 Almudena Val-Blasco1†
Almudena Val-Blasco1† Jose A. Navarro-García2‡Maria Tamayo3†‡Maria J. Piedras4‡
Jose A. Navarro-García2‡Maria Tamayo3†‡Maria J. Piedras4‡ Patricia Prieto3†‡
Patricia Prieto3†‡ Carmen Delgado3†
Carmen Delgado3† Gema Ruiz-Hurtado2Laura Rozas-Romero1Marta Gil-Fernández1
Gema Ruiz-Hurtado2Laura Rozas-Romero1Marta Gil-Fernández1 Carlos Zaragoza5†
Carlos Zaragoza5† Lisardo Boscá3†
Lisardo Boscá3† María Fernández-Velasco1†*
María Fernández-Velasco1†*- 1Innate Immune Response Group, Instituto de Investigación Hospital Universitario La Paz, La Paz University Hospital, Madrid, Spain
- 2Cardiorenal Translational Laboratory and Hypertension Unit, Institute of Research i+12, Hospital Universitario 12 de Octubre, Madrid, Spain
- 3Departamento de Bioquímica, Facultad de Medicina, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas, Madrid, Spain
- 4Department of Anatomy, Faculty of Health Sciences, Francisco de Vitoria University (UFV), Pozuelo de Alarcón, Spain
- 5Unidad de Investigación Cardiovascular, Universidad Francisco de Vitoria, Hospital Universitario Ramón y Cajal (IRYCIS), CIBERCV, Madrid, Spain
Heart failure (HF) is a complex syndrome characterized by cardiac dysfunction, Ca2+ mishandling, and chronic activation of the innate immune system. Reduced cardiac output in HF leads to compensatory mechanisms via activation of the adrenergic nervous system. In turn, chronic adrenergic overstimulation induces pro-arrhythmic events, increasing the rate of sudden death in failing patients. Nucleotide-binding oligomerization domain-containing protein 1 (NOD1) is an innate immune modulator that plays a key role in HF progression. NOD1 deficiency in mice prevents Ca2+ mishandling in HF under basal conditions, but its role during β-adrenergic stimulation remains unknown. Here, we evaluated whether NOD1 regulates the β-adrenergic modulation of Ca2+ signaling in HF. Ca2+ dynamics were examined before and after isoproterenol perfusion in cardiomyocytes isolated from healthy and from post-myocardial infarction (PMI) wild-type (WT) and Nod1-/- mice. Isoproterenol administration induced similar effects on intracellular [Ca2+]i transients, cell contraction, and sarcoplasmic reticulum (SR)-Ca2+ load in healthy WT and Nod1-/- cells. However, compared with WT-PMI cells, isoproterenol exposure induced a significant increase in the [Ca2+]i transients and cell contraction parameters in Nod1-/--PMI cells, which mainly due to an increase in SR-Ca2+ load. NOD1 deficiency also prevented the increase in diastolic Ca2+ leak (Ca2+ waves) induced by isoproterenol in PMI cells. mRNA levels of β1 and β2 adrenergic receptors were significantly higher in Nod1-/--PMI hearts vs WT-PMI hearts. Healthy cardiomyocytes pre-treated with the selective agonist of NOD1, iE-DAP, and perfused with isoproterenol showed diminished [Ca2+]i transients amplitude, cell contraction, and SR-Ca2+ load compared with vehicle-treated cells. iE-DAP-treated cells also presented increased diastolic Ca2+ leak under β-adrenergic stimulation. The selectivity of iE-DAP on Ca2+ handling was validated by pre-treatment with the inactive analog of NOD1, iE-Lys. Overall, our data establish that NOD1 deficiency improves the β-adrenergic modulation of Ca2+ handling in failing hearts.
Introduction
Heart failure (HF) is a complex clinical disorder characterized by the inability of the heart to deliver blood and nutrients to metabolic tissues. Chronic HF is a progressive disease, with high morbidity and mortality, and poses a significant economic burden on the healthcare system. Given its poor prognosis, the identification of molecular pathways contributing to HF is a major research goal.
Compensatory activation of the adrenergic nervous system is a pathophysiological response to HF progression, which functions to maintain cardiac homeostasis through activation of neural hormones, mainly catecholamines (Gordan et al., 2015). Accordingly, sympathetic hyperactivity is a hallmark of HF, activating β-adrenergic receptors to increase heart rate and cardiac contractility via excitation–contraction (EC) coupling, in an attempt to counteract the decreased cardiac output. Chronic exposure of the heart to elevated levels of catecholamines may, however, ultimately lead to the desensitization of β-adrenergic receptors, exacerbating the loss of cardiac function and increasing the risk for triggered arrhythmias and sudden death (Feldman et al., 2005; Brum et al., 2006; Venetucci et al., 2008).
There is a growing body of evidence suggesting a link between the innate immune response and HF progression (Lin and Knowlton, 2014; Mann, 2015). Whether the myocardial inflammatory response impairs cardiac damage is a question of broad significance. Recent studies indicate that some receptors of the innate immune system, including nucleotide-binding oligomerization domain-like receptors (NLRs), play significant roles in the host response after cardiac damage (Bullón et al., 2016; Monnerat et al., 2016; Val-Blasco et al., 2017a). Nucleotide-binding oligomerization domain (NOD) receptors are NLR family members that recognize conserved motifs of bacterial peptidoglycans in many Gram-negative bacteria (Caruso et al., 2014). Beyond their role as microbial pattern recognition receptors, it has recently been described that NODs can also be activated by non-infectious factors such as endoplasmic reticulum stress (Keestra-Gounder et al., 2016). Ligand activation of NOD receptors induces a conformational change in the protein, leading to self-oligomerization and promoting the recruitment of its adaptor, receptor-interacting protein 2 (RIP2), which activates nuclear factor-κB and triggers the inflammatory response (Park et al., 2007). NOD-containing protein 1 (NOD1) is expressed in the heart, and is functional in resident fibroblasts and cardiomyocytes (Fernández-Velasco et al., 2012; Delgado et al., 2015). Several studies have demonstrated a role for NOD1 in the progression of cardiovascular diseases, including atherosclerosis and diabetic cardiomyopathy (Prieto et al., 2014; Kanno et al., 2015; Val-Blasco et al., 2017b). We have recently shown that NOD1 is upregulated in both mouse and human failing myocardium, and its genetic deletion or pharmacological blockade prevents cardiac dysfunction and deleterious remodeling in failing hearts principally by preventing the HF-related Ca2+ mishandling (Val-Blasco et al., 2017a). Conversely, activation of NOD1 promotes a dysregulation of the intracellular Ca2+ dynamics, similar to those observed in HF (Delgado et al., 2015).
Ca2+ plays a key role in cardiac EC coupling. During EC coupling, an action potential leads to a small increase in intracellular Ca2+ via activated sarcolemmal L-type Ca channels (ICaL), and this is amplified by a greater release of Ca2+ from the sarcoplasmic reticulum (SR) by ryanodine receptors (RyR2), increasing the intracellular Ca2+ concentration ([Ca2+]i) – a process known as Ca2+-induced Ca2+ release. This elevation in [Ca2+]i results in myofilament activation and cell contraction. During relaxation, Ca2+ is removed from the cytosol primarily by two mechanisms: Ca2+ re-uptake by the SR-ATPase (SERCA2a) and Na+/Ca2+ exchanger activation.
Excitation–contraction coupling is modulated by the sympathetic nervous system through activation of the β-adrenergic receptor coupled to Gs-type G-proteins, promoting an elevation in the intracellular concentration of cAMP (Bers, 2002). In turn, cAMP activates protein kinase A (PKA), resulting in the phosphorylation of several EC coupling-Ca2+ transporters such as ICaL, RyR2, and phospholamban, among others (Mayourian et al., 2018).
Several lines of evidence have established a direct relationship between Ca2+ dysregulation and HF progression (Gómez et al., 1997; Heinzel et al., 2011; Ruiz-Hurtado et al., 2015). Depressed systolic Ca2+ release, the decline in the SR-Ca2+ load, and the increased Ca2+ diastolic leak seem to be key players in HF progression, and these changes worsen under chronic β-adrenergic activation. We recently showed that NOD1 modulates intracellular Ca2+ handling in human and experimental failing hearts (Val-Blasco et al., 2017a); however, the role of NOD1 in the modulation of Ca2+ handling under β-adrenergic stimulation has not been investigated. Using a mouse HF model, we determined whether the deficiency of NOD1 impairs the modulation of Ca2+ dynamics under β-adrenergic stimulation.
Materials and Methods
Myocardial Infarction Model
Male Nod1-/- mice on a C57BL/6J (6B; 129P2-Nod1tm1Nnz/J) background were used in this study (Chamaillard et al., 2003). As controls, we used wild-type (WT) C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME, United States). Experiments involving mice were carried out in compliance with Spanish and European guidelines (2010/63/EU) regarding animal policy and welfare recommendations. The present study was performed in male mice because they have been reported to be more prone than female to develop cardiac injury and Ca2+ mishandling after isoproterenol administration in experimental models of HF (Cross et al., 2002).
Mice were anesthetized by intraperitoneal (i.p.) injection of a mixture of ketamine (Imalgene®, 70 mg/kg) and xylazine (Rompun®, 10 mg/kg). Unconscious mice were shaved in the anterior region of the neck and the chest, intubated by tracheostomy, and connected to a small animal ventilator (MiniVent type 845, Harvard Apparatus) for artificial ventilation at 150 strokes/minute and 230 μL stroke volume. A 2–3 mm incision parallel to the lower costal edge was made and the left pectoralis major muscle was dissociated until the ribs were exposed. Left thoracotomy was performed between the third and fourth ribs to visualize the anterior surface of the heart and left lung. A 1-mm-thick piece of gelatin sponge (Spongostan®), slightly moistened with saline, was inserted through the hollow thoracotomy to protect the lung. The ribs were separated with an eyelid-retractor and a branch of the left coronary artery was ligated with a blue polypropylene monofilament surgical non-absorbable suture 6/0. The Spongostan® sponge was then removed and the chest was closed with braided silk non-absorbable suture 4/0. Respiratory stimulation was performed to return the heart to spontaneous breathing. For postoperative analgesia, buprenorphine (Buprex®, 0.05 mg/kg) was applied subcutaneously. Mice were kept on a warm electric blanket until spontaneous recovery. Sham-operated mice were used as the control group and underwent the same procedure, but without coronary ligation.
Myocyte Isolation
Adult male mice (2 months of age) were anesthetized with sodium pentobarbital (100 mg/kg i.p.) and heparinized (4 UI/g i.p.). Hearts were rapidly removed and cannulated via the ascending aorta for Langendorff perfusion. Retrograde perfusion was initiated with a free calcium Tyrode solution containing 0.2 mM EGTA over 2–3 min at room temperature, and subsequently with Tyrode solution containing CaCl2 (0.1 mM) and type II collagenase (1 mg/mL; Worthington Biochemical, Lakewood, NY, United States). Successful digestion was assumed when the flux rate increased and the heart color changed from red to pallor. The heart was then taken off the apparatus, and the ventricles were removed, minced into small pieces, and mechanically dissociated in a thermostatic bath at 37°C in the enzymatic solution. The cardiomyocyte cell suspension was filtered through a nylon mesh (250 μm) and centrifuged at 300 rpm for 3 min. The cell pellet was suspended in Tyrode solution containing CaCl2 (0.5 mM) and centrifuged as before. Finally, the cells were suspended in Tyrode solution containing CaCl2 (1 mM). The Tyrode solution contained (in mM): 130 NaCl, 5.4 KCl, 0.5 MgCl2, 25 HEPES, 0.4 NaH2PO4, and 22 glucose, which was adjusted to pH 7.4 with NaOH.
Confocal Microscopy
Local increases in intracellular Ca2+ concentration, [Ca2+]i transients, and Ca2+ waves were analyzed in intact isolated cardiomyocytes loaded with the fluorescent Ca2+ dye Fluo-3AM (5 μmol/L, Invitrogen). [Ca2+]i transients were recorded in cells electrically excited at 2 Hz by field stimulation using two parallel platinum electrodes. Ca2+ waves were acquired in quiescent myocytes. SR Ca2+ load was assessed by rapid caffeine application (10 mM) in cells previously excited at 2 Hz.
Images were acquired by confocal microscopy (Meta Zeiss LSM 710, 40× oil immersion objective with a 1.2 NA), by scanning the cell with an argon laser every 1.54 ms. Fluo-3AM was excited at 488 nm and emitted fluorescence was collected at >505 nm. Data analysis was performed with homemade routines using software written on the IDL platform (Research System Inc., Boulder, CO, United States), designed by Dr. AM Gómez (Inserm, UMR-S 1180). Images were corrected for background fluorescence. The fluorescence values (F) were normalized by the basal fluorescence (F0) to obtain the fluorescence ratio (F/F0).
Drugs and Treatments
Isolated cardiomyocytes were perfused with 10-8 M isoproterenol (Sigma-Aldrich, Madrid, Spain) for 1–5 min and intracellular Ca2+ dynamics were recorded. In some experiments, cells were pretreated with the selective NOD1 agonist C12-iE-DAP (iE-DAP, InvivoGen, San Diego, CA, United States), which has significant cell membrane permeability and high potency (Fernández-Velasco et al., 2012; Delgado et al., 2015; Val-Blasco et al., 2017a), or iE-Lys (Invivogen), an inactive analog of NOD1. Nodinitib-1 (Cayman) was used as a selective NOD1 inhibitor and mice were injected IP with 5 μmol/L nodinitib-1 or vehicle (<0.01% DMSO) three times weekly for 6 weeks.
Real-Time PCR
Frozen heart tissue was pulverized to powder and then homogenized in TRIZOL Reagent® solution (Ambion) with a Polytron system (MWR). RNA (250 ng) was reverse transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368813). This template cDNA was used in the qPCR reaction with Power SYBR Green PCR Master mix (Applied Biosystems #4367659) and specific primers in a 7900HT Fast Real Time PCR system (Applied Biosystems). 18s-RNA was used as a housekeeping gene. The primer sequences (5′-3′) for β1 and β2 adrenergic receptors and 18s-RNA were as follows:
β1-F CGCTGATCTGGTCATGGGAT
β1-R GAAGAAGGAGCCGTACTCCC
β2-F AATAGCAACGGCAGAACGGA
β2-R TCACAAAGCCTTCCATGCCT
18s-F CCAGTAAGTGCGGGTCATAAGC
18s-R CCTCACTAAACCATCCAATCGG
Statistical Analysis
Results are reported as mean ± SEM. Statistical analysis was performed using two-way analysis of variance, paired (two-sided) Student’s t test or χ2 test, as appropriate. All statistical analyses were performed with Origin 8.0 software (OriginLab, Northampton, MA, United States). Significance was set at P < 0.05.
Results
Hypertrophy Development in Mice Subjected to Myocardial Infarction
Compared with healthy WT mice, post-myocardial infarction (PMI)-mice 6 weeks after surgery showed significantly increased heart weight, heart weight/body weight ratio, and heart weight/tibia length ratio, consistent with the development of cardiac hypertrophy (Table 1). By contrast, Nod1-/--PMI mice showed similar heart weight, heart weight/body weight, and heart weight/tibia length values to Nod1-/- mice (Table 1). These data indicate that deficiency of NOD1 attenuates cardiac hypertrophy in our PMI mouse model.
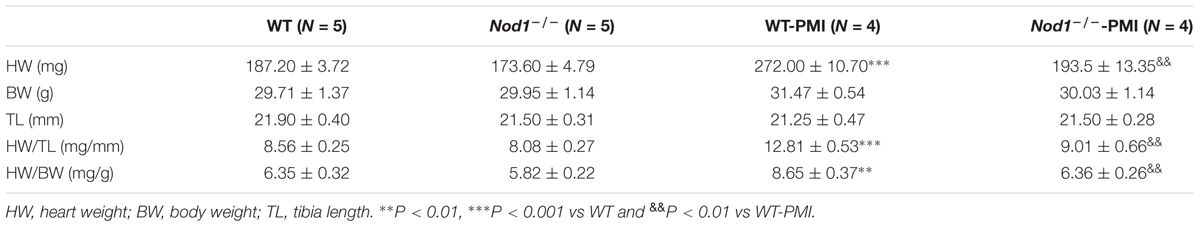
TABLE 1. Cardiac properties of wild-type and Nod1-deficent mice.
Deficiency of NOD1 Improves the β-Adrenergic Modulation of Systolic Ca2+ Release and Sarcoplasmic Reticulum Ca2+ Load in PMI Cardiomyocytes
We first analyzed whether deficiency of NOD1 determined the β-adrenergic response to Ca2+ handling in healthy myocytes. Isoproterenol perfusion induced a similar increase in [Ca2+]i transients amplitude (Figure 1A), cell contraction (Figure 1B), and SR-Ca2+ load (Figure 1C) in WT and Nod1-/- cardiomyocytes.
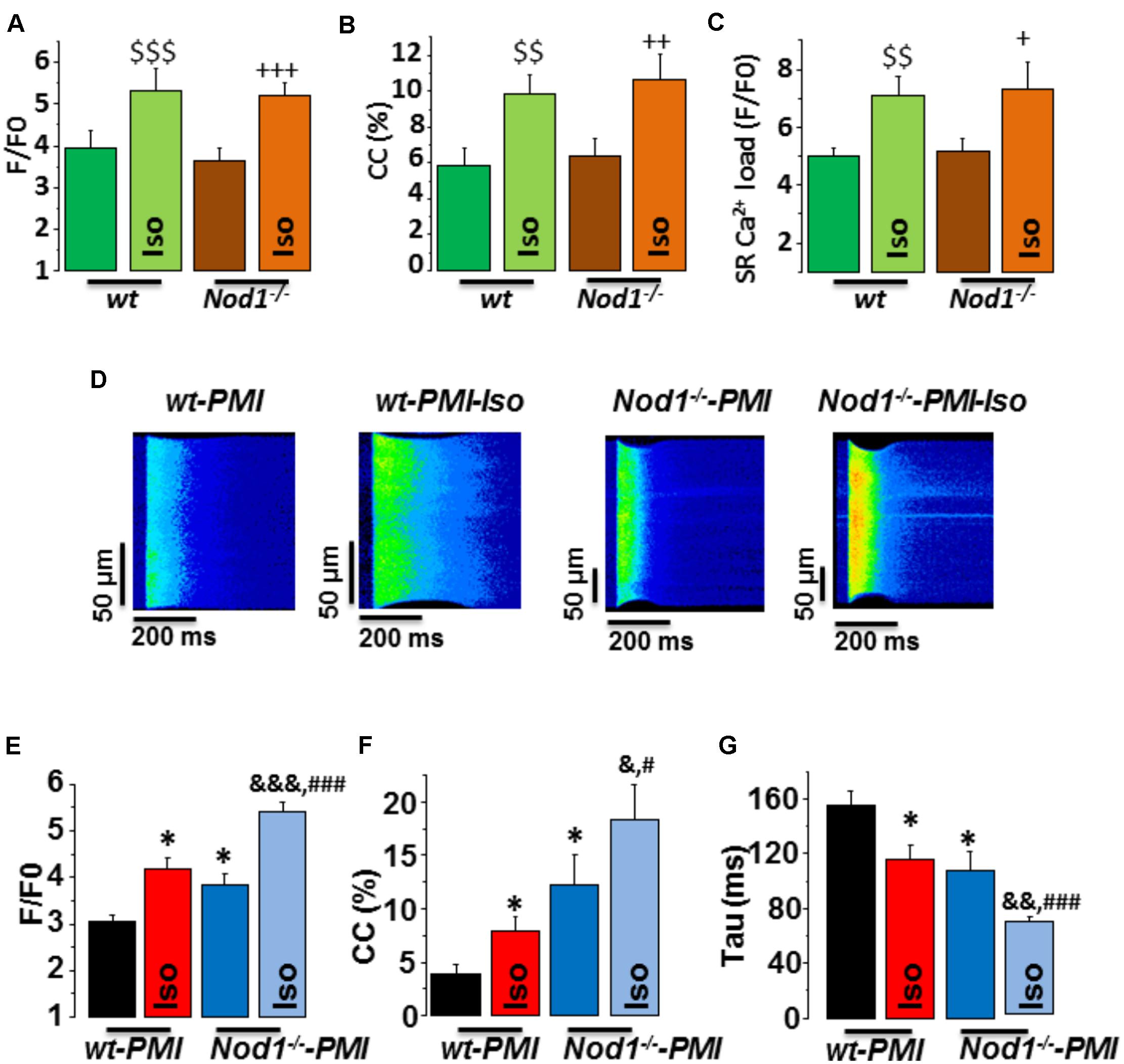
FIGURE 1. Deficiency of NOD1 improves the modulation of systolic Ca2+ handling and SR-Ca2+ load by β-adrenergic stimulation in cardiomyocytes from failing mice. (A–C) Mean values of amplitude of [Ca2+]i transients (F/F0, A), cell contractions (CC, %, B), and caffeine-evoked [Ca2+]i transients amplitude (SR-Ca2+ load, F/F0, C) in WT (n = 13 cells/5 mice) and NOD1-/- cardiomyocytes (n = 14 cells/4 mice) with and without 10-8 M isoproterenol (Iso). (D) Representative line-scan confocal images of [Ca2+]i transients obtained from WT-PMI and NOD1-/--PMI cardiomyocytes with and without Iso perfusion. (E,F) Mean values of peak fluorescence [Ca2+]i transients (F/F0, E), cell contraction (CC, %, F), and decay time of [Ca2+]i transients (tau, ms, G) in WT-PMI (n = 12 cells/4 mice) and NOD1-/--PMI (n = 13 cells/4 mice) cardiomyocytes with and without Iso. Histograms represent the mean ± SEM: $$P < 0.01, $$$P < 0.001 vs WT; +P < 0.05 vs NOD1-/-, ++P < 0.01 vs NOD1-/-, +++P < 0.001 vs NOD1-/-; ∗P < 0.05 vs WT-PMI; &P < 0.05, &&P < 0.01, &&&P < 0.001 vs NOD1-/--PMI; and #P < 0.05, ###P < 0.001 vs WT-PMI treated with Iso.
Given that β-adrenergic regulation of Ca2+ dynamics plays a key role in HF-related cardiac dysfunction, we next questioned whether NOD1 deficiency impacted isoproterenol-induced modulation of Ca2+ handling in cardiomyocytes after PMI. Figure 1D shows representative line-scan Ca2+ fluorescence images obtained after electric field stimulation at 2 Hz in all groups. Under β-adrenergic stimulation, both WT-PMI and Nod1-/--PMI cardiomyocytes exhibited increased [Ca2+]i transients (Figure 1E), augmented cell contraction (Figure 1F), and faster time decay of [Ca2+]i transients (Figure 1G), but all these changes were significantly greater in Nod1-/--PMI cells. These results reveal that, compared with WT-PMI cells, Nod1-/--PMI myocytes show a significant improvement in Ca2+ mishandling, which was mostly due to the increased amplitude of [Ca2+]i transients (Figures 1D,E) and also higher cell contraction parameters (Figure 1F). In this line, mean values of the amplitude of [Ca2+]i transients (F/F0) in cardiomyctes obtained under isoproterenol administration were 5.31 ± 0.52 (n/N = 13/5) in WT, 5.18 ± 0.32 (n/N = 14/4) in Nod1-/-, 4.18 ± 0.22 (n/N = 12/4) in WT-PMI, and 5.41 ± 0.21 (n/N = 13/4) in Nod1-/--PMI (Figures 1A,E). Thus, [Ca2+]i transients obtained in the Nod1-/--PMI group during isoproterenol perfusion were similar to those of equivalent healthy WT and Nod1-/- cardiomyocytes. Consistent with this finding, treatment of WT-PMI mice with the selective NOD1 inhibitor nodinitib-1 resulted in higher amplitude isoproterenol-induced [Ca2+]i transients in isolated cardiomyocytes compared with cells isolated from vehicle-treated mice (Supplementary Figure S1).
Next, to question whether changes in [Ca2+]i transients during β-adrenergic stimulation in PMI cardiomyocytes were related to modifications in SR-Ca2+ load, we measured caffeine-evoked [Ca2+]i transients in all groups. Figure 2A shows examples of line-scan images of caffeine-evoked [Ca2+]i transients obtained in each group. Mean values of the amplitude of caffeine-evoked [Ca2+]i transients (F/F0) in cardiomyocytes during isoproterenol exposure was 7.07 ± 0.69 (n/N = 9/3) in WT, 7.34 ± 0.93 (n/N = 10/4) in Nod1-/- (Figure 1C), 5.77 ± 0.46 (n/N = 13/4) in WT-PMI, and 7.40 ± 0.50 (n/N = 12/4) in Nod1-/--PMI (Figure 2B). Thus, the amplitude of [Ca2+]i transients triggered by caffeine was significantly higher in Nod1-/--PMI mice than in WT-PMI mice, both in the absence or the presence of isoproterenol (Figure 2B). Additionally, the high values of SR-Ca2+ load in the Nod1-/--PMI group treated with isoproterenol were similar to those found in healthy WT and Nod1-/- cells treated with isoproterenol.
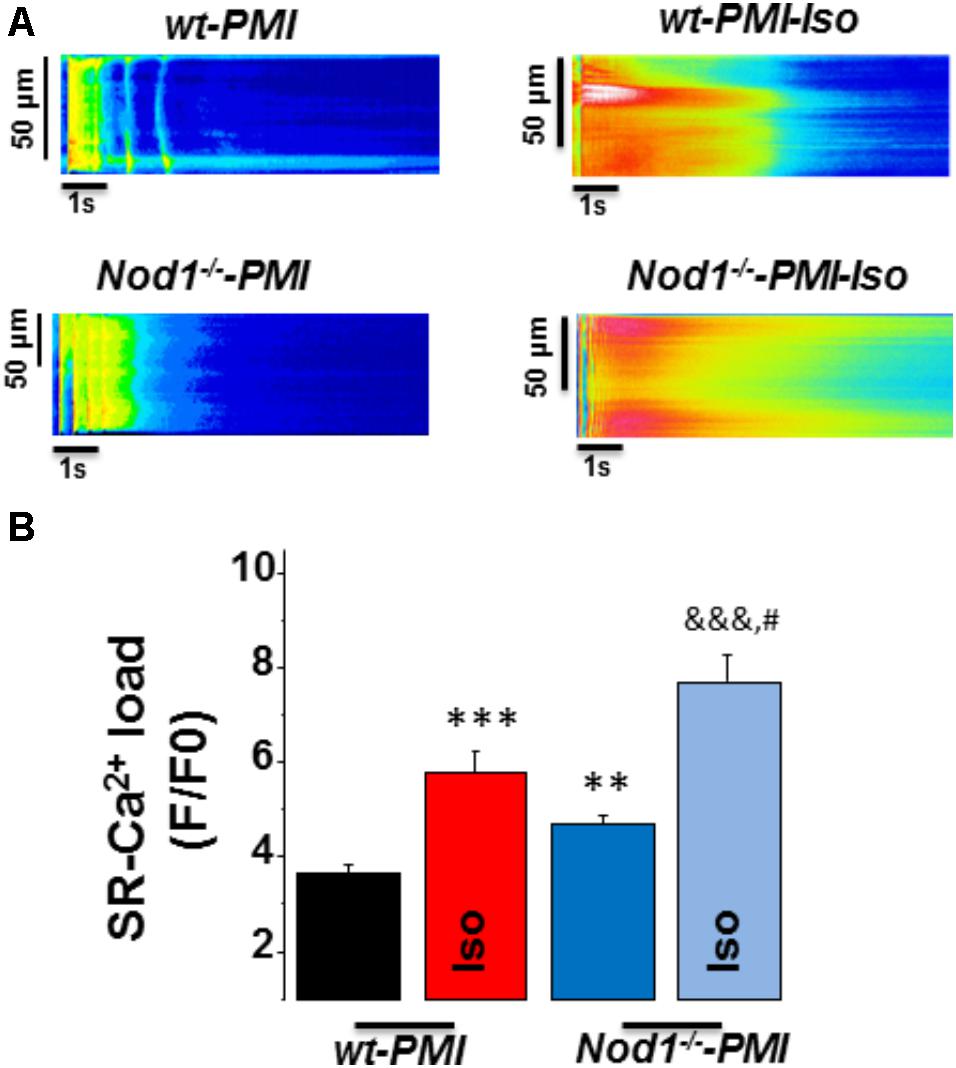
FIGURE 2. Deficiency of NOD1 increases the β-adrenergic response on SR-Ca2+ load in cardiomyocytes from failing mice. (A) Representative line-scan confocal images of caffeine-evoked [Ca2+]i transients obtained from WT-PMI and NOD1-/--PMI cardiomyocytes with and without Iso perfusion. (B) Mean values of caffeine-evoked [Ca2+]i transients amplitude (SR-Ca2+ load, F/F0) in WT-PMI (n = 23 cells/4 mice), NOD1-/--PMI (n = 19 cells/4 mice), WT-PMI (n = 13 cells/4 mice), and NOD1-/--PMI (n = 12 cells/4 mice) cardiomyocytes treated with Iso. Histograms represent the mean ± SEM: ∗∗P < 0.01, ∗∗∗P < 0.001 vs WT-PMI; &&&P < 0.001 vs NOD1-/--PMI; and #P < 0.05 vs WT-PMI cardiomyocytes treated with Iso.
Taken together, β-adrenergic stimulation induced better systolic Ca2+ release and cell contractility in Nod1-/--PMI cells than in WT-PMI cells. These effects correlated with the significant increase in the SR-Ca2+ load induced by isoproterenol in Nod1-/--PMI cells.
Deficiency of NOD1 Prevents the Increased Diastolic Ca2+ Release Induced by β-Adrenergic Stimulation in PMI Cardiomyocytes
Since SR-Ca2+ load impairment is closely related to changes in diastolic Ca2+ release, we analyzed spontaneous Ca2+ waves in quiescent cells.
Figure 3A shows a representative line-scan image of a Ca2+ wave recording from a WT-PMI myocyte. Ca2+ wave occurrence analysis (Figure 3B) revealed no statistically significant changes between WT-PMI (14.60%) and Nod1-/--PMI (10.50%) cells under basal conditions. By contrast, both groups showed a significant elevation in the Ca2+ wave rate after isoproterenol perfusion, but this was significantly higher in WT-PMI cardiomyocytes perfused with isoproterenol (Figure 3). Accordingly, the percentage of cells with Ca2+ waves during isoproterenol exposure was 34.07% in WT-PMI cells and 15.65% in Nod1-/--PMI cells (P < 0.01).
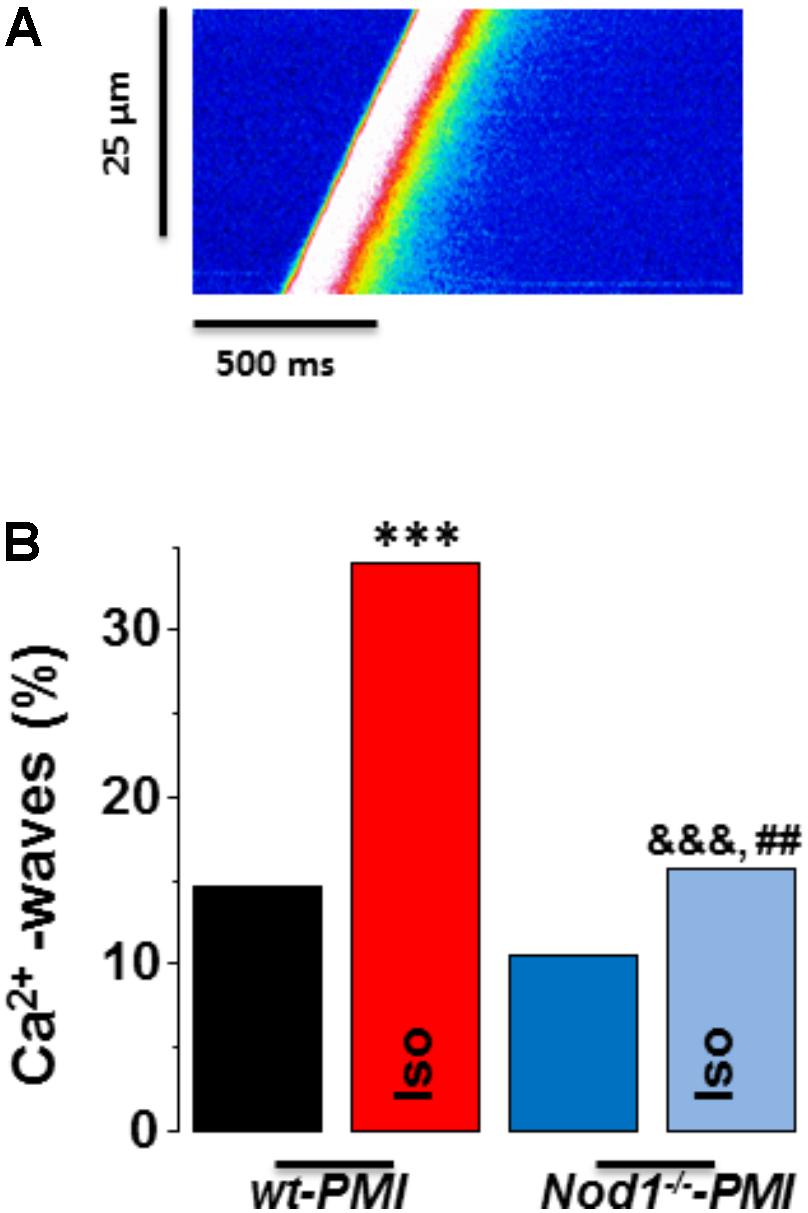
FIGURE 3. Deficiency of NOD1 prevents the increase in diastolic Ca2+ release induced by isoproterenol in cardiomyocytes from failing mice. (A) A representative line-scan image of a Ca2+ wave recording from a WT-PMI myocyte. (B) The mean data for Ca2+ wave occurrence from WT-PMI (n = 9 cells/3 mice) and NOD1-/--PMI (n = 7 cells/3 mice) cardiomyocytes with and without Iso. Histograms represent the mean ± SEM: ∗P < 0.05, ∗∗∗P < 0.001, vs WT-PMI; &P < 0.05, &&&P < 0.001 vs NOD1-/--PMI; and ##P < 0.05 vs WT-PMI cardiomyocytes treated with Iso.
These results indicate that deficiency of NOD1 prevents the increase of Ca2+ diastolic leak induced by β-adrenergic stimulation in PMI cardiomyocytes, thus providing an explanation for the improvement in the SR-Ca2+ load and the resulting better systolic Ca2+ release in Nod1-/--PMI cells under isoproterenol administration.
mRNA Levels of the β1 and β2 Adrenergic Receptors Are Elevated in Nod1-/--PMI Hearts
Analysis of the mRNA levels of β1 and β2 adrenergic receptors in hearts from WT-PMI and Nod1-/--PMI mice revealed significantly increased expression of both receptors in Nod1-/--PMI hearts (Figures 4A,B). These increased β1 and β2 adrenergic receptor levels can explain at least in part the improvement in Ca2+ regulation observed in the Nod1-/--PMI group under isoproterenol administration.
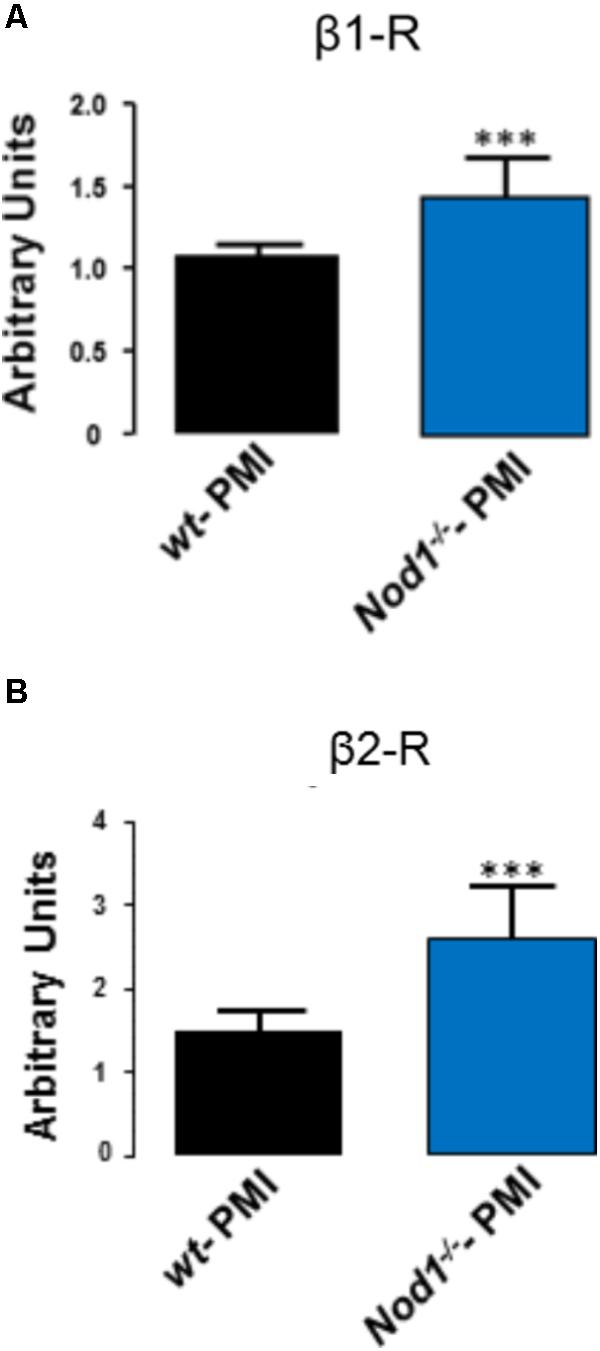
FIGURE 4. Hearts from Nod1-/--PMI mice have elevated mRNA levels of β1 and β2 adrenergic receptors. Histograms illustrate mRNA levels of β1 (A) and β2 (B) adrenergic receptors normalized to 18s RNA levels obtained in WT-PMI (N = 6) and Nod1-/--PMI hearts (N = 6). Values are expressed in arbitrary units. ∗∗∗P < 0.001 vs WT-PMI.
Selective Activation of NOD1 Compromises the β-Adrenergic Regulation of Ca2+ Handling in Wild-Type Cardiomyocytes
We next addressed whether the selective activation of NOD1 impairs the β-adrenergic regulation of Ca2+ handling in healthy cardiomyocytes. To do this, we pretreated WT cardiomyocytes for 1 h with the NOD1 ligand C12-iE-DAP (iEDAP; 20 μg/mL) and then we examined intracellular Ca2+ dynamics in the absence or presence of isoproterenol perfusion. Under isoproterenol stimulation, iE-DAP-treated cells showed reduced amplitude of [Ca2+]i transients (Figures 5A,B), cell contraction (Figure 5C), and SR-Ca2+ load (Figure 5D) compared with vehicle-treated cardiomyocytes. Treatment of cells with the inactive analog of IE-DAP, iE-Lys (20 μg/mL), resulted in a similar isoproterenol response for Ca2+ handling to vehicle-treated cells, demonstrating the selective effect of iE-DAP on Ca2+ handling under β-adrenergic stimulation (Figures 5A–D).
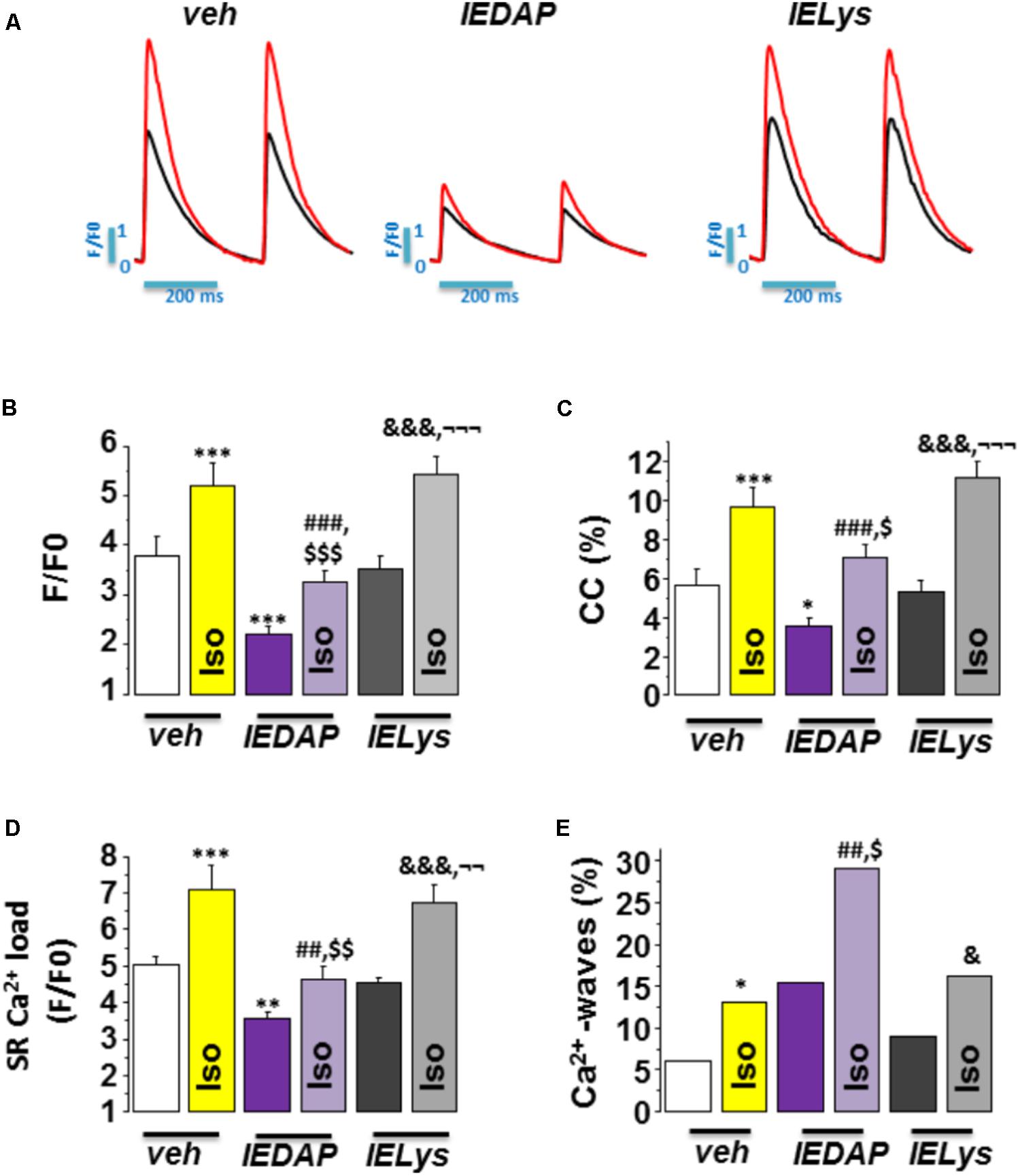
FIGURE 5. Effect of Isoproterenol on [Ca2+]i transients, SR-Ca2+ load, and diastolic Ca2+ release in wild-type cardiomyocytes treated with C12-iE-DAP. (A) Representative [Ca2+]i transients recorded in cardiomyocytes treated with vehicle (left), C12-iEDAP (20 μg/mL IEDAP, middle), and iE-LYS (20 μg/mL IELYS, right), with (red) or without (black) Iso. Mean values of (B) peak fluorescence [Ca2+]i transients, (C) cell contraction, (D) caffeine-evoked [Ca2+]i transients amplitude (SR-Ca2+ load), and (E) Ca2+ wave occurrence in cardiomyocytes treated with vehicle (veh; n = 15 cells/7 mice), C12-iE-DAP (IEDAP n = 16 cells/6 mice), or with iE-Lys (IELYS, n = 14 cells/5 mice) in the absence or the presence of Iso. Histograms represent the mean ± SEM: ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs veh; ##P < 0.01, ###P < 0.001 vs IEDAP; &P < 0.05, &&&P < 0.001 vs IELYS; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs veh plus Iso; and ¬¬P < 0.01, ¬¬¬P < 0.001 vs IEDAP
plus Iso.
Finally, we investigated whether the selective activation of NOD1 impairs diastolic Ca2+ release under β-adrenergic stimulation. As shown in Figure 5E, isoproterenol perfusion promoted a significant increase in Ca2+ waves in WT cells pretreated with iE-DAP as compared with vehicle- or iE-Lys-treated cells. Importantly, the percentage of Ca2+ waves in cells that were pretreated with iE-DAP and under isoproterenol perfusion was very similar to that found in WT-PMI cardiomyocytes under the same conditions (34.07% in WT-PMI cells and 29.20% in cells treated with iE-DAP; compare Figures 3C, 5E). Supporting these data, iE-DAP administration failed to increase the percentage of Ca2+-wave-positive Nod1-/- cardiomyocytes (data not shown).
Overall, these results indicate that NOD1 activation limits the β-adrenergic regulation of Ca2+ handling due to impairment of systolic Ca2+ release ([Ca2+]i transients) and SR-Ca2+ load, which are highly conditioned by the increased diastolic Ca2+ leak.
Discussion
Over the last decade, a large number of studies have shown that compensatory upregulation of the adrenergic nervous system response is a key factor in disease progression in chronic failing hearts. During the early stages of HF, there is a steady activation of the sympathetic nervous system to maintain cardiac output (Lymperopoulos, 2013), whereas in later stages, continuous adrenergic stimulation triggers negative feedback regulation of β-receptor activity, causing a downregulation in the number of receptors and also functional impairment, a process termed receptor desensitization (Böhm et al., 1990; Madamanchi, 2007), ultimately contributing to toxicity and worsening of cardiac outcomes.
Some innate immune mediators, such as NLRs, play an important role in the host response to cardiac damage. Among them, NOD1 is expressed and functional in the heart (Fernández-Velasco et al., 2012; Delgado et al., 2015) and is upregulated in failing hearts both in a mouse HF model and in failing human myocardium (Val-Blasco et al., 2017a). Genetic deletion of NOD1 prevents HF-related cardiac dysfunction through modulation of EC coupling, chiefly by improving systolic Ca2+ release and by maintaining Ca2+ SR load, which are both compromised in HF (Val-Blasco et al., 2017a).
Our present results show that deficiency of NOD1 does not affect the regulation of Ca2+ handling by β-adrenergic stimulation under physiological conditions, as systolic Ca2+ release and SR-Ca2+ load were similar between WT and Nod1-/- cardiomyocytes. In the failing heart, however, deficiency of NOD1 prevents the depression of [Ca2+]i transients and cell contractility due to maintained SR Ca2+ load. Accordingly, during β-adrenergic stimulation, deficiency of NOD1 leads to a significant improvement in systolic Ca2+ release, cell contraction parameters, and SR Ca2+ load in PMI cardiomyocytes, with levels mirroring those found in isoproterenol-stimulated WT and healthy Nod1-/-cells. These results are interesting given that the decreased β-adrenergic function in HF plays a key role in the compromised cardiac contraction.
Several groups have shown that other pro-inflammatory mediators including TNFα, CXR4 or IL-1β impair the β-adrenergic response and reduce cardiac contractility (Gulick et al., 1989; Finkel et al., 1992; Schulz et al., 1995; Murray and Freeman, 1996; Pyo et al., 2006), and short-term treatment of rat cardiomyocytes with the pro-inflammatory cytokines IL-1α and TNFα blocks isoproterenol-induced increases in cell contractility (Bick et al., 1997). These findings are in agreement with an early report implicating IL-1β in uncoupling β-adrenergic responses in cardiomyocytes (Gulick et al., 1989). Consistent with these data, our study shows that a selective NOD1 agonist induces a minor but significant effect on cell contractility in isoproterenol-treated cardiomyocytes. NOD1 activation also reduced the β-adrenergic modulation of systolic Ca2+ release due to reduced SR-Ca2+ load, and by increasing the diastolic Ca2+ leak. These results can be explained by the fact that NOD1 activation in cardiomyocytes induces the over-phosphorylation of RyR2, promoting the open state of the receptor and inducing Ca2+ release from the SR to the cytoplasm, increasing the diastolic Ca2+ leak (waves), and compromising the SR-Ca2+ load (Delgado et al., 2015; Val-Blasco et al., 2017a).
In many cases, increased diastolic Ca2+ release is the cause of depressed SR-Ca2+ load in HF. Our data show that during β-adrenergic stimulation, diastolic Ca2+ release is significantly increased in failing WT cells, whereas deficiency of NOD1 prevents the increase in the HF-related diastolic Ca2+ waves. These data go some way to explain the improvement in the SR-Ca2+ load and the resulting improved systolic Ca2+ release and cell contractility in Nod1-/--PMI cells under isoproterenol stimulation.
Nod1-/--PMI hearts showed physiological levels of phospho-RyR2 (Val-Blasco et al., 2017a), thus contributing to block Ca2+ diastolic leak in Nod1-/--PMI cardiomyocytes. Aberrant diastolic Ca2+ leak in many cases such as HF leads to arrhythmogenesis and cardiac dysfunction (Reiken et al., 2003; Ai et al., 2005; van Oort et al., 2010; Marks, 2013; Terentyev et al., 2014). RyR-derived Ca2+ leak can cause arrhythmias by Ca2+ waves that trigger delays after depolarizations (Lehnart et al., 2006). Pro-arrhythmogenic events are common in failing patients, particularly under β-adrenergic stimulation, and around 30–50% of these individuals die from sudden cardiac death, mostly associated with ventricular arrhythmias. Our data suggest that blocking NOD1 can be a new tool to prevent ventricular arrhythmias and cardiac dysfunction resulting from Ca2+ mishandling in HF.
In line with our results, elevated levels of the inflammatory cytokines TNFα and IL-1β have been reported in both tissue and plasma from HF patients with arrhythmias (Levine et al., 1990; Testa et al., 1996; Aukrust et al., 2004). Moreover, other inflammatory mediators such as Toll-like receptors (TLRs) have been implicated in the generation of ventricular and atrial arrhythmias (Katoh et al., 2014; Monnerat-Cahli et al., 2014; Gurses et al., 2016). Importantly, TLRs seem to play a role in the regulation of the β-adrenergic response. Accordingly, Kizaki et al. (2008) demonstrated that TLR4 stimulation induces a down-regulation of β2 receptors in macrophages (Wang et al., 2009). In this regard, our results demonstrate that hearts from Nod1-/--PMI mice have higher mRNA levels of β1 and β2 adrenergic receptors than WT-PMI hearts, suggesting a possible mechanism for the larger systolic Ca2+ release and SR Ca2+ reuptake in by Nod1-/--PMI mice. β-adrenergic receptors are activated through heterotrimeric G proteins and subsequent activation of the adenylyl cyclase, which modulates the activity of several proteins essential for EC Ca2+ coupling, such as ICaL, RyR2, and phospholamban (Mayourian et al., 2018). Therefore, the elevated β-adrenergic receptor levels in Nod1-/--PMI cells may increase the efficiency of cardiac EC coupling through the augmented function of ICaL, RyR, and SERCA, improving both systolic Ca2+ release and SR Ca2+ loading in failing hearts.
One of the most common treatments to manage β-adrenergic impairment in HF patients is the use of β-blockers. It has been demonstrated that β-blocker treatment reduces the detrimental effects of catecholamine stimulation, such as toxicity, pathological elevated heart rate, and adverse remodeling and arrhythmia development in failing patients. Improving β-adrenergic regulation of Ca2+ handling by blocking NOD1 can be a new tool to improve the cardiac function in failing hearts.
Conclusion
Our study demonstrates that deficiency of NOD1 improves the β-adrenergic modulation of Ca2+ handling in isolated cells from failing mice. Therefore, NOD1 emerges as a new potential target in the treatment of cardiac dysfunction and ventricular arrhythmias associated with HF.
Author Contributions
MF-V conceived, designed, and discussed the experiments and wrote the paper. AV-B, JN-G, MT, PP, CZ, MP, LR-R, and MG-F performed the experiments. MF-V, AV-B, JN-G, CD, GR-H, and PP analyzed the data. MF-V, LB, GR-H, and CD contributed reagents, materials, and analysis tools.
Funding
MF-V is Miguel Servet II researcher of ISCIII (MSII16/00047 Carlos III Health Institute). This work was supported by grants PI14/01078, CP15/00129, PI17/01093, and PI17/01344 from ISCIII, Fondo Europeo de Desarrollo Regional (FEDER), Ministerio de Economía y Competitividad of Spain (SAF2014-57190-R; SAF2017-84777-R), FSE, and CIBER-CV, a network funded by ISCIII.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the technical assistance of L. Martin, V. Terrón, D. Navarro, and L. Sánchez. We would like to thank Dr. Gabriel Núñez (University of Michigan, United States) for kindly providing Nod1-/- mice and Dr. Kenneth McCreath for editorial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.00702/full#supplementary-material
References
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Aukrust, P., Yndestad, A., Damås, J. K., and Gullestad, L. (2004). Inflammation and chronic heart failure-potential therapeutic role of intravenous immunoglobulin. Autoimmun. Rev. 3, 221–227. doi: 10.1016/S1568-9972(03)00103-4
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bick, R. J., Liao, J. P., King, T. W., LeMaistre, A., McMillin, J. B., and Buja, L. M. (1997). Temporal effects of cytokines on neonatal cardiac myocyte Ca2+ transients and adenylate cyclase activity. Am. J. Physiol. 272, H1937–H1944. doi: 10.1152/ajpheart.1997.272.4.H1937
Böhm, M., Ungerer, M., and Erdmann, E. (1990). Beta adrenoceptors and m-cholinoceptors in myocardium of hearts with coronary artery disease or idiopathic dilated cardiomyopathy removed at cardiac transplantation. Am. J. Cardiol. 66, 880–882. doi: 10.1016/0002-9149(90)90376-C
Brum, P. C., Hurt, C. M., Shcherbakova, O. G., Kobilka, B., and Angelotti, T. (2006). Differential targeting and function of alpha2A and alpha2C adrenergic receptor subtypes in cultured sympathetic neurons. Neuropharmacology 51, 397–413. doi: 10.1016/j.neuropharm.2006.03.032
Bullón, P., Alcocer-Gómez, E., Carrión, A. M., Marín-Aguilar, F., Garrido-Maraver, J., Román-Malo, L., et al. (2016). AMPK phosphorylation modulates pain by activation of NLRP3 Inflammasome. Antioxid. Redox Signal. 24, 157–170. doi: 10.1089/ars.2014.6120
Caruso, R., Warner, N., Inohara, N., and Núñez, G. (2014). NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908. doi: 10.1016/j.immuni.2014.12.010
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., et al. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707. doi: 10.1038/ni945
Cross, H. R., Murphy, E., and Steenbergen, C. (2002). Ca(2+) loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 283, H481–H489. doi: 10.1152/ajpheart.00790.2001
Delgado, C., Ruiz-Hurtado, G., Gómez-Hurtado, N., González-Ramos, S., Rueda, A., Benito, G., et al. (2015). NOD1, a new player in cardiac function and calcium handling. Cardiovasc. Res. 106, 375–386. doi: 10.1093/cvr/cvv118
Feldman, D. S., Carnes, C. A., Abraham, W. T., and Bristow, M. R. (2005). Mechanisms of disease: beta-adrenergic receptors-alterations in signal transduction and pharmacogenomics in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2, 475–483. doi: 10.1038/ncpcardio0309
Fernández-Velasco, M., Prieto, P., Terrón, V., Benito, G., Flores, J. M., Delgado, C., et al. (2012). NOD1 activation induces cardiac dysfunction and modulates cardiac fibrosis and cardiomyocyte apoptosis. PLoS One 7:e45260. doi: 10.1371/journal.pone.0045260
Finkel, M. S., Oddis, C. V., Jacob, T. D., Watkins, S. C., Hattler, B. G., and Simmons, R. L. (1992). Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257, 387–389. doi: 10.1126/science.1631560
Gómez, A. M., Valdivia, H. H., Cheng, H., Lederer, M. R., Santana, L. F., Cannell, M. B., et al. (1997). Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276, 800–806. doi: 10.1126/science.276.5313.800
Gordan, R., Gwathmey, J. K., and Xie, L. H. (2015). Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 7, 204–214. doi: 10.4330/wjc.v7.i4.204
Gulick, T., Chung, M. K., Pieper, S. J., Lange, L. G., and Schreiner, G. F. (1989). Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc. Natl. Acad. Sci. U.S.A. 86, 6753–6757. doi: 10.1073/pnas.86.17.6753
Gurses, K. M., Kocyigit, D., Yalcin, M. U., Canpinar, H., Yorgun, H., Sahiner, M. L., et al. (2016). Monocyte toll-like receptor expression in patients with atrial fibrillation. Am. J. Cardiol. 117, 1463–1467. doi: 10.1016/j.amjcard.2016.02.014
Heinzel, F. R., MacQuaide, N., Biesmans, L., and Sipido, K. (2011). Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J. Mol. Cell. Cardiol. 50, 390–400. doi: 10.1016/j.yjmcc.2010.11.008
Kanno, S., Nishio, H., Tanaka, T., Motomura, Y., Murata, K., Ihara, K., et al. (2015). Activation of an innate immune receptor, Nod1, accelerates atherogenesis in Apoe-/- mice. J. Immunol. 194, 773–780. doi: 10.4049/jimmunol.1302841
Katoh, S., Honda, S., Watanabe, T., Suzuki, S., Ishino, M., Kitahara, T., et al. (2014). Atrial endothelial impairment through Toll-like receptor 4 signaling causes atrial thrombogenesis. Heart Vessels 29, 263–272. doi: 10.1007/s00380-013-0369-3
Keestra-Gounder, A. M., Byndloss, M. X., Seyffert, N., Young, B. M., Chávez-Arroyo, A., Tsai, A. Y., et al. (2016). NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397. doi: 10.1038/nature17631
Kizaki, T., Izawa, T., Sakurai, T., Haga, S., Taniguchi, N., Tajiri, H., et al. (2008). Beta2-adrenergic receptor regulates Toll-like receptor-4-induced nuclear factor-kappaB activation through beta-arrestin 2. Immunology 124, 348–356. doi: 10.1111/j.1365-2567.2007.02781.x
Lehnart, S. E., Terrenoire, C., Reiken, S., Wehrens, X. H., Song, L. S., Tillman, E. J., et al. (2006). Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 103, 7906–7910. doi: 10.1073/pnas.0602133103
Levine, B., Kalman, J., Mayer, L., Fillit, H. M., and Packer, M. (1990). Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 323, 236–241. doi: 10.1056/NEJM199007263230405
Lin, L., and Knowlton, A. A. (2014). Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 100, 1–8. doi: 10.1016/j.lfs.2014.01.062
Lymperopoulos, A. (2013). Physiology and pharmacology of the cardiovascular adrenergic system. Front. Physiol. 4:240. doi: 10.3389/fphys.2013.00240
Madamanchi, A. (2007). Beta-adrenergic receptor signaling in cardiac function and heart failure. Mcgill J. Med. 10, 99–104.
Mann, D. L. (2015). Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ. Res. 116, 1254–1268. doi: 10.1161/CIRCRESAHA.116.302317
Marks, A. R. (2013). Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest. 123, 46–52. doi: 10.1172/JCI62834
Mayourian, J., Ceholski, D. K., Gonzalez, D. M., Cashman, T. J., Sahoo, S., Hajjar, R. J., et al. (2018). Physiologic, pathologic, and therapeutic paracrine modulation of cardiac excitation-contraction coupling. Circ. Res. 122, 167–183. doi: 10.1161/CIRCRESAHA.117.311589
Monnerat, G., Alarcón, M. L., Vasconcellos, L. R., Hochman-Mendez, C., Brasil, G., Bassani, R. A., et al. (2016). Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 7:13344. doi: 10.1038/ncomms13344
Monnerat-Cahli, G., Alonso, H., Gallego, M., Alarcón, M. L., Bassani, R. A., Casis, O., et al. (2014). Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. J. Mol. Cell. Cardiol. 76, 116–125. doi: 10.1016/j.yjmcc.2014.08.012
Murray, D. R., and Freeman, G. L. (1996). Tumor necrosis factor-alpha induces a biphasic effect on myocardial contractility in conscious dogs. Circ. Res. 78, 154–160. doi: 10.1161/01.RES.78.1.154
Park, J. H., Kim, Y. G., McDonald, C., Kanneganti, T. D., Hasegawa, M., Body-Malapel, M., et al. (2007). RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 178, 2380–2386. doi: 10.4049/jimmunol.178.4.2380
Prieto, P., Vallejo-Cremades, M. T., Benito, G., González-Peramato, P., Francés, D., Agra, N., et al. (2014). NOD1 receptor is up-regulated in diabetic human and murine myocardium. Clin. Sci. 127, 665–677. doi: 10.1042/CS20140180
Pyo, R. T., Sui, J., Dhume, A., Palomeque, J., Blaxall, B. C., Diaz, G., et al. (2006). CXCR4 modulates contractility in adult cardiac myocytes. J. Mol. Cell. Cardiol. 41, 834–844. doi: 10.1016/j.yjmcc.2006.08.008
Reiken, S., Gaburjakova, M., Guatimosim, S., Gomez, A. M., D’Armiento, J., Burkhoff, D., et al. (2003). Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 278, 444–453. doi: 10.1074/jbc.M207028200
Ruiz-Hurtado, G., Li, L., Fernández-Velasco, M., Rueda, A., Lefebvre, F., Wang, Y., et al. (2015). Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J. Gen. Physiol. 146, 295–306. doi: 10.1085/jgp.201511366
Schulz, R., Panas, D. L., Catena, R., Moncada, S., Olley, P. M., and Lopaschuk, G. D. (1995). The role of nitric oxide in cardiac depression induced by interleukin-1 beta and tumour necrosis factor-alpha. Br. J. Pharmacol. 114, 27–34. doi: 10.1111/j.1476-5381.1995.tb14901.x
Terentyev, D., Rees, C. M., Li, W., Cooper, L. L., Jindal, H. K., Peng, X., et al. (2014). Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ. Res. 115, 919–928. doi: 10.1161/CIRCRESAHA.115.305146
Testa, M., Yeh, M., Lee, P., Fanelli, R., Loperfido, F., Berman, J. W., et al. (1996). Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 28, 964–971. doi: 10.1016/S0735-1097(96)00268-9
Val-Blasco, A., Piedras, M. J., Ruiz-Hurtado, G., Suarez, N., Prieto, P., Gonzalez-Ramos, S., et al. (2017a). Role of NOD1 in heart failure progression via regulation of Ca2+ handling. J. Am. Coll. Cardiol. 69, 423–433. doi: 10.1016/j.jacc.2016.10.073
Val-Blasco, A., Prieto, P., Gonzalez-Ramos, S., Benito, G., Vallejo-Cremades, M. T., Pacheco, I., et al. (2017b). NOD1 activation in cardiac fibroblasts induces myocardial fibrosis in a murine model of type 2 diabetes. Biochem. J. 474, 399–410. doi: 10.1042/BCJ20160556
van Oort, R. J., McCauley, M. D., Dixit, S. S., Pereira, L., Yang, Y., Respress, J. L., et al. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298
Venetucci, L. A., Trafford, A. W., O’Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Keywords: β-adrenergic response, heart failure, NOD1, Ca2+ handling, innate immune system
Citation: Val-Blasco A, Navarro-García JA, Tamayo M, Piedras MJ, Prieto P, Delgado C, Ruiz-Hurtado G, Rozas-Romero L, Gil-Fernández M, Zaragoza C, Boscá L and Fernández-Velasco M (2018) Deficiency of NOD1 Improves the β-Adrenergic Modulation of Ca2+ Handling in a Mouse Model of Heart Failure. Front. Physiol. 9:702. doi: 10.3389/fphys.2018.00702
Received: 01 March 2018; Accepted: 22 May 2018;
Published: 14 June 2018.
Edited by:
Tarik Smani, Universidad de Sevilla, SpainReviewed by:
Marcelo Catalan, Arturo Prat University, ChileJian Wu, University of Southern California, United States
Copyright © 2018 Val-Blasco, Navarro-García, Tamayo, Piedras, Prieto, Delgado, Ruiz-Hurtado, Rozas-Romero, Gil-Fernández, Zaragoza, Boscá and Fernández-Velasco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Fernández-Velasco, mvelasco@iib.uam.es; maria.fernandez@idipaz.es
†CIBER-CV from ISCIII
‡These authors have contributed equally to this work.