 Xianxian Yu1,2†
Xianxian Yu1,2† Xiaoshan Duan1,2†
Xiaoshan Duan1,2† Rui Zhang1
Rui Zhang1 Xuehao Fu1,2
Xuehao Fu1,2 Lingling Ye1,2
Lingling Ye1,2 Hongzhi Kong1
Hongzhi Kong1 Guixia Xu1*
Guixia Xu1* Hongyan Shan1*
Hongyan Shan1*- 1State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 2University of Chinese Academy of Sciences, Beijing, China
AP1/FUL, SEP, AGL6, and FLC subfamily genes play important roles in flower development. The phylogenetic relationships among them, however, have been controversial, which impedes our understanding of the origin and functional divergence of these genes. One possible reason for the controversy may be the problems caused by changes in the exon-intron structure of genes, which, according to recent studies, may generate non-homologous sites and hamper the homology-based sequence alignment. In this study, we first performed exon-by-exon alignments of these and three outgroup subfamilies (SOC1, AG, and STK). Phylogenetic trees reconstructed based on these matrices show improved resolution and better congruence with species phylogeny. In the context of these phylogenies, we traced evolutionary changes of exon-intron structures in each subfamily. We found that structural changes have occurred frequently following gene duplication and speciation events. Notably, exons 7 and 8 (if present) suffered more structural changes than others. With the knowledge of exon-intron structural changes, we generated more reasonable alignments containing all the focal subfamilies. The resulting trees showed that the SEP subfamily is sister to the monophyletic group formed by AP1/FUL and FLC subfamily genes and that the AGL6 subfamily forms a sister group to the three abovementioned subfamilies. Based on this topology, we inferred the evolutionary history of exon-intron structural changes among different subfamilies. Particularly, we found that the eighth exon originated before the divergence of AP1/FUL, FLC, SEP, and AGL6 subfamilies and degenerated in the ancestral FLC-like gene. These results provide new insights into the origin and evolution of the AP1/FUL, FLC, SEP, and AGL6 subfamilies.
Introduction
MADS-box genes encode a family of transcription factors that have been found in plants, animals, and fungi (Theissen et al., 2000; Becker and Theissen, 2003; Ferrario et al., 2004; Causier et al., 2010; Rijpkema et al., 2010). In plants, the best-studied MADS-box genes are those involved in the specification of floral meristem and floral organ identities. Protein products of these genes are characterized by existence of four regions: the MADS (M) domain, the intervening (I) region, the keratin-like (K) domain, and the C-terminal (C) region (Theissen et al., 1996; Nam et al., 2003). Extensive phylogenetic studies have revealed that these MADS-box genes belong to eight different subfamilies or lineages: APETALA1 (AP1)/FRUITFULL (FUL), APETALA3 (AP3), PISTILLATA (PI), AGAMOUS (AG), SEEDSTICK (STK), SEPALLATA1 (SEP1), SEPALLATA3 (SEP3), and AGAMOUS-LIKE6 (AGL6) (reviewed in Theissen et al., 2000; Becker and Theissen, 2003; Nam et al., 2003). Among these, the evolutionary histories of the AP3, PI, AG, and STK subfamilies are relatively clear and can be traced back to the most recent common ancestor (MRCA) of extant seed plants (Aoki et al., 2004; Kramer et al., 2004; Dreni and Kater, 2013; Dreni et al., 2013). The relationships among the remainder four subfamilies, however, are still controversial, although the sisterhood of SEP1 and SEP3 (collectively called SEP) has got consistent support. In some studies, SEP was resolved as the sister of AP1/FUL (Carlsbecker et al., 2003; Litt and Irish, 2003; Kim et al., 2005; Futamura et al., 2008; Li et al., 2010), whereas in others, it forms a sister to AGL6 (Kofuji et al., 2003; Nam et al., 2003; Parenicova, 2003; Zahn et al., 2005; Litt, 2007; Amborella Genome Project, 2013; Kim et al., 2013; Ruelens et al., 2013; Ubi et al., 2013; Wong et al., 2013; Yockteng et al., 2013). Interestingly, if the former scenario is correct, then it implies that both AP1/FUL and SEP have originated before the diversification of angiosperms; otherwise, it implies that both AP1/FUL and SEP have existed in the MRCA of extant seed plants but have been independently lost in the lineage leading to extant gymnosperms. The observation that the FLOWERING LOCUS C (FLC) may be the real sister of AP1/FUL (Ruelens et al., 2013) further complicated the issue, making it necessary to re-investigate the relationships among the aforementioned gene subfamilies.
Many factors, such as biased sampling, long-branch attraction, and heterogenous substitution rates, can lead to skewed topology of a phylogenetic tree (Kong et al., 2004; Leebens-Mack et al., 2005). However, the most important factor is the reliability of the alignment used for phylogeny estimation. Since using only conserved regions would reduce resolution, most studies include as many as possible alignable sites. Yet, it has recently been revealed that changes in the exon-intron structure of genes (i.e., structural changes, which may be caused by exon/intron gain/loss, exonization/pseudoexonization, and intraexonic insertion/deletion; Roy and Gilbert, 2005; Xu et al., 2012; Long et al., 2013) may hamper the homology-based alignment because they may lead to the addition of nonhomologous sequence or removal of homologous nucleotide. Since almost all studies only used coding sequences (CDS) or protein sequences to generate their alignment, nonhomologous sites caused by structural changes could be forced to align together. In the MADS-box gene family, structural changes have been shown to be rather common and can indeed cause shifts of reading frame (Litt and Irish, 2003; Vandenbussche et al., 2003a; Litt, 2007; Shan et al., 2007; Xu and Kong, 2007; Liu et al., 2011; Xu et al., 2012). A good example comes from comparing the three core eudicots lineages of the AP1/FUL subfamily: euFUL, AGL79 (also called core eudicot FUL-like), and euAP1 (Litt and Irish, 2003; Litt, 2007; Shan et al., 2007). Proteins encoded by the first two lineages have a paleoAP1 motif at the C-terminal region, the first six amino acids of which were also defined as FUL-like motif in some studies (Litt and Irish, 2003; Litt, 2007) and show high similarity with part of AGL6 II and SEP II motifs. The euAP1 lineage, however, encodes for a quite different C-terminal region with two different motifs: a transcription activation domain and a euAP1 motif, the final four amino acids of which were also called farnesylation motif (Litt and Irish, 2003; Litt, 2007). Detailed investigation revealed that the novel sequence was generated by a 1-bp deletion in exon 8 of the ancestral euAP1 gene (Litt and Irish, 2003; Vandenbussche et al., 2003a; Litt, 2007; Shan et al., 2007). Similarly, an 8-bp insertion (Vandenbussche et al., 2003a) or a 1-bp deletion (Kramer et al., 2006) in the last exon has likely given rise to a new euAP3 motif in the euAP3 lineage of the AP3 subfamily. During phylogenetic reconstruction of the AP1/FUL, SEP, AGL6, and FLC subfamilies, however, none of the previous studies considered exon-intron structural changes when generating the final alignment, which may explain why different studies have obtained slightly different topologies.
In this article, we first investigated structural changes during the evolution of these and related subfamilies such as SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1), AG, and STK. We found that structural changes have occurred frequently in these subfamilies and could indeed affect phylogenetic estimation and the understanding of gene evolution. With the knowledge of structural changes, we generated more reasonable alignments containing all the focal subfamilies. All the resulting trees support the sisterhood of AP1/FUL and FLC, with SEP and AGL6 being successive sisters to them. In the context of this new topology, we discussed the contribution of structural changes to the origin and functional diversification of different subfamilies.
Materials and Methods
Sequence Retrieval and Classification
The protein, coding, and genomic (if available) sequences of focal MADS-box genes were retrieved by BLAST searches against the GenBank (http://www.ncbi.nlm.nih.gov), FGP (http://fgp.bio.psu.edu), Phytozome (http://phytozome.jgi.doe.gov), Amborella Genome Database (http://www.amborella.org), TAIR (https://www.arabidopsis.org), MPOB (http://genomsawit.mpob.gov.my), and PlantGDB (http://www.plantgdb.org) databases, with multiple sequences being used as queries. The resulting dataset was then trimmed by the following strategies. First, CDSs shorter than 400 bp were excluded, because they are not very informative or accurate. Second, all but one of the multiple highly similar (i.e., >95% identical at the CDS level) sequences from the same species were eliminated, because they represent alleles of the same gene. Third, for genes with alternative splicing, only the transcript showing the least structural divergence from closely related homologs was adopted. And fourth, poorly annotated sequences from whole-genome sequenced species were excluded. As a result, 792 sequences were retained for further analyses.
To assign the retained sequences into different subfamilies, we built a preliminary phylogenetic tree (using the same methods described below) with shared regions (Dataset S1). The matrix for every subfamily has a broad taxonomic coverage, including sequences from early-diverging angiosperms, monocots, magnoliids, basal eudicots, core eudicots, and gymnosperm species (if applicable). Detailed information of genes included in this study was listed in Table S1.
Sequence Alignment and Phylogenetic Reconstruction
For each subfamily, protein sequences were initially aligned using ClustalX 1.83 with default options (Thompson et al., 1997), and its corresponding codon-based CDS alignment was generated by the PAL2NAL program (http://www.bork.embl.de/pal2nal/). A preliminary tree was constructed with the CDS alignment excluding poorly aligned regions (i.e., columns). The sequences in both protein and CDS alignments were then reordered according to their phylogenetic placements as well as the phylogenetic relationships among species. By comparing closely related sequences, we were able to determine homologous sites and refine the alignments. Considering the effect of structural changes on the reliability of alignment, we marked the exon-intron boundaries for genes with structural annotation (from genome-sequenced species) and carefully checked the alignments of neighboring sequences exon by exon. Special attention was paid to the exons that showed considerable divergence in sequences or lengths, in which structural changes have likely occurred. To improve the alignment quality, a pairwise alignment was performed by using both focal exons and their flanking noncoding sequences. Referring to these results, the CDS alignment can be adjusted with confidence, which were carried out in MEGA 6.0 (Tamura et al., 2013). Since our alignments involved human judgment and might be arbitrary, we also generated an amino acid alignment using Probalign (Roshan and Livesay, 2006) for each subfamily and its corresponding codon-based CDS alignment. Eventually, the CDS alignments excluding nonhomologous and highly divergent regions/sites were used for phylogenetic analyses.
To estimate the phylogenetic relationships among different subfamilies, we generated a combined matrix using the “profile-profile alignment” method in Muscle 3.6 (Edgar, 2004), followed by manual adjustments as described above. To maximize the reliability of our phylogenetic analyses, we created three different alignments (I, II, and III). For alignment I, all the 792 sequences were included. Alignment II contained 498 sequences with the exclusion of genes or gene lineages that experienced structural changes shortly after gene duplications. More stringently, in alignment III, we only included 57 exemplars from basal angiosperms, basal eudicots and gymnosperms (if applicable), which showed less structural divergence during evolution (for details, see results). Because no FLC-like gene has ever been identified from basal angiosperms and basal eudicots (Ruelens et al., 2013; this study), FLC-like genes from core eudicot species and Musa were used for this subfamily. For all the alignments, only homologous sites and regions were used for phylogenetic analyses (Dataset S9).
Phylogenetic relationships of genes within each subfamily were revealed by the maximum-likelihood (ML) method, which was performed on the DNA matrix with PhyML (version 2.4) (Guindon and Gascuel, 2003). The most appropriate molecular evolution model (GTR+I+Γ) was selected, following the estimate with MODELTEST version 3.06 (Posada and Crandall, 1998). A BIONJ tree was used as a starting point for ML searches (Guindon and Gascuel, 2003), and bootstrap analyses were performed with 100 replicates. In addition to the ML method, we also performed Bayesian inference (BI; Ronquist et al., 2012) for alignments I, II, and III to confirm the phylogenetic relationships among the AP1/FUL, SEP, AGL6, and FLC subfamilies. We ran four chains, sampling one tree every 1000 generations for 15,000,000 generations using GTR+I model (starting with a random tree). The first 25% trees were considered burn-in and discarded from further analysis.
Determination of Exon-Intron Structural Changes
To understand the history of structural changes, we first determined the causal of each gap in the alignment and then tried to trace the origin of each gap on the phylogenetic tree. Gaps located at one or both sides of an exon could be caused by exonization/pseudoexonization or exon gain/loss events. The former could be inferred when exonic sequence of one gene was alignable with intronic or intergenic sequence of the other gene. The latter is the phenomenon when an entire exon of one gene could not be aligned to any region (including noncoding sequences) of the other. Gaps within an exon are usually caused by intraexonic insertions/deletions. We mapped the occurrence and the causal of each gap on the phylogenetic tree and deduced at which branch they have happened according to the maximum parsimony principle. In addition to the above mechanisms, intron gain/loss is also responsible for structural changes as previously reported (Xu et al., 2012), which was regarded when one exon of a certain gene could be perfectly aligned with two neighboring exons of the other gene. Different from other mechanisms, no gaps could be found in the alignment if intron gain/loss has happened, but it could lead to the difference in exon numbers. Therefore, the evolutionary history of intron gain/loss was also inferred. With the knowledge of these exon-intron structural changes, we estimated the exon-intron structures of the various ancestral genes in the MRCAs of extant core eudicots, Ranunculales, magnoliids, monocots, angiosperms, and gymnosperms (if applicable).
Results
Structural Changes within the AP1/FUL Subfamily
A total of 209 genes were used for the structural analysis of AP1/FUL subfamily members. By performing exon-by-exon alignment, we generated a dataset consisting of 711 nucleotide sites, among which 607 were phylogenetically informative (Dataset S1). The topology of the final phylogenetic tree was largely consistent with previous studies and not sensitive to missing data (Litt and Irish, 2003; Preston and Kellogg, 2006; Shan et al., 2007; Xu and Kong, 2007; Litt and Kramer, 2010; Pabón-Mora et al., 2013). Nonetheless, the resolution was slightly improved and the positions of most genes were better congruent with angiosperm phylogeny. In contrast, the dataset created based on an alignment produced by Probalign only included 696 sites, among which 591 were informative (Dataset S2). Moreover, in the resulting phylogenetic tree, the positions of some major plant groups were discordant with angiosperm phylogeny (Dataset S2). Similar results were obtained when other MADS-box gene subfamilies were analyzed (Datasets S3–S8). This suggests that phylogenetic estimation can indeed be improved when structural changes were taken into consideration during alignment.
In the context of the improved phylogeny, we attempted to trace the evolutionary changes in the exon-intron structure of AP1/FUL subfamily members. We found that the AP1/FUL-like genes generally consist of eight exons, among which the first six have been highly conserved. In contrast, exons 7 and 8 vary greatly in length (from 77 to 209 bp for exon 7 and 34 to 148 bp for exon 8), suggestive of dramatic structural changes (Figure S1). Detailed comparisons revealed that intraexonic insertion/deletion occurred more frequently than exonization/psedoexonization in this subfamily, and that structural changes were not distributed evenly among branches. For example, an average of 2 insertion/deletion events was detected in the Solanaceae euFUL-like genes (Figure 1A), while at least 8 structural change events were observed for each of the OsMADS15 lineage members (Figure 1B).
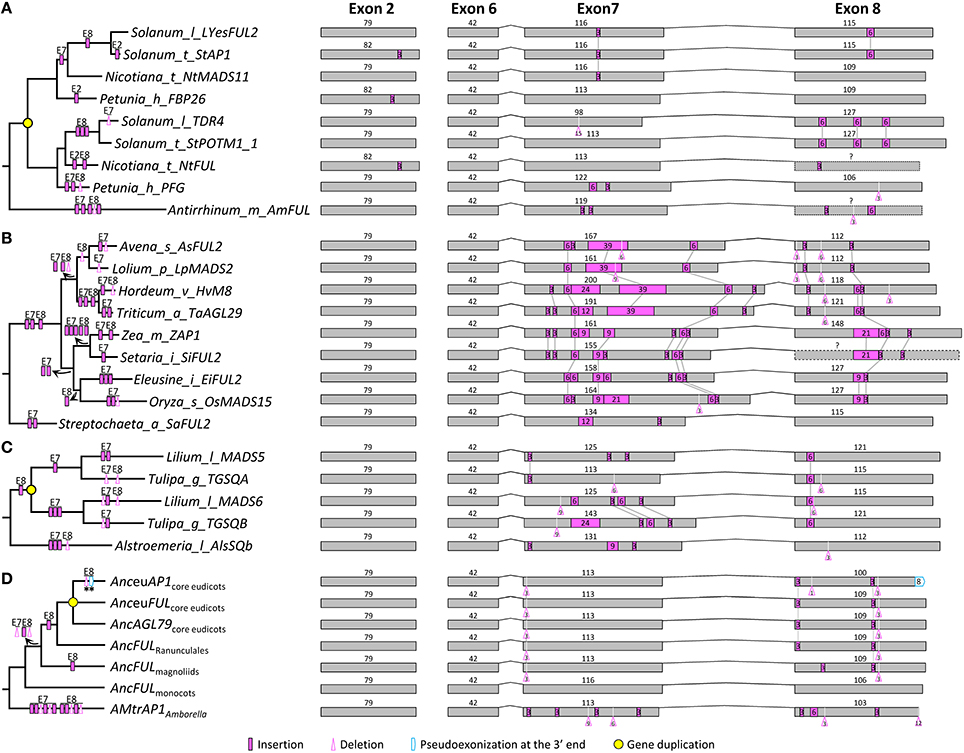
Figure 1. Evolution of exon-intron structure in the AP1/FUL subfamily. (A–C) Representative structural change events occurred in the euFUL lineage of Solanaceae (A), the OsMADS15 lineage of Poaceae (B), and AP1/FUL-like genes of Liliales (C). (D) Exon-intron structural changes at several key nodes on the AP1/FUL phylogenetic tree. “Anc” (for Ancestor) is prefixed to the name of each gene lineage. Details are shown in Figure S1. Exons and introns are represented by boxes and curved lines, respectively. The length of each exon is shown above the box. Shared structural change events are linked by gray lines. Different mechanisms responsible for structural changes are marked on corresponding branches of the phylogenetic tree. Stars indicate structural changes involving non-triplet sequences.
We also found many structural change events shared by certain plant groups or major gene lineages. For example, in exon 7, a 3-bp deletion was detected in all OsMADS14/15 members of monocots, and two independent 3-bp insertions were found in the OsMADS14 and OsMADS15 lineages of Poaceae (Figure S1). In exon 8, one 3-bp insertion near the 5′ boundary was shared by all the sampled eudicot members (Figure S1), suggestive of an ancient structural change event occurred before the diversification of eudicots. There are also multiple cases where structural changes have caused divergence of duplicate genes. For instance, in the OsMADS18/20 lineage of monocots, a gene duplication event resulted in the creation of two sublineages in Liliaceae (Figure 1C). The ancestor of one sublineage has experienced a 3-bp insertion in exon 7, while that of the other sublineage has undergone three insertions of different lengths in the same exon. Consistent with previous studies (Litt and Irish, 2003; Vandenbussche et al., 2003a; Shan et al., 2007), we also detected a 1-bp deletion in exon 8 of all examined euAP1-like genes, which led to pseudoexonization of the last 8 nucleotides (Figure 1D). With the knowledge of these structural changes, we inferred that the AP1/FUL-like gene in the MRCA of extant angiosperms is composed of eight exons with the lengths of 185, 79, 65, 100, 42, 42, 113, and 106 bp, respectively.
Structural Changes within the SEP Subfamily
We obtained 119 SEP1- and 87 SEP3-like genes to analyze exon-intron structural changes in the SEP subfamily. According to a previous study (Zahn et al., 2005) and this study, SEP1-like genes contain three major lineages in both core eudicots (i.e., SEP1/2, FBP9, and SEP4) and grasses (i.e., OsMADS1, OsMADS5, and OsMADS34; Figure S2). Except for SEP1/2-like genes in Brassicaceae and EgAGL2-5 in Elaeis guineensis, all these genes have eight exons. For the Brassicaceae SEP1/2-like genes, the fifth exon (84 bp) could be aligned perfectly to the fifth (42 bp) plus the sixth (42 bp) exon of other genes, suggestive of an intron loss event that occurred before the diversification of Brassicaceae (Figure 2A). Like the situation in the AP1/FUL subfamily, structural change events were mostly observed in the seventh and eighth exons, but the occurrence frequency was much lower (Figure 2 and Figure S2). A large number of structural changes could be found before the diversification of certain plant groups. For example, one 3-bp insertion in exon 2, one 15-bp insertion in exon 7, and two insertions (3 and 6 bp, respectively) in exon 8 of SEP1/2-like genes have likely occurred in the MRCA of Brassicaceae and Cleomaceae (Figure 2A and Figure S2). The longest insertion (66 bp) was observed in exon 7 of the SEP4 gene of Capsella rubella, adjacent to which was an extra 33-bp insertion that has occurred in the ancestor of this and two other related species (Brassica rapa and Arabidopsis; Figure 2B). There were also evidences showing the contribution of structural changes to the divergence of duplicate genes. For instance, maize has a pair of duplicate genes (ZmM24 and ZmM31) in the OsMADS34 lineage. A 3-bp deletion happened in exon 8 of ZmM24, making the lengths of this exon different between them (Figure 2C). In addition to recent duplicates, structural changes in more ancient duplicates were also detected. One 3-bp deletion event in exon 2 of the OsMADS1 lineage, as well as one 45-bp pseudoexonization event in exon 8 of the OsMADS5 lineage, has likely taken place before the diversification of grasses (Figure S2). Within the SEP1 clade, no structural change event has likely occurred before the origins of major plant groups (i.e., monocots, magnoliids, and core eudicots; Figure 2D and Figure S2). Based on this information, we inferred that the SEP1-like gene in the MRCA of extant angiosperms contains eight exons, with the lengths of 185, 79, 62, 100, 42, 42, 137, and 85 bp, respectively.

Figure 2. Evolution of exon-intron structure in the SEP1 subfamily. (A–C) Representative structural change events occurred in SEP1/2 (A) and SEP4 (B) lineages of Brassicaceae, and the OsMADS34 lineage of Poaceae (C). (D) Exon-intron structural changes at several key nodes on the SEP1 phylogenetic tree. Details are shown in Figure S2. The symbols describing structural changes are the same as those in Figure 1.
The phylogenetic tree of SEP3-like genes indicates no major gene duplication event (Figure S3). All of the 87 genes have eight exons. For exons 1, 4, 5, and 6, the lengths are largely conserved (185, 100, 42, and 42 bp, respectively) with a few exceptions (Figure S3). Exons 2, 3, 7, and 8, in contrast, vary remarkably in length, suggestive of multiple structural changes (Figure 3 and Figure S3). For exon 2, independent exonization events were observed in several taxa, such as Fabaceae, Brassicaceae, and Eupomatia, among others (Figures 3A,B, and Figure S3). In exon 3, a 9-bp exonization event was detected in members of Asparagales, Commelianales, and Poales, suggestive of an early structural change event during the evolution of monocots. Still in this exon, a more ancient exonization (6 bp) event was found before the divergence of Chloranthaceae (Figure S3). In exon 7, the MRCA of eudicots has experienced a 3-bp deletion event, while that of grasses has undergone two independent insertion events (Figures 3B,C and Figure S3). The earliest structural change event was a 9-bp deletion in exon 8, which happened after the divergence of Amborella trichopoda (hereafter called Amborella; Figure 3D). Taking into account of all the structural change events, we estimated that the SEP3-like gene in the MRCA of extant angiosperms contains eight exons, the lengths of which are 185, 79, 62, 100, 42, 42, 140, and 85 bp, respectively.
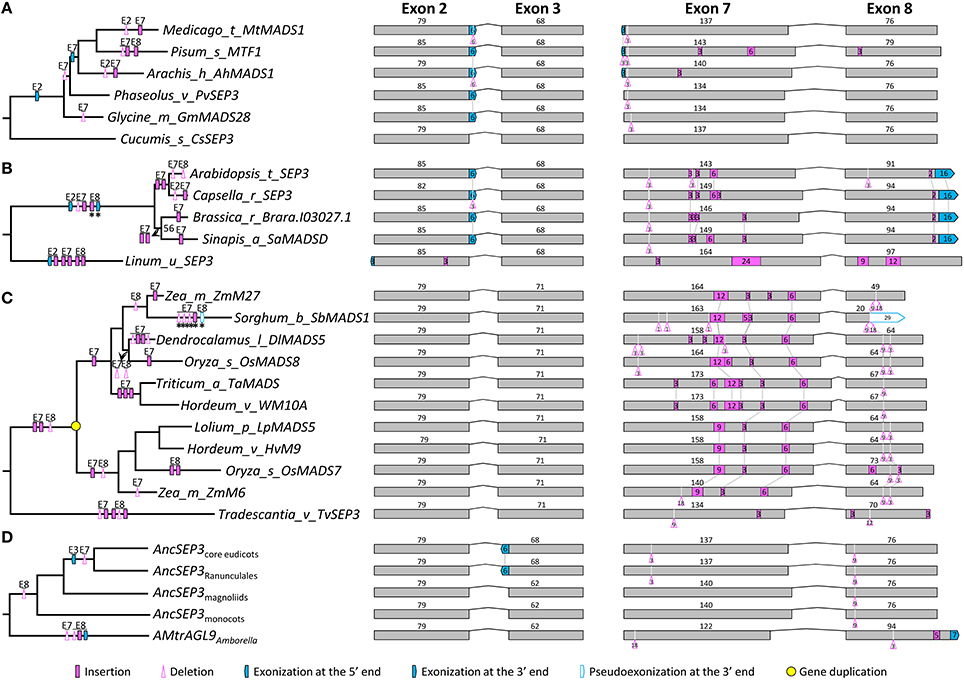
Figure 3. Evolution of exon-intron structure in the SEP3 subfamily. (A–C) Representative structural change events occurred in SEP3-like genes of Fabaceae (A), Brassicaceae (B), and Poaceae (C). (D) Exon-intron structural changes at several key nodes on the SEP3 phylogenetic tree. Details are shown in Figure S3. The symbols describing structural changes are the same as those in Figure 1.
Structural Changes in the AGL6 Subfamily
Within the AGL6 subfamily, 119 genes from angiosperms and 13 from gymnosperms were used for structural change analyses. The topology of the AGL6 gene tree was similar to previous studies (Li et al., 2010; Kim et al., 2013). All the sampled genes except for ZfAGL6a in Zamia fischeri possess eight exons (Figure S4). The lengths of exons 1, 3, 4, and 5 (182, 62, 100, and 42 bp, respectively) are largely the same with exceptions in only five genes. In exon 2, other than a 3-bp deletion event occurred before the diversification of core eudicots, multiple independent insertion events were detected in several taxa, such as Brassicaceae and Ranunculaceae (Figures 4A,B, and Figure S4). In exon 6, a 21-bp exonization event occurred in the MRCA of asterids (Figure S4). Like the situation in the above two subfamilies, exons 7 and 8 were subject to multiple structural change events. In exon 7, major events include a 6-bp insertion in the MRCA of extant gymnosperms, a 3-bp insertion and three independent 3-bp deletions in the MRCA of extant angiosperms, a 3-bp insertion in the MRCA of Ranunculales, a 6-bp insertion in the MRCA of core eudicots, a 3-bp insertion and a 3-bp deletion in the MRCA of rosids, a 3-bp insertion and a 3-bp deletion in the MRCA of Asteraceae, a 6-bp insertion in the MRCA of Brassicaceae, and two 3-bp insertions in the MRCA of Poaceae (Figures 4A,D and Figure S4). In exon 8, independent insertion/deletion events were observed prior to the origins of eudicots, Asteraceae, and Poaceae, respectively (Figure S4). Structural divergence after gene duplication was also not a rare case in this subfamily. For example, OsMADS6 and OsMADS17 are two lineages generated by the pre-Poaceae gene duplication event, subsequent to which the former lineage went through two insertions in each of exon 7 and exon 8, while the latter experienced a 3-bp insertion in exon 2 and two 3-bp insertions in exon 8 (Figure 4C). Independent insertion/deletion events were also found in the duplicate lineages (Gg1 and Gg2) of gymnosperms (Figure 4D and Figure S4; Li et al., 2010). Considering all these structural change events, we inferred that the AGL6-like gene in the MRCA of extant angiosperms contains eight exons, with the lengths of 182, 79, 62, 100, 42, 42, 134, and 85 bp, respectively.
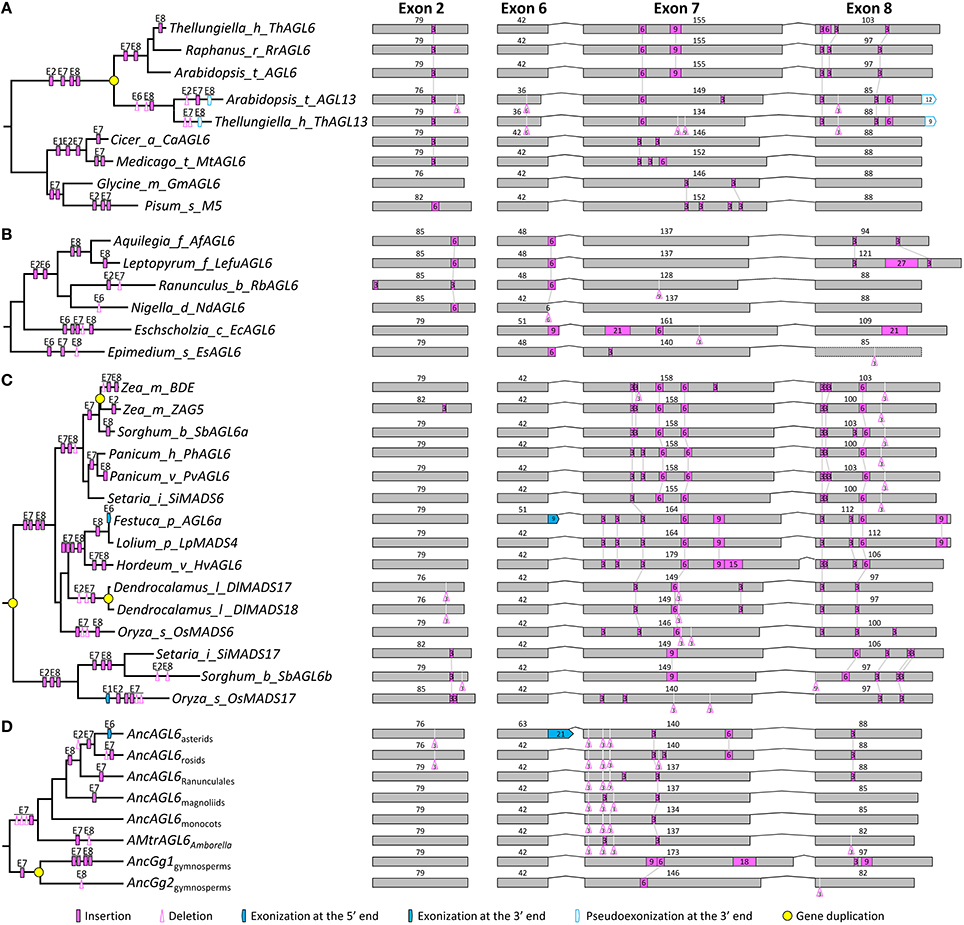
Figure 4. Evolution of exon-intron structure in the AGL6 subfamily. (A–C) Representative structural change events occurred in AGL6-like genes of Brassicaceae (A), Ranunculaceae (B), and Poaceae (C). (D) Exon-intron structural changes at several key nodes on the AGL6 phylogenetic tree. Details are shown in Figure S4. The symbols describing structural changes are the same as those in Figure 1.
Structural Changes within the FLC Subfamily
A recent study showed that FLC-like genes form a sister group to the AP1/FUL subfamily, and are closely related to the SEP and AGL6 subfamilies (Ruelens et al., 2013). By carefully examining the sequences and deeply mining all available plant genomic data, we found that, as Ruelens et al. (2013) revealed, FLC-like genes could only be identified in core eudicots, Poaceae, and Musa (Musaceae). These findings suggest that FLC-like genes may have been lost independently in several lineages of angiosperms (Ruelens et al., 2013). Our phylogenetic tree showed that the FLC-like genes form two clades. One clade contains genes from core eudicots, including FLC and MAF1/2/3/4/5 lineages generated by a pre-Brassicaceae gene duplication event; the other is composed of monocot genes, including OsMADS51 and OsMADS37 lineages produced by a pre-Poaceae gene duplication event. Unlike the aforementioned subfamilies, the core eudicot FLC-like genes have seven exons and exons 1, 4, 5, and 6 (185, 100, 42, and 42 bp, respectively) are evolutionarily conserved. In contrast, most monocot genes possess only five exons (Figure S5). Given the dramatic divergence of exon-intron structures of the OsMADS37-lineage genes, they were excluded from further analysis.
In the context of the phylogeny, we traced the history of structural changes in this subfamily. We found that some structural change events were shared by core eudicot genes or Brassicaceae genes. In Poaceae, multiple structural change events are likely to have happened in the ancestor of the OsMADS51 lineage. For example, an intron loss event was detected in exon 5 because it could be aligned to the fifth and sixth exons of core eudicot genes. The last exon, which is the counterpart of the seventh exon in genes from core eudicots, probably has been lost; however, due to rapid sequence evolution of this subfamily, the underlying mechanism is hard to determine. Other relatively trivial structural change events include a 3-bp insertion and a 3-bp deletion in exon 1, a 3-bp insertion and a 15-bp deletion in exon 3, and a 3-bp deletion in exon 4 (Figure S5). Based on these analyses, we inferred that the FLC-like gene in the MRCA of extant angiosperms has lost an exon and thus contains seven exons, with the lengths of 185, 79, 68, 100, 42, 42, and 105 bp, respectively.
Structural Changes within the SOC1, AG, and STK Subfamilies
Structural changes of the outgroup subfamilies (SOC1, AG, and STK subfamilies) were also examined, which show relatively close relationships with the AP1/FUL, SEP, AGL6, and FLC subfamilies (Kim et al., 2005, 2013; Amborella Genome Project, 2013; Ruelens et al., 2013). SOC1 subfamily members are present in both angiosperms and gymnosperms. All genes from monocots form a monophyletic clade with moderate bootstrap support (72%), with Poaceae genes falling into three lineages (WSOC1, TaAGL7, and TaAGL23). Within core eudicots, another three lineages, each containing genes from rosids and asterids, may have been generated by the γ genome triplication event (Tang et al., 2008). Here we named them euSOC1, AGL42/71/72, and AGL14/19 after the homologs in Arabidopsis (Figure S6). All except for three SOC1-like genes (i.e., Brara.I00679.1 in Brassica rapa, SOC1 in Linum usitatissimus, and CsSOC1B in Cucumis sativus) are composed of seven exons. For the first six exons, only a few structural change events were detected, which sparsely distributed across the angiosperm clade. Most structural changes were found in exon 7, including multiple insertion/deletion and exonization/psuedoexonization events (Figure S6). Taken together, we inferred that the SOC1-like gene in the MRCA of extant seed plants likely contains seven exons, with the lengths of 182, 82, 62, 100, 42, 42, and 132 bp, respectively.
The phylogenetic relationships of the AG and STK subfamilies were largely consistent with a previous study (Zahn et al., 2006), with the majority of genes containing seven exons (Figure S7). Structural analyses revealed several major structural changes in the AG subfamily, such as a 3-bp insertion in exon 7 after the divergence of Amborella and a 6-bp insertion in exon 7 before the diversification of eudicots. In the STK subfamily, one 3-bp exonization event in exon 3 and two separate insertions in exon 7 have occurred in the MRCA of monocots (Figure S7). Tracing back to the MRCA of extant seed plants, we concluded that the ancestral AG/STK-like gene contains seven exons, with the lengths of 182, 82, 62, 100, 42, 42, and 159 bp, respectively.
Phylogenetic Relationships and Structural Differences among Subfamilies
To resolve the relationships among all focal subfamilies, we constructed phylogenetic trees with three different matrices (alignments I, II, and III) (see Section Materials and Methods; Dataset S9). Topologies of all three trees were largely consistent, but the nodal supports at key nodes increased as more structurally diverged sequences were removed (Figure S8 and Figure 5). In the first tree, which was constructed using the matrix composed of all 792 sequences (alignment I), AP1/FUL and FLC are sisters, with 57% ML bootstrap support (BP) and 0.99 Bayesian posterior probabilities (PP), and SEP is the sister to them (50% BP and 0.97 PP). AGL6 shows a sister relationship with the abovementioned three subfamilies (89% BP and 1.00 PP; Figure S8A). Considering that duplicate genes usually show accelerated evolutionary rate and more frequent structural changes that may screw the phylogeny, we next removed duplicated genes that diverged greatly in structure and generated a second matrix (alignment II). The tree built using this matrix gained increased supports for almost all of the abovementioned nodes (Figure S8B). To further improve the resolution, we selected genes (alignment III) with more conserved exon-intron structure from the second matrix and constructed the third tree. All focal nodes were strongly supported in both ML and BI trees (Figure 5).
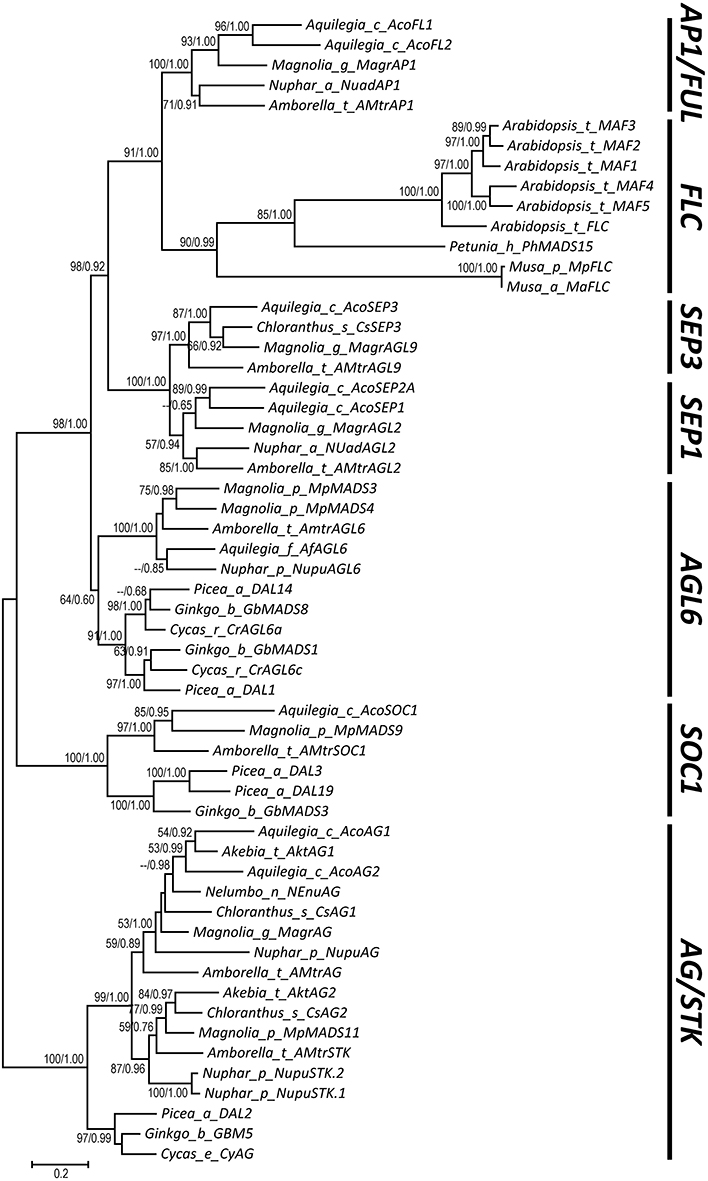
Figure 5. A phylogenetic tree showing relationships of the AP1/FUL, FLC, SEP, and AGL6 subfamilies. The bootstrap values (>50%) obtained from maximum likelihood analysis and the posterior probabilities (>0.5) estimated by Bayesian inference are shown next to the nodes.
Based on our alignment and the topology of the resultant phylogenetic trees, we traced the evolutionary changes of exon-intron structures in these subfamilies. As described earlier, in the MRCA of extant angiosperms or seed plants (if applicable), the AP1/FUL, SEP, and AGL6 genes all possess eight exons, while the FLC, AG/STK, and SOC1 genes all contain seven exons (Figure 6). Unambiguous homologous relationships of exon 1 to exon 6 could be determined based on conservation of the encoded amino acid sequences, i.e., the MADS domain, I region, and K domain. Structural change events were found in exons 1, 2, 3, 7, and 8, some of which were shared by different subfamilies and consistent with their phylogenetic relationships (Figure 6). In exon 1, Kim et al. (2013) found a 3-bp gap in all AGL6-like genes but not in the AP1/FUL and SEP subfamilies. Here we found that this gap also appears in genes of AG/STK and SOC1 subfamilies, suggesting a 3-bp insertion in the ancestor of AP1/FUL, FLC, and SEP subfamily genes (Figure 6). In exon 2, a 3-bp deletion has likely occurred in the ancestor of AP1/FUL, FLC, SEP, and AGL6 subfamily genes. The length of exon 3 in all except for the AP1/FUL and FLC subfamilies is 62 bp. A 3-bp insertion plus an independent 3-bp exonization have resulted in an exon of 65 bp in the ancestor of the AP1/FUL subfamily and 68 bp in that of the FLC subfamily.
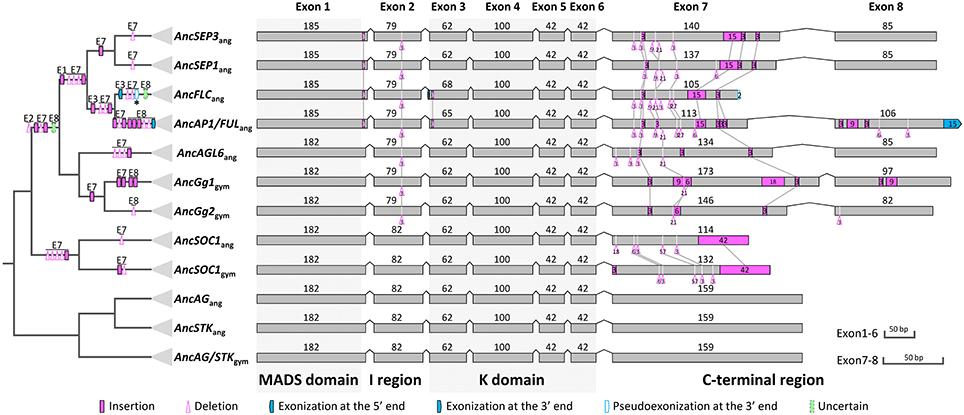
Figure 6. Evolution of exon-intron structures of the AP1/FUL, FLC, SEP, and AGL6 subfamilies. The simplified tree is from Figure 5 and Figure S8. Show here is the ancestral exon-intron structure of each subfamily in the MRCA of extant angiosperms and in the MRCA of extant gymnosperms (if applicable). The MADS domain, I region, K domain, and C-terminal region are indicated below exons, and the MADS and K domains are highlighted with gray boxes. “ang” is the abbreviation for “angiosperms,” and “gym” for “gymnosperms.” The symbols describing structural changes are the same as those in Figure 1.
In all these subfamilies, exons 7 and 8 (if present), which encode(s) for the C-terminal region, is highly variable but contains short, relatively conserved, lineage-specific motifs. We found that in exon 7, the AG II motif (Kramer et al., 2004) was alignable to the SEP I motif (Zahn et al., 2005), the FUL motif (Shan et al., 2007), and the AGL6 I motif (Ohmori et al., 2009), and that the last four amino acids (LxxG) are quite conserved. This suggests that the seventh exons of different subfamilies (Figure S9) are homologous. In this exon, two 3-bp insertions and one 21-bp deletion have occurred before the divergence of AP1/FUL, FLC, SEP, and AGL6 subfamilies. Three deletions with lengths of 3-, 3-, and 9-bp, respectively, as well as a 15-bp insertion were shared by the AP1/FUL, FLC and SEP subfamilies. The ancestor of AP1/FUL and FLC subfamily genes has likely experienced two deletion events. A 3-bp insertion shared by the SEP subfamily genes was also observed (Figure 6). These shared structural change events provide further support for the phylogenetic relationships among the four subfamilies.
Exon 8 is specific for the AP1/FUL, SEP, and AGL6 subfamilies. Based on our phylogeny, it is highly likely that this exon originated before the divergence of these subfamilies. To figure out the mechanisms responsible for the evolutionary changes of this exon, we further searched putatively homologous sequences of this exon at the downstream 200 kb intergenic region of representative genes from the FLC, SOC1, AG, and STK subfamilies. However, due to the relatively long divergence time, we could not find any alignable region. Thus it is hard to determine whether this exon was generated by exonization or exon gain in the ancestor of the four subfamilies. Likewise, it is difficult to determine how this exon was lost in the FLC-like genes. More interestingly, we found that the ancestor of the AP1/FUL subfamily has experienced an exonization event at the 3′ boundary of exon 8. As we mentioned earlier, except for euAP1 proteins, all the other members of this subfamily encode for a paleoAP1 motif (Vandenbussche et al., 2003a; Shan et al., 2007), the first six amino acids of which is defined as FUL-like motif (Litt and Irish, 2003; Litt, 2007) and could be aligned to the C-terminal ends of the SEP and AGL6 proteins (Figure S10). To understand the origin of the extra 5 amino acids in the paleoAP1 motif, we tried to align the coding sequence of this region to the 3′ untranslated regions of SEP and AGL6 subfamily genes. The resultant alignment (Figure S10) suggested that two point mutations (T–C and A–C) may have broken the original stop codon in the ancestor of the AP1/FUL subfamily, thereby leading to exonization of the next in-frame 15 bp and thus addition of new amino acids in the protein product (Figure S10). Intriguingly, the Amborella AMtrAP1 does not contain the extra 5 amino acids. Further investigation showed that this may have been caused by independent insertions and point mutations because the corresponding region in this species does not show much similarity with other AP1/FUL-like genes, or with SEP or AGL6 subfamily members.
Discussion
Prevalence and Functional Impacts of Exon-Intron Structural Changes
Although previous studies have reported structural changes in MADS-box genes (Litt and Irish, 2003; Vandenbussche et al., 2003a; Kramer et al., 2006; Shan et al., 2007; Xu and Kong, 2007; Xu et al., 2012; Fourquin et al., 2013), it is ours that first trace the evolution of them in several subfamilies. By conducting such a detailed analysis, we found that: (1) structural changes are highly prevalent during the evolution of MADS-box genes, which contributed to the divergence of genes within and among subfamilies; (2) as has been shown in previous studies (Xu and Kong, 2007; Xu et al., 2009, 2012; Liu et al., 2011), structural changes could be achieved by three types of mechanisms, i.e., exon/intron gain/loss, exonization/pseudoexonizaiton, and intraexonic insertion/deletion; (3) although structural changes can occur in every exon, most of them took place in exons or the part of an exon that encodes for the I region or the C-terminal region; (4) most structural changes were fixed in a specific gene or species, but some important ones were preserved over long evolutionary time. Clearly, these results provide a comprehensive and updated insight into the significant role that structural changes have played in the diversification of gene families.
The frequent occurrence of structural changes in the C-terminal region is not surprising because it has long been demonstrated that this region varies considerably in length and sequence among MADS-box proteins. However, highly variable as it is, this region contains quite conserved motifs. Structural changes rarely occurred in these motifs, but when they did, they could occasionally cause the formation of new motifs (Litt and Irish, 2003; Vandenbussche et al., 2003a; Kramer et al., 2006; Litt, 2007; Shan et al., 2007). One typical example is the generation of the euAP3 motif by either insertion of eight nucleotides (Vandenbussche et al., 2003a) or deletion of one nucleotide (Vandenbussche et al., 2003a) in an ancestral paleoAP3-motif encoding gene. Another example is the generation of two new motifs in euAP1 proteins by 1-bp deletion (Litt and Irish, 2003; Vandenbussche et al., 2003a; Kramer et al., 2006; Litt, 2007; Shan et al., 2007). The above examples both involve out-of-frame insertions/deletions, which are generally deleterious. However, when occurring in duplicate genes, the presence of a redundant copy could compensate for the possible loss of function caused by frameshift mutations, enabling these mutations to lead to functional divergence (Raes and Van de Peer, 2005). As a previous study suggested, this might be the main pattern for novel motif generation in transcription factor families (Vandenbussche et al., 2003a). Interestingly, we found that other structural change mechanisms could also contribute to the generation of novel motifs. For example, the paleoAP1 motif was created by degeneration of the original stop codon and exonization of adjacent 15 nucleotides. More dramatically, the eighth exon, part of which encodes for conserved motifs in the AP1/FUL, SEP, and AGL6 subfamilies, was likely generated by an exonization or exon gain event. These new motifs, which have been highly conserved for a remarkably long evolutionary time, are likely of extraordinary importance and could be a good starting point for functional studies.
Currently, there are only limited data on the functions of several C-terminal motifs and the results are conflicting. For example, one study showed that the euAP3 motif endowed euAP3-like proteins with new functions in specifying perianth structures in core eudicots (Lamb and Irish, 2003); whereas two other studies demonstrated that this motif was dispensable for floral organ identity determination (Piwarzyk et al., 2007; Su et al., 2008). The transactivation domain could indeed confer activation capability to euAP1-like proteins of Arabidopsis, radish (Raphanus sativus), and tobacco (Nicotiana tabacum and Nicotiana sylvestris; Cho et al., 1999). However, a couple of functional studies showed that euFUL and FUL-like proteins were able to substitute for AP1, indicating that the C-terminal motifs may not be essential for the functions of euAP1-like proteins (Gocal et al., 2001; Jang et al., 2002; Chen et al., 2008). Also, Krizek and Meyerowitz (1996) presented evidence that the C-terminal domains of AP1 and AG are not necessary for functional specificity. These opposing results may have been caused by different experimental methods, or possible redundancy of these proteins in high-order complexes (Litt and Kramer, 2010). Further investigations are needed in the future to address this question.
Effects of Structural Changes on Alignment and Phylogenetic Relationships among the AP1/FUL, SEP, AGL6, and FLC Subfamilies
A reliable alignment is extremely important for the accuracy of phylogenetic estimation. Sequence similarity is empirically considered as a hint for homology; however, when evolutionary time is too long, it would be quite difficult to draw an unambiguous conclusion. In the present study, we demonstrated that structural changes are common during the evolution of a gene subfamily, and would directly or indirectly disrupt the homology of corresponding sites or regions in a couple of ways. First, insertion/deletion or exonization/pseudoexonization of non-triplet sequences would lead to shifts of reading frame and thus destroy homology of the downstream coding region. Second, independent changes at the same position in different species may be aligned together and thus erroneously produce nonhomologous sites in the matrix. We found quite a few such cases, one of which is several independent exonization events in exon 2 of core eudicot SEP3-like genes (Figure S3). Third, when a certain position is a hot spot for insertion/deletion, it would be hard to determine whether corresponding sites are homologous or not. This phenomenon has been observed frequently in grass genes (Figures S1, S3–S4, S6–S7). Finally, a structural change event may occur within a codon, and thus the homology is interrupted. Multiple cases have been found in this study, such as independent exonization events at the 5′ end of exon 8 in some genes of the AP1/FUL and SEP subfamilies (Figures S1–S3).Therefore, with the accessibility of more complete genome sequences, it is feasible to generate a more reasonable alignment by referring to exon-intron structure information.
In this study, with the knowledge of structural changes in each subfamily, we refined our alignments and estimated phylogenetic relationships of the AP1/FUL, FLC, SEP, and AGL6 subfamilies. Our tree showed that SEP is sister to the monophyletic group formed by AP1/FUL and FLC, and that AGL6 is the sister to the three abovementioned subfamilies. The topology is different from the one reported by Ruelens et al. (2013), in which SEP and AGL6 are sister to each other and together they are nested with the lineage formed by AP1/FUL and FLC. Based on their phylogenetic tree and syntenic evidence, Ruelens et al. (2013) proposed that the ancestor of AP1/FUL, FLC, SEP, and AGL6 subfamily genes experienced a tandem duplication event in the MRCA of extant seed plants, creating the ancestor of SEP and AGL6, and the ancestor of AP1/FUL and FLC. Then the former went through a duplication event and generated ancestral SEP and AGL6 genes. The segment containing the ancestral SEP and the ancestor of AP1/FUL and FLC was then lost in the MRCA of extant gymnosperms. However, according to our phylogenetic tree and taking the syntenic evidence into account, we hypothesize that the ancestor of AGL6, SEP, AP1/FUL, and FLC has experienced a duplication event in the MRCA of extant seed plants, generating the ancestral AGL6 and the ancestor of SEP, AP1/FUL, and FLC. The latter was then lost in the MRCA of extant gymnosperms but went through a tandem duplication event prior to the origin of angiosperms, bringing forth the ancestral SEP and the ancestor of AP1/FUL and FLC. Then the two genes underwent a whole genome duplication event in the MRCA of extant angiosperms and created SEP1 and SEP3, and AP1/FUL and FLC, respectively. Our hypothesis is equally parsimonious with that of Ruelens et al. (2013) and the phylogenetic tree also showed stronger supports at key nodes than previous studies (Carlsbecker et al., 2003; Kim et al., 2005; Futamura et al., 2008; Li et al., 2010). Moreover, structural changes shared by different subfamilies provide extra evidence for our topology (Figure 6). The gradual improvement of nodal supports with successive removal of structurally diverged sequences suggests that structural changes could indeed influence sequence alignment and then phylogenetic estimation, which need to be carefully considered when studying the evolution of a certain gene family.
Structural Diversification Is Associated with Functional Divergence among Subfamilies
Our results showed that structural changes have taken place in all the focal subfamilies but with different extents. The divergence pattern is significantly associated with their functions. For example, SEP-like genes have experienced much less structural changes than the AP1/FUL, FLC, and AGL6 subfamily genes during evolution. Accumulating evidences have shown that the SEP subfamily members play conserved and vital roles in specifying floral organ identities of angiosperms. Silencing or mutation of SEP-like genes in different species, such as Arabidopsis SEP1/2/3/4, petunia FBP2/FBP5, tomato TM5/TM29, Nigella damascena NdSEP1/2/3, and rice OsMADS1/5/7/8, can lead to the transition of floral organs to sepal-, bract-, or leaf-like organs (Pnueli et al., 1994; Pelaz et al., 2001; Ampomah-Dwamena et al., 2002; Ferrario et al., 2003; Vandenbussche et al., 2003b; Ditta et al., 2004; Cui et al., 2010; Wang et al., 2015). Biochemical data revealed that the SEP-like proteins are able to form quaternary complexes with other floral MADS-box proteins in many species, such as Arabidopsis, petunia, Gerbera hybrida, Vitis vinifera, and rice (Honma and Goto, 2001; Ferrario et al., 2003; Ruokolainen et al., 2010; Seok et al., 2010; Smaczniak et al., 2012; Mellway and Lund, 2013). Recently, we reported that heterodimers between the SEP-like proteins and other floral MADS-box proteins can be formed in early diverging angiosperms, such as Amborella and Nuphar pumila (Amborella Genome Project, 2013; Li et al., 2015). Moreover, by conducting yeast two-hybrid assays with resurrected proteins of the MRCA of extant angiosperms, we found that the ancestral SEP-like proteins have broad interactions with other ancestral floral MADS-box proteins (Li et al., 2015). Therefore, it is highly likely that the SEP-like gene in the MRCA of extant angiosperms has obtained the function of determining floral organ identities and the ability to mediate the formation of floral quartets, which has been retained during the evolution due to their stable gene structures and conserved sequence features.
Unlike SEP, the AP1/FUL and FLC subfamilies have undergone severe rounds of structural divergence since the duplication of the ancestral gene. In addition to the insertion/deletion events that occurred in the ancestor of AP1/FUL and FLC, dramatic exon-intron structural changes, including exon loss, exonization, pseudoexonization, insertions, and deletions, have taken place in the respective ancestors of FLC and AP1/FUL. Divergence in gene structure of these two subfamilies resulted in shorter FLC-like proteins, but longer AP1/FUL-like proteins. Consistent with this, members of these two subfamilies tend to perform different functions in floral development. As has been reported, some FLC subfamily members act as floral repressors responsive to vernalization (Michaels and Amasino, 1999; Sheldon et al., 2006), while the AP1/FUL-like genes mainly function as positive regulators in determining the identities of inflorescences, floral meristems, and floral organs, and controlling the development of compound leaves and fruits (Irish and Sussex, 1990; Huijser et al., 1992; Gu et al., 1998; Pabón-Mora et al., 2012, 2013; Burko et al., 2013). Intriguingly, some AP1/FUL subfamily members are also involved in vernalization, such as WAP1 in wheat (Triticum aestivum; Danyluk et al., 2003; Murai et al., 2003; Trevaskis et al., 2003; Yan et al., 2003; Kim et al., 2009). However, since members of other MADS-box gene subfamilies, such as STMADS11-like genes in grasses (Kane et al., 2005), are also identified as vernalization repressors, this type of function may have evolved multiple times independently. Frequent structural changes happened in the AP1/FUL subfamily may also be the cause of functional divergence between AP1/FUL and SEP subfamilies. We have recently revealed that the ancestral AP1/FUL protein lost the ability to interact with the AG and STK proteins in the MRCA of extant angiosperms (Li et al., 2015). This suggests that the two gene subfamilies have diverged at the early stage of angiosperm evolution, and that the functions of AP1/FUL-like genes further diversified during evolution due to the accumulation of more gene structural changes.
Different from the SEP, AP1/FUL, and FLC subfamilies, the AGL6 subfamily originated before the diversification of extant seed plants, and experienced one round of gene duplication event in the MRCA of extant gymnosperms. In angiosperms, AGL6-like genes show various functions. For example, one of the Arabidopsis AGL6-like genes, AGL6, is responsible for the regulation of lateral organ development, flowering time, and circadian clock (Koo et al., 2010; Yoo et al., 2011; Huang et al., 2012, 2013), but the other one, AGL13, is involved in male and female gametophyte morphogenesis (Hsu et al., 2014). The AGL6-like gene in a basal eudicot species, Nigella damascena, acts as an A-function gene to determine the sepal and petal identities (Wang et al., 2015). In Zingiberales (monocot plants), the AGL6-like genes may regulate stamen morphology (Yockteng et al., 2013). Interestingly, in several angiosperm species, AGL6-like genes, such as PhAGL6 of petunia (Rijpkema et al., 2009), BEARDED-EAR (BDE) of maize (Thompson et al., 2009), and OsMADS6 of rice (Ohmori et al., 2009), function redundantly with SEP-like genes. In this article, we found that frequent structural change events have taken place during the evolution of angiosperm AGL6-like genes. Presumably, the unstable gene structures, plus regulatory divergence, have contributed to the functional diversification of angiosperm AGL6-like genes. Although some structural divergence events have also been revealed in the ancestor of angiosperm AGL6-like genes and the respective ancestors of gymnosperm Gg1 and Gg2 lineages, it seems that these ancestral proteins have similar interaction patterns. For instance, in gymnosperms, the AGL6-like proteins of Gnetum gnemon, GGM9 and GGM11, can interact with proteins of the AP3/PI and AG/STK subfamilies, and may have the ability to mediate multimeric protein complex formation (Wang et al., 2010). In the MRCA of extant angiosperms, AGL6 has relatively high possibility to interact with other floral proteins, similar to SEP (Li et al., 2015). Therefore, it is very likely that the quaternary complexes mediated by AGL6 have existed in the MRCA of extant seed plants (Wang et al., 2010). With the origin of SEP and the formation of obligate heterodimers between AP3 and PI in the MRCA of extant angiosperms (Melzer et al., 2014; Li et al., 2015), the multimerization of floral MADS-box proteins becomes equally dependent on SEP or AGL6. Afterwards, due to quick divergence of ancestral SEP and AGL6 genes in exon-intron structure, together with point mutations and changes in expression regulation, the SEP-like proteins become major mediators of floral quartets in extant angiosperms. Overall, the evolution of the SEP, AP1/FUL, FLC, and AGL6 subfamilies are complicated; their differences in exon-intron structures are only one aspect of their divergence. More studies are needed to clarify the functional diversification of these genes.
Author Contributions
XY, XD, RZ, XF, and LY analyzed data; XY, XD, RZ, GX, and HS wrote the paper; GX, HS, and HK designed the research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kong lab members for helpful discussions, and anonymous reviewers for their constructive comments. This work was supported by National Natural Science Foundation of China (Grants 31125005, 31422006, and 31570225) and the Specialized Fund from the CAS Youth Innovation Promotion Association to HS and GX.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00598
Table S1. Genes used in this study.
Dataset S1. The phylogenetic tree used for gene classification.
Dataset S2. Matrices of the AP1/FUL subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. The resulting tree is shown in Figure S1. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S3. Matrices of the SEP1 subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S4. Matrices of the SEP3 subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S5. Matrices of the AGL6 subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S6. Matrices of the FLC subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S7. Matrices of the SOC1 subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S8. Matrices of the AG/STK subfamily. (A) The matrix for phylogenetic analysis in consideration of exon-intron structural changes, in which only alignable sites are included. (B) The matrix based on an alignment generated by Probalign and the resulting tree. (C) The matrix for exon-intron structural change analysis.
Dataset S9. Matrices for phylogenetic construction and exon-intron structural change inference among subfamilies. (A) The matrix of alignment I and the resulting maximum likelihood tree. (B) The matrix of alignment II and the resulting maximum likelihood tree. The simplified trees are shown in Figure S8. (C) The matrix of alignment III. The resulting tree is shown in Figure 5. (D) The matrix for exon-intron structural change analysis among different subfamilies.
Figure S1. Evolution of exon-intron structure in the AP1/FUL subfamily. (A) A maximum-likelihood tree of the AP1/FUL subfamily, with higher-than-50% bootstrap values indicated for each node. Different mechanisms responsible for structural changes are marked on corresponding branches of the phylogenetic tree. Stars indicate structural changes involving non-triplet sequences. (B) Schematic representation of exon-intron structural changes. Exons and introns are represented by boxes and curved lines, respectively. Exon length is shown above the box, and intron length (if available) is indicated below the curved lines. Shared structural change events are linked by gray lines.
Figure S2. Evolution of exon-intron structure in the SEP1 subfamily. (A) A maximum-likelihood tree of the SEP1 subfamily. (B) Schematic representation of exon-intron structural changes. The symbols describing structural changes are the same as those in Figure S1.
Figure S3. Evolution of exon-intron structure in the SEP3 subfamily. (A) A maximum-likelihood tree of the SEP3 subfamily. (B) Schematic representation of exon-intron structural changes. The symbols describing structural changes are the same as those in Figure S1.
Figure S4. Evolution of exon-intron structure in the AGL6 subfamily. (A) A maximum-likelihood tree of the AGL6 subfamily. (B) Schematic representation of exon-intron structural changes. The symbols describing structural changes are the same as those in Figure S1.
Figure S5. Evolution of exon-intron structure in the FLC subfamily. (A) A maximum-likelihood tree of the FLC subfamily. (B) Schematic representation of exon-intron structural changes. Note that due to the dramatic sequence divergence of OsMADS37-like genes after gene duplication, the mechanisms underlying structural changes are difficult to determine. For these genes, only the exon-intron structures are shown. The symbols describing structural changes are the same as those in Figure S1.
Figure S6. Evolution of exon-intron structure in the SOC1 subfamily. (A) A maximum-likelihood tree of the SOC1 subfamily. (B) Schematic representation of exon-intron structural changes. The symbols describing structural changes are the same as those in Figure S1.
Figure S7. Evolution of exon-intron structure in the AG/STK subfamily. (A) A maximum-likelihood tree of the AG/STK subfamily. (B) Schematic representation of exon-intron structural changes. The symbols describing structural changes are the same as those in Figure S1.
Figure S8. Simplified phylogenetic trees showing relationships of the AP1/FUL, FLC, SEP, and AGL6 subfamilies, constructed based on alignments I (A) and II (B). The bootstrap values (>50%) obtained from maximum likelihood analysis and the posterior probabilities (>0.5) estimated by Bayesian inference are shown next to the nodes.
Figure S9. Alignment of amino acids encoded by exon 7 of representatives of the AP1/FUL, SEP, AGL6, and AG/STK subfamilies. Subfamily-specific motifs are highlighted by red boxes.
Figure S10. Creation of the paleoAP1 motif. Both nucleotide (A) and amino acid alignments (B) of the paleoAP1 motif in the sampled AP1/FUL-like genes and its corresponding regions in representatives of SEP-and AGL6-like genes are shown. On top of the alignments, an asterisk or a number indicates every ten nucleotides or amino acids. In (A), coding sequences and 3′ untranslated regions are represented by uppercase and lowercase letters, respectively. In (B), the paleoAP1 motif is boxed. Stars in the amino acid sequence correspond to stop codons.
References
Amborella Genome Project (2013). The Amborella genome and the evolution of flowering plants. Science 342:1241089. doi: 10.1126/science.1241089
Ampomah-Dwamena, C., Morris, B. A., Sutherland, P., Veit, B., and Yao, J. L. (2002). Down-regulation of TM29, a tomato SEPALLATA homolog, causes parthenocarpic fruit development and floral reversion. Plant Physiol. 130, 605–617. doi: 10.1104/pp.005223
Aoki, S., Uehara, K., Imafuku, M., Hasebe, M., and Ito, M. (2004). Phylogeny and divergence of basal angiosperms inferred from APETALA3- and PISTILLATA-like MADS-box genes. J. Plant Res. 117, 229–244. doi: 10.1007/s10265-004-0153-7
Becker, A., and Theissen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489. doi: 10.1016/S1055-7903(03)00207-0
Burko, Y., Shleizer-Burko, S., Yanai, O., Shwartz, I., Zelnik, I. D., Jacob-Hirsch, J., et al. (2013). A role for APETALA1/FRUITFULL transcription factors in tomato leaf development. Plant Cell 25, 2070–2083. doi: 10.1105/tpc.113.113035
Carlsbecker, A., Sundström, J., Tandre, K., Englund, M., Kvarnheden, A., Johanson, U., et al. (2003). The DAL10 gene from Norway spruce (Picea abies) belongs to a potentially gymnosperm-specific subclass of MADS-box genes and is specifically active in seed cones and pollen cones. Evol. Dev. 5, 551–561. doi: 10.1046/j.1525-142X.2003.03060.x
Causier, B., Schwarz-Sommer, Z., and Davies, B. (2010). Floral organ identity: 20 years of ABCs. Semin. Cell Dev. Biol. 21, 73–79. doi: 10.1016/j.semcdb.2009.10.005
Chen, M. K., Lin, I. C., and Yang, C. H. (2008). Functional analysis of three lily (Lilium longiflorum) APETALA1-like MADS box genes in regulating floral transition and formation. Plant Cell Physiol. 49, 704–717. doi: 10.1093/pcp/pcn046
Cho, S., Jang, S., Chae, S., Chung, K. M., Moon, Y. H., An, G., et al. (1999). Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol. Biol. 40, 419–429. doi: 10.1023/A:1006273127067
Cui, R., Han, J., Zhao, S., Su, K., Wu, F., Du, X., et al. (2010). Functional conservation and diversification of class E floral homeotic genes in rice (Oryza sativa). Plant J. 61, 767–781. doi: 10.1111/j.1365-313X.2009.04101.x
Danyluk, J., Kane, N. A., Breton, G., Limin, A. E., Fowler, D. B., and Sarhan, F. (2003). TaVRT-1, a putative transcription factor associated with vegetative to reproductive transition in cereals. Plant Physiol. 132, 1849–1860. doi: 10.1104/pp.103.023523
Ditta, G., Pinyopich, A., Robles, P., Pelaz, S., and Yanofsky, M. F. (2004). The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 14, 1935–1940. doi: 10.1016/j.cub.2004.10.028
Dreni, L., and Kater, M. M. (2013). MADS reloaded: evolution of the AGAMOUS subfamily genes. New Phytol. 201, 717–732. doi: 10.1111/nph.12555
Dreni, L., Osnato, M., and Kater, M. M. (2013). The ins and outs of the rice AGAMOUS subfamily. Mol. Plant 6, 650–664. doi: 10.1093/mp/sst019
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ferrario, S., Immink, R. G., and Angenent, G. C. (2004). Conservation and diversity in flower land. Curr. Opin. Plant Biol. 7, 84–91. doi: 10.1016/j.pbi.2003.11.003
Ferrario, S., Immink, R. G., Shchennikova, A., Busscher-Lange, J., and Angenent, G. C. (2003). The MADS-box gene FBP2 is required for SEPALLATA function in petunia. Plant Cell 15, 914–925. doi: 10.1105/tpc.010280
Fourquin, C., del Cerro, C., Victoria, F. C., Vialette-Guiraud, A., de Oliveira, A. C., and Ferrándiz, C. (2013). A change in SHATTERPROOF protein lies at the origin of a fruit morphological novelty and a new strategy for seed dispersal in Medicago genus. Plant Physiol. 162, 907–917. doi: 10.1104/pp.113.217570
Futamura, N., Totoki, Y., Toyoda, A., Igasaki, T., Nanjo, T., Seki, M., et al. (2008). Characterization of expressed sequence tags from a full-length enriched cDNA library of Cryptomeria japonica male strobili. BMC Genomics 9:383. doi: 10.1186/1471-2164-9-383
Gocal, G. F., King, R. W., Blundell, C. A., Schwartz, O. M., Andersen, C. H., and Weigel, D. (2001). Evolution of floral meristem identity genes. Analysis of Lolium temulentum genes related to APETALA1 and LEAFY of Arabidopsis. Plant Physiol. 125, 1788–1801. doi: 10.1104/pp.125.4.1788
Gu, Q., Ferrándiz, C., Yanofsky, M. F., and Martienssen, R. (1998). The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125, 1509–1517.
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Honma, T., and Goto, K. (2001). Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. doi: 10.1038/35054083
Hsu, W., Yeh, T., Huang, K., Li, J., Chen, H., and Yang, C. (2014). AGAMOUS-LIKE13, a putative ancestor for the E functional genes, specifies male and female gametophyte morphogenesis. Plant J. 77, 1–15. doi: 10.1111/tpj.12363
Huang, X., Ding, J., Effgen, S., Turck, F., and Koornneef, M. (2013). Multiple loci and genetic interactions involving flowering time genes regulate stem branching among natural variants of Arabidopsis. New Phytol. 199, 843–857. doi: 10.1111/nph.12306
Huang, X., Effgen, S., Meyer, R. C., Theres, K., and Koornneef, M. (2012). Epistatic natural allelic variation reveals a function of AGAMOUS-LIKE6 in axillary bud formation in Arabidopsis. Plant Cell 24, 2364–2379. doi: 10.1105/tpc.112.099168
Huijser, P., Klein, J., Lönnig, W., Meijer, H., Saedler, H., and Sommer, H. (1992). Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 11, 1239.
Irish, V. F., and Sussex, I. M. (1990). Function of the apetala-1 gene during Arabidopsis floral development. Plant Cell 2, 741–753. doi: 10.1105/tpc.2.8.741
Jang, S., An, K., Lee, S., and An, G. (2002). Characterization of tobacco MADS-box genes involved in floral initiation. Plant Cell Physiol. 43, 230–238. doi: 10.1093/pcp/pcf015
Kane, N. A., Danyluk, J., Tardif, G., Ouellet, F., Laliberté, J. F., Limin, A. E., et al. (2005). TaVRT-2, a member of the StMADS-11 clade of flowering repressors, is regulated by vernalization and photoperiod in wheat. Plant Physiol. 138, 2354–2363. doi: 10.1104/pp.105.061762
Kim, D. H., Doyle, M. R., Sung, S., and Amasino, R. M. (2009). Vernalization: winter and the timing of flowering in plants. Annu. Rev. Cell Dev. Biol. 25, 277–299. doi: 10.1146/annurev.cellbio.042308.113411
Kim, S., Koh, J., Yoo, M. J., Kong, H., Hu, Y., Ma, H., et al. (2005). Expression of floral MADS-box genes in basal angiosperms: implications for the evolution of floral regulators. Plant J. 43, 724–744. doi: 10.1111/j.1365-313X.2005.02487.x
Kim, S., Soltis, P. S., and Soltis, D. E. (2013). AGL6-like MADS-box genes are sister to AGL2-like MADS-box genes. J. Plant Biol. 56, 315–325. doi: 10.1007/s12374-013-0147-x
Kofuji, R., Sumikawa, N., Yamasaki, M., Kondo, K., Ueda, K., Ito, M., et al. (2003). Evolution and divergence of the MADS-box gene family based on genome-wide expression analyses. Mol. Biol. Evol. 20, 1963–1977. doi: 10.1093/molbev/msg216
Kong, H., Leebens-Mack, J., Ni, W., and Ma, H. (2004). Highly heterogeneous rates of evolution in the SKP1 gene family in plants and animals: functional and evolutionary implications. Mol. Biol. Evol. 21, 117–128. doi: 10.1093/molbev/msh001
Koo, S. C., Bracko, O., Park, M. S., Schwab, R., Chun, H. J., Park, K. M., et al. (2010). Control of lateral organ development and flowering time by the Arabidopsis thaliana MADS-box Gene AGAMOUS-LIKE6. Plant J. 62, 807–816. doi: 10.1111/j.1365-313X.2010.04192.x
Kramer, E. M., Jaramillo, M. A., and Di Stilio, V. S. (2004). Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS-box genes in angiosperms. Genetics 166, 1011–1023. doi: 10.1534/genetics.166.2.1011
Kramer, E. M., Su, H. J., Wu, C. C., and Hu, J. M. (2006). A simplified explanation for the frameshift mutation that created a novel C-terminal motif in the APETALA3 gene lineage. BMC Evol. Biol. 6:30. doi: 10.1186/1471-2148-6-30
Krizek, B. A., and Meyerowitz, E. M. (1996). Mapping the protein regions responsible for the functional specificities of the Arabidopsis MADS domain organ-identity proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 4063–4070. doi: 10.1073/pnas.93.9.4063
Lamb, R. S., and Irish, V. F. (2003). Functional divergence within the APETALA3/PISTILLATA floral homeotic gene lineages. Proc. Natl. Acad. Sci. U.S.A. 100, 6558–6563. doi: 10.1073/pnas.0631708100
Leebens-Mack, J., Raubeson, L. A., Cui, L., Kuehl, J. V., Fourcade, M. H., Chumley, T. W., et al. (2005). Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one's way out of the Felsenstein zone. Mol. Biol. Evol. 22, 1948–1963. doi: 10.1093/molbev/msi191
Li, H., Liang, W., Jia, R., Yin, C., Zong, J., Kong, H., et al. (2010). The AGL6-like gene OsMADS6 regulates floral organ and meristem identities in rice. Cell Res. 20, 299–313. doi: 10.1038/cr.2009.143
Li, L., Yu, X., Guo, C., Duan, X., Shan, H., Zhang, R., et al. (2015). Interactions among proteins of floral MADS-box genes in Nuphar pumila (Nymphaeaceae) and the most recent common ancestor of extant angiosperms help understand the underlying mechanisms of the origin of the flower. J. Syst. Evol. 53, 285–296. doi: 10.1111/jse.12148
Litt, A. (2007). An evaluation of A-function: evidence from the APETALA1 and APETALA2 gene lineages. Int. J. Plant Sci. 168, 73–91. doi: 10.1086/509662
Litt, A., and Irish, V. F. (2003). Duplication and diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: implications for the evolution of floral development. Genetics 165, 821–833.
Litt, A., and Kramer, E. M. (2010). The ABC model and the diversification of floral organ identity. Semin. Cell Dev. Biol. 21, 129–137. doi: 10.1016/j.semcdb.2009.11.019
Liu, Y., Guo, C., Xu, G., Shan, H., and Kong, H. (2011). Evolutionary pattern of the regulatory network for flower development: insights gained from a comparison of two Arabidopsis species. J. Syst. Evol. 49, 528–538. doi: 10.1111/j.1759-6831.2011.00158.x
Long, M., VanKuren, N. W., Chen, S., and Vibranovski, M. D. (2013). New gene evolution: little did we know. Annu. Rev. Genet. 47, 307–333. doi: 10.1146/annurev-genet-111212-133301
Mellway, R. D., and Lund, S. T. (2013). Interaction analysis of grapevine MIKCc-type MADS transcription factors and heterologous expression of putative véraison regulators in tomato. J. Plant Physiol. 170, 1424–1433. doi: 10.1016/j.jplph.2013.05.010
Melzer, R., Härter, A., Rümpler, F., Kim, S., Soltis, P. S., Soltis, D. E., et al. (2014). DEF-and GLO-like proteins may have lost most of their interaction partners during angiosperm evolution. Ann. Bot. 114, 1431–1443. doi: 10.1093/aob/mcu094
Michaels, S. D., and Amasino, R. M. (1999). FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. doi: 10.1105/tpc.11.5.949
Murai, K., Miyamae, M., Kato, H., Takumi, S., and Ogihara, Y. (2003). WAP1, a wheat APETALA1 homolog, plays a central role in the phase transition from vegetative to reproductive growth. Plant Cell Physiol. 44, 1255–1265. doi: 10.1093/pcp/pcg171
Nam, J., dePamphilis, C. W., Ma, H., and Nei, M. (2003). Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Mol. Biol. Evol. 20, 1435–1447. doi: 10.1093/molbev/msg152
Ohmori, S., Kimizu, M., Sugita, M., Miyao, A., Hirochika, H., Uchida, E., et al. (2009). MOSAIC FLORAL ORGANS1, an AGL6-like MADS-box gene, regulates floral organ identity and meristem fate in rice. Plant Cell 21, 3008–3025. doi: 10.1105/tpc.109.068742
Pabón-Mora, N., Ambrose, B. A., and Litt, A. (2012). Poppy APETALA1/FRUITFULL orthologs control flowering time, branching, perianth identity, and fruit development. Plant Physiol. 158, 1685–1704. doi: 10.1104/pp.111.192104
Pabón-Mora, N., Sharma, B., Holappa, L. D., Kramer, E. M., and Litt, A. (2013). The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. Plant J. 74, 197–212. doi: 10.1111/tpj.12113
Parenicova, L. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis new openings to the MADS world. Plant Cell 15, 1538–1551. doi: 10.1105/tpc.011544
Pelaz, S., Tapia-López, R., Alvarez-Buylla, E. R., and Yanofsky, M. F. (2001). Conversion of leaves into petals in Arabidopsis. Curr. Biol. 11, 182–184. doi: 10.1016/S0960-9822(01)00024-0
Piwarzyk, E., Yang, Y., and Jack, T. (2007). Conserved C-terminal motifs of the Arabidopsis proteins APETALA3 and PISTILLATA are dispensable for floral organ identity function. Plant Physiol. 145, 1495–1505. doi: 10.1104/pp.107.105346
Pnueli, L., Hareven, D., Broday, L., Hurwitz, C., and Lifschitz, E. (1994). The TM5 MADS-box gene mediates organ differentiation in the three inner whorls of tomato flowers. Plant Cell 6, 175–186. doi: 10.1105/tpc.6.2.175
Posada, D., and Crandall, K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Preston, J. C., and Kellogg, E. A. (2006). Reconstructing the evolutionary history of paralogous APETALA1/FRUITFULL-like genes in grasses (Poaceae). Genetics 174, 421–437. doi: 10.1534/genetics.106.057125
Raes, J., and Van de Peer, Y. (2005). Functional divergence of proteins through frameshift mutations. Trends Genet. 21, 428–431. doi: 10.1016/j.tig.2005.05.013
Rijpkema, A. S., Vandenbussche, M., Koes, R., Heijmans, K., and Gerats, T. (2010). Variations on a theme: changes in the floral ABCs in angiosperms. Semin. Cell Dev. Biol. 21, 100–107. doi: 10.1016/j.semcdb.2009.11.002
Rijpkema, A. S., Zethof, J., Gerats, T., and Vandenbussche, M. (2009). The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J. 60, 1–9. doi: 10.1111/j.1365-313X.2009.03917.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Roshan, U., and Livesay, D. R. (2006). Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics 22, 2715–2721. doi: 10.1093/bioinformatics/btl472
Roy, S. W., and Gilbert, W. (2005). Rates of intron loss and gain: implications for early eukaryotic evolution. Proc. Natl. Acad. Sci. U.S.A. 102, 5773–5778. doi: 10.1073/pnas.0500383102
Ruelens, P., de Maagd, R. A., Proost, S., Theissen, G., Geuten, K., and Kaufmann, K. (2013). FLOWERING LOCUS C in monocots and the tandem origin of angiosperm-specific MADS-box genes. Nat. Commun. 4:2280. doi: 10.1038/ncomms3280
Ruokolainen, S., Ng, Y. P., Albert, V. A., Elomaa, P., and Teeri, T. H. (2010). Large scale interaction analysis predicts that the Gerbera hybrida floral E function is provided both by general and specialized proteins. BMC Plant Biol. 10:129. doi: 10.1186/1471-2229-10-129
Seok, H. Y., Park, H. Y., Park, J. I., Lee, Y. M., Lee, S. Y., An, G., et al. (2010). Rice ternary MADS protein complexes containing class B MADS heterodimer. Biochem. Biophys. Res. Commun. 401, 598–604. doi: 10.1016/j.bbrc.2010.09.108
Shan, H., Zhang, N., Liu, C., Xu, G., Zhang, J., Chen, Z., et al. (2007). Patterns of gene duplication and functional diversification during the evolution of the AP1/SQUA subfamily of plant MADS-box genes. Mol. Phylogenet. Evol. 44, 26–41. doi: 10.1016/j.ympev.2007.02.016
Sheldon, C. C., Jean Finnegan, E., Dennis, E. S., and James Peacock, W. (2006). Quantitative effects of vernalization on FLC and SOC1 expression. Plant J. 45, 871–883. doi: 10.1111/j.1365-313X.2006.02652.x
Smaczniak, C., Immink, R. G., Muiño, J. M., Blanvillain, R., Busscher, M., Busscher-Lange, J., et al. (2012). Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. U.S.A. 109, 1560–1565. doi: 10.1073/pnas.1112871109
Su, K., Zhao, S., Shan, H., Kong, H., Lu, W., Theissen, G., et al. (2008). The MIK region rather than the C-terminal domain of AP3-like class B floral homeotic proteins determines functional specificity in the development and evolution of petals. New Phytol. 178, 544–558. doi: 10.1111/j.1469-8137.2008.02382.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Theissen, G., Becker, A., Di Rosa, A., Kanno, A., Kim, J. T., Munster, T., et al. (2000). A short history of MADS-box genes in plants. Plant Mol. Biol. 42, 115–149. doi: 10.1023/A:1006332105728
Theissen, G., Kim, J. T., and Saedler, H. (1996). Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43, 484–516. doi: 10.1007/BF02337521
Thompson, B. E., Bartling, L., Whipple, C., Hall, D. H., Sakai, H., Schmidt, R., et al. (2009). bearded-ear encodes a MADS-box transcription factor critical for maize floral development. Plant Cell 21, 2578–2590. doi: 10.1105/tpc.109.067751
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Trevaskis, B., Bagnall, D. J., Ellis, M. H., Peacock, W. J., and Dennis, E. S. (2003). MADS box genes control vernalization-induced flowering in cereals. Proc. Natl. Acad. Sci. U.S.A. 100, 13099–13104. doi: 10.1073/pnas.1635053100
Ubi, B. E., Saito, T., Bai, S., Nishitani, C., Ban, Y., Ikeda, K., et al. (2013). Characterization of 10 MADS-box genes from Pyrus pyrifolia and their differential expression during fruit development and ripening. Gene 528, 183–194. doi: 10.1016/j.gene.2013.07.018
Vandenbussche, M., Theissen, G., Van de Peer, Y., and Gerats, T. (2003a). Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 31, 4401–4409. doi: 10.1093/nar/gkg642
Vandenbussche, M., Zethof, J., Souer, E., Koes, R., Tornielli, G. B., Pezzotti, M., et al. (2003b). Toward the analysis of the petunia MADS-box gene family by reverse and forward transposon insertion mutagenesis approaches: B, C, and D floral organ identity functions require SEPALLATA-like MADS-box genes in petunia. Plant Cell 15, 2680–2693. doi: 10.1105/tpc.017376
Wang, P., Liao, H., Zhang, W., Yu, X., Zhang, R., Shan, H., et al. (2015). Flexibility in the structure of spiral flowers and its underlying mechanisms. Nat. Plants 2, 15188. doi: 10.1038/nplants.2015.188
Wang, Y., Melzer, R., and Theissen, G. (2010). Molecular interactions of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the evolutionary origin of ‘floral quartets’. Plant J. 64, 177–190. doi: 10.1111/j.1365-313X.2010.04325.x
Wong, C. E., Singh, M. B., and Bhalla, P. L. (2013). Novel members of the AGAMOUS LIKE 6 subfamily of MIKCC-type MADS-box genes in soybean. BMC Plant Biol. 13:105. doi: 10.1186/1471-2229-13-105
Xu, G., Guo, C., Shan, H., and Kong, H. (2012). Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. U.S.A. 109, 1187–1192. doi: 10.1073/pnas.1109047109
Xu, G., and Kong, H. (2007). Duplication and divergence of floral MADS-box genes in grasses: evidence for the generation and modification of novel regulators. J. Integr. Plant Biol. 49, 927–939. doi: 10.1111/j.1744-7909.2007.00502.x
Xu, G., Ma, H., Nei, M., and Kong, H. (2009). Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc. Natl. Acad. Sci. U.S.A. 106, 835–840. doi: 10.1073/pnas.0812043106
Yan, L., Loukoianov, A., Tranquilli, G., Helguera, M., Fahima, T., and Dubcovsky, J. (2003). Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. U.S.A. 100, 6263–6268. doi: 10.1073/pnas.0937399100
Yockteng, R., Almeida, A. M., Morioka, K., Alvarez-Buylla, E. R., and Specht, C. D. (2013). Molecular evolution and patterns of duplication in the SEP/AGL6-like lineage of the Zingiberales: a proposed mechanism for floral diversification. Mol. Biol. Evol. 30, 2401–2422. doi: 10.1093/molbev/mst137
Yoo, S. K., Hong, S. M., Lee, J. S., and Ahn, J. H. (2011). A genetic screen for leaf movement mutants identifies a potential role for AGAMOUS-LIKE 6 (AGL6) in circadian-clock control. Mol. Cells 31, 281–287. doi: 10.1007/s10059-011-0035-5
Zahn, L. M., Kong, H., Leebens-Mack, J. H., Kim, S., Soltis, P. S., Landherr, L. L., et al. (2005). The evolution of the SEPALLATA subfamily of MADS-box genes: a preangiosperm origin with multiple duplications throughout angiosperm history. Genetics 169, 2209–2223. doi: 10.1534/genetics.104.037770
Zahn, L. M., Leebens-Mack, J. H., Arrington, J. M., Hu, Y., Landherr, L. L., dePamphilis, C. W., et al. (2006). Conservation and divergence in the AGAMOUS subfamily of MADS-box genes: evidence of independent sub- and neofunctionalization events. Evol. Dev. 8, 30–45. doi: 10.1111/j.1525-142X.2006.05073.x
Keywords: APETALA1/FRUITFULL, SEPALLATA, AGAMOUS-LIKE6, FLOWERING LOCUS C, exon-intron structural change
Citation: Yu X, Duan X, Zhang R, Fu X, Ye L, Kong H, Xu G and Shan H (2016) Prevalent Exon-Intron Structural Changes in the APETALA1/FRUITFULL, SEPALLATA, AGAMOUS-LIKE6, and FLOWERING LOCUS C MADS-Box Gene Subfamilies Provide New Insights into Their Evolution. Front. Plant Sci. 7:598. doi: 10.3389/fpls.2016.00598
Received: 01 October 2015; Accepted: 18 April 2016;
Published: 02 May 2016.
Edited by:
Verónica S. Di Stilio, University of Washington, USAReviewed by:
Stefan Gleissberg, Gleissberg.org, USAAmy Litt, University of California, Riverside, USA
Ji Yang, Fudan University, China
Copyright © 2016 Yu, Duan, Zhang, Fu, Ye, Kong, Xu and Shan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guixia Xu, xuguixia1982@ibcas.ac.cn;
Hongyan Shan, shanhongyan@ibcas.ac.cn
†These authors have contributed equally to this work.