. (C,D) CNV in group 1 and group 2, including deletion and duplication. (E,F) SV in group 1 and group 2, including insertion and deletion. (G) Average number of nucleotide differences per site (pi); blue refers to group 1, orange to group 2. Group 1 contains P. hopeiensis HB-2, group 2 contains P. hopeiensis HB-1.
Population Structure of Pyrus
After filtering, the high-quality SNPs identified on 17 chromosomes (Supplementary Figure 1) were used as markers in the subsequent principal component analysis (PCA), population structure and evolutionary tree analysis. The neighbor-joining tree showed that the P. hopeiensis populations were arrayed on two different branches (Figure 7A). The P. hopeiensis HB-1 population was on one branch, and the P. hopeiensis HB-2 population on the other branch. Two P. betulifolia plotted together, and three P. bretschneideri ‘Guali' plotted together. Moreover, PWH06, PWH07, PWH09, PWH10, PWH13 wild pears, and P. hopeiensis HB-1 were close to one another on the neighbor-joining tree. PWH17 and P. hopeiensis HB-2 were close. The PCA separated individuals with different genetic backgrounds and grouped individuals with similar genetic backgrounds. The PCA results was similar to that of the neighbor-joining tree. Individuals of P. hopeiensis HB-1 and P. hopeiensis HB-2 were clustered together in the PCA plot (Figure 7B). The population structure showed that the cross-validation error rate was smallest when the kinship coefficient (K) = 3 (Supplementary Figure 2). And when K = 3, the genetic backgrounds between P. hopeiensis HB-1 and P. hopeiensis HB-2 populations were identical and the genes penetration between PWH01, PWH03, PWH09, PWH12, PWH14, PWH15, PWH17, PWH21, PWH22, PWH23, and P. hopeiensis were found (Figure 7C). The results that were not fully congruent with the neighbor-joining tree and PCA plots. Supplementary Figure 3 showed the outcomes when K= 5–9.
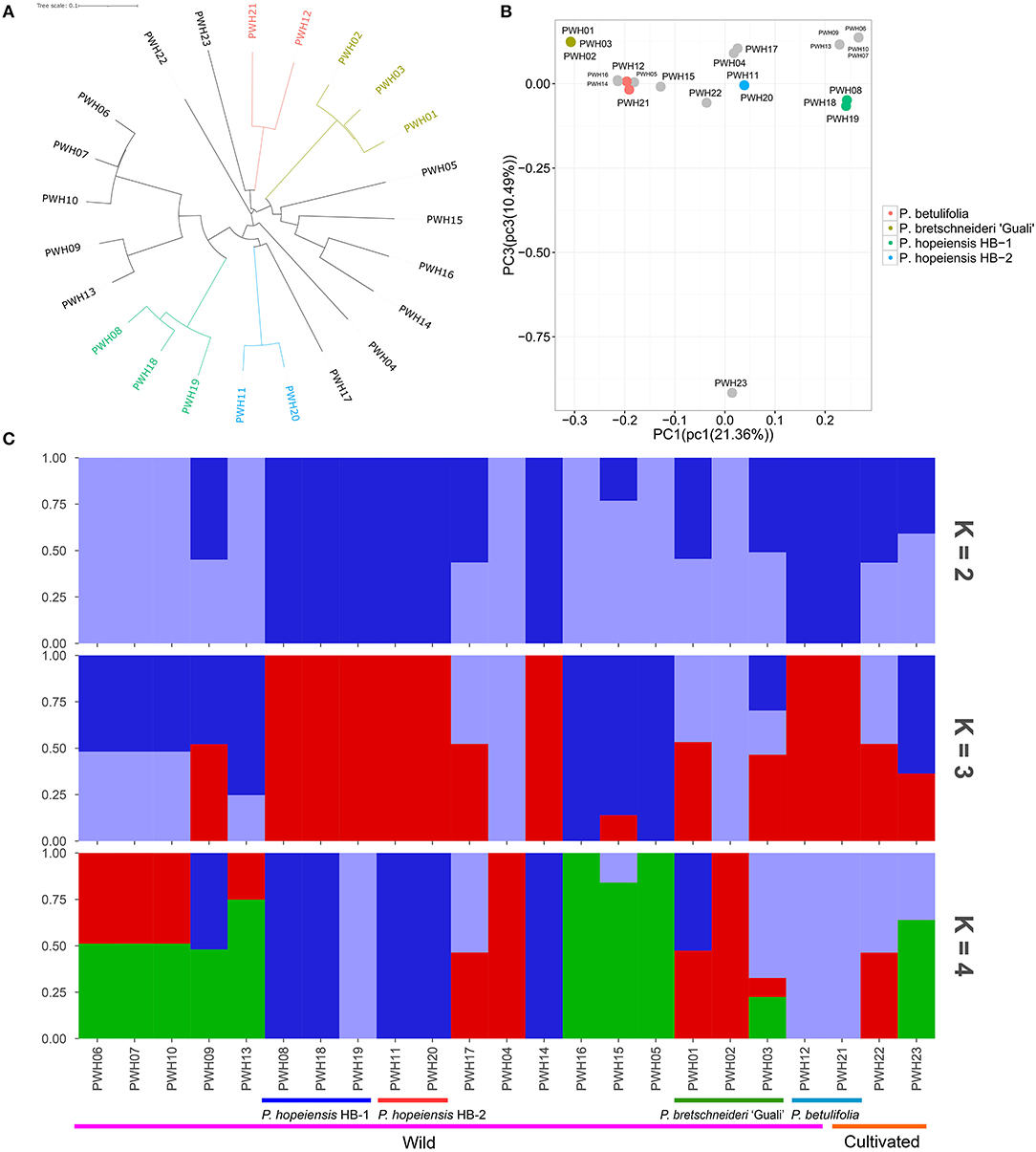
Figure 7. Population structure. (A) Neighbor-joining phylogenetic tree constructed using SNP data. (B) Principal component analysis (PCA) of the 23 Pyrus accessions. (C) Population structure (K = 2–4) of the 23 Pyrus accessions.
Identification of Positive Selective Sweeps
The Fst (Figure 8A) and π ratio (Figure 8B) were used to calculate the positive selection area between the P. hopeiensis population and the P. bretschneideri “Guali” population. The Fst value calculations obtained 2,916 positive selection genes, and 2,921 positive selection genes were detected by the π ratio. In addition, regions with significantly elevated Fst values (Fst > 0.45) and regions with elevated π ratios (π ratio > 1.07) were selected. A total of 381 overlapping genes including SAUR72, IAA20, HSFA2, and RKP genes with strong selection signals (Figure 8C) (Supplementary Table 7). The Fst values and the π ratios of the candidate region exceeded those of the whole genome site. Figure 8D showed the results of the Fst and π ratio selective elimination analysis for P. hopeiensis.
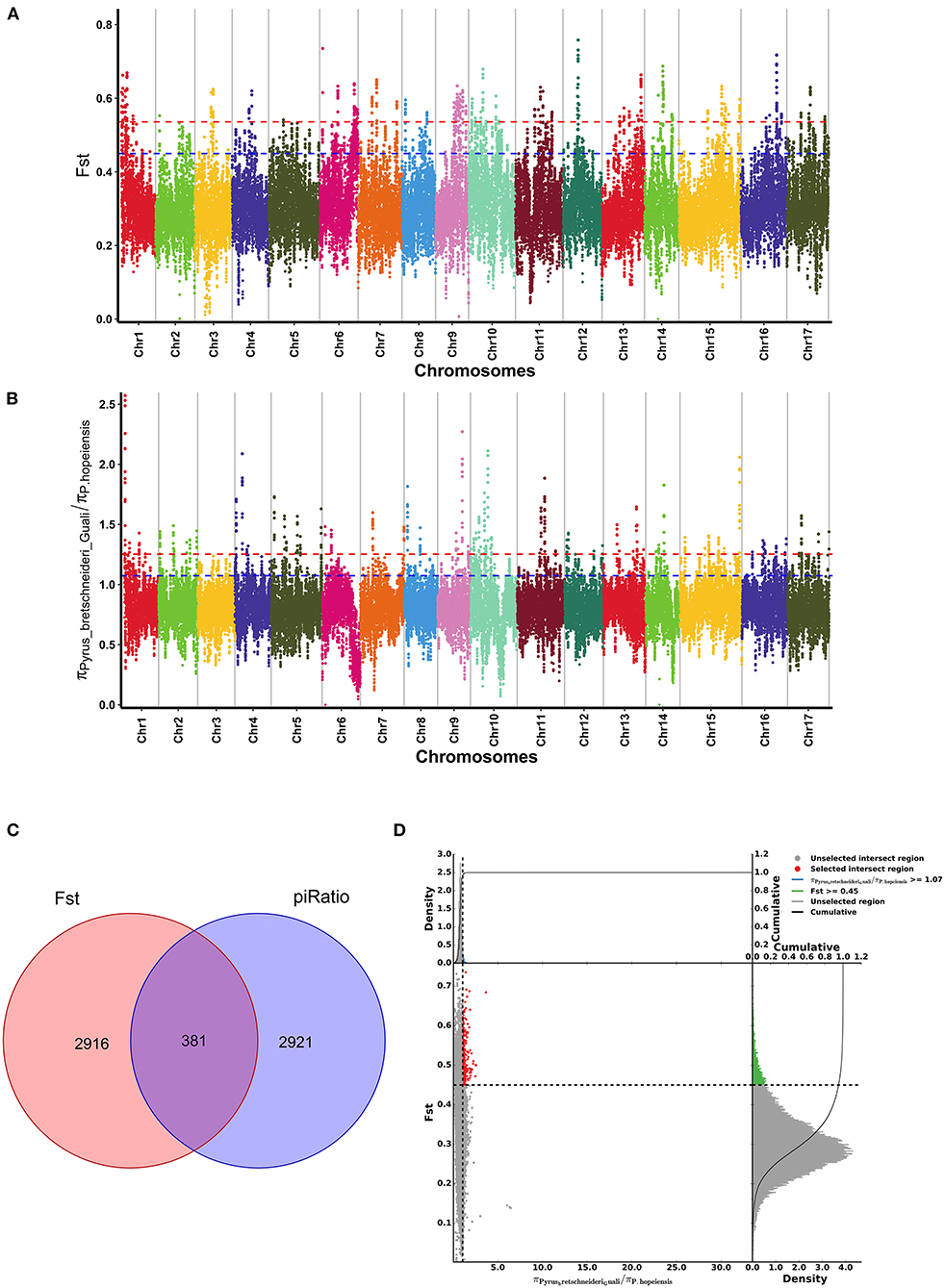
Figure 8. Genome-wide selection scan of Pyrus hopeiensis. (A,B) Manhattan plot of the genome-wide distribution of Fst values and π ratios in P. hopeiensis using a 100-kb window size and a 10-kb step size. (C) Venn diagram showing the gene overlap among Fst values and π ratios in the significant selection region. (D) Selective sweep analysis of P. hopeiensis based on Fst values and π ratios. The transverse coordinates are π ratios and the longitudinal coordinates are Fst values. The dot plot in the center presents the corresponding Fst values and π ratios in different windows. The red region indicates the top 5% of regions selected by π and Fst. The area below the black curve indicates the enriched area.
Gene Functional Enrichment Analysis
The genomic regions between P. hopeiensis and P. bretschneideri “Guali” were compared to identify signatures of positive selection within P. hopeiensis following environmental and artificial selection pressures. Gene function annotations were performed on 381 genes selected by Fst values and π ratios. The GO entries were enriched in 345 biological processes, of which 45 were significantly enriched, including glycogen metabolic process, energy reserve metabolic processes, and cellular glucan metabolic processes (Supplementary Table 8). Among the 66 enriched cell components, seven were significantly enriched, including intrinsic component of membranes, the histone acetyltransferase complex, and the protein acetyltransferase complex (Supplementary Table 9). Among the 92 enriched molecular functions, 24 were significantly enriched, including cellulose synthase activity, transition metal ion binding, and metal ion binding (Supplementary Table 10). GO Term (level 2) analysis showed that these candidate genes were enriched in 15 biological processes, nine cell components, and four molecular functions (Figure 9A; Supplementary Table 11). Metabolic process GO:0008152 had the most enriched genes (55) in the biological processes, followed by cellular process GO:0009987 with 49 genes, and single-organism process GO:0044699 with 35 genes. In the molecular function category, catalytic activity GO:0003824 had the most enriched genes (50), followed by binding GO:0005488 with 32 genes. And in the cell components category, there were 20 genes that were enriched in membrane GO: 0016020, cell GO: 0005623, and cell part GO: 0044464.
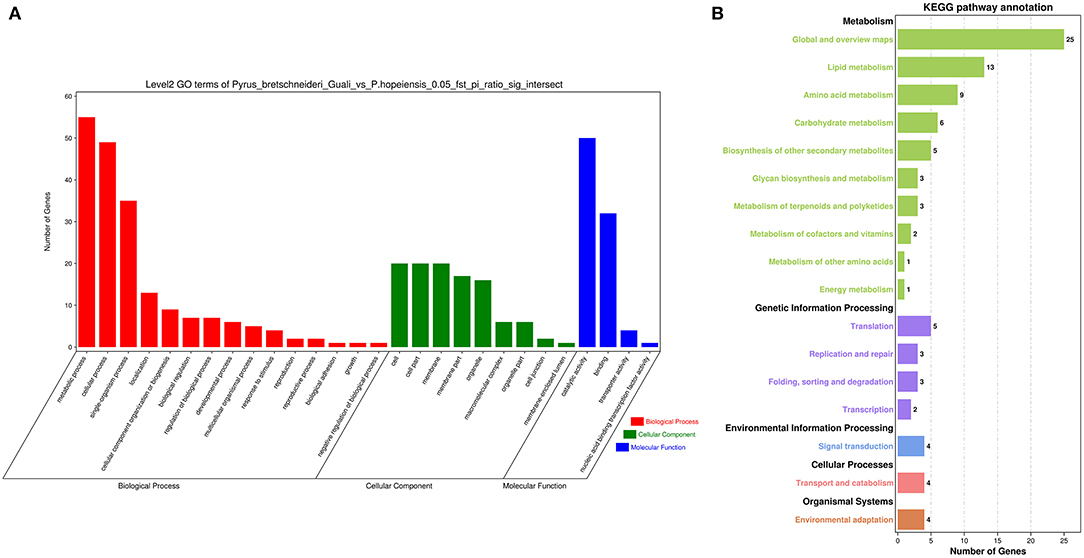
Figure 9. Statistical analysis of enrichment in P. hopeiensis. (A) GO analysis and (B) KEGG pathway analysis.
To better understand the signal pathways of candidate genes in the P. hopeiensis population, KEGG pathway enrichment analysis were performed on the selected candidate genes. In total, 54 pathways were enriched in the P. hopeiensis population (Supplementary Table 12). Among the pathways, four were significantly enriched pathways, including lysine degradation (five genes), sphingolipid metabolism (four genes), other glycan degradation (three genes), and betaine biosynthesis (two genes). These genes were classified into 17 pathways based on the annotation of KEGG class B (Figure 9B). The global and overview maps pathway had the most enriched genes (25 gene), followed by lipid metabolism (13 genes).
Discussion
Pears originated in China, and the wild resources exist widely (Rubtsov, 1944). The protection, development and rational use of rare and endangered wild plants have become elements of the core content for protecting biodiversity. At present, the habitat conditions for P. hopeiensis are sub-optimal, and wild resources are being severely damaged. Changli in Hebei Province has a long history of fruit tree cultivation. Local people reclaim terrain on the nearby mountains for planting fruit trees. The local flower industry is also relatively well-developed, and the area under flower gardens has gradually expanded, invading the native habitat of P. hopeiensis. As the fruits of P. hopeiensis are small, bitter and inedible, villagers have used the nearby P. hopeiensis population primarily as rootstocks or they have felled the trees to expand the agricultural areas. Moreover, the P. hopeiensis population grew mostly on cliffs, in ravines, and in sandy areas, where their roots were largely exposed. Large numbers of climbing woody plants occurred around P. hopeiensis trees in the ravine, and these represent a competitive threat to the survival of the trees. Many mountainous landscapes occur in Changli and the local people have often planted Pinus species for greening on these mountains, and the procedure has degraded conditions required for the survival of P. hopeiensis. In addition, low temperatures can seriously impact the yield and distribution of pears. In recent years, temperatures have been unstable in diverse locations and pear blossoms had suffered severe freezing damage. These factors are all major threats to the expansion and survival of the P. hopeiensis population. If the wild resources of P. hopeiensis are not protected in a timely manner, the risk of extirpating the wild resources of this species will be extremely high. It is imperative to protect and restore the wild resources of P. hopeiensis. Thus, we have implemented protective measures for the species. These measures included intensifying publicity efforts to increase local residents' awareness of the need to protect P. hopeiensis, establishing P. hopeiensis protection communities for in-situ conservation, promoting scientific research on P. hopeiensis ecology and reproductive biology to provide basic data for ex-situ conservation, strengthening environmental protection in the distribution area of P. hopeiensis, and establishing and improving the management of P. hopeiensis archives.
In our study, 23 Pyrus accessions were re-sequenced. And the mutation types such as SNP, InDel, SV and CNV were detected and annotated. Among them, SNPs and InDels were the most abundant form of genetic variation in plant genomes (Rafalski, 2002; Montanari et al., 2019), and they were widely used in the scientific community. SNP markers were frequently used in plant genetic diversity research, the construction of high-density genetic linkage maps, and the identification of loci or genes related to complex traits (Chen et al., 2014; Winfield et al., 2016). A total of 107,133,072 SNPs and 141,866,26 insertions or deletions were detected, and most of these variants were located in intergenic regions or introns (Xanthopoulou et al., 2019; Li et al., 2020). The average dN/dS ratio of these Pyrus accessions was 1.044, and the numbers of synonymous SNPs exceeded the number of non-synonymous SNPs, similar to previous studies (Cheng et al., 2019; Li et al., 2019; Cui et al., 2020). However, the ratio was lower than those reported for wild soybean (1.36) (Lam et al., 2010), sweet cherry (1.78) (Xanthopoulou et al., 2020) or grapevine (1.17) (Liang et al., 2019). In addition, the heterozygous rates of different Pyrus species were varied, ranging from 0.19 to 0.53%, and averaging of 0.44%. The heterozygous rate of these Pyrus species was much lower than those of P. betulifolia (1.54%) (Dong et al., 2020), (P. ussuriensis × communis) × spp. “Zhongai1” (1.45%) (Ou et al., 2019) and P. bretschneideri “Dangshangsu” (1.02%) (Wu et al., 2013).
In the period of 2016–2018, many field surveys about the numbers and distribution of P. hopeiensis trees during different periods of flowering and fruiting were carried out. More than 100 specimens were found in the hills area near Xingshuyuan Village, and most of those in woodlands, farmlands or bordering other facilities were scattered bushes. Most specimens were shrubs produced by roots sprouting and only 10 full trees were found. The maximum age of the trees was about 50 years. Besides, five strains of P. hopeiensis found on Changyushan were identified using SSR molecular markers and no differences among them were found. However, there were differences in morphology and SSR markers between P. hopeiensis specimens from Xingshuyuan and Changyushan. Therefore, we grouped specimens in the population occurring in Xingshuyuan within the category P. hopeiensis HB-1, and the population on Changyushan within the category P. hopeiensis HB-2. In 2019, the wild pears collected in the region where we found P. hopeiensis were used for whole genome resequencing and the SNPs obtained were used for population structure analysis. The phylogenetic tree and the PCA analysis were congruent, indicating that P. hopeiensis specimens from Xingshuyuan and Changyushan were distributed on different phylogenetic tree branches, and the two groups were belonging to different genotypes. Moreover, the results showed that PWH06, PWH07, PWH09, PWH10, PWH13 were closely associated with P. hopeiensis HB-1, while PWH17 was more closely associated with P. hopeiensis HB-2. Based on this information and details of tree morphology, it speculated that P. hopeiensis may be a hybrid population, which was consistent with the opinion of Yu (1979), who suggested that P. hopeiensis is a natural hybrid secondary species. The population structure analysis showed that when K = 3, there were genes penetration between P. bretschneideri “Guali” (PWH01, PWH03), P. betulifoliars (PWH12, PWH21), PWH09, PWH14, PWH17, P. ussuriensis Maxin. cv. Jingbaili (PWH22), Pyrus communis L. cv. Early Red Comice (PWH23), and P. hopeiensis. These findings differed somewhat from the results of the neighbor-joining tree and PCA. However, when K = 5, P. hopeiensis and P. betulifolia could be distinguished, although gene penetration also existed. P. ussuriensis Maxin. cv. Jingbaili is among the varieties in the P. ussuriensis Maxim system. Gene penetration occurred between P. ussuriensis Maxin. cv. Jingbaili and P. hopeiensis, consistent with the view that P. ussuriensis Maxim may have been a participant in the hybrid origin of P. hopeiensis. Besides, P. hopeiensis also occurs in Laoshan, Shandong Province. The cluster analysis of Liang et al. (2015) showed that P. hopeiensis samples from Laoshan and Changli had crossed with one another. There was no direct correlation between genetic distance and the geographic location of P. hopeiensis populations. Gene exchange among P. hopeiensis populations was frequent. The results also showed that the genetic distances between P. hopeiensis, P. ussuriensis Maxim and P. phaeocarpa were all smaller than the distances from other Pyrus taxa, indicating that close relationships between these three species. P. betulifoliar and P. phaeocarpa were also close. These molecular level data support Yu's speculation about the relationship between the three species (P. hopeiensis, P. ussuriensis Maxim and P. phaeocarpa). The close relationship between P. betulifolia and P. phaeocarpa may account for the genetic exchange between P. hopeiensis and P. betulifolia that we detected. Furthermore, P. hopeiensis bred mainly by natural hybridization, which also provided potential for gene penetration between P. hopeiensis and other congeners.
Many species have experienced strong positive selection in the long process of evolution. These species exhibit selected cancellation signals. Two methods (Fst and π ratio) were used to analyze the entire genome of P. hopeiensis and P. bretschneideri “Guali,” and the important selected signals of P. hopeiensis relative to P. bretschneideri 'Guali'. A total of 381 overlapping genes including SAUR72, IAA20, HSFA2, and RKP genes with strong selected signals were detected. AUX/IAA (auxin/indole-3-acetic acid), GH3 (Gretchen Hagen 3), and SAUR (Small auxin up-regulated RNA) are the early auxin-responsive gene families in plants. Among them, SAUR is the largest gene family and plays a critical role in response to signals such as drought, low temperature and diseases (Ren and Gray, 2015). Low temperature stress can increase the expression of six SAUR genes (PtSAUR12, PtSAUR34, PtSAUR54, PtSAUR67, PtSAUR91, and PtSAUR97) in poplar trees (Hu et al., 2018). HSF (Heat shock transcription factor) is an important factor in the regulation of plant stress resistance, and plays a key role in plant cold stress response, heat resistance, drought tolerance, and salt stress (Andrási et al., 2021). The overexpressed HbHsfA1 and HbHsfB1 may be candidates to improve cold stress tolerance of rubber trees (Deng et al., 2018). Moreover, the KEGG analysis showed that these genes were significantly enriched in four pathways, including lysine degradation, sphingolipid metabolism, other glycan degradation, and betaine biosynthesis. Among these pathways, functional studies of sphingolipid metabolism-related genes indicated that sphingolipids have important roles in plant growth, development and response to biotic and abiotic stress (Wu et al., 2015; Li et al., 2016; Zheng et al., 2018). Sphingolipids are mainly responsible for promoting the active response of plants to various adversity coercive forces, such as drought, freezing damage, and cold damage (Dunn et al., 2004), by enhancing the stability of the plant plasma membrane and the vacuolar membrane. Betaine (GB) is a N-methylamino acid and it is a type of quaternary ammonium compound. It is ubiquitous in bacteria, algae, animals and a variety of plants (Zuo, 2019). Most plants can detect the accumulation of betaine when subjected to adversity stress, and this has an important role in resistance to external environmental stress. Betaine improves salt tolerance, cold tolerance and frost resistance in plants, and reduces the damage caused by drought stress. Under drought and salt stress, betaine can be used as a molecular chaperone to protect the activity of intracellular proteins and metabolic enzymes while participating in the processes of energy metabolism and improving the efficiency of photosynthesis (Yancey et al., 1982). GB reduced the malondialdehyde (MDA) content of sweet tea leaves under drought stress, increased the quantity of osmotic adjustment substances and the relative water content in sweet tea and it also regulated antioxidant enzyme activity (Li et al., 2017). Moreover, betaine increased the abilities of corn (Zhao et al., 2018), pomegranate fruits (Molaei et al., 2021) and other species to resist low temperature stress. Therefore, combined with phenotypic, it speculated that P. hopeiensis may have a better ability of cold tolerance, which provided a basis for the future research.
Conclusions
A preliminary investigation of the distribution, numbers of survivors, habitat, and reproductive methods on P. hopeiensis population were first conducted. The number of P. hopeiensis specimens was low and the distribution was limited. More than 100 P. hopeiensis HB-1 individuals were found in Xingshuyuan village, and five P. hopeiensis HB-2 individuals were found in Changyushan village. The habitat of P. hopeiensis was degraded and P. hopeiensis were found mostly on cliffs with bare roots. It reproduced mainly by natural hybridization and by root tiller production under natural conditions. The whole genome re-sequencing of 23 Pyrus accessions were performed and a lot of mutations of SNPs, InDels, CNVs, and SVs were detected. The SNP and InDel mutation data are valuable resources for studying species evolution, domestication and trait discovery. Using a combination of morphological data for Pyrus species and the population structure constructed by SNPs, it suggested that P. hopeiensis was a natural hybrid secondary species, consistent with previous studies. Because the natural hybridization under natural conditions was a major means of propagation in P. hopeiensis, there existed a wide range of gene exchanges between P. hopeiensis and local Pyrus species. The positive selection region of P. hopeiensis and the functional enrichment of these positive selection genes were analyzed. And the KEGG significant enrichment analysis showed that those genes that were subject to positive selection were annotated to pathways that played an important role in plant resistance to external environmental stress, including sphingolipid metabolism and betaine. This study had implications for the evolution and classification of P. hopeiensis and provided a foundation for breeders aiming to develop improved pear varieties with good phenotypic traits and increased productivity using the abundant germplasm resources.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI SRA BioProject, accession no: PRJNA724820.
Author Contributions
MY conceived and designed the experiments and revised the manuscript. YL, SW, and YZ collected the samples and analyzed the sequence data. YL and JZ drafted the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the National Key Research and Development Plan Research on protection and restoration of typical small populations of wild plants (Grant No. 2016YFC0503106). This study was supported by The Master Candidate Innovation Capacity Training Funding Project of Hebei Education Department (CXZZBS2020092).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We were grateful to/thank Guangzhou Genedenovo Biotechnology Co., Ltd for assisting in bioinformatics analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.668796/full#supplementary-material
References
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. doi: 10.1101/gr.114876.110
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Andrási, N., Pettkó-Szandtner, A., and Szabados, L. (2021). Diversity of plant heat shock factors: regulation, interactions, and functions. J. Exp. Bot. 72, 1558–1575. doi: 10.1093/jxb/eraa576
Chen, H., Xie, W., He, H., Yu, H., Chen, W., Li, J., et al. (2014). A highdensity SNP genotyping array for rice biology and molecular breeding. Mol. Plant 7, 541–553. doi: 10.1093/mp/sst135
Chen, K., Wallis, J. W., McLellan, M. D., Larson, D. E., Kalicki, J. M., Pohl, C. S., et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. doi: 10.1038/nmeth.1363
Cheng, Y., Jiang, S., Zhang, X., He, H., and Liu, J. (2019). Whole-genome re-sequencing of corylus heterophylla blank-nut mutants reveals sequence variations in genes associated with embryo abortion. Front. Plant Sci. 10:1465. doi: 10.3389/fpls.2019.01465
Cui, J., Yang, Y., Luo, S., Wang, L., Huang, R., Wen, Q., et al. (2020). Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Hortic. Res. 7:85. doi: 10.1038/s41438-020-0305-5
Deng, X., Wang, J., Wang, J., and Tian, W. (2018). Two HbHsfA1 and HbHsfB1 genes from the tropical woody plant rubber tree confer cold stress tolerance in Saccharomyces cerevisiae. Braz. J. Bot. 41, 711–724. doi: 10.1007/s40415-018-0485-5
Dong, X., Wang, Z., Tian, L., Zhang, Y., Qi, D., Huo, H., et al. (2020). De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 18:13226. doi: 10.1111/pbi.13226
Dunn, T. M., Lynch, D. V., Michaelson, L. V., and Napier, J. A. (2004). A post-genomic approach to understanding sphingolipid metabolism in Arabidopsis thaliana. Ann. Bot. 93, 483–497. doi: 10.1093/aob/mch071
Hu, W. F., Yan, H. W., Luo, S. S., Pan, F., Wang, Y., and Xiang, Y. (2018). Genome-wide analysis of poplar SAUR gene family and expression profiles under cold, polyethylene glycol, and indole-3-acetic acid treatments. Plant Physiol. Biochem. 128, 50–65. doi: 10.1016/j.plaphy.2018.04.021
Hudson, R. R., Slatkin, M., and Maddison, W. P. (1992). Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589. doi: 10.1093/genetics/132.2.583
Jiang, X. F., Chu, Q. G., and Zhang, C. S. (1992). Studies on the classification and evolution of the genus pyrus in china. J. Laiyang Agr. Coll. 1, 18–21. (in Chinese).
Jin, X., He, M., Ferguson, B., Meng, Y., Ouyang, L., Ren, J., et al. (2012). An effort to use human-based exome capture methods to analyze chimpanzee and macaque exomes. PLoS ONE 7:e40637. doi: 10.1371/annotation/450f85d8-03c6-4bd4-aa8e-b5b5894b4593
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Li, A. M., Zhang, L., Zhang, C., Zhao, P. B., Gou, W., Chen, F. C., et al. (2017). Effects of fulvic acid and betaine pretreatment on physiological characteristics and photosynthesis of malus hupehensis under drought stress. Acta Bot. Boreal. Occident. Sin. 37, 307–314. (in Chinese).
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J., Yin, J., Rong, C., Li, K. E., Wu, J. X., Huang, L. Q., et al. (2016). Orosomucoid proteins interact with the small subunit of serine palmitoyltransferase and contribute to sphingolipid homeostasis and stress responses in Arabidopsis. Plant Cell 28, 3038–3051. doi: 10.1105/tpc.16.00574
Li, Y., Cao, K., Zhu, G. G., Fang, W. C., Chen, C. W., Wang, X. W., et al. (2019). Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome Biol. 20:36. doi: 10.1186/s13059-019-1648-9
Li, Y., Zhang, J., Li, L., Gao, L., Xu, J., and Yang, M. (2018). Structural and comparative analysis of the complete chloroplast genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and three other Pyrus Species. Int. J. Mol. Sci. 19:3262. doi: 10.3390/ijms19103262
Li, Y., Zhu, F. L., Zheng, X. W., Hu, M. L., Dong, C., Diao, Y., et al. (2020). Comparative population genomics reveals genetic divergence and selection in lotus, Nelumbo nucifera. BMC Genomics 21:146. doi: 10.1186/s12864-019-6376-8
Liang, T. T., Ma, Y., and Zang, D. K. (2015). Genetic diversity analysis of wild germplasm resource of pyrus based on SRAP markers. North Hortic. 13, 1–5. (in Chinese).
Liang, Z., Duan, S., Sheng, J., Zhu, S., Ni, X., Shao, J., et al. (2019). Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses. Nat. Commun. 10:1190. doi: 10.1038/s41467-019-09135-8
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. doi: 10.1038/ng.3117
Linsmith, G., Rombauts, S., Montanari, S., Deng, C. H., Celton, J., Guérif, P., et al. (2019). Pseudo-chromosome–length genome assembly of a double haploid “Bartlett” pear (Pyrus communis L.). GigaScience 8:138. doi: 10.1093/gigascience/giz138
Molaei, S., Rabiei, V., Soleimani, A., and Razavi, F. (2021). Exogenous application of glycine betaine increases the chilling tolerance of pomegranate fruits cv. Malase Saveh during cold storage. J. Food Process. Preserv. 45:e15315. doi: 10.1111/jfpp.15315
Montanari, S., Bianco, L., Allen, B. J., Martínez-García, P. J., Bassil, N. V., Postman, J., et al. (2019). Development of a highly efficient Axiom™ 70 K SNP array for Pyrus and evaluation for high-density mapping and germplasm characterization. BMC Genomics 20:331. doi: 10.1186/s12864-019-5712-3
Ou, C., Wang, F., Wang, J., Li, S., Zhang, Y., Fang, M., et al. (2019). A de novo genome assembly of the dwarfing pear rootstock Zhongai 1. Sci. Data 6:281. doi: 10.1038/s41597-019-0291-3
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Pu, F. S., Lin, S. H., Song, W. Q., Chen, R. Y., and Li, X. L. (1985). Studies on karyotypes of pyrus in China (1). J. Wuhan Bot. Res. 4, 381–387. (in Chinese).
Rafalski, A. (2002). Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 5, 94–100. doi: 10.1016/S1369-5266(02)00240-6
Ren, H., and Gray, W. M. (2015). SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 8, 1153–1164. doi: 10.1016/j.molp.2015.05.003
Rubtsov, G. A. (1944). Geographical distribution of the genus Pyrus and trends and factors in its evolution. Am. Nat. 78, 358–366. doi: 10.1086/281206
Team, R. C. (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wang, L., Li, X. G., Xue, H. B., Wang, L., and Li, J. (2016). Mapping QTLs for fruit related traits in pear. Acta Hortic. Sin. 43, 2431–2441. (in Chinese). doi: 10.16420/j.issn.0513-353x.2016-0367
Winfield, M. O., Allen, A. M., Burridge, A. J., Barker, G. L. A., Benbow, H. R., Wilkinson, P. A., et al. (2016). High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206. doi: 10.1111/pbi.12485
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408. doi: 10.1101/gr.144311.112
Wu, J. X., Li, J., Liu, Z., Yin, J., Chang, Z. Y., Rong, C., et al. (2015). The Arabidopsis ceramidase AtACER functions in disease resistance and salt tolerance. Plant J. 81, 767–780. doi: 10.1111/tpj.12769
Xanthopoulou, A., Manioudaki, M., Bazakos, C., Kissoudis, C., Farsakoglou, A. M., Karagiannis, E., et al. (2020). Whole genome re-sequencing of sweet cherry (Prunus avium L.) yields insights into genomic diversity of a fruit species. Hortic. Res. 7:60. doi: 10.1038/s41438-020-0281-9
Xanthopoulou, A., Montero-Pau, J., Mellidou, I., Kissoudis, C., Blanca, J., Pic,ó, B., et al. (2019). Whole-genome resequencing of Cucurbita pepo morphotypes to discover genomic variants associated with morphology and horticulturally valuable traits. Hortic. Res. 6:94. doi: 10.1038/s41438-019-0176-9
Yancey, P. H., Clark, M. E., Hand, S. C., Bowlus, R. D., and Somero, G. N. (1982). Living with water stress: evolution of osmolyte systems. Science 217:1214. doi: 10.1126/science.7112124
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yu, D. J. (1979). Taxonomy of Chinese Fruit Trees. Beijing: China Agriculture Press, 122–147. (in Chinese).
Yu, D. J. (1984). Taxonomy of Deciduous Fruit Trees. Qinzhou: Shanghai Scientific and Technical Publishers, 53–54. (in Chinese).
Zhang, X. L., Aishajiang, M., Xue, H. B., Yan, P., and Wan, J. X. (2018). Recent advances in research on the genetic linkage maps in pears. J. Fruit Sci. 35, 1–8. (in Chinese). doi: 10.13925/j.cnki.gsxb.2018.S.01
Zhao, X. Q., Peng, Y. L., Fang, P., Wu, B. Y., and Yan, H. P. (2018). Mitigation effect analysis of different exogenous regulatory substances on maize under low-temperature stress. Agric. Res. Arid Areas 36, 184–193. (in Chinese).
Zheng, P., Wu, J. X., Sahu, S. K., Zeng, H. Y., Huang, L. Q., Liu, Z., Xiao, S., and Yao, N. (2018). Loss of alkaline ceramidase inhibits autophagy in Arabidopsis and plays an important role during environmental stress response. Plant Cell Environ. 41, 37–849. doi: 10.1111/pce.13148
Keywords: Pyrus hopeiensis, SNPs, whole genome re-sequencing, PCA, Fst and π ratio, KEGG
Citation: Li Y, Zhang J, Wang S, Zhang Y and Yang M (2021) The Distribution and Origins of Pyrus hopeiensis-“Wild Plant With Tiny Population” Using Whole Genome Resequencing. Front. Plant Sci. 12:668796. doi: 10.3389/fpls.2021.668796
Received: 17 February 2021; Accepted: 28 April 2021;
Published: 17 June 2021.
Edited by:
Yunpeng Cao, Central South University Forestry and Technology, ChinaReviewed by:
Yongping Cai, Anhui Agricultural University, ChinaLihu Wang, Hebei University of Engineering, China
Copyright © 2021 Li, Zhang, Wang, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minsheng Yang, yangms100@126.com
†These authors have contributed equally to this work and share first authorship