Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics
 Vladimir N. Uversky
Vladimir N. Uversky- 1Department of Molecular Medicine, USF Health Byrd Alzheimer's Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 2Laboratory of New Methods in Biology, Institute for Biological Instrumentation, Russian Academy of Sciences, Pushchino, Russia
Recognition of the natural abundance and functional importance of intrinsically disordered proteins (IDPs), and protein hybrids that contain both intrinsically disordered protein regions (IDPRs) and ordered regions, is changing protein science. IDPs and IDPRs, i.e., functional proteins and protein regions without unique structures, can often be found in all organisms, and typically play vital roles in various biological processes. Disorder-based functionality complements the functions of ordered proteins and domains. However, by virtue of their existence, IDPs/IDPRs, which are characterized by remarkable conformational flexibility and structural plasticity, break multiple rules established over the years to explain structure, folding, and functionality of well-folded proteins with unique structures. Despite the general belief that unique biological functions of proteins require unique 3D-structures (which dominated protein science for more than a century), structure-less IDPs/IDPRs are functional, being able to engage in biological activities and perform impossible tricks that are highly unlikely for ordered proteins. With their exceptional spatio-temporal heterogeneity and high conformational flexibility, IDPs/IDPRs represent complex systems that act at the edge of chaos and are specifically tunable by various means. In this article, some of the wonders of intrinsic disorder are discussed as illustrations of their “mysterious” (meta)physics.
Introduction
Despite the classical structure-function paradigm (which is typically envisioned as the “lock-and-key” model, in which a unique biological function of a protein is defined by its specific, highly structured state determined by the amino acid sequence [1]) that dominated scientific minds for more than a century, many protein functions do not require a unique structure. The carriers of such structure-independent functions are intrinsically disordered proteins (IDPs) or protein hybrids that contain both intrinsically disordered protein regions (IDPRs) and ordered domains. IDPs and IDPRs can often be found in nature, so much so that noticeable amounts of both can be found in all proteomes of organisms in all kingdoms of life, and all viral proteomes analyzed so far [2–8]. Computational research clearly indicated not only that IDPs/IDPRs are commonly present in all living organisms [3, 5, 6, 8–23], but also established that the abundance of disorder increases proportionally with organism complexity [3, 5, 6, 8, 24]. According to various computational analyses, the presumed portion of sequences with predicted long IDPRs (≥30 residues) is roughly the same in bacteria and archaea, but is drastically higher in eukaryota [2, 6, 8, 17, 25]. The latter estimation is based on the increased roles of cellular signaling in eukaryotes that often relies on IDPs/IDPRs [7, 26–32]. Furthermore, only a small fraction of proteins whose crystal structures are known and recorded in the Protein Data Bank (PDB) do not possess any disorder [33, 34]. Furthermore, the presence of structures in PDB does not rule out disordered proteins, since, for example, an IDP/IDPR can undergo induced folding at binding to a specific partner (another protein, nucleic acid, small molecules, etc.), which will make it well-ordered.
The goal of this article is to reflect on some of the fascinating features of IDPs/IDPRs and to show that their ability to break rules is extending far beyond the aforementioned disconnection between structure and function. To address this, sections below discuss some of the “wonders” of intrinsic disorder rooted in very peculiar and mysterious (meta)physics of intrinsic disorder. Several of the concepts of strange biophysics of intrinsic disorder were already considered in earlier publications [35–37], and will be briefly discussed here. More attention will be paid to some newer disorder-related concepts and paradigms.
Although one could consider the use of terms such as “mysterious” and “metaphysics” or “improbable” and “impossible” for the description of the “strangeness” of IDPs/IDPRs as excessive language, the majority of structural and functional features of IDPs/IDPRs described in this article do not align with the traditional viewpoints of a classical structural biologist or a protein physicist trained within the frames of “lock-and-key” model. As many researchers currently working on IDPs originally came from these or similar backgrounds, it seems reasonable to assume that at least some of them will share such an emotional take on the “strangeness” of IDPs. Furthermore, the use of these terms does not only represent a reflection of the “strangeness” of IDPs, but also echoes a psychological barrier that one needs to pass while moving from the classical structure-function paradigm (where one gene encodes for a unique amino acid sequence that folds into a unique 3D structure responsible for the unique biological function of a given protein) to the structure-function continuum concept (where a single gene can produce multiple proteins with many functions).
Mysterious (Meta)Physics of Intrinsic Disorder
Prevalence of Exclusivity/Exceptionality
The discovery of the wide penetrance of IDPs/IDPRs into all proteomes of all living organisms and viruses came as a big surprise to the vast majority of structural biologists. In fact, although biologically active proteins without unique structures were systematically observed years before the discovery of the protein intrinsic disorder phenomenon, they were originally considered as rare exceptions from the “lock-and-key” rule and were therefore mostly ignored [4]. However, almost immediately after recognition of the fact that IDPs/IDPRs constitute a new class of biologically active proteins, it became clear that, instead of being exceptionally unique and rare exceptions, these proteins/regions are highly prevalent in nature, thereby defining the “prevalence of exclusivity/exceptionality” or the “exceptional abundance of exceptions” concept. In fact, in a brief historical overview of the temporal developments of understanding of the natural abundance of IDPs/IDPRs, it was pointed that this process was very rapid [38]. The very first collection of IDPRs included 67 disordered regions longer than seven residues found in 61 PDB proteins, that were grouped into sets of short, medium, and long IDPRs, containing 7–21, 22–44, and 45 or more residues, respectively [39]. Soon after, a set of 91 IDPs with extended disorder (or natively unfolded proteins) was assembled [40]. Consequently, the list of experimentally validated IDPs was extended first to 157 proteins [41] and later to more than 200 proteins containing at least one IDPR of 30 consecutive residues or longer [7, 29, 30]. A recent literature analysis showed that there are approximately 1,150 non-redundant proteins in the list of validated IDPSs [42].
The development of tools for reliable prediction of intrinsic disorder predisposition based on sequence data has presented the opportunity to evaluate the commonness of IDPs/IDPRs at the proteome level. Already, the first application of one of the such tools to the Swiss Protein Database produced a very “big catch”: long IDPRs of at least 40 consecutive residues were predicted to be present in over 15,000 proteins, and particularly high disorder scores were found for more than 1,000 proteins [43]. Analysis of proteins encoded by 31 genomes from 3 kingdoms of life showed that eukaryotes typically have a higher disorder score than either archaea or prokaryotes, since 52–67% of eukaryotic proteins have long IDPRs as compared to 26–51 and 16–45% proteins with such long IDPRs in archaea and bacteria, respectively [8, 24]. The natural abundance of intrinsic disorder was later supported by multiple comprehensive computational studies [3, 6, 17, 24, 44, 45], which are illustrated by Figure 1, where the correlation between the intrinsic disorder content and proteome size is shown for 3,484 viral, archaeal, bacterial, and eukaryotic species [3]. All these data clearly indicated that IDPs/IDPRs are not rare exceptions, but are rather a rule.
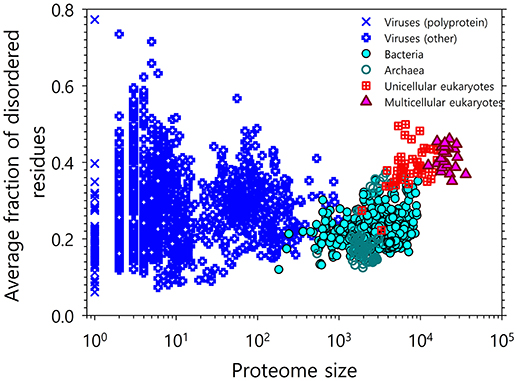
Figure 1. Correlation between the intrinsic disorder content and proteome size for 3,484 species from viruses, archaea, bacteria, and eukaryotes. Each symbol indicates a species. There are six groups of species in total: viruses expressing one polyprotein precursor (open blue circles), other viruses (blue crosses), bacteria (green circles), archaea (red open circles), unicellular eukaryotes (brown open squares), and multicellular eukaryotes (yellow triangles). Each viral polyprotein was analyzed as a single polypeptide chain, without parsing it into the individual proteins before predictions. The proteome size is the number of proteins in the proteome of that species shown in the log base. The average fraction of disordered residues is calculated by averaging the fraction of disordered residues of each sequence over the all sequences of that species. Disorder prediction is evaluated by PONDR® VSL2B. This plot is based on the data published in [3].
Finally, although enzymes were mostly excluded from the IDP/IDPR realm due to the structural requirements for catalysis for a long time, a recent analysis revealed that the lengths of IDPRs in enzymes are similar to the lengths of IDPRs found in non-enzymes, have unique functions that parallel functions of IDPRs in non-enzymes, and are present in quantities that are precisely related to the enzyme type and length [46]. Furthermore, it was also shown that eukaryotic enzymes typically contained more disorder than their prokaryotic counterparts, suggesting that at the proteome level, disorder is correlated with functional and organismic complexity, which also holds true for enzymes [46]. Based on these observations it is possible to conclude that in the protein world, disorder is everywhere.
Complexity of Simplicity
On multiple levels, ranging from amino acid composition to amino acid sequence, to structural content and spatial organization, ordered proteins, and domains are obviously much more complex than IDPs and IDPRs. It seems that this global, multi-level simplicity of IDPs/IDPRs is rooted in the peculiarities of their amino acid sequences, which are known to be depleted in order-promoting residues (Trp, Cys, Tyr, Ile, Phe, Val, Asn, and Leu) and enriched in disorder-promoting residues (Arg, Pro, Gln, Gly, Glu, Ser, Ala, and Lys) (see Figure 2A) [4, 49, 51–56] and commonly contain repeats [57, 58]. Therefore, IDPs/IDPRs are characterized by the reduced informational content of their amino acid sequences, and their amino acid alphabet is decreased in comparison with the alphabet utilized in the amino acid sequences of ordered domains and proteins [59]. Furthermore, since they do not need to gain any sort of ordered structure, IDPs/IDPRs have a noticeably greater sequence space than that of ordered proteins [35]. Therefore, the sequence simplicity associated with intrinsic disorder is translated into an immensely expanded sequence space, which, in its turn, gives rise to the enormous structural complexity of these proteins and regions [59]. The expanded sequence space of IDPs/IDPRs with extended disorder is illustrated by Figure 2B, where the mouse proteome is presented in a form of the “modified” charge-hydropathy plot [59]. Comparing the areas accessible to extended IDPs and soluble compact proteins clearly indicates that the sequence space accessible to compact proteins is significantly smaller than the space accessible to IDPs. This difference is even bigger in reality, since the area attributed to compact proteins also technically includes compact IDPs [24, 40].
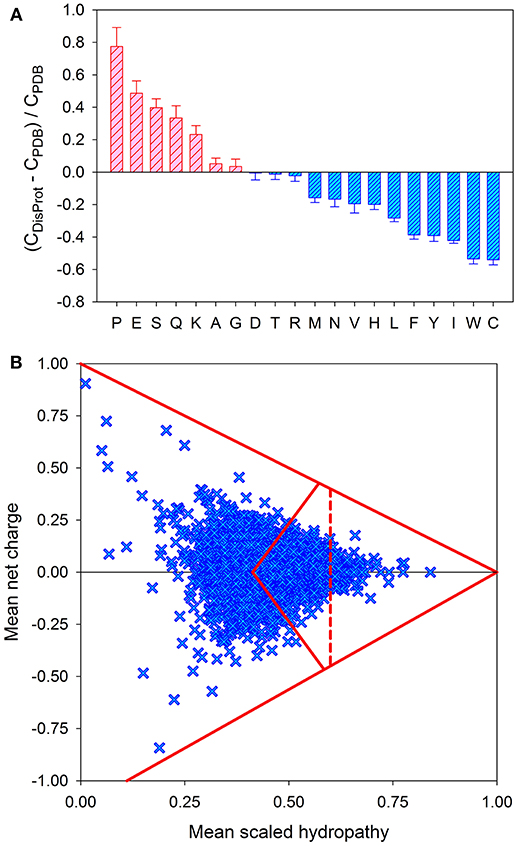
Figure 2. Peculiarities of the amino acid sequences of IDPs. (A) Amino acid determinants defining structural and functional differences between the ordered and intrinsically disordered proteins. Fractional difference in the amino acid composition (compositional profile) between the typical IDPs from the DisProt database [47] and a set of completely ordered proteins [48] calculated for each amino acid residue. The fractional difference was evaluated as (CDisProt-CPDB)/CPDB, where CDisProt is the content of a given amino acid in a DisProt database [47] and CPDB is the corresponding content in the dataset of fully ordered proteins from PDB select 25 [48]. Positive bars correspond to residues found more abundantly in IDPs, whereas negative bars show residues in which IDPs are depleted. Amino acid types were ranked according to their decreasing disorder-promoting potential [49]. (B) Evaluation of the charge-hydropathy space available for mouse proteins. In this plot, each protein is represented by a single point calculated based on its mean net charge and mean net hydropathy. This plot differs from the traditional charge-hydropathy plot (CH-plot) [40] by showing both positive and negative mean net charge values instead of the absolute mean net charges. The area accessible to sequences encoding ordered and disordered proteins are defined by several boundaries: (i) the known boundary separating compact proteins and extended IDPs (<R> = 2.785 <H> − 1.151, where <R> and <H> correspond to the absolute mean charge and mean hydropathy, respectively [40]); (ii) the mirror image of this boundary (<R> = −2.785 <H> + 1.151, where <R> and <H> correspond to the mean charge and mean hydropathy, respectively), which is included to consider negatively charged proteins; (iii) two boundaries showing logical limits of the CH-space (<R> = −1.125 + 1.125 <H> and <R> = 1.00–<H>), evaluated for a series of hypothetical polypeptides containing different proportions of Ile (which is, according to the Kyte and Doolittle scale, the most hydrophobic residue with the normalized hydropathy of 1 [50] and a negatively charged Asp (which is characterized by the normalized Kyte and Doolittle hydropathy of 0.1111 [50]) or a positively charged Arg (which is characterized by the normalized Kyte and Doolittle hydropathy of 0.0 [50]); and (iv) the boundary line within the area corresponding to compact proteins to separate soluble and membrane proteins, since proteins whose hydropathy in the normalized Kyte and Doolittle scale exceeds 0.7 are unlikely to be soluble.
Similarly, despite (or due to) their inability to fold into unique structures in isolation and despite (or due to) their simplified spatial organization and globally reduced structural content, IDPs are characterized by exceptional spatiotemporal heterogeneity, where parts of an IDP are dis/ordered to varying degrees at one moment in time, but can change state at a future point in time [35, 59]. Therefore, IDPs are not homogeneous, but represent a very complex mixture of a broad variety of partially foldable, potentially foldable, differently foldable, or completely unfoldable segments [35, 59]. This behavior of an IDP as a highly frustrated system that does not possess a singular folded state is reflected in its free energy landscape, which is relatively flat and simple, lacks a deep energy minimum that can be found within ordered proteins, and appears as a “hilly plateau,” where the “hills” correspond to forbidden conformations [60–62]. Such a simplified and flattened energy landscape is extremely sensitive to different environmental changes that can modify the landscape in several different ways, lowering some energy minima while raising some energy barriers. This explains the conformational plasticity of an IDP/IDPR, its extreme sensitivity to changes in the environment, its ability to interact with multiple different partners, and consequently to fold in different ways [35]. Therefore, this exceptional spatio-temporal heterogeneity of IDPs/IDPRs, where functional elements have varying degrees of disorder and have unique responses to environmental changes, is directly related the astonishing multifunctionality of disordered proteins that are able to control, regulate, interact with, as well as be controlled and regulated by, a plethora of structurally unrelated partners. In summary, the multilevel structural, spatiotemporal, and sequential simplicity of IDPs/IDPRs and their simplified energy landscapes define the structural heterogeneity and exceptional functional complexity of disordered proteins and regions.
Charges Rule
As was already indicated, the (non)foldability of proteins is encoded in their amino acid sequences [4, 7, 40, 56, 63, 64]. In fact, similar to ordered (foldable) proteins, whose unique biologically active structures are formed based on the information included into their amino acid sequences, the ability to not fold and still be functional in the absence of unique structures is also encoded in the specific features of the amino acid sequences of IDPs/IDPRs. For extended IDPs, these features include the presence of multiple uncompensated charged groups (which are usually negative) that define the high net charges of many IDPs at neutral pH and their extreme pI values [40, 65–67], combined with a low content of hydrophobic amino acid residues [40, 66, 67]. These features suggest that a combination of relatively high net charge with low mean hydropathy serves as a prerequisite for the absence of compact structure in proteins under physiological conditions [40]. Furthermore, consideration of just two global physico-chemical parameters of a polypeptide chain, namely its absolute mean charge and mean hydropathy, is often sufficient to differentiate between extended (non-compact) IDPs and structured (compact) proteins [24, 40]. This observation is illustrated by Figure 3A, where a charge-hydropathy phase space of ordered and extendedly disordered proteins is shown. Here, blue squares and red circles correspond to compact (ordered) proteins and IDPs with extended disorder (coil-like and pre-molten globule-like), respectively. The area accessible to sequences encoding extended IDPs is depicted as a light pink pentagon, while the area for ordered (compact) proteins is shown as a light cyan triangle. These areas are based on two boundaries: the boundary showing the logical limits of the CH-space (<R> = 1.125–1.125 <H> [35]), and the known boundary separating compact proteins and extended IDPs (<R> = 2.785 <H> − 1.151, where <R> and <H> correspond to the absolute mean chare and mean hydropathy, respectively [40]). As was pointed out earlier, the former boundary can be defined based on the analysis of a series of hypothetical polypeptides that have varying amounts of a hydrophobic residue isoleucine. Using the Kyte and Doolittle hydropathy scale, this residue is defined as the most hydrophobic residue with a normalized hydropathy of 1 [50], and a charge residue aspartic acid characterized by the normalized Kyte and Doolittle hydropathy of 0.1111 [50]. From a physical viewpoint, the lack of compact structure in a protein with low hydrophobicity and high net charge makes perfect sense, since strong charge-charge repulsion in such a polypeptide is not compensated by strong enough hydrophobic attraction [4, 35, 40, 63, 64].
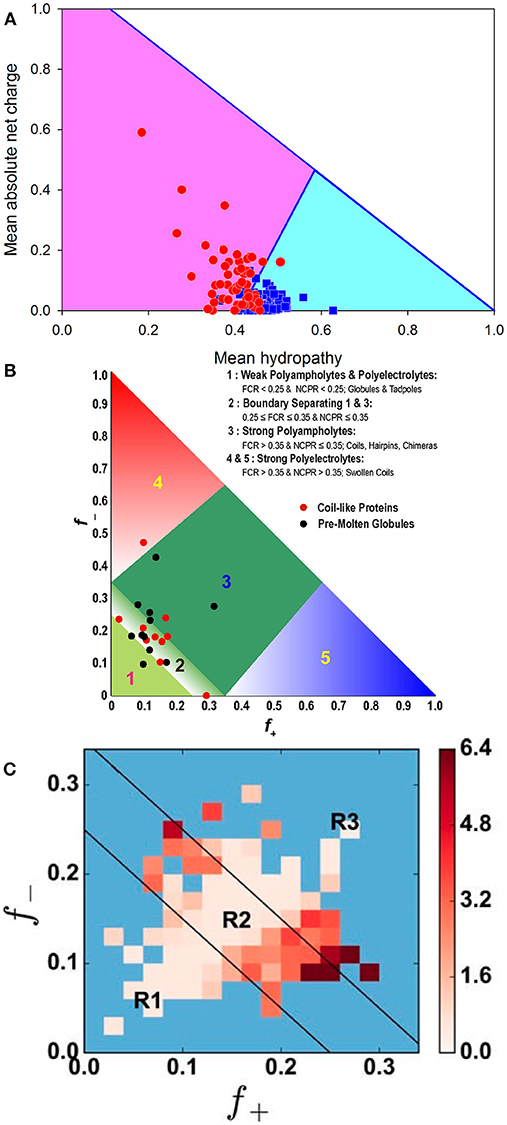
Figure 3. Illustrating roles of charges in conformational control of IDPs. (A) Evaluation of the charge-hydropathy space available for compact proteins (blue squares) and extended IDPs (red circles). The area accessible to sequences encoding compact proteins is shown as a light cyan triangle, whereas the area accessible to sequences encoding IDPs is depicted as light pink pentagon. These two areas are defined by two boundaries, the known boundary separating compact proteins and extended IDPs (<R> = 2.785 <H>-1.151, where <R> and <H> correspond to the absolute mean chare and mean hydropathy, respectively [40]), and the boundary showing logical limits of the CH-space (<R> = 1.125–1.125 <H>). Reprinted from Uversky [35]. (B) Diagram-of-states for IDPs based on the peculiarities of the distribution of charged residues within the amino acid sequences. Region 1 corresponds to either weak polyampholytes or weak polyelectrolytes that form globule or tadpole-like conformations. Region 3 corresponds to strong polyampholytes that form distinctly non-globular conformations, such as coil-like, hairpin-like, or hybrids. A boundary region labeled two separates regions 1 and 3, and the conformations within this region are likely to represent a continuum of possibilities between the types of conformations adopted by sequences in regions 1 and 3. Sequences with compositions corresponding to regions 4 and 5 are strong polyelectrolytes with FCR >0.35 and NCPR >0.3. These sequences are expected to sample coil-like conformations. The legend summarizes statistics for different regions based on sequences drawn from the DisProt database. The figure includes annotation by properties of sequences that have been designated as being “coils” or “pre-molten-globules” based on measurements of their hydrodynamic radii [64]. This plot was adopted from [68]. (C) Predicted heat map of IDPs from DisProt that shows the average x for DisProt proteins with positive (f+) and negative charge fractions (f−) corresponding to their bin location. Darker colors represent bins characterized by higher x(f+, f−) values. Blue bins correspond to locations within the f+–f− phase space that contain fewer than two proteins [69]. Black lines show boundaries between the R1, R2, and R3 regions in the diagram-of-states [68]. Reprinted from Firman and Ghosh [69].
Besides net charge per residue (NCPR), which obviously represents a rather coarse description of a polypeptide chain, the level of compaction of a disordered protein can also be affected by the fraction of charged residues (FCR), as well as the distribution patterns of charges. This is due to the fact that a large fraction of IDPs/IDPRs are polyampholytes containing both positively and negatively charged residues [47, 68, 70]. In other words, the fractions of both positive (f+) and negative charges (f−) can be used for conformational classification IDPs, generating a specific diagram-of-states [68, 71] and can even be used for gaining some knowledge on the IDP functionality [72]. These two parameters, f+ and f−, can also be used to define both NCPR and FCR as follows: NCPR = | f+ – f − | and FCR = (f+ + f−) [68]. It was emphasized that, depending on their content of charged residues, polyampholytes can be classified as weak (FCR < 0.3) or strong (FCR ≥ 0.3), and can also be neutral (NCPR ~ 0) or have a net charge [68]. Furthermore, conformational properties of polyampholytic IDPs can be determined based on the FCR and sequence-specific distributions of oppositely charged residues [68]. Here, the charge distribution can be denoted as κ, with 0 ≤ κ ≤ 1, where a κ value of 0 corresponds to a sequence with a uniform charge distribution, whereas increasing κ values represent less uniformly-distributed charges in a protein, leading to the appearance of patches with high FCR values containing large fractions of similarly charged residues. Proteins with lower κ values are akin to self-avoiding random walks (i.e., have random coil-like structural organization), while proteins with higher κ values have more collapsed, hairpin-like structures [68]. More generally, Figure 3B shows that based on this diagram-of-states, conformations of IDPs can be classified into five specific groups: (R1) weak polyampholytes and polyelectrolytes, (R2) the region between R1 and R3, (R3) strong polyampholytes, and (R4 and R5) strong polyelectrolytes, associated with typical conformations of IDPs/IDPRs [68, 71]. Importantly, a complementary analysis of the peculiarities of distribution of hydrophobic residues within the amino acid sequences showed that, unlike folded and soluble globular proteins, IDPs/IDPRs are characterized by more diversified distributions of hydrophobicity, and that this diversity (or deviation of distribution from uniformity) can be measured by Gini index, which is a tool to estimate distribution uniformity, or by Shannon entropy [73].
Furthermore, it was recently established that even consideration of f+, f−, and FCR is not sufficient for accurate evaluation of the hydrodynamic dimensions of IDPs, since structurally characterized IDPs with similar f+ and f− values have a broad variation of their dimensions [69]. Therefore, when the same charge composition of a polypeptide chain is retained, the actual positioning of the charged residues within the amino acid sequence (or sequence charge decoration metric) may have a profound effect on the polypeptide conformation triggering the coil to globule transition [69, 74]. In fact, when the R1, R2, and R3 regions of the aforementioned diagram-of-states were subdivided into a smaller grid spacing of f+ and f− with Δf+ = Δf− = 0.02, and when the corresponding average values of the chain expansion parameter x at T = 300 K were computed for all proteins within a given bin, and such bin-dependent protein-averaged size x(f+, f−) was plotted as a heat map, the resulting plot (see Figure 3C) clearly showed the presence of significant inter-bin variations in protein hydrodynamic dimensions [69]. Here, darker colors represent bins characterized by higher x(f+, f−) values. Figure 3C shows that the diagonal of the heat map contains proteins with small x values corresponding to the situation where f+ ≈ f−, whereas bins located away from the diagonal contain proteins characterized by much larger volumes due to their higher net charges [69]. These observations indicate that the structural properties and conformational behavior of an IDP are controlled not only by the actual number of positively and negatively charged residues (i.e., by its net charge per residue) or the peculiarities of global distribution of charges within the polypeptide chain, but also by the distinctiveness of charge placement in the primary sequence (i.e., sequence charge decoration metric). Furthermore, in light of this sensitivity of a polypeptide chain to the peculiarities of placement of charged residues within the amino acid sequence, the structure- and function-modulating roles of posttranslational modifications (PTMs) leading to the sequence-specific changes in the charge state of a protein become evident [69]. Curiously, it was also indicated that IDPs have specific “hot spots,” modifying which can cause maximal changes in protein conformation and regulate transitions between the extended and collapsed states and vice versa [69].
Born to be Wild: Extended IDPs/IDPRs do Not Behave as the “Well-Bred Kids” (Ordered Globular Proteins) do
Due to the specific peculiarities of their amino acid sequences (high net charge and low hydrophobicity), conformationally, extended IDPs do not follow the rules established for ordered globular proteins and domains that are known to lose specific structure and biological function at various denaturing conditions (e.g., extreme temperatures or pH). In fact, extended IDPs, such as α-synuclein [75], caldesmon 636-771 fragment [76], phosphodiesterase γ-subunit [77], the receptor extracellular domain of nerve growth factor [78], αs-casein [79], and several other IDPs are known to gain more ordered structure based on temperature, and are usually more structured at higher temperatures (due to the enhancement of the hydrophobic interaction), while being more disordered at lower temperatures [35, 80].
Similarly, many extended IDPs, such as pig calpastatin domain I [81], naturally occurring human peptide LL-37 [82], prothymosin α [83], histidine rich protein II [84], human α-synuclein [75], and several others, undergo partial folding at extremely acidic and/or alkaline conditions as a result of neutral pH minimizing their large net charge, leading to the minimization of the intramolecular charge/charge repulsion and thereby permitting hydrophobicity-driven folding to a more ordered/collapsed conformation [80].
Obviously, these considerations should be protracted from IDPs to the extended IDPRs commonly present in various hybrid proteins. Furthermore, it is likely that the peculiarities of the amino acid composition of a given IDP/IDPR or even the particularity of distribution of charges in its sequence could define the degree of IDP/IDPR responsiveness to changes in its environment. In other words, differently disordered parts of protein molecules can differently respond to a similar environmental cue, providing another level of complexity for these highly heterogeneous entities.
Structural Heterogeneity and Multifunctionality of IDPs
A very specific feature of IDPs/IDRs is the fact that these biologically active proteins/regions fail to form specific 3D structures and exist as highly dynamic structural ensembles, either at the secondary or at the tertiary level [7, 27, 32, 40, 56, 85, 86]. It is recognized now that IDPs/IDRs may possess extended disorder (where intrinsic disorder is present in a form of random coil or pre-molten globule), and collapsed disorder (where the intrinsic disorder is present in a molten globular form) [7, 86, 87]. Furthermore, proteins may also have regions of semi-disorder; i.e., fragments that have the same predicted probability to be ordered or disordered, and that play vital roles in protein aggregation and participate in protein-protein interactions, being able to undergo binding-induced folding [88]. More generally, IDPs/IDPRs are characterized by a very complex and heterogeneous spatiotemporal structural organization, possessing foldons (independent foldable units of a protein), unfoldons (ordered regions that have to undergo an order-to-disorder transition to become functional), semi-foldons (regions that are always in a semi-folded form), inducible foldons (disordered regions that can fold at least in part due to the interaction with binding partners), and non-foldons (non-foldable protein regions), [35, 89–91]. This intricate structural, mosaic-like “anatomy” of IDPs/IDPRs defines their unique molecular “physiology,” where differently (dis)ordered structural elements might have well-defined and specific functions [92], thereby defining a possibility of a protein molecule to be multifunctional and be involved in regulation of, interaction with, and be controlled by multiple structurally unrelated partners.
Overall, IDPs/IDPRs are complex systems with sophisticated structurally and functionally heterogeneous organization. They are uniquely placed at the core of the structure-function continuum concept, where instead of the classical (but heavily oversimplified) “one gene–one protein–one structure–one function” view, the actual protein structure-function relationship is described by the more convoluted “one-gene–many-proteins–many-functions” model [92, 93]. Therefore, intrinsic disorder represents a cornerstone of the proteoform concept [94] introduced to explain an important observation that the complexity of a biological system is primarily determined not by the genome size, but by its proteome size [95]. In fact, the number of functionally different proteins is known to dramatically exceed the number of protein-encoding genes (e.g., the human genome is approaching 20,700 genes [96], but the actual number of functionally different proteins is in the range of a few million). The increased size of a functional proteome over a corresponding genome is determined by multiple factors, ranging from the allelic variations and various pre-translational mechanisms affecting genes (e.g., production of numerous mRNA variants by the alternative splicing and mRNA editing) to numerous chemical changes induced in proteins by various post-translational modifications (PTMs) [97–101]. As a result, a single gene can efficiently encode for a set of distinct protein molecules, giving rise to the aforementioned proteoform concept [94]. Furthermore, in addition various genetic and non-genetic factors increasing chemical variability of a polypeptide chain, the protein structural diversity can be further increased by intrinsic disorder and functioning [93].
I Like to Move It, Move It: Structural and Functional Dynamics of IDPs
It is clear that internal dynamics is crucial for the biological activity of many (if not all) proteins, indicating that protein functionality requires at least some degree of conformational flexibility and structural dynamics [102–106]. In fact, even in ordered proteins, functional dynamics involve movements of not only individual amino acid residues or groups of amino acids in an active site relative to each, but also displacements of entire domains, and these function-related movements that happen in a wide spread of time-scales, ranging from femtoseconds to seconds, are required for the facilitation of catalytic activity [103, 104]. Furthermore, enzymes abundantly (in fact, as amply as non-enzymes) contain lengthy IDPRs with functions similar to those of IDPRs in non-enzymes (i.e., recognition, regulation, signaling, and control) [46]. Finally, it was pointed out that enzymes might possess evolutionary conserved dynamic traits, where, for example, RNase homologs with different biological functions, such as angiogenesis, anti-pathogenicity, immunosuppressivity, and highly efficient RNA cleavage, are not only grouped into evolutionarily distinct functional sub-families, but also show remarkable intra-subfamily conservation of dynamical properties evaluated by the quantitative characterization of the slowest modes of corresponding molecular dynamics (MD) simulations [107] that are not directly related to the conserved or semi-conserved features in the corresponding disorder profiles showing distribution of the per-residue disorder predisposition of these proteins [108]. These observations suggest that even for ordered globular proteins, in addition to the well-recognized structural conservation, which is a functionally important feature, functionality may also be linked to the evolutionarily conserved dynamical traits [107], which are only partially related to the conserved intrinsic disorder predisposition [108]. Therefore, such conservation of structural dynamics represents an important constituent of the evolutionary conservation of a functional protein fold [107, 108].
However, IDPs/IDPRs are different from ordered proteins and domains in terms of the amplitudes, time scales, and spatio-temporal coordination of their functional and structural dynamics. As a matter of fact, it is recognized now that the full structural description of IDPs/IDPRs is only feasible via the utilization of experimental [e.g., NMR-based chemical shifts (CSs)], residual dipolar couplings (RDCs), 15N R2 relaxation rates, paramagnetic relaxation enhancement (PRE) distance restraints, J-couplings, pulsed field gradient (PFG)-derived Rh values, and 1H-15N heteronuclear nuclear Overhauser effects (hetNOEs), complemented by SAXS [109, 110] and computational ensemble approaches (such as molecular dynamics simulations, or generation of a large number of conformations and selection of a subset that fits the experimental constrains [60, 111–113] that describe the structure of an IDP/IDPR as an ensemble of conformations and that can generate detailed information on short- and long-range contacts and backbone flexibility [114]. Because of their heteropolymeric nature, IDPs/IDPRs are rarely completely unstructured (i.e., behave as polymeric random coils) but often contain regions of transiently populated elements of secondary structure (so-called pre-structured motifs or PreSMos), which commonly serve as targets for their interaction partners to facilitate binding in a kinetically efficient manner [91, 92, 115–122]. Because of the recognition of the crucial importance of structural dynamics and flexibility for the functionality of IDPs and IDPRs, this field of protein science is currently undergoing a phase of explosive development, which precludes comprehensive consideration of this subject in a short overview article. Therefore, interested readers should look for dedicated review articles, of which there are many.
Interaction Professionals: A Multitude of Intrinsic Disorder-Based Binding Modes
Over the years, understanding protein interactability has undergone a dramatic shift from a highly static to a very dynamic view. Everything started with the “lock-and-key” model of protein functionality that implied the crucial need of a spatially and temporally fixed structure of an active site [1, 123]. In this model, functional protein interaction with its partners resembles fitting a specifically shaped key into a unique lock, where only the substrate with the correct shape/size (key) is able to fit into the specifically shaped active site (key-hole) of the particular protein (lock), and where a high degree of geometrical precision in the substrate-protein interface complementarity defines their specificity of interaction [124]. Later, to account for changes in protein structure during the interaction process, the “induced fit” model was proposed [102], where a protein possesses some partial flexibility, and substrate binding induces some changes in the shape of the active site [125]. The next step was recognition of the presence of the “preexisting population, conformational selection, and population shift” mechanism [126–130], implying that, due to some flexibility, the native protein is better described as a conformational ensemble rather than as a unique rigid conformation [131], and further suggesting that interaction with a binding partner is based on the selection of the most binding-compatible conformation from this ensemble, thereby leading to shifting conformational equilibrium toward such an interaction-prone member of the conformational ensemble [131]. Finally, consideration of intrinsic disorder has opened seemingly unlimited opportunities of how a protein can feel and react to its environment, and how it can interact with its binding partners [91, 118].
In fact, despite their lack of stable structure, many IDPs/IDPRs are promiscuous binders that are never-nude, being always-complexed, invariably interacting with various partners via multiple binding scenarios [4, 7, 35, 117], and forming static, semi-static, and fuzzy or dynamic complexes [91, 118]. IDPs/IDPRs are capable of semi-static and dynamic polyvalent interactions [119], where one protein's multiple binding sites are simultaneously bound to another protein's receptors [132]. Some examples of such behaviors include bivalent and polybivalent scaffolds formed by two monovalent IDPs via interaction with a dimeric protein with two symmetrical binding sites or by self-association [133], IDP/IDPR wrapping around the binding partner [118], or highly dynamic shuffle complexes, where several binding sites of one protein are involved in concomitant, non-cooperative, tri-partite interaction with binding sites of another protein, thereby functioning similarly to holding a hot potato [134].
Many IDPs/IDPRs can fold to their partners at binding [29, 30, 91, 92, 115–135], with the degree of such binding-induced folding being different in various systems, thereby forming complexes with broad structural and functional heterogeneity [91, 118]. IDPs/IDPRs can act as molecular glue or mortar [29] by filling the gaps and cracks between the structural elements of a binding partner [136]. They can also serve as molecular epoxy, where partners undergo at least partial binding-induced folding at the complex formation [137–139]. Intrinsic disorder forms the basis of the dynamic “on-off” switch-type interactions commonly found in signaling networks, where IDPs/IDPRs bind partners with low affinity and high specificity [4, 140, 141]. Many IDPs/IDPRs serve as morphing shape-changers that are able to differently fold as a result of binding to different partners [56, 87, 135, 142–144], with the binding region of such a morphing IDP adopting completely different structures in the rigidified assemblies formed by the binding to divergent partners [28, 116, 117, 145, 146].
Some IDPs/IDPRs form fuzzy complexes, where significant disorder remains at least outside the binding interface [76, 147–153]. In such fuzzy complexes, under-folded bound regions are used as alternative contacts for specific partners and can also be fine-tuned by PTMs [147]. Furthermore, preservation of intrinsic disorder within dynamic signaling complexes provides disorder carriers with the capability to be engaged in entropic chain activities, which rely entirely on an extended random-coil conformation of a polypeptide that is able to carry out its function while staying flexible [4, 154]. Such dynamic signaling complexes with entropic chain activities represent stochastic machines [155] that contain structured domains connected by long flexible linkers [146, 155–157], and which work by stochastic, uncoordinated movements of the long disordered linkers (rather than by coordinated conformational changes) that move constantly and chaotically, but eventually enable productive functionality [155].
Although in the multitude of the aforementioned binding scenarios protein functionality seems to commonly originate from disorder-to-order transitions, many functions of ordered proteins were shown to rely on local or even global functional unfolding [89]. This regulated unfolding [158] or conditional disorder has an induced nature [159] and transient character [160], and may be initiated by several environmental factors, ranging from changes in temperature, pH, mechanical force, redox potential, or light exposure, to specific interactions with ligands, membranes, nucleic acids, or other proteins, to the release of autoinhibition, to various posttranslational modifications [89]. It was pointed out that such ordered proteins with dormant disorder serve as an important illustration of the global importance of intrinsic disorder for protein function.
Recently, another important property of IDPs/IDPRs pertaining to their foldability-interactability relationship was uncovered, namely the ability of IDPs/IDPRs to form tight complexes (with the affinity approaching picomolar levels [161]) without gaining any ordered structure and retaining long-range flexibility and highly dynamic character [161–163]. Formation of such extremely disordered complexes with ultrahigh affinity was described for oppositely charged proteins, and was proposed to rely on long-range electrostatic attraction between the charge-rich regions of the polypeptide chains, instead of being dependent on some structurally defined binding sites or some specific interactions between individual residues [161].
Therefore, IDPs/IDPRs clearly act as interaction specialists with an astonishing arsenal of binding modes, utilizing which can lead to the formation of a broad variety of complexes with remarkable structural and functional diversification.
Emergent Behavior of IDPs/IDPRs
Being a complex, “edge of the chaos” system, IDPs/IDPRs are characterized by emergent behavior based on the intricate self-organization processes, leading to the appearance of unanticipated novel structures, patterns, and properties [35]. One of the well-studied examples of such self-organized emergent behavior is given by the spatiotemporal oscillations of the Min protein system (MinD, MinC, and MinE) that moves from pole to pole of the rod-shaped Escherichia coli cells, and which is related to the spatial regulation of the positioning of the cytokinetic Z ring that determines the division plane at the middle of the cell [164, 165]. Min system oscillates in vivo with the intrinsic wavelength comparable to the size of the E. coli cell [166]. This is illustrated by Figure 4A representing a model of the pole-to-pole oscillations of these proteins in a rod-shaped system with a length of 3.8 μm [167]. Oscillations in the Min system originates from the ATP-dependent interactions of Min proteins with each other and with the cytoplasmic membrane [164], where the MinC/MinD oscillations between the ends of the cell are regulated by MinE acting as the “Tarzan of the jungle” while interacting with MinD [169]. A molecular mechanism of action of Min system is illustrated by Figure 4B, showing that the major regulator of oscillations, the MinE homodimer, is present in two functional forms, either a latent 6β-stranded conformation that diffuses in the cytoplasm or an active 4β-stranded form bound to the membrane and MinD [168]. The latent 6β-stranded form itself exists in two rapidly interconverting states, where the cryptic membrane-targeting sequences (MTSs) present at the N-tails of each protomer are dynamically associated with the β-sheet leading to the closed and open forms, with the open form being able to reversibly interact with the membrane via the released MTSs [168]. If the loop region connecting MTS with the remaining part of MinE in the membrane-bound open form of the 6β-stranded conformation encounters the membrane-bound MinD dimer, this sensor region undergoes a disorder-to-order transition, and a nascent α-helix is formed, which then propagates into the β1-strand leading to the dissociation of this region from the MinE dimer interface. In the resulting 4β-stranded form, this new long α-helix protrudes from the surface of MinE dimer and is further stabilized by binding to the dimeric interface of MinD (see Figure 4C). Formation of the MinE-MinD complex leads to the stimulation of the MinD ATPase activity, ATP hydrolysis, MinD dimer dissociation, and the release of the MinE dimer. These released 4β-stranded active forms of MinE are either handed off to another membrane-bound MinD dimer, or dissociate from the membrane and fold back to the 6β-stranded structure, where the six-stranded β-sheet is sandwiched between the MTS on one side, and a coiled coil formed by two long antiparallel α-helices on the opposite side [170]. The probability of MinE to be handed off to the neighboring MinD dimer is rather high due to the high MinD density on the membrane [168, 169]. Figure 4D represents the PONDR® FIT-generated disorder profile of the E. coli MinE and shows that this protein is predicted to possess a noticeable amount of intrinsic disorder, especially in its N-tail containing the aforementioned MTS and conformational switch. This observation indicates that the intrinsically disordered nature of this region might be related to its capability to undergo structural transitions required for MinD binding [35].
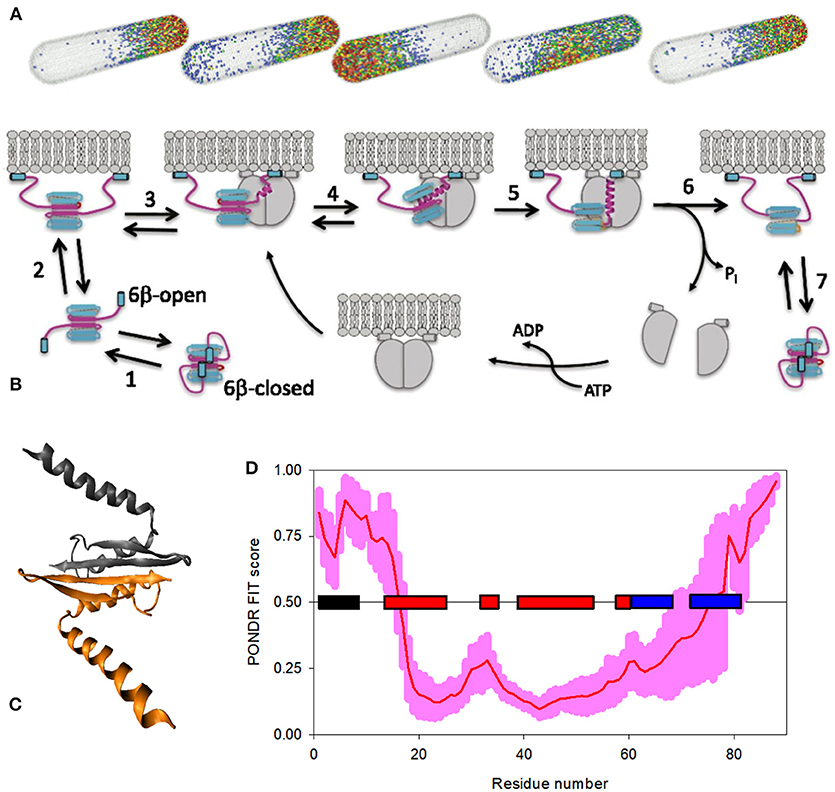
Figure 4. Bacterial Min proteins, as an illustration of a system with disorder-based emergent behavior. (A) A model showing the pole-to-pole oscillations of the members of MinE system in a rod-shaped structure with the length of 3.8 μm [167]. Modified with permission from [167]. (B) Model describing activation of MinE by interaction with MinD [168]. Here, the latent 6β-stranded form of MinE exists in two conformations, closed and open, which are in the rapid exchange (step 1). Open form can reversibly interact with the membrane via its MTSs (step 2). Once the loop region connecting MTS with the remaining MinE body encounters MinD, it undergoes a disorder-to-order transition, forming a nascent α-helix (step 3). Next, the long α-helix is formed by conversion of the β1 strand into a helical state, resulting in the creation of an active 4β-stranded form caused by the dissociation of this N-terminal region from the MinE dimer interface (step 4). The subsequent stabilization of this contact α-helix is achieved by its binding to the MinD dimeric interface, leading to the formation of MinE-MinD complex (step 5), leading to the stimulation of the MinD ATPase activity, ATP hydrolysis, and the release and dissociation of MinD dimer (step 6). If the MinE does not encounter another MinD, it spontaneously returns to the latent form (step 7). Therefore, following the dissociation of MinD, the fate of MinE depends on two competing reactions: it is either handed off to another MinD, or dissociates from the membrane and snaps back to the 6β-stranded structure. A higher density of MinD on the membrane favors the former [168, 169]. (C) Crystal structure of the MinD-bound MinE dimer from E. coli (PDB ID: 3R9J). (D) PONDR® FIT-generated disorder profile in the E. coli MinE protein. The light pink shadow shows the distribution of statistical errors of the PONDR® FIT prediction. MTS and the elements of MinE secondary structure, α-helices, and β-strands, are correspondingly shown as black, red, and blue bars located in the middle of the plot.
The aforementioned oscillating behavior of Min system on bacterial and artificial membranes can be reproduced by MinD and MinE via MinD-driven recruitment of MinE to the membrane [171–173]. In fact, on the supported lipid bilayers in vitro, these Min proteins self-organize to form mesoscale patterns of traveling protein surface waves emerging from the repetitive binding-detaching cycles of proteins to the membrane [174, 175]. Besides the Min system, prokaryotes contain several other members of the WAKA protein family (Walker A cytomotive ATPase; also knows as ParA), which also possess oscillatory behavior and are related to regulation of development, spatial regulation of cell division, and segregation of chromosomes and plasmids [176–179]. For example, the intracellular localization of E.coli plasmids carrying par2 locus is determined by the interplay of the DNA binding protein ParB, the oscillating ATPase ParA, and the specific cis-acting DNA regions to which ParB binds [179]. Here, in the presence of ParB and aforementioned cis-acting DNA regions, ParA is able to form oscillating spiral-shaped structures, whereas in the absence of ParB and DNA, ParA was engaged in the formation of stationary helices that were extended from one end of the nucleoid to the other [179]. Furthermore, liquid-liquid phase separation-driven formation of various proteinaceous membrane-less organelles (PMLOs), which are commonly found in both prokaryotic and eukaryotic cells, formation of large protein clusters on the membrane surface, and liquid-gel phase transitions might represent other examples of the emergent behavior of intrinsically disordered proteins. These three scenarios are briefly considered below.
Liquids in Liquid
On the other side of the spectrum of disorder-based protein complexes described above are proteinaceous membrane-less organelles (PMLOs), also known as non-membranous cytoplasmic/nucleoplasmic granules, or cellular/nuclear micro-domains, or intracellular/ intranuclear bodies, that are commonly found in the nucleus and cytoplasm of various cells [180–184]. The dimensions of PMLOs are cell size–dependent, and they contain proteins and RNA (and/or DNA) [185]. These very large (detectable by light microscope), highly dynamic (but stable), and liquid-like assemblages are formed via the intracellular liquid-liquid demixing phase separations [186–189] and originate due to the colocalization of molecules at high concentrations within a small cellular or nuclear micro-domain [190, 191], leading to the liquid-liquid phase transitions (LLPTs) or the intracellular liquid-liquid demixing phase separation [185, 186, 188]. Since PMLOs are not covered by the membranes, their components are involved in direct contact and exchange with the nucleoplasm or cytoplasm [190, 191]. Furthermore, PMLOs are characterized by a liquid-like behavior, being capable of dripping, wetting, and relaxing into spherical structures upon fusion [192–195]. These liquid-droplet phases of the nucleoplasm/cytoplasm [192–197] possess relatively low intrinsic density and viscosity that are comparable to those of the cytoplasm or nucleoplasm [198, 199].
Formation of PMLOs may be initiated by a number of factors, including changes in the concentrations of specific small molecules or salts, changes in the concentration of proteins undergoing phase separation, changes in the pH and/or temperature of the solution, changes in osmolarity, by the binding of these proteins to some definite partners, by various PTMs and alternative splicing of the phase-forming proteins, or by changes in other environmental conditions that affect the protein-protein or protein-nucleic acid interactions [185, 188, 200–202]. PMLOs, of which are many [181, 182] (see Figure 5), represent an intricate form of the disorder-based protein complexes [92, 182, 183, 187, 204, 205], which serve as important illustrations of emerging behavior, and which are highly dynamic in nature and are typically formed without any noticeable structural changes in the proteins undergoing LLPTs [206]. The structural integrity and biogenesis of PMLOs are both exclusively determined by protein–protein, protein–RNA, and/or protein–DNA interactions [207, 208], and the process of PMLO formation is highly controlled, completely reversible, and strongly condition-dependent [181, 182].
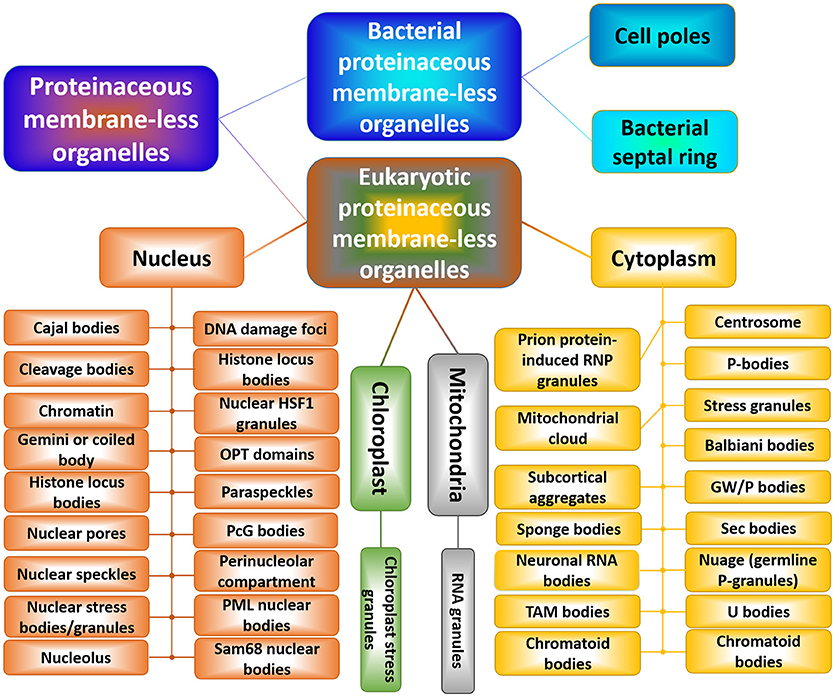
Figure 5. Schematic representation of the horde of cytoplasmic, nuclear, mitochondrial, and chloroplast PMLOs in eukaryotes and bacterial PMLOs. This figure was adopted with permission from Zaslavsky and Uversky [203].
Proteins capable of biological phase separation are characterized by structural/sequence modularity, invariably contain high levels of intrinsic disorder, and as a result of their highly disordered nature, are able to participate in weak multivalent interactions [92, 182, 183, 204, 205]. These weak multivalent interactions range from the heterologous electrostatic attraction between the oppositely charged biological polymers, such as oppositely charged proteins or positively charged nucleic acids and proteins, to homologous interactions of the same protein molecules containing repetitive donor and acceptor domains/regions (e.g., multiple stretches of positively- and negatively-charged residues) needed for the multivalent binding [181, 182]. In addition to specific environmental cues, the PMLO biogenesis is regulated via various PTMs and alternative splicing of the phase-forming proteins [188, 209]. Since many of the PMLO resident proteins are IDPs, and since the formation of all the PMLOs analyzed so far relies on IDPs/IDPRs, it is clear that intrinsic disorder is needed for PMLO biogenesis [188, 210].
Reversible Hydrogels
In addition to liquid-liquid phase transitions, some proteins can (at least in vitro) undergo liquid-gel phase separation leading to the formation of hydrogels [211]. Here, instead of liquid-like PMLOs, the resulting hydrogels cannot flow under steady-state conditions [212–214], likely because they are composed of uniformly polymerized amyloid-like fibers [213]. Importantly, fibers found in these hydrogels are different from the pathological fibrils associated with numerous human diseases in at least two ways. In fact, hydrogels are highly dynamic systems that can be reversibly formed and disassembled in the response to some environmental cue (e.g., addition of specific small molecule or phosphorylation of a protein undergoing phase separation), and they are formed via heterotypic polymerization [213]. This is in stark contrast to pathological amyloids and amyloid-like fibrils, which are characterized by an extremely high conformational stability (once formed, they almost never disintegrate), and formation of which typically represents a homotypic polymerization reaction that specifically engages molecules of the same protein. Importantly, similar to the LLPT, the dynamic liquid-gel phase separation depends on multivalent interactions between proteins with low complexity (LC) domains, many of which are known to be intrinsically disordered. Examples of systems undergoing dynamic liquid-hydrogel phase separation include heterotypic polymerization of the LC domain of the fused in sarcoma (FUS) RNA-binding protein with RNA [213]; polymerization of mutant FUS forms associated with amyotrophic lateral sclerosis [215]; RNA-dependent hydrogel formation of the LC domains of RBM3, hnRNPA1, hnRNPA2, CIRBP, and the yeast Sup35 protein [213, 216]; RNA-dependent hydrogel formation of the LC domains of Ewings sarcoma (EWS) and TAF15 proteins [212]; and Phe and Gly (FG)-rich repeats of nucleoporins, e.g., yeast nucleoporin Nsp1p [214].
Micrometer-Scale Signaling Zones at the Membrane Surface
Although the aforementioned liquid-liquid and liquid-gel phase transitions were described in three-dimensional solutions, it was pointed out that the dynamic interactions between the multivalent cytoplasmic tails of transmembrane proteins and their multivalent binding partners can trigger the formation of large (at least micron-sized) two-dimensional protein clusters on the membrane surface [217]. This possibility was illustrated by the system that included the phosphorylated cytoplasmic domain of Nephrin and its intracellular targets, Nck and N-WASP [217]. Although in a three-dimensional solution, these three proteins phase separated into dynamic micron sized liquid droplets when critical protein concentration (that depended on the valency and affinity of interacting species) in solution was achieved [197], attachment of phosphorylated Nephrin to supported lipid bilayers of DOPC in the presence of Nck and N-WASP resulted in the formation at membranes of the micron-sized concentrated puncta containing all three proteins [217]. Furthermore, these phase-separated two-dimensional protein clusters were able to successfully promote actin filament assembly via the Arp2/3 complex recruited to the membrane through binding N-WASP, and were themselves remodeled by the resultant filament network [217]. These observations suggest that the multivalent protein interactions leading to phase separation can be responsible for regulation and control of some signaling pathways via generation of spatially organized micron-scale protein clusters [217].
These observations indicated that multivalency-induced polymerization and phase separation can occur in three-dimensional solutions and in two-dimensional systems. Importantly, computational analysis revealed that all three members of this system contain high levels of intrinsic disorder. In fact, more than 60% of residues in the C-terminal cytoplasmic tail of human Nephrin (PMID: O60500, amino acids 1077–1241) are predicted to be disordered. In one study, a rat neural Wiskott-Aldrich syndrome protein (N-WASP, PMID: O08816) construct containing residues 183–193 fused to 273–501 was predicted to be completely disordered. Finally, more than 35% of human cytoplasmic protein NCK1 (PMID: P16333) with C139S, C232A, C266S, and C340S mutations are predicted to be disordered as well.
Stability of Instability
A typical globular/ordered protein is known to have a unique 3D-structure, which is determined by its amino acid sequence [218] and stabilized by a set of specific non-covalent interactions (conformational forces) of different physico-chemical nature, such as electrostatic interactions, hydrophobic interactions, hydrogen bonds, and van der Waals interactions. This unique structure can be destroyed by a variety of conditions [219–222], resulting in a random coil-like conformation at the most denaturing conditions (e.g., high concentrations of strong denaturants, such as guanidinium chloride or urea). The unfolding of a typical globular protein represent a highly cooperative process described by a sharp sigmoidal curve, and represents a case of the intramolecular all-or-none transition. As was already discussed, highly biased amino acid sequences of IDPs/IDPRs define some unusual conformational responses to changes in their environment (e.g., they partially fold at some extreme conditions, such as high temperature or high/low pH). Also, the denaturant-induced unfolding of molten globular IDPs (i.e., compact disordered proteins with high secondary structure content) is described by a shallow sigmoidal curve representing a low cooperativity transition [80, 223], whereas denaturant-induced unfolding of IDPs with extended disorder and low content of residual structure (e.g., native pre-molten globules and native coils) typically represent non-cooperative monotonous feature-less curves, indicating that IDPs/IDPRs are characterized by low conformational stability [80]. This structural instability of IDPs/IDPRs is further supported by their well-known exceptional sensitivity to proteolytic degradation [224–233].
Although IDPs/IDPRs lack stable well-folded 3D-structures, are typified by non-cooperative unfolding behavior, and possess high proteolytic sensitivity, they are characterized by exceptional endurance, being able to sustain exposure to extremely harsh environmental conditions, keep their functionality under such extreme conditions, or almost instantaneously regain functional state after returning to normal conditions [35, 37]. This ability of IDPs/IDPRs to be unharmed by prolonged exposure to harsh environments is obviously due to their lack of ordered structure, since one cannot break what's already broken [83].
Tunable Chaos
Being complex biological systems which are seemingly positioned at the edge of chaos, IDPs are not completely random entities, since they evolved to have some adjustable, controllable, regulable, tunable, and, oftentimes, very specific properties required for their biological functions. Furthermore, nature itself is using various means (such as different posttranslational modifications, mutations, alternative splicing, and interaction with numerous binding partners of different physico-chemical nature) to tune functional properties of IDPs [91]. The complexity of intrinsic disorder is further enhanced by the fact that different parts of an IDP are not always completely independent, but can “feel” and respond to various neighboring parts. As a result, different regions of an IDP can undergo unintentional induced changes that represent a kind of side-effect of the desired changes of the target regions.
Concluding Remarks
Modern protein science is reshaped by the discovery of natural abundance of functional IDPs and hybrid proteins with functional IDPRs. These proteins and regions are very different from ordered proteins and domains at multiple levels. IDPs/IDPRs have recognizable amino acid sequences with multiple biases, which makes them easily predictable. They are characterized by the exceptional spatio-temporal heterogeneity, which is related to their remarkable multifunctionality. From the viewpoint of order-based functionality, IDPs/IDPRs represent a complete disaster. They contradict to the basic logics of the “lock-and-key”-centered protein functionality and break multiple rules devised by the researchers studying structure, folding, and functions of ordered proteins. Their characteristic features, such as intricate conformational behavior, strong and multifaceted response to the environmental cues, exceptional structural and functional complexity, extreme tunability, ability to gain some obscure and highly diversified “poses” at binding partners, ability to fold differently at interaction with different binding partners, or only partially fold at interaction with their partners preserving noticeable levels of flexibility or even bind tightly without any binding-induced folding, and their emergent behavior, all are rooted in some specific peculiarities of their amino acid sequences. An accurate description of the multilevel complexity of IDPs/IDPRs requires the development of new rules, which are also needed for solving various disorder-related mysteries. Future research will shed more light on these highly abundant, exceptionally important, but still poorly understood members of the protein world.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am thankful to Alexey Uversky for careful reading and editing of this manuscript.
References
1. Fischer E. Einfluss der configuration auf die wirkung der enzyme. Ber Dt Chem Ges. (1894) 27:2985–93.
2. Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, Wang K, et al. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci. (2015) 72:137–51. doi: 10.1007/s00018-014-1661-9
3. Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3,500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. (2012) 30:137–49. doi: 10.1080/07391102.2012.675145
4. Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta (2010) 1804:1231–64. doi: 10.1016/j.bbapap.2010.01.017
5. Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. (2010) 2010:568068. doi: 10.1155/2010/568068
6. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. (2004) 337:635–45. doi: 10.1016/j.jmb.2004.02.002
7. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, et al. Intrinsically disordered protein. J Mol Graph Model. (2001) 19:26–59. doi: 10.1016/S1093-3263(00)00138-8
8. Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. (2000) 11:161–71.
9. Kahali B, Ghosh TC. Disorderness in Escherichia coli proteome: perception of folding fidelity and protein-protein interactions. J Biomol Struct Dyn. (2013) 31:472–6. doi: 10.1080/07391102.2012.706071
10. Di Domenico T, Walsh I, Tosatto SC. Analysis and consensus of currently available intrinsic protein disorder annotation sources in the MobiDB database. BMC Bioinform. (2013) 14(Suppl. 7):S3. doi: 10.1186/1471-2105-14-S7-S3
11. Pancsa R, Tompa P. Structural disorder in eukaryotes. PLoS ONE (2012) 7:e34687. doi: 10.1371/journal.pone.0034687
12. Midic U, Obradovic Z. Intrinsic disorder in putative protein sequences. Proteome Sci. (2012) 10(Suppl. 1):S19. doi: 10.1186/1477-5956-10-S1-S19
13. Korneta I, Bujnicki JM. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput Biol. (2012) 8:e1002641. doi: 10.1371/journal.pcbi.1002641
14. Hegyi H, Tompa P. Increased structural disorder of proteins encoded on human sex chromosomes. Mol Biosyst. (2012) 8:229–36. doi: 10.1039/c1mb05285c
15. Schad E, Tompa P, Hegyi H. The relationship between proteome size, structural disorder and organism complexity. Genome Biol. (2011) 12:R120. doi: 10.1186/gb-2011-12-12-r120
16. Dyson HJ. Expanding the proteome: disordered and alternatively folded proteins. Q Rev Biophys. (2011) 44:467–518. doi: 10.1017/S0033583511000060
17. Xue B, Williams RW, Oldfield CJ, Dunker AK, Uversky VN. Archaic chaos: intrinsically disordered proteins in Archaea. BMC Syst Biol. (2010) 4(Suppl. 1):S1. doi: 10.1186/1752-0509-4-S1-S1
18. Tompa P, Kalmar L. Power law distribution defines structural disorder as a structural element directly linked with function. J Mol Biol. (2010) 403:346–50. doi: 10.1016/j.jmb.2010.07.044
19. Pentony MM, Jones DT. Modularity of intrinsic disorder in the human proteome. Proteins (2010) 78:212–21. doi: 10.1002/prot.22504
20. Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. (2009) 34:53–9. doi: 10.1016/j.tibs.2008.10.009
21. Shimizu K, Toh H. Interaction between intrinsically disordered proteins frequently occurs in a human protein-protein interaction network. J Mol Biol. (2009) 392:1253–65. doi: 10.1016/j.jmb.2009.07.088
22. Krasowski MD, Reschly EJ, Ekins S. Intrinsic disorder in nuclear hormone receptors. J Proteome Res. (2008) 7:4359–72. doi: 10.1021/pr8003024
23. Tompa P, Dosztanyi Z, Simon I. Prevalent structural disorder in E. coli and S cerevisiae proteomes J Proteome Res. (2006) 5:1996–2000. doi: 10.1021/pr0600881
24. Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry (2005) 44:1989–2000. doi: 10.1021/bi047993o
25. Na I, Redmon D, Kopa M, Qin Y, Xue B, Uversky VN. Ordered disorder of the astrocytic dystrophin-associated protein complex in the norm and pathology. PLoS ONE (2013) 8:e73476. doi: 10.1371/journal.pone.0073476
26. Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. (2005) 6:197–208. doi: 10.1038/nrm1589
27. Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. (2002) 27:527–33. doi: 10.1016/S0968-0004(02)02169-2
28. Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. (2002) 12:54–60. doi: 10.1016/S0959-440X(02)00289-0
29. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry (2002) 41:6573–82. doi: 10.1021/bi012159+
30. Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. (2002) 62:25–49. doi: 10.1016/S0065-3233(02)62004-2
31. Dunker AK, Obradovic Z. The protein trinity – linking function and disorder. Nat Biotechnol. (2001) 19:805–6. doi: 10.1038/nbt0901-805
32. Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. (1999) 293:321–31. doi: 10.1006/jmbi.1999.3110
33. Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins (2003) 53:566–72. doi: 10.1002/prot.10532
34. DeForte S, Uversky VN. Resolving the ambiguity: making sense of intrinsic disorder when PDB structures disagree. Protein Sci. (2016) 25:676–88. doi: 10.1002/pro.2864
35. Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta (2013) 1834:932–51. doi: 10.1016/j.bbapap.2012.12.008
36. Uversky VN. Under-folded proteins: conformational ensembles and their roles in protein folding, function, and pathogenesis. Biopolymers (2013) 99:870–87. doi: 10.1002/bip.22298
37. Uversky VN. Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J Biol Chem. (2016) 291:6681–8. doi: 10.1074/jbc.R115.685859
38. Uversky VN. Paradoxes and wonders of intrinsic disorder: prevalence of exceptionality. Intrins Disord Proteins (2015) 3:e1065029. doi: 10.1080/21690707.2015.1065029
39. Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Dunker AK. Identifying disordered regions in proteins from amino acid sequence. In IEEE International Conference on Neural Networks. Piscataway, NJ: IEEE Service Center (1997). p. 90–5. doi: 10.1109/ICNN.1997.611643
40. Uversky VN, Gillespie JR, Fink AL. Why are natively unfolded proteins unstructured under physiologic conditions? Proteins (2000) 41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7
41. Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins (2003) 52:573–84. doi: 10.1002/prot.10437
42. DeForte S, Uversky VN. Intrinsically disordered proteins in PubMed: what can the tip of the iceberg tell us about what lies below? RSC Adv. (2016) 6:11513–21. doi: 10.1039/c5ra24866c
43. Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, et al. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput. (1998) 1998:437–48.
44. Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, Tosatto SC. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics (2015) 31:201–8. doi: 10.1093/bioinformatics/btu625
45. Burra PV, Kalmar L, Tompa P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS ONE (2010) 5:e12069. doi: 10.1371/journal.pone.0012069
46. DeForte S, Uversky VN. Not an exception to the rule: the functional significance of intrinsically disordered protein regions in enzymes. Mol Biosyst. (2017) 13:463–9. doi: 10.1039/c6mb00741d
47. Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, et al. DisProt: The database of disordered proteins. Nucleic Acids Res. (2007) 35:D786–93. doi: 10.1093/nar/gkl893
48. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
49. Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. (2007) 92:1439–56. doi: 10.1529/biophysj.106.094045
50. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. (1982) 157:105–32.
51. Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. (2008) 15:956–63. doi: 10.2174/092986608785849164
52. Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinform. (2007) 8:211. doi: 10.1186/1471-2105-8-211
53. Williams RM, Obradovic Z, Mathura V, Braun W, Garner EC, Young J, et al. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. (2001) 2001:89–100. doi: 10.1142/9789814447362_0010
54. Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins (2001) 42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3
55. Garner E, Cannon P, Romero P, Obradovic Z, Dunker AK. Predicting disordered regions from amino acid sequence: common themes despite differing structural characterization. Genome Inform Ser Workshop Genome Inform. (1998) 9:201–13.
56. Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, et al. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. (1998) 1998:473–84.
57. Jorda J, Xue B, Uversky VN, Kajava AV. Protein tandem repeats - the more perfect, the less structured. FEBS J. (2010) 277:2673–82. doi: 10.1111/j.1742-464X.2010.07684.x
58. Darling AL, Uversky VN. Intrinsic disorder in proteins with pathogenic repeat expansions. Molecules (2017) 22:27. doi: 10.3390/molecules22122027
59. Uversky VN. Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrins Disord Proteins (2016) 4:e1135015. doi: 10.1080/21690707.2015.1135015
60. Fisher CK, Stultz CM. Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol. (2011) 21:426–31. doi: 10.1016/j.sbi.2011.04.001
61. Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol. (2010) 102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003
62. Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. (2008) 37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924
63. Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. (2002) 11:739–56. doi: 10.1110/ps.4210102
64. Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. (2002) 269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x
65. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PTJr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry (1996) 35:13709–15. doi: 10.1021/bi961799n
66. Gast K, Damaschun H, Eckert K, Schulze-Forster K, Maurer HR, Muller-Frohne M, et al. Prothymosin alpha: a biologically active protein with random coil conformation. Biochemistry (1995) 34:13211–8. doi: 10.1021/bi00040a037
67. Hemmings HC Jr, Nairn AC, Aswad DW, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. II. Purification and characterization of the phosphoprotein from bovine caudate nucleus. J Neurosci. (1984) 4:99–110.
68. Das RK, Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci USA. (2013) 110:13392–7. doi: 10.1073/pnas.1304749110
69. Firman T, Ghosh K. Sequence charge decoration dictates coil-globule transition in intrinsically disordered proteins. J Chem Phys. (2018) 148:123305. doi: 10.1063/1.5005821
70. Piovesan D, Tabaro F, Micetic I, Necci M, Quaglia F, Oldfield CJ, et al. DisProt 7.0: a major update of the database of disordered proteins. Nucleic Acids Res. (2017) 45:D219–27. doi: 10.1093/nar/gkw1056
71. Mao AH, Crick SL, Vitalis A, Chicoine CL, Pappu RV. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc Natl Acad Sci USA. (2010) 107:8183–8. doi: 10.1073/pnas.0911107107
72. Das RK, Ruff KM, Pappu RV. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr Opin Struct Biol. (2015) 32:102–12. doi: 10.1016/j.sbi.2015.03.008
73. Carugo O. Hydrophobicity diversity in globular and nonglobular proteins measured with the Gini index. Protein Eng Des Sel. (2017) 30:781–4. doi: 10.1093/protein/gzx060
74. Sawle L, Ghosh K. A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J Chem Phys. (2015) 143:085101. doi: 10.1063/1.4929391
75. Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. (2001) 276:10737–44. doi: 10.1074/jbc.M010907200
76. Permyakov SE, Millett IS, Doniach S, Permyakov EA, Uversky VN. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins (2003) 53:855–62. doi: 10.1002/prot.10481
77. Uversky VN, Permyakov SE, Zagranichny VE, Rodionov IL, Fink AL, Cherskaya AM, et al. Effect of zinc and temperature on the conformation of the gamma subunit of retinal phosphodiesterase: a natively unfolded protein. J Proteome Res. (2002) 1:149–59. doi: 10.1021/pr0155127
78. Timm DE, Vissavajjhala P, Ross AH, Neet KE. Spectroscopic and chemical studies of the interaction between nerve growth factor (NGF) and the extracellular domain of the low affinity NGF receptor. Protein Sci. (1992) 1:1023–31.
79. Kim TD, Ryu HJ, Cho HI, Yang CH, Kim J. Thermal behavior of proteins: heat-resistant proteins and their heat-induced secondary structural changes. Biochemistry (2000) 39:14839–46. doi: 10.1021/bi001441y
80. Uversky VN. Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. (2009) 28:305–25. doi: 10.1007/s10930-009-9201-4
81. Konno T, Tanaka N, Kataoka M, Takano E, Maki M. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim Biophys Acta (1997) 1342:73–82.
82. Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. (1998) 273:3718–24.
83. Uversky VN, Gillespie JR, Millett IS, Khodyakova AV, Vasiliev AM, Chernovskaya TV, et al. Natively unfolded human prothymosin alpha adopts partially folded collapsed conformation at acidic pH. Biochemistry (1999) 38:15009–16. doi: 10.1021/bi990752+
84. Lynn A, Chandra S, Malhotra P, Chauhan VS. Heme binding and polymerization by Plasmodium falciparum histidine rich protein II: influence of pH on activity and conformation. FEBS Lett. (1999) 459:267–71.
85. Uversky VN, Dunker AK. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol Rep. (2013) 5:1. doi: 10.3410/B5-1
86. Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handjournal of Protein Folding. Weinheim: Wiley-VCH, Verlag GmbH & Co. KGaA (2005). p. 271–353.
87. Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. (2003) 60:1852–71. doi: 10.1007/s00018-003-3096-6
88. Zhang T, Faraggi E, Li Z, Zhou Y. Intrinsically semi-disordered state and its role in induced folding and protein aggregation. Cell Biochem Biophys. (2013) 67:1193–205. doi: 10.1007/s12013-013-9638-0
89. Jakob U, Kriwacki R, Uversky VN. Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev. (2014) 114:6779–805. doi: 10.1021/cr400459c
90. Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. (2013) 22:693–724. doi: 10.1002/pro.2261
91. Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. (2013) 19:4191–213. doi: 10.2174/1381612811319230005
92. Uversky VN. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. (2015) 282:1182–9. doi: 10.1111/febs.13202
93. Uversky VN. p53 proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci. (2016) 17:1874. doi: 10.3390/ijms17111874
94. Smith LM, Kelleher NL Consortium for Top Down P. Proteoform: a single term describing protein complexity. Nat Methods (2013) 10:186–7. doi: 10.1038/nmeth.2369
95. Schluter H, Apweiler R, Holzhutter HG, Jungblut PR. Finding one's way in proteomics: a protein species nomenclature. Chem Cent J. (2009) 3:11. doi: 10.1186/1752-153X-3-11
96. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature (2012) 489:57–74. doi: 10.1038/nature11247
97. Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteom. (2005) 4:1920–32. doi: 10.1074/mcp.M500279-MCP200
98. Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, Sun Z, Huang CY, et al. The state of the human proteome in 2012 as viewed through peptideatlas. J Proteome Res. (2013) 12:162–71. doi: 10.1021/Pr301012j
99. Farrah T, Deutsch EW, Omenn GS, Sun Z, Watts JD, Yamamoto T, et al. State of the human proteome in 2013 as viewed through peptideatlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven human proteome project. J Proteome Res. (2014) 13:60–75. doi: 10.1021/Pr4010037
100. Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature (2014) 509:570–81. doi: 10.1038/Nature13302
101. Reddy PJ, Ray S, Srivastava S. The quest of the human proteome and the missing proteins: digging deeper. Omics J Integr Biol. (2015) 19:276–82. doi: 10.1089/omi.2015.0035
102. Koshland DE Jr. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. (1958) 44:98–104.
103. Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. (2005) 127:15248–56. doi: 10.1021/ja055251s
104. Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature (2005) 438:117–21. doi: 10.1038/nature,04105
105. Frauenfelder H, Chen G, Berendzen J, Fenimore PW, Jansson H, McMahon BH, et al. A unified model of protein dynamics. Proc Natl Acad Sci USA. (2009) 106:5129–34. doi: 10.1073/pnas.0900336106
106. Kohen A. Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc Chem Res. (2015) 48:466–73. doi: 10.1021/ar500322s
107. Narayanan C, Bernard DN, Bafna K, Gagne D, Chennubhotla CS, Doucet N, et al. Conservation of dynamics associated with biological function in an enzyme superfamily. Structure (2018) 26:426–36.e3. doi: 10.1016/j.str.2018.01.015
108. Uversky VN. Conserved functional dynamics: i like to move it, move it! Structure (2018) 26:371–73. doi: 10.1016/j.str.2018.02.010.
109. Schneider R, Huang JR, Yao M, Communie G, Ozenne V, Mollica L, et al. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol Biosyst. (2012) 8:58–68. doi: 10.1039/c1mb05291h
110. Uversky VN, Dunker AK. Multiparametric analysis of intrinsically disordered proteins: looking at intrinsic disorder through compound eyes. Anal Chem. (2012) 84:2096–104. doi: 10.1021/ac203096k
111. Stanley N, Esteban-Martín S, De Fabritiis G. Progress in studying intrinsically disordered proteins with atomistic simulations. Prog Biophys Mol Biol. (2015) 119:47–52. doi: 10.1016/j.pbiomolbio.2015.03.003
112. Schor M, Mey ASJS, MacPhee CE. Analytical methods for structural ensembles and dynamics of intrinsically disordered proteins. Biophys Rev. (2016) 8:429–39. doi: 10.1007/s12551-016-0234-6
113. Best RB. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr Opin Struct Biol. (2017) 42:147–54. doi: 10.1016/j.sbi.2017.01.006
114. Fu B, Vendruscolo M. Structure and dynamics of intrinsically disordered proteins. Adv Exp Med Biol. (2015) 870:35–48. doi: 10.1007/978-3-319-20164-1_2
115. Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry (2005) 44:12454–70. doi: 10.1021/bi050736e.
116. Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics (2008) 9 (Suppl. 1):S1. doi: 10.1186/1471-2164-9-S1-S1
117. Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. (2014) 83:553–84. doi: 10.1146/annurev-biochem-072711-164947
118. Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. (2011) 40:1623–34. doi: 10.1039/c0cs00057d
119. Uversky VN. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. (2015) 589(19 Pt A):2498–506. doi: 10.1016/j.febslet.2015.06.004
120. Szöllosi D, Horváth T, Han KH, Dokholyan NV, Tompa P, Kalmár L, et al. Discrete molecular dynamics can predict helical prestructured motifs in disordered proteins. PLoS ONE (2014) 9:e95795. doi: 10.1371/journal.pone.0095795
121. Kim DH, Han KH. PreSMo target-binding signatures in intrinsically disordered proteins. Mol Cells (2018) 41:889–99. doi: 10.14348/molcells.2018.0192
122. Milles S, Salvi N, Blackledge M, Jensen MR. Characterization of intrinsically disordered proteins and their dynamic complexes: from in vitro to cell-like environments. Prog Nucl Magn Reson Spectrosc. (2018) 109:79–100. doi: 10.1016/j.pnmrs.2018.07.001
123. Lemieux UR, Spohr U. How Emil Fischer was led to the lock and key concept for enzyme specificity. Adv Carbohydrate Chem Biochem. (1994) 50:1–20.
124. Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci USA. (2000) 97:8868–73. doi: 10.1073/pnas.160259697
125. Betts MJ, Sternberg MJ. An analysis of conformational changes on protein-protein association: implications for predictive docking. Protein Eng. (1999) 12:271–83. doi: 10.1093/protein/12.4.271
126. Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci USA. (2005) 102:18908–13. doi: 10.1073/pnas.0507603102
127. Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. (1999) 8:1181–90. doi: 10.1110/ps.8.6.1181
128. Tsai CJ, Ma B, Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci USA. (1999) 96:9970–2.
129. Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. (1999) 12:713–20.
130. Careri G, Fasella P, Gratton E. Statistical time events in enzymes: a physical assessment. CRC Crit Rev Biochem. (1975) 3:141–64.
131. Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science (1991) 254:1598–603. doi: 10.1126/science.1749933
132. Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. (1998) 37:2755–94.
133. Barbar E, Nyarko A. Polybivalency and disordered proteins in ordering macromolecular assemblies. Semin Cell Dev Biol. (2015) 37:20–5. doi: 10.1016/j.semcdb.2014.09.016
134. Hendus-Altenburger R, Pedraz-Cuesta E, Olesen CW, Papaleo E, Schnell JA, Hopper JT, et al. The human Na+/H+ exchanger 1 is a membrane scaffold protein for extracellular signal-regulated kinase 2. BMC Biol. (2016) 14:31. doi: 10.1186/s12915-016-0252-7
135. Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. (2009) 19:31–8. doi: 10.1016/j.sbi.2008.12.003
136. Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science (2000) 289:905–20. doi: 10.1126/science.289.5481.905
137. Gunasekaran K, Tsai CJ, Nussinov R. Analysis of ordered and disordered protein complexes reveals structural features discriminating between stable and unstable monomers. J Mol Biol. (2004) 341:1327–41. doi: 10.1016/j.jmb.2004.07.002
138. Xu D, Tsai CJ, Nussinov R. Mechanism and evolution of protein dimerization. Protein Sci. (1998) 7:533–44. doi: 10.1002/pro.5560070301
139. Teschke CM, King J. Folding and assembly of oligomeric proteins in Escherichia coli. Curr Opin Biotechnol. (1992) 3:468–73.
140. Schulz GE. Nucleotide Binding Proteins. In: Balaban M, editors. Molecular Mechanism of Biological Recognition. New York, NY: Elsevier; North-Holland Biomedical Press (1979). p. 79–94.
141. Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. (2002) 323:573–84. doi: 10.1016/S0022-2836(02)00969-5
142. Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science (1993) 262:1718–21. doi: 10.1126/science.8259515
143. Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. (1996) 93:11504–9.
144. Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets: the roles of intrinsic disorder in protein interaction networks. FEBS J. (2005) 272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x
145. Hsu WL, Oldfield CJ, Xue B, Meng J, Huang F, Romero P, et al. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. (2013) 22:258–73. doi: 10.1002/pro.2207
146. Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, et al. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. (2003) 22:494–501. doi: 10.1093/emboj/cdg068
147. Sharma R, Raduly Z, Miskei M, Fuxreiter M. Fuzzy complexes: specific binding without complete folding. FEBS Lett. (2015) 589(19 Pt A):2533–42. doi: 10.1016/j.febslet.2015.07.022
148. Fuxreiter M, Tompa P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol. (2012) 725:1–14. doi: 10.1007/978-1-4614-0659-4_1
149. Fuxreiter M. Fuzziness: linking regulation to protein dynamics. Mol Biosyst. (2012) 8:168–77. doi: 10.1039/c1mb05234a
150. Hazy E, Tompa P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem (2009) 10:1415–9. doi: 10.1002/cphc.200900205
151. Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. (2008) 33:2–8. doi: 10.1016/j.tibs.2007.10.003
152. Sigalov AB, Zhuravleva AV, Orekhov VY. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie (2007) 89:419–21. doi: 10.1016/j.biochi.2006.11.003
153. Sigalov A, Aivazian D, Stern L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry (2004) 43:2049–61. doi: 10.1021/bi035900h
154. Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recogn. (2005) 18:343–84. doi: 10.1002/jmr.747
155. Xue B, Romero PR, Noutsou M, Maurice MM, Rudiger SG, William AMJr, et al. Stochastic machines as a colocalization mechanism for scaffold protein function. FEBS Lett. (2013) 587:1587–91. doi: 10.1016/j.febslet.2013.04.006
156. Xing Y, Takemaru K, Liu J, Berndt JD, Zheng JJ, Moon RT, et al. Crystal structure of a full-length beta-catenin. Structure (2008) 16:478–87. doi: 10.1016/j.str.2007.12.021
157. Zhang Y, Qiu WJ, Chan SC, Han J, He X, Lin SC. Casein kinase I and casein kinase II differentially regulate axin function in Wnt and JNK pathways. J Biol Chem. (2002) 277:17706–12. doi: 10.1074/jbc.M111982200
158. Mitrea DM, Kriwacki RW. Regulated unfolding of proteins in signaling. FEBS Lett. (2013) 587:1081–8. doi: 10.1016/j.febslet.2013.02.024
159. Bardwell JC, Jakob U. Conditional disorder in chaperone action. Trends Biochem Sci. (2012) 37:517–25. doi: 10.1016/j.tibs.2012.08.006
160. Creamer TP. Transient disorder: calcineurin as an example. Intrin Disorder Proteins (2013) 1:e26412. doi: 10.4161/idp.26412
161. Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature (2018) 555:61–6. doi: 10.1038/nature25762
162. Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recogn. (2010) 23:105–16. doi: 10.1002/jmr.961
163. Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci USA. (2007) 104:9650–5. doi: 10.1073/pnas.0702580104
164. Lutkenhaus J. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu Rev Biochem. (2007) 76:539–62. doi: 10.1146/annurev.biochem.75.103004.142652
165. Loose M, Kruse K, Schwille P. Protein self-organization: lessons from the min system. Annu Rev Biophys. (2011) 40:315–36. doi: 10.1146/annurev-biophys-042910-155332
166. Raskin DM, de Boer PA. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc Natl Acad Sci USA. (1999) 96:4971–6. doi: 10.1073/pnas.96.9.4971
167. Bonny M, Fischer-Friedrich E, Loose M, Schwille P, Kruse K. Membrane binding of MinE allows for a comprehensive description of Min-protein pattern formation. PLoS Comput Biol. (2013) 9:e1003347. doi: 10.1371/journal.pcbi.1003347
168. Park KT, Villar MT, Artigues A, Lutkenhaus J. MinE conformational dynamics regulate membrane binding, MinD interaction, and Min oscillation. Proc Natl Acad Sci USA. (2017) 114:7497–504. doi: 10.1073/pnas.1707385114
169. Park KT, Wu W, Battaile KP, Lovell S, Holyoak T, Lutkenhaus J. The Min oscillator uses MinD-dependent conformational changes in MinE to spatially regulate cytokinesis. Cell (2011) 146:396–407. doi: 10.1016/j.cell.2011.06.042
170. Ghasriani H, Ducat T, Hart CT, Hafizi F, Chang N, Al-Baldawi A, et al. Appropriation of the MinD protein-interaction motif by the dimeric interface of the bacterial cell division regulator MinE. Proc Natl Acad Sci USA. (2010) 107:18416–21. doi: 10.1073/pnas.1007141107
171. Kruse K. A dynamic model for determining the middle of Escherichia coli. Biophys J. (2002) 82:618–27. doi: 10.1016/S0006-3495(02)75426-X
172. Meinhardt H, de Boer PA. Pattern formation in Escherichia coli: a model for the pole-to-pole oscillations of Min proteins and the localization of the division site. Proc Natl Acad Sci USA. (2001) 98:14202–7. doi: 10.1073/pnas.251216598
173. Howard M, Rutenberg AD, de Vet S. Dynamic compartmentalization of bacteria: accurate division in E. coli. Phys Rev Lett. (2001) 87(27 Pt 1):278102. doi: 10.1103/PhysRevLett.87.278102
174. Loose M, Fischer-Friedrich E, Ries J, Kruse K, Schwille P. Spatial regulators for bacterial cell division self-organize into surface waves in vitro. Science (2008) 320:789–92. doi: 10.1126/science.1154413
175. Loose M, Fischer-Friedrich E, Herold C, Kruse K, Schwille P. Min protein patterns emerge from rapid rebinding and membrane interaction of MinE. Nat Struct Mol Biol. (2011) 18:577–83. doi: 10.1038/nsmb.2037
176. Badrinarayanan A, Le TB, Laub MT. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. (2015) 31:171–99. doi: 10.1146/annurev-cellbio-100814-125211
177. Toro E, Shapiro L. Bacterial chromosome organization and segregation. Cold Spring Harb Perspect Biol. (2010) 2:a000349. doi: 10.1101/cshperspect.a000349
178. Ebersbach G, Briegel A, Jensen GJ, Jacobs-Wagner C. A self-associating protein critical for chromosome attachment, division, and polar organization in caulobacter. Cell (2008) 134:956–68. doi: 10.1016/j.cell.2008.07.016
179. Ebersbach G, Gerdes K. Bacterial mitosis: partitioning protein ParA oscillates in spiral-shaped structures and positions plasmids at mid-cell. Mol Microbiol. (2004) 52:385–98. doi: 10.1111/j.1365-2958.2004.04002.x
180. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. (2017) 18:285–98. doi: 10.1038/nrm.2017.7
181. Uversky VN. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol. (2017) 44:18–30. doi: 10.1016/j.sbi.2016.10.015
182. Uversky VN. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv Colloid Interface Sci. (2017) 239:97–114. doi: 10.1016/j.cis.2016.05.012
183. Gomes E, Shorter J. The molecular language of membraneless organelles. J Biol Chem. (2018). doi: 10.1074/jbc.TM118.001192. [Epub ahead of print].
184. Wheeler RJ, Hyman AA. Controlling compartmentalization by non-membrane-bound organelles. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170193. doi: 10.1098/rstb.2017.0193
185. Brangwynne CP. Phase transitions and size scaling of membrane-less organelles. J Cell Biol. (2013) 203:875–81. doi: 10.1083/jcb.201308087
186. Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. (2014) 30:39–58. doi: 10.1146/annurev-cellbio-100913-013325.
187. Toretsky JA, Wright PE. Assemblages: functional units formed by cellular phase separation. J Cell Biol. (2014) 206:579–88. doi: 10.1083/jcb.201404124
188. Uversky VN, Kuznetsova IM, Turoverov KK, Zaslavsky B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. (2015) 589:15–22. doi: 10.1016/j.febslet.2014.11.028
189. Chong PA, Forman-Kay JD. Liquid-liquid phase separation in cellular signaling systems. Curr Opin Struct Biol. (2016) 41:180–6. doi: 10.1016/j.sbi.2016.08.001
190. Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature (2000) 404:604–9. doi: 10.1038/35007077
191. Pederson T. Protein mobility within the nucleus – what are the right moves? Cell (2001) 104:635–8. doi: 10.1016/S0092-8674(01)00258-6
192. Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell (2013) 152:791–805. doi: 10.1016/j.cell.2013.01.033
193. Feric M, Brangwynne CP. A nuclear F-actin scaffold stabilizes ribonucleoprotein droplets against gravity in large cells. Nat Cell Biol. (2013) 15:1253–9. doi: 10.1038/ncb2830
194. Brangwynne CP, Mitchison TJ, Hyman AA. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci USA. (2011) 108:4334–9. doi: 10.1073/pnas.1017150108
195. Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science (2009) 324:1729–32. doi: 10.1126/science.1172046
196. Aggarwal S, Snaidero N, Pahler G, Frey S, Sanchez P, Zweckstetter M, et al. Myelin membrane assembly is driven by a phase transition of myelin basic proteins into a cohesive protein meshwork. PLoS Biol. (2013) 11:e1001577. doi: 10.1371/journal.pbio.1001577
197. Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature (2012) 483:336–40. doi: 10.1038/nature10879
198. Updike DL, Hachey SJ, Kreher J, Strome S. P granules extend the nuclear pore complex environment in the C. elegans germ line. J Cell Biol. (2011) 192:939–48. doi: 10.1083/jcb.201010104
199. Handwerger KE, Cordero JA, Gall JG. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol Biol Cell. (2005) 16:202–11. doi: 10.1091/mbc.E04-08-0742
200. Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. (2015) 11:899–904. doi: 10.1038/Nphys3532
201. Zhu L, Brangwynne CP. Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol. (2015) 34:23–30. doi: 10.1016/j.ceb.2015.04.003
202. Holehouse AS, Pappu RV. Functional implications of intracellular phase transitions. Biochemistry (2018) 57:2415–23. doi: 10.1021/acs.biochem.7b01136
203. Zaslavsky BY, Uversky VN. In aqua veritas: the indispensable yet mostly ignored role of water in phase separation and membrane-less organelles. Biochemistry (2018) 57:2437–51. doi: 10.1021/acs.biochem.7b01215
204. Lin Y, Currie SL, Rosen MK. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J Biol Chem. (2017) 292:19110–20. doi: 10.1074/jbc.M117.800466
205. Wei MT, Elbaum-Garfinkle S, Holehouse AS, Chen CC, Feric M, Arnold CB, et al. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat Chem. (2017) 9:1118–25. doi: 10.1038/nchem.2803
206. Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell (2015) 57:936–47. doi: 10.1016/j.molcel.2015.01.013
207. Dundr M, Misteli T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol. (2010) 2:a000711. doi: 10.1101/cshperspect.a000711
208. Mao YS, Zhang B, Spector DL. Biogenesis and function of nuclear bodies. Trends Genet (2011) 27:295–306. doi: 10.1016/j.tig.2011.05.006
209. Rhoads SN, Monahan ZT, Yee DS, Shewmaker FP. The role of post-translational modifications on prion-like aggregation and liquid-phase separation of FUS. Int J Mol Sci. (2018) 19:e886. doi: 10.3390/ijms19030886
210. Darling AL, Liu Y, Oldfield CJ, Uversky VN. Intrinsically disordered proteome of human membrane-less organelles. Proteomics (2018) 18:e1700193. doi: 10.1002/pmic.201700193
211. Mitrea DM, Kriwacki RW. Phase separation in biology; functional organization of a higher order. Cell Commun Signal. (2016) 14:1. doi: 10.1186/s12964-015-0125-7
212. Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell (2013) 155:1049–60. doi: 10.1016/j.cell.2013.10.033
213. Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell (2012) 149:753–67. doi: 10.1016/j.cell.2012.04.017
214. Frey S, Richter RP, Gorlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science (2006) 314:815–7. doi: 10.1126/science.1132516
215. Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell (2015) 162:1066–77. doi: 10.1016/j.cell.2015.07.047
216. Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell (2015) 163:123–33. doi: 10.1016/j.cell.2015.09.015
217. Banjade S, Rosen MK. Phase transitions of multivalent proteins can promote clustering of membrane receptors. Elife (2014) 3:e04123. doi: 10.7554/eLife.04123
218. Anfinsen CB, Haber E, Sela M, White FH Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci USA. (1961) 47:1309–14.
220. Neurath H, Greenstein JP, Putnam FW, Erickson JO. The chemistry of protein denaturation. Chem. Rew. (1944) 34:157–265.
221. Mirsky AE, Pauling L. On the structure of native, denatured and coagulated proteins. Proc Natl Acad Sci USA. (1936) 22:439–47.
222. Anson ML, Mirsky AE. The effect of denaturation on the viscosity of protein systems. J Gen Physiol. (1932)15:341–50.
223. Neyroz P, Zambelli B, Ciurli S. Intrinsically disordered structure of Bacillus pasteurii UreG as revealed by steady-state and time-resolved fluorescence spectroscopy. Biochemistry (2006) 45:8918–30. doi: 10.1021/bi060227s
224. Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. (2004) 51:299–321.
225. Iakoucheva LM, Kimzey AL, Masselon CD, Bruce JE, Garner EC, Brown CJ, et al. Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci. (2001) 10:560–71. doi: 10.1110/ps.29401
226. Balazs A, Csizmok V, Buday L, Rakacs M, Kiss R, Bokor M, et al. High levels of structural disorder in scaffold proteins as exemplified by a novel neuronal protein, CASK-interactive protein1. FEBS J. (2009) 276:3744–56. doi: 10.1111/j.1742-4658.2009.07090.x
227. Brocca S, Samalikova M, Uversky VN, Lotti M, Vanoni M, Alberghina L, et al. Order propensity of an intrinsically disordered protein, the cyclin-dependent-kinase inhibitor Sic1. Proteins (2009) 76:731–46. doi: 10.1002/prot.22385
228. Nocula-Lugowska M, Rymarczyk G, Lisowski M, Ozyhar A. Isoform-specific variation in the intrinsic disorder of the ecdysteroid receptor N-terminal domain. Proteins (2009) 76:291–308. doi: 10.1002/prot.22342
229. Suskiewicz MJ, Sussman JL, Silman I, Shaul Y. Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. (2011) 20:1285–97. doi: 10.1002/pro.657
230. Johnson DE, Xue B, Sickmeier MD, Meng J, Cortese MS, Oldfield CJ, et al. High-throughput characterization of intrinsic disorder in proteins from the protein structure initiative. J Struct Biol. (2012) 180:201–15. doi: 10.1016/j.jsb.2012.05.013
231. Tsvetkov P, Myers N, Moscovitz O, Sharon M, Prilusky J, Shaul Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol Biosyst. (2012) 8:368–73. doi: 10.1039/c1mb05283g
232. Gardner KA, Moore DA, Erickson HP. The C-terminal linker of Escherichia coli FtsZ functions as an intrinsically disordered peptide. Mol Microbiol. (2013) 89:264–75. doi: 10.1111/mmi.12279
Keywords: intrinsically disordered protein (IDP), intrinsically disordered region, spatio–temporal heterogeneity, structural flexibility, binding promiscuity, emergent behavior, edge of chaos
Citation: Uversky VN (2019) Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 7:10. doi: 10.3389/fphy.2019.00010
Received: 28 July 2018; Accepted: 17 January 2019;
Published: 07 February 2019.
Edited by:
Michele Vendruscolo, University of Cambridge, United KingdomReviewed by:
Paolo De Los Rios, École Polytechnique Fédérale de Lausanne, SwitzerlandHaiguang Liu, Beijing Computational Science Research Center, China
Copyright © 2019 Uversky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vladimir N. Uversky, vuversky@health.usf.edu