 Yiling Cao1
Yiling Cao1 Ji-Hong Lin
Ji-Hong Lin Chun Zhang
Chun Zhang- 1Department of Nephrology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 25th Medical Department, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
Diabetic nephropathy (DN) is a major cause of morbidity and mortality in diabetes and is the most common cause of end stage renal disease (ESRD). Renal fibrosis is the final pathological change in DN. It is widely believed that cellular phenotypic switching is the cause of renal fibrosis in diabetic nephropathy. Several types of kidney cells undergo activation and differentiation and become reprogrammed to express markers of mesenchymal cells or podocyte-like cells. However, the development of targeted therapy for DN has not yet been identified. Here, we discussed the pathophysiologic changes of DN and delineated the possible origins that contribute to myofibroblasts and podocytes through phenotypic transitions. We also highlight the molecular signaling pathways involved in the phenotypic transition, which would provide valuable information for the activation of phenotypic switching and designing effective therapies for DN.
1 Introduction
With rapid urbanization and significant changes in lifestyles, diabetes mellitus (DM) rapidly become a global health burden, which impacts approximately 9% of the population worldwide (Domingo-Lopez et al., 2022). The disease is characterized by chronic hyperglycemia due to inadequate insulin secretion or reduced insulin sensitivity (American Diabetes Association Professional Practice, 2022). The morbidity and mortality of diabetes are mostly related to the development of diabetes-associated complications affecting the kidney and other organ systems. Diabetic nephropathy (DN) usually occurs in patients with DM without long-term adequate glycemic control (Lin et al., 2018). About one-third of patients with type 1 DM and half with type 2 DM will develop chronic kidney disease, which is clinically defined by the presence of impaired renal function or elevated urinary albumin excretion, or both (Akhtar et al., 2020). DN is a chronic progressive disorder and is the leading cause of end stage kidney disease in the United States and most developed countries, representing in some parts of the world more than 50% of subjects requiring kidney replacement therapy (Collins et al., 2014; Umanath and Lewis, 2018). The kidneys undergo several changes and result in a clinical presentation including persistent albuminuria, hypertension, and progressive reductions in glomerular filtration rate (GFR) (Umanath and Lewis, 2018; Sugahara et al., 2021).
The most typical morphological change of DN is the excessive deposition of extracellular matrix (ECM) proteins in the mesangium and basement membrane of the glomerulus and the renal tubulointerstitium, leading to glomerulosclerosis and interstitial fibrosis (Mason and Wahab, 2003). Renal fibrosis is the final pathological change in DN (Humphreys, 2018). It is widely accepted that the activated myofibroblasts are the principal effector cells that are responsible for the excess deposition of interstitial ECM under pathological conditions, but their origin in the kidney remains uncertain (Humphreys, 2018; Yuan et al., 2019). The phenotype switching of fibroblasts to secrete and remodel the extracellular matrix is one of the origins of myofibroblasts. However, emerging evidence suggests that diabetes also alters the phenotype of normal, non-fibroblast kidney cells, such as podocytes, mesangial cells, and tubular epithelial cells. As a result, differentiated kidney cells become reprogrammed to secrete and accumulate extracellular matrix (Simonson, 2007). Moreover, major advances have been achieved in parietal epithelial cells (PECs) in recent years. PECs have been considered to have the capacity to transdifferentiate toward podocytes in DN, which may serve as a potential therapeutic target to replace podocytes. Identification of the mechanisms underlying these processes is necessary for the development of future therapies. This review aims to further understand the transdifferentiation processes in the DN and provide ideas for designing new prevention and therapeutics. The regulation of phenotypic transitions in the development process of DN should yield productive clues for therapies.
2 Pathophysiology of DN
DN usually manifests a clinical syndrome including increased urine albumin excretion (>300 mg/day), persistent reduction in GFR, and increased blood pressure (Sugahara et al., 2021). The cause of DN is the high blood glucose that destroys the blood vessels in the kidneys resulting in its dysfunction (Gajjala et al., 2015). The pathogenesis of diabetic nephropathy has been deeply investigated, and the roles of various mechanisms have been described, which include the effect of high glucose, inflammation, oxidative stress, renin-angiotensin system activation, and endoplasmic reticulum stress, etc (Rask-Madsen and King, 2013; Gujarati et al., 2021). These changes lead to various cellular responses, the expression of secretory factors and extracellular matrix accumulation (Maezawa et al., 2015). Hyperglycemia can cause several morphological changes that involve all sections of the kidney. Early alterations include the development of glomerular hyperfiltration and hypertrophy (DeFronzo et al., 2021). As the disease progresses, ECM, such as collagen I and IV and fibronectin (FN) accumulation, mesangial cells proliferation, podocyte foot process effacement and number decrease, basement membrane thickening, tubular atrophy and peritubular capillary rarefaction, culminating in interstitial fibrosis and glomerulosclerosis. Clinical manifestations show increased urinary albumin excretion, progressive loss of renal function and ultimately kidney failure (Carew et al., 2012; Umanath and Lewis, 2018).
The major structural abnormality of the glomerulus is mesangial matrix expansion. This is initially due to increased mesangial cell proliferation and matrix deposition (Thomas and Ford Versypt, 2022). As the disease progresses, matrix accumulation is the predominant mesangial change. Glomerular basement membrane (GBM) is diffuse thickening and filled with extracellular matrix components, which gradually occludes the glomerular capillaries, thus reducing the area available for glomerular filtration and leading to proteinuria or macromolecular leakiness (Fioretto and Mauer, 2007; Vasanth Rao et al., 2019; Akhtar et al., 2020). Diffuse GBM thickening has been demonstrated in almost all diabetic patients (Zhang et al., 2018a). Epithelial podocyte foot processes, together with the fenestrated endothelium and the GBM form the glomerular filtration barrier (Zhou and Liu, 2015). Podocytes wrap the glomerular capillaries and are attached to the GBM. Podocyte detachment and foot process effacement owing to cytoskeletal changes and podocyte phenotypic alterations is the principal cause of albuminuria in DN (Li et al., 2008; Wang et al., 2019). Extensive podocyte damage will eventually result in podocyte loss and a decrease in density in the development of DN (Tharaux and Huber, 2012). During the phenotypic transition, this architecture of podocytes is disrupted and the actin cytoskeleton is redistributed, which induced structural alterations to the slit diaphragm and foot process effacement (Garg and Holzman, 2012). The podocyte detachment causes the areas of bare GBM. The exposed GBM region would subsequently attach to the PECs and the Bowman’s capsule, which along with ECM deposition subsequently leads to glomerulosclerosis (Li et al., 2007; Mora-Fernandez et al., 2014).
Tubulointerstitial fibrosis is a consistent feature of DN, not just the consequence of glomerular injury (Tonolo and Cherchi, 2014). The pathological changes show tubular hypertrophy, increased apoptosis, thickening of the tubular basement membrane, interstitial inflammatory cell infiltration and finally tubular atrophy and tubulointerstitial fibrosis (Liu et al., 2019). Tubulointerstitial fibrosis features interstitial matrix deposition, inflammation, fibroblast activation, microvascular rarefaction, and tubular cell loss (Peng et al., 2022). Hyperglycemia causes renal tubular epithelial cells to transform into mesenchymal cells, leading to increased expression of FN and α-smooth muscle actin (α-SMA) (Hung et al., 2021). Injured tubular epithelial cells (TECs) could also release many effectors like inflammatory cytokines and chemokines on surrounding cells such as fibroblasts, causing them to transform into myofibroblasts and produce ECM components. The progression of glomerulosclerosis and tubulointerstitial fibrosis ultimately causes a deterioration of renal dysfunction in DN.
3 Phenotypic transitions in DN
Hyperglycemia causes long-term renal injury and increased deposition of ECM, which constitute the main drivers of renal fibrosis. Although many types of cells can produce ECM, it is widely accepted that myofibroblasts play a major role in the synthesis and secretion of ECM under pathological conditions (Duffield, 2014). A recent study demonstrated that the majority of scar tissue in human and mouse kidney fibrosis originates from platelet-derived growth factor receptor α (Pdgfrα+)/platelet-derived growth factor receptor alpha β (Pdgfrβ+) dual-positive fibroblasts and myofibroblasts (Kuppe et al., 2021). Myofibroblast is characterized by its signature protein α-SMA and has a greater capacity for generating collagen fibers (Liu, 2011). However, the origin of myofibroblasts in DN remains controversial (Loeffler and Wolf, 2015). With much research in the past years focused on the origin of myofibroblasts, there are several known sources of myofibroblasts, including resident mesenchymal cells (fibroblasts and pericytes), the transition from resident endothelial cells (ECs) or TECs through endothelial-mesenchymal transition (EndoMT) and epithelial-mesenchymal transition (EMT) (Schrimpf et al., 2012; Srivastava et al., 2019). A study in 2013 used multiple genetically engineered mice to track the source of myofibroblasts. It was found that the source of myofibroblasts is 50% arising from the proliferation of resident fibroblasts, 35% through differentiation from bone marrow, 5% from EMT, and 10% from EndoMT (LeBleu et al., 2013). A new study employed single-cell RNA-seq revealing pericytes and fibroblasts as the major cellular sources of myofibroblasts, while tubular epithelia, endothelium and monocytes only exhibited minor ECM expression (Kuppe et al., 2021).
EMT, reactivated in wound healing, fibrosis and cancer progression, is a complex set of phenotypical changes induced in epithelial cells that lose the hallmark epithelial characteristics including cell-cell basement membrane contacts, structural polarity and reorganizing their cytoskeleton, which changes the signaling program that defines cell shape and reprogrammes gene expression (Lamouille et al., 2014). EMT has been classified into three distinct classes: Type I EMT is associated with embryogenesis and organ development; type II EMT is induced in the context of inflammation and fibrosis, and type III EMT is associated with cancer progression (Zeisberg and Neilson, 2009; Carew et al., 2012). Type II EMT is commonly defined as the ability of epithelial cells to undergo dedifferentiation in fibrosis. Overexpression of many EMT inducers increases the expression of important transcription factors like Snail family transcriptional repressor (Snail), Twist family BHLH Transcription Factor (Twist) and Zinc finger E-Box binding homeobox (ZEB) that regulate EMT (Barrallo-Gimeno and Nieto, 2005; Feng et al., 2022).
EndoMT is a subtype of EMT. ECs compose the internal surface of the vessels and play an essential role in vascular homeostasis. EndoMT plays an influential role in fibrogenesis in many tissues, such as the kidney, liver heart and lung, and increases myofibroblasts and ECM. Similar to EMT, EndoMT defines as a complex biological process in which ECs lose their endothelial markers, adhesion and apical-basal polarity and transit into mesenchymal cell type under certain conditions, leading to vascular rarefaction, organ fibrosis and dysfunction (Zeisberg et al., 2008; Dejana et al., 2017).
Podocytes, as special glomerular epithelial cells, can obtain a mesenchymal property in a high-glucose condition, which is also called EMT (Yamaguchi et al., 2009). In addition to the aforementioned cells, PECs have been receiving great attention as progenitor cells of podocytes. Activated PECs are one of the possible sources of podocytes through phenotypic transitions at the vascular pole and increase the expression and secretion of ECM proteins in DN (Appel et al., 2009).
4 Cellular transdifferentiation in the glomerulus
4.1 Podocyte-mesenchymal transition in DN
Glomerular visceral epithelial cells, also called podocytes, are terminally differentiated cells that circle the basement membrane of the glomerular capillary. As terminal differentiation cells, podocytes cannot regenerate when they suffer from injury (Faul et al., 2007). Podocyte foot processes interdigitate with each other as well as with neighboring podocytes to form a filtration slit called a slit diaphragm (Greka and Mundel, 2012). Several proteins associated with the slit diaphragm, such as nephrin, podocin, CD2-associated protein (Cd2-ap), and zonula occludens-1 (ZO-1), are of critical importance in establishing the glomerular filtration barrier (Zhou and Liu, 2015). As the most important component of the renal filtration barrier, podocytes are vulnerable to hyperglycemia and play a crucial role in pathological mechanisms underlying DN. Studies in vivo and in vitro found that podocytes exposed to high glucose concentration could undergo mesenchymal transition, which aggravated podocytes’ structure and function injury (Yamaguchi et al., 2009; Zhang et al., 2012). In this process, podocytes appear to lose their epithelial polarity, alteration of the cell-cell junctions represented by a loss of the slit diaphragm, and rearrangement of the actin cytoskeleton, which finally causes foot process effacement. Podocytes lose specific markers expression of nephrin, podocin, ZO-1 and P-cadherin (Shankland, 2006) (Figure 1). It has been established that mutations of these proteins lead to foot process effacement and podocyte dysfunction resulting in defective glomerular filtration along with the increase in proteinuria. By contrast, the expression levels of mesenchymal cell marker proteins, such as fibroblast-specific protein-1 (FSP-1), α-SMA, desmin, matrix metalloproteinase 9 (MMP9), and expression of the main transcription factors: Snail1 and Snail2 are increased (Yamaguchi et al., 2009). These changes lead to podocyte dysfunction, filtration barrier damage, and protein leakage, thereby ultimately promoting glomerular sclerosis and the progress of DN (Yamaguchi et al., 2009). These phenotypic phenomena are consistent with the process of EMT (Liu, 2010).
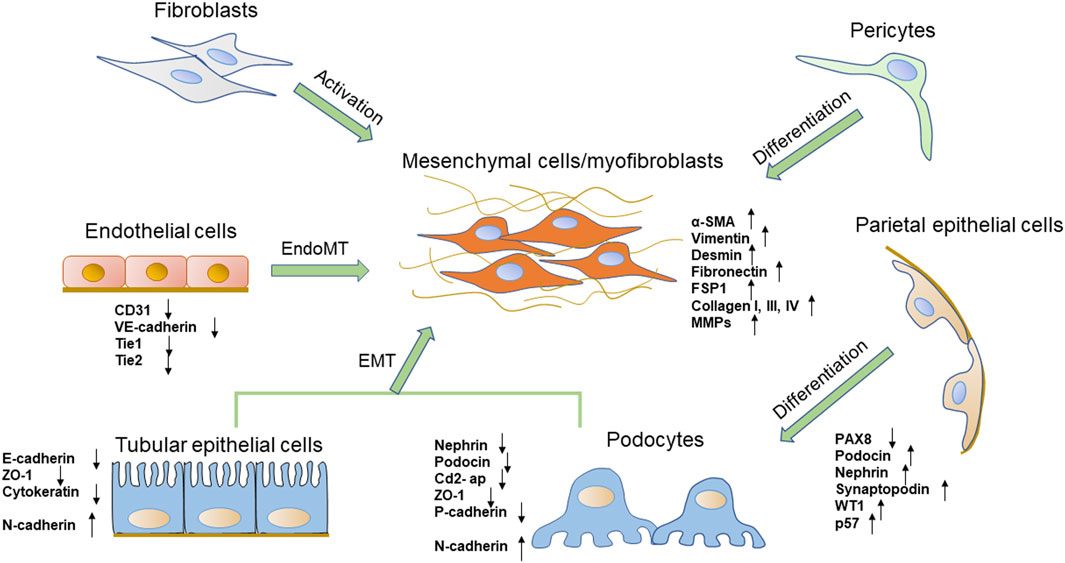
FIGURE 1. The phenotypic transitions of several types of renal cells and the expressions of markers. The origin of the myofibroblasts includes several types of cells, such as fibroblasts, pericytes, tubular epithelial cells, and endothelial cells. Podocytes also obtain a mesenchymal property through mesenchymal transition. PECs are one of the possible sources of podocytes.
However, it should be noted that the phenotype transition of podocytes does not satisfy all the criteria for EMT. First of all, one of the criteria for EMT is a cadherin switch from E-cadherin to N-cadherin. But podocytes express P-cadherin instead of E-cadherin and undergo a cadherin switch from P-cadherin to N-cadherin, which precludes them from matching this definition of EMT. Besides, no evidence showed that podocytes expressed markers of EMT like heat shock protein 47 (HSP47) and discoidin or domain receptor 2 (DDR2) in the literature (Zeisberg and Neilson, 2009; Galichon et al., 2011; May et al., 2014; Kim et al., 2017).
4.2 PECs transdifferentiation in DN
Recent studies have suggested that PECs are directly involved in the pathogenesis of certain glomerular diseases. PECs, which line the inner surface of Bowman’s capsule, resemble squamous epithelial cells and form primary cilia. PECs and podocytes arise from the same ancestral cell but unlike podocytes, PECs maintain the ability to proliferate under physiological conditions (Shankland et al., 2014). They act as a selective permeability barrier to the urinary filtrate and prevent proteins in the glomerular ultrafiltrate in the Bowman’s space from entering the extraglomerular space (Ohse et al., 2009). Tight junctions are present between adjacent PECs in normal glomeruli, The junction proteins including claudin-1, ZO-1 and occludin form an impermeable barrier. PECs could increase the potential for proliferation, migration, production of extracellular matrix and chemokines and cytokines, and expression of certain markers (Holderied et al., 2015; Lazareth et al., 2020). Researchers have found that PECs serve as potential reparative and regenerative precursors for podocytes when podocytes are lost (Appel et al., 2009; Kietzmann et al., 2015). PECs proliferate with aging and differentiate into podocytes to supplement podocyte deletion. They could co-express PEC and podocyte-specific proteins, acquire ultrastructural features of podocytes and migrate from Bowman’s capsule to the capillary tuft (Kaverina et al., 2020). Activated PECs can express specific podocytes markers, like podocin, nephrin, synaptopodin, Wilms’ tumor 1 (WT1), p57 and they still express the activation marker for PECs, CD44, but no longer express the PEC marker paired box 8 (PAX8) and lose the capacity to proliferate (Figure 1) (Schulte et al., 2014; Kaverina et al., 2020). Several studies have shown the importance of the activation of PECs as progenitor cells to regenerate podocytes in human and mouse DN models (Pichaiwong et al., 2013; Andeen et al., 2015). Activated and proliferated PECs were observed in diabetic rats and were positively correlated with the extent of proteinuria and podocyte loss (Zhao et al., 2011). In another DN model, authors have identified that glomerular PECs could function as a possible source of regenerating podocyte progenitor for podocyte restoration, which may reverse or prevent DN (Pichaiwong et al., 2013; Luna-Antonio et al., 2017). The mechanism of PECs activation and PECs transition is rarely understood. Notch signaling pathway and extracellular signal-regulated kinase (ERK) signaling pathway may involve in the transition from PECs to podocytes (Sweetwyne and Susztak, 2013; Urushihara et al., 2020). Wnt/β-catenin signaling pathway, which is a widely recognized pathway that mediates cell differentiation, is also likely involved in PECs transition (Ni et al., 2021). However, more researches need to be employed for a better understanding of the potential role of activated PECs in DN.
5 Cellular transdifferentiation in the tubulointerstitium
5.1 EMT of TECs in DN
EMT of renal TECs is one of the major pathogeneses of renal interstitial fibrosis. TECs are postulated to contribute to the phenotypic change in myofibroblasts through the process of EMT (Liu, 2010; Galichon and Hertig, 2011). During that process, injured TECs are activated and lose their epithelial phenotype and acquire new characteristic features of mesenchymal cells. The processes are summarized as four key events. First, TECs lose epithelial cell junctions and cell polarity. Then, the epithelial genes such as E-cadherin, ZO-1 are suppressed, and mesenchymal markers including α-SMA, vimentin, FSP1, type I collagen and FN are activated (Liu et al., 2018a) (Figure 1). E-cadherin plays a crucial role in maintaining the structural integrity of renal epithelia and its polarization, and the downregulation of the E-cadherin expression is an important change in EMT (Kalluri and Neilson, 2003). Next, the cytoskeletal architecture reorganizes and changes cellular morphology. Finally, they obtain the ability to secrete factors like matrix metalloproteinases (MMPs) to disrupt the basement membrane and enhanced invasive behavior (Yang and Liu, 2001; Lamouille et al., 2014).
TECs and myofibroblasts are normally separated by a tubular basement membrane, which prevents contact between TECs and interstitial matrix components (Fan et al., 1999). The completion of an EMT is signaled by the degradation of the underlying basement membrane and TECs increase motility, through which they cross the barrier and migrates into the interstitium (Kalluri and Weinberg, 2009; Das et al., 2019). However, studies have questioned the role of EMT in renal fibrosis based on evidence that epithelial cells actively cross the tubular basement membrane and could not find a direct contribution of epithelial cells to the myofibroblast population (Humphreys et al., 2010; LeBleu et al., 2013; Lovisa et al., 2016). Recent studies offered new insights into the potential role of tubular EMT in renal fibrosis. They found a large population of differentiated TECs co-expressed epithelial and mesenchymal markers and remained associated with their basement membrane after renal injury (LeBleu et al., 2013; Lovisa et al., 2016). This type of renal epithelial cell is considered to have undergone incomplete EMT called partial EMT. Partial EMT is in agreement with earlier studies (Kriz et al., 2011; Hajarnis et al., 2018). Although TECs remain attached to the basement membrane during fibrosis rather than cross the basement membrane to join the myofibroblast pool, evidence has demonstrated partial EMT is sufficient to induce tubular function impairment. The expression of TECs fundamental proteins such as E-cadherin and ZO-1 reduces, which impairs TECs polarity and junction, and by triggering cell cycle arrest impairing the repair of the damaged tissue. Partial EMT also modifies the epithelial secretome profile thereby promoting the secretion of profibrogenic cytokines and fibrosis (Grande et al., 2015; Lovisa et al., 2015).
5.2 Fibroblast differentiation in DN
Fibroblasts, the main cells of the renal interstitial space, are important for maintaining the structural integrity of kidneys by maintaining constituent ECM. About 50% of myofibroblasts in unilateral ureteral obstruction (UUO) are shown to be originated from resident fibroblasts via proliferation (LeBleu et al., 2013). Aberrant activation of fibroblasts and transformation into myofibroblasts contribute to progressive fibrotic events, which are the main cellular source of the ECM and contribute significantly to the pathogenesis of kidney fibrosis (Chen et al., 2019). Activated fibroblasts acquire smooth muscle features including expression of desmin and α-SMA (Figure 1) (Gabbiani, 2003; Hinz et al., 2007). Fibroblasts are activated by inflammatory cytokines, growth factors, hypoxia and mechanical forces to proliferate and produce ECM components like collagen I, III, IV and fibronectin (Kessler et al., 2001). Injured TECs are potent producers of growth factors and cytokines such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), hedgehog, and Wnt ligands, which could cause fibroblasts to transform into myofibroblasts (Dussaule et al., 2011; Meran and Steadman, 2011). TGF-β-mediated fibroblast activation is thought to be the major driver of kidney fibrosis in DN (Desmouliere et al., 1993; Gerrits et al., 2020).
6 Cellular transdifferentiation from microvessels
ECs and pericytes are the two main cellular constituents in renal microvessels. Both are of great interest for research on phenotypic transitions in the development of DN.
6.1 EndoMT in DN
The kidney is a highly vascularized organ, characterized by a remarkable diversity of EC populations (Verma and Molitoris, 2015). The glomerular ECs contribute to the glomerular filtration barrier and support podocyte structure, whereas ECs of the microvasculature around kidney tubules contribute to tubular reabsorption (Jourde-Chiche et al., 2019). When renal ECs are injured, the production of nitric oxide (NO) decreases and vasodilation function is impaired, which decreases renal blood flow and initiation of EndoMT (Basile et al., 2018). EndoMT, which happens in tubulointerstitial vessels as well as glomerular ECs, appears to play a significant role in the development and progression of DN (Lu et al., 2021). In 2008, a study by Zeisberg et al. (2008) firstly identified the crucial role of EndoMT in DN. Studies demonstrated a remarkable ratio of myofibroblasts that could co-expressed endothelial markers and myofibroblast markers in streptozotocin (STZ)-induced DN, indicating the endothelial origin of myofibroblasts. They declared that the 30–50% activated fibroblast cells were derived from ECs. Another study investigated the role of EndoMT in mice and demonstrated that 10–24% of renal interstitial myofibroblasts emerged via EndoMT in STZ-induced DN mice (Song et al., 2019). However, a study in 2013 revealed that only 10% of myofibroblasts were of endothelial origin following kidney injury (LeBleu et al., 2013). EndoMT is also observed co-expression of endothelial and mesenchymal markers in glomeruli of DN patients. During EndoMT, renal ECs gradually lose their specific endothelial cell markers, such as platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31), VE-cadherin, tyrosine kinase with immunoglobulin like and EGF like domains 1 (Tie1) and TEK Receptor Tyrosine Kinase (Tie2), while acquiring mesenchymal or myofibroblastic phenotype genes and proteins, such as α-SMA, N-cadherin, FSP-1 and increase the production of ECM proteins (Figure 1) (Sanchez-Duffhues et al., 2018). Meanwhile, ECs lose endothelial characteristics such as increased vascular permeability, polarization and cell-cell connections. Thus, these cells get a high migration potential and promote the development of renal fibrosis (Kizu et al., 2009). Dysfunctional glomerular ECs could communicate with adjacent podocytes and mesangial cells via secreting effectors, which aggravates glomerular injury and creates a vicious cycle that leads to proteinuria in diabetic patients (Fu et al., 2015).
6.2 Pericytes differentiation in DN
Pericytes are present in the microvasculature, at terminal arterioles, capillaries and post-capillary venules. They share a common basement membrane with ECs (Stratman et al., 2009; van Dijk et al., 2015). In the kidney, pericytes are located in the tubular system called peritubular pericytes, and in renal glomeruli called mesangial cells (also described as pericyte-like cells) (Schlondorff, 1987; Geevarghese and Herman, 2014). Mesangial cells play an important role in supporting glomerular capillary network. They display pericyte functions by regulating glomerular blood flow and controlling glomerular filtration rate due to their contractility (Kramann and Humphreys, 2014). Like pericytes, mesangial cells express PDGFR-β and generate mesangial basement membrane molecules, such as collagen IV and FN (Isono et al., 2002). PDGFR-β has a critical role in pericyte activation, proliferation, differentiation and communication with ECs (Leveen et al., 1994). According to experimental and clinical studies, mesangial cells are considered to be an important precursor cell type of myofibroblasts in DN (Simonson, 2007) (Figure 1). High glucose enhances mesangial cells to undergo proliferation and alters the normal phenotype to express α-SMA and other growth factors, causing mesangial matrix expansion (Koya et al., 2000; Isono et al., 2002).
In the tubulointerstitial system, peritubular pericytes are essential for peritubular capillary integrity as well as tubular integrity (Kida et al., 2013). Under pathological conditions, peritubular pericytes are activated and are the key precursor of myofibroblasts. Pericytes subsequently migrate from the peritubular capillaries into the interstitium and differentiation into myofibroblasts resulting in scar formation, progressive fibrosis and deterioration of renal function (Lin et al., 2008; Humphreys et al., 2010). Meanwhile, pericytes differentiation into myofibroblasts also loses vessel stabilizing abilities leading to peritubular capillary disintegration and rarefaction after the injury (Schrimpf et al., 2012). Pericytes also synthesize erythropoietin (EPO) in normal kidneys, while myofibroblasts lose the ability to produce EPO, leading to renal anemia and thereby enhanced tissue hypoxia (Chang et al., 2016; Souma et al., 2016). Studies showed inhibition of PDGFR signaling prevented the proliferation and differentiation of pericytes and reduced interstitial kidney fibrosis. Therefore, targeting pericytes may provide a novel strategy to prevent kidney fibrosis (Chen et al., 2011). However, the studies involved in the role of pericytes in DN are limited. Future studies should continue identifying the specific contribution of pericytes to DN and renal fibrosis (Hayes, 2019).
7 Molecular pathways mediating phenotypic transitions in DN
Various molecular pathways in phenotypic transitions in renal diseases have been studied. But in DN, research is still limited (Table 1). TGF-β plays a significant role in phenotypic transitions of renal cells (Figure 2). In elevated glucose levels, TGF-β increases in podocytes, TECs, ECs and pericytes to induce mesenchymal transitions via several SMAD family member (Smad)-dependent and Smad-independent signal transduction pathways (Hills and Squires, 2010; Xie et al., 2015; Wu et al., 2017; Dong et al., 2019a). In the classic TGF-β/Smad signaling pathway, TGF-β binds to its receptor TGF-βR1, and then Smad2/3 is phosphorylated and combines with Smad4 to form a Smad complex, followed by translocation into the nucleus and activation of transcription of pro-fibrotic genes like α-SMA, connective tissue growth factor (CTGF), MMP-9 and Snail. Several research studies have been conducted focusing on the regulation of TGF-β/Smad signaling pathway (Lan, 2012). The negative regulator ski-related protein N (SnoN), Sirtuin 6 (Sirt6) prevented nuclear translocation or deacetylated Smad3. Many micro-RNA like mir-17, mir-30c and circRNA could inhibit TGF-β/Smad signaling pathway (Zhao et al., 2017; Srivastava et al., 2019; Fu et al., 2021). Like EMT, the TGF-β/Smad-dependent signaling pathway has also become a hot topic in EndoMT research (Li et al., 2010). TGF-β/Smad-dependent signaling pathway induces specific target genes, including Snail1, Snail2 and Twist, leading to the initiation of EndoMT (Song et al., 2019; Gwon et al., 2020). Inhibition of EndoMT by blockade of TGF-β/Smad signaling could reduce renal fibrosis, and retard the progression of nephropathy in STZ-induced DN (Shi et al., 2020). However, Smad-independent pathways of TGF-β signaling involved in phenotypic transitions include ras homolog gene family member A (RhoA) (Bijkerk et al., 2012; Zhang et al., 2013), extracellular signal-regulated kinase (ERK) (Zhou et al., 2010; Li et al., 2017a; Dong et al., 2019b), p38 mitogen-activated protein kinase (MAPK) (Bakin et al., 2002; Li et al., 2017a; Deng et al., 2020; Li et al., 2022), and phopshatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) (Lee and Han, 2010; Lu et al., 2019), Ras (Su et al., 2020), integrin-linked kinase (ILK) (Du et al., 2016; Qi et al., 2018; Li et al., 2019a), nuclear factor kappa B (NF-κB) (Li et al., 2012; Wei et al., 2021) and β-catenin (Wang et al., 2011). For example, ERK could disassemble adherens junctions and promote cell motility by controlling the expression of E-cadherin and ZO-1 (Xie et al., 2004). C-reactive protein (CRP) promoted EMT only in the presence of TGF-β via ERK1/2 signaling (Zhang et al., 2019). Heparanase could also promote EndoMT via activating ERK signaling (Chang et al., 2022). Methyltransferase Like 14 (METTL14) regulated PI3K/Akt signaling pathway via phosphatase and tensin homolog (PTEN), which affected EMT of renal tubular cells in DN (Xu et al., 2021). The Smad-dependent and Smad-independent pathways can coordinate in the regulation of phenotypic transitions.
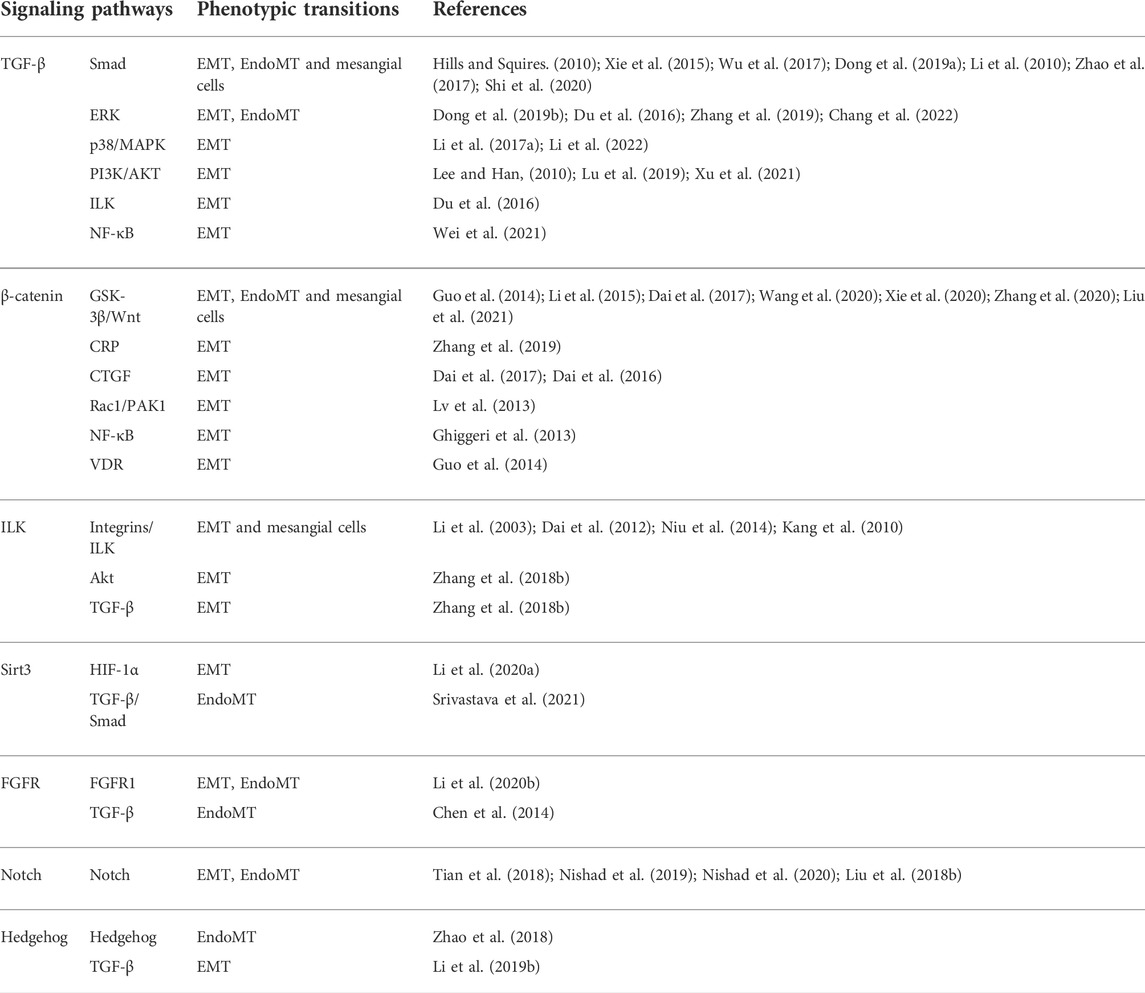
TABLE 1. The major signaling pathways involved in phenotypic transitions in DN.
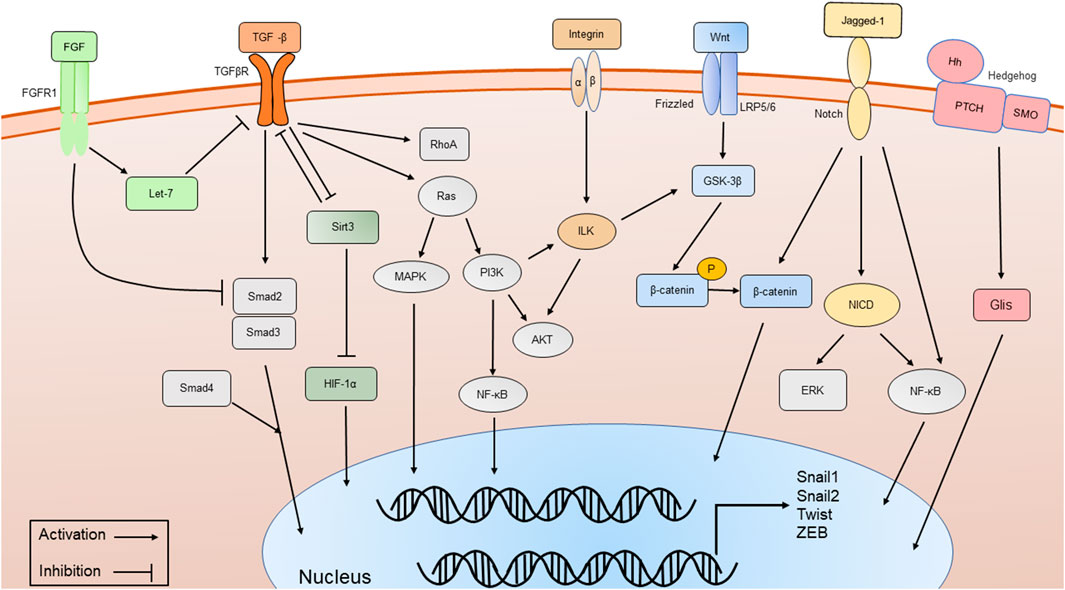
FIGURE 2. The major signaling pathways involved in phenotypic transitions, including TGF-β/Smad-dependent and Smad-independent signal transduction pathways, Wnt/β-catenin signaling pathway, Integrin/ILK signaling pathway, Notch signaling pathway, Sirt3 signaling pathway, FGFR1 and Hedgehog signaling pathway.
Another key molecule is named β-catenin (Figure 2). Wnt/β-catenin signaling pathway expression is increased in podocytes, TECs and ECs of hyperglycemic patients, as well as mouse models and plays a critical role in integrating cell differentiation (Lee and Han, 2010; Li et al., 2015; Dai et al., 2017). β-catenin assembles in the cytoplasm and translocates to the nucleus, regulates the expression of Wnt target genes and enhances myofibrogenesis. Suppressing the Wnt/β-catenin signaling pathway has been shown to restrain renal fibrosis. Many compounds like Panax Notoginseng and Astragaloside IV have been proven to exert renal protection by inhibiting the Wnt/β-catenin signaling pathways in STZ-induced animal models (Wang et al., 2020; Xie et al., 2020; Zhang et al., 2020). Glycogen synthase kinase 3β (GSK-3β) showed an increased expression and promoted the EMT of podocytes in high glucose by classical GSK- 3β/Wnt/β-catenin (Guo et al., 2014). CRP, CTGF and RAS-related C3 botulinum toxin substrate 1 (Rac1)/p21-activated kinase 1 (PAK1), NF-κB signaling could promote podocyte EMT by regulating the nuclear translocation of β-catenin under a high-glucose condition (Ghiggeri et al., 2013; Lv et al., 2013; Dai et al., 2016; Zhang et al., 2019). In contrast, Caudal-type homeobox transcription factor 2 (CDX2) could improve renal EMT during DN by suppressing the Wnt/β-catenin signaling pathway (Liu et al., 2021). Vitamin D receptor (VDR) competed with β-catenin in combination with transcription factor 4 (TCF4) and inhibited the activity of β-catenin. In the high-glucose environment, VDR decreased and aggravated EMT (Guo et al., 2014).
Integrin-linked kinase (ILK) is an intracellular serine/threonine kinase that interacts with integrins and activates several downstream effectors such as Akt and GSK-3β, which are involved in the regulation of cell adhesion, cell shape changes, and deposition of the ECM[(Naves et al., 2012; Zhang et al., 2018b)]. Integrins/ILK signaling pathway (Figure 2) has been proven to induce EMT in TECs and podocytes (Li et al., 2003; Dai et al., 2012; Niu et al., 2014). As a protein kinase, ILK directly phosphorylates several physiologically important downstream effector kinases including p38 MAPK, Akt and GSK-3β, leading to the stabilization and accumulation of β-catenin (Hannigan et al., 2005). The inhibition of ILK ameliorated proteinuria or attenuates EMT (Li et al., 2009; Kang et al., 2010). The effect of ILK also has been identified as partially regulated by TGF-β/Smad signaling pathway. Inhibition of ILK could block podocytes EMT induced by TGF-β (Li et al., 2003). Hesperetin produced protective effects in podocyte EMT possibly by suppressing TGF-β/ILK/Akt signaling in DN (Zhang et al., 2018b).
Mitochondrial deacetylase sirtuin 3 (Sirt3) and fibroblast growth factor receptors (FGFRs) may be the key signaling pathways that inhibit EMT and EndoMT (Figure 2) (Li et al., 2017b; Li et al., 2020a; Srivastava et al., 2021). Sirt3 deficiency induces hypoxia inducible factor 1α (HIF-1α) overaccumulation, which results in kidney fibrosis via induction of the EMT (Higgins et al., 2007; Sun et al., 2009). Sodium-glucose Cotransporter-2 (SGLT2) inhibitor empagliflozin has direct effects on tubule metabolic pathways associated with the inhibition of aberrant glycolysis and restoration of Sirt3 expression, resulting in suppression of the EMT (Li et al., 2020a). Studies showed endothelial Sirt3 pathway regulated glucose and lipid metabolism and related EndoMT processes by maintaining control of TGF-β/Smad3 signaling in the kidney of diabetic mice. Sirt3 knockout in mice resulted in increased levels of EndoMT and reactive oxygen species (ROS), and promoted renal dysfunction, while in Sirt3 knock-in EC-specific transgenic mice, renal fibrosis and EndoMT, as well as oxidative stress, were ameliorated (Srivastava et al., 2021). Studies observed that fibroblast growth factors (FGF)/FGFR1 signaling was downregulated in the kidney of DN (Baelde et al., 2004; Liang et al., 2018). FGF/FGFR1 signaling has been confirmed to suppress TGF-β signaling in ECs (Chen et al., 2014). Diabetic FGFR1 knockout mice have significantly higher EndoMT and EMT levels and severe organ fibrosis (Li et al., 2020b).
Notch signaling has been shown to mediate EMT and EndoMT in DN (Asfahani et al., 2018; Tian et al., 2018; Nishad et al., 2019). Notch receptors on the cell surface bind various ligands, including Jagged-1, resulting in the cleavage of Notch receptor by proteases and notch intracellular domain (NICD) translocates to the nucleus to regulate the expression of transcription factors like Snails (Hayward et al., 2008). Growth hormone induced Notch signaling in podocytes and contributed to proteinuria through EMT as well as renal fibrosis (Nishad et al., 2019). Advanced glycation end-products (AGEs) could activate Notch signaling in podocytes and provoke EMT (Nishad et al., 2020). Notch also indirectly regulates EMT through various signaling pathways, including NF-κB and β-catenin, and through the action of various regulatory miRNAs (Yang et al., 2011) (Figure 2). Overexpression of Notch in ECs resulted in the loss of VE-cadherin and subsequent EndoMT (Timmerman et al., 2004). Over-expression of NUMB endocytic adaptor protein (Numb) inhibited the expression of Notch signaling within EMT of DN (Liu et al., 2018b). Notch signaling activation mediated TGF-β-induced EMT in both human and rat TECs in cultured (Bielesz et al., 2010). MMP-9 played an important role in TGF-β-induced EndoMT via upregulation of Notch signaling in glomerular ECs (Zhao et al., 2016).
Hedgehog signaling pathway (HH) is a well-known pathway in EMT (Gonzalez and Medici, 2014). Members of the Hedgehog family contain sonic hedgehog (Shh), Indian hedgehog (Ihh), and desert hedgehog (Dhh). The HH pathway consists of two transmembrane proteins, patched (Ptch) and smoothened (Smo) (Figure 2). Active Smo promotes transcription factors GLI family zinc finger (Glis) nuclear translocation. Mesenchymal stem cells reversed the renal injury and EMT process by blocking NF-κB/hedgehog pathways (Xiao et al., 2020). Hyperglycemia elevated renal hedgehog-interacting protein (Hhip) gene expression subsequently activated the TGFβ1/Smad2/3 cascade and promotes EndoMT (Zhao et al., 2018; Zhao et al., 2021). PTEN could induce partial EMT through Hedgehog signaling pathway in DN mice (Li et al., 2019b).
8 Conclusion and perspective
DN is closely related to renal function loss in patients and is one of the major causes of end stage renal disease throughout the world. Renal cellular phenotypic transitions have become one of the most fascinating topics in the studies of organ fibrosis in recent years. In this review, we discussed the cellular phenotypic transitions in the kidney. TECs, ECs, fibroblasts and pericytes are important precursors of myofibroblasts. Podocytes also undergo similar phenotypic changes to EMT under high glucose conditions, which promote the development of glomerulosclerosis. PECs proliferate and differentiate into podocytes when podocyte loss occurs during injury. Most of the known molecular mechanisms and downstream interactions between TGF-β/Smad classic pathway and multiple other pathways of phenotypic transitions in DN are summarized. However, the understanding of some processes like pericytes to myofibroblasts, PECs to podocytes, or EndoMT in DN has just begun. Various molecular mechanisms remain unclear in DN. More work in the future should focus on deeply exploring the role of these cells and strive to find potential new targets for the treatment of renal fibrotic diseases, which is of great significance for the development of DN treatment.
Author contributions
YC prepared and wrote the original version of the manuscript. J-HL, H-PH, and CZ reviewed and edited the manuscript. All authors have read and agreed to the final version of the manuscript.
Funding
This study was financially supported by the National Natural Science Foundation of China (81961138007, 81974096, 81770711, 81873602, 81800610 and 81974097).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akhtar, M., Taha, N. M., Nauman, A., Mujeeb, I. B., and Al-Nabet, A. (2020). Diabetic kidney disease: Past and present. Adv. Anat. Pathol. 27 (2), 87–97. doi:10.1097/PAP.0000000000000257
American Diabetes Association Professional Practice, C. (2022). 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2022. Diabetes Care 45, S17–S38. doi:10.2337/dc22-S002
Andeen, N. K., Nguyen, T. Q., Steegh, F., Hudkins, K. L., Najafian, B., and Alpers, C. E. (2015). The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int. 88 (5), 1099–1107. doi:10.1038/ki.2015.273
Appel, D., Kershaw, D. B., Smeets, B., Yuan, G., Fuss, A., Frye, B., et al. (2009). Recruitment of podocytes from glomerular parietal epithelial cells. J. Am. Soc. Nephrol. 20 (2), 333–343. doi:10.1681/ASN.2008070795
Asfahani, R. I., Tahoun, M. M., Miller-Hodges, E. V., Bellerby, J., Virasami, A. K., Sampson, R. D., et al. (2018). Activation of podocyte Notch mediates early Wt1 glomerulopathy. Kidney Int. 93 (4), 903–920. doi:10.1016/j.kint.2017.11.014
Baelde, H. J., Eikmans, M., Doran, P. P., Lappin, D. W., de Heer, E., and Bruijn, J. A. (2004). Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am. J. Kidney Dis. 43 (4), 636–650. doi:10.1053/j.ajkd.2003.12.028
Bakin, A. V., Rinehart, C., Tomlinson, A. K., and Arteaga, C. L. (2002). p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell. Sci. 115, 3193–3206. doi:10.1242/jcs.115.15.3193
Barrallo-Gimeno, A., and Nieto, M. A. (2005). The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 132 (14), 3151–3161. doi:10.1242/dev.01907
Basile, D. P., Collett, J. A., and Yoder, M. C. (2018). Endothelial colony-forming cells and pro-angiogenic cells: Clarifying definitions and their potential role in mitigating acute kidney injury. Acta Physiol. 222 (2), e12914. doi:10.1111/apha.12914
Bielesz, B., Sirin, Y., Si, H., Niranjan, T., Gruenwald, A., Ahn, S., et al. (2010). Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Invest. 120 (11), 4040–4054. doi:10.1172/JCI43025
Bijkerk, R., de Bruin, R. G., van Solingen, C., Duijs, J. M., Kobayashi, K., van der Veer, E. P., et al. (2012). MicroRNA-155 functions as a negative regulator of RhoA signaling in TGF-beta-induced endothelial to mesenchymal transition. Microrna 1 (1), 2–10. doi:10.2174/2211536611201010002
Carew, R. M., Wang, B., and Kantharidis, P. (2012). The role of EMT in renal fibrosis. Cell. Tissue Res. 347 (1), 103–116. doi:10.1007/s00441-011-1227-1
Chang, K., Xie, Q., Niu, J., Gu, Y., Zhao, Z., Li, F., et al. (2022). Heparanase promotes endothelial-to-mesenchymal transition in diabetic glomerular endothelial cells through mediating ERK signaling. Cell. Death Discov. 8 (1), 67. doi:10.1038/s41420-022-00858-0
Chang, Y. T., Yang, C. C., Pan, S. Y., Chou, Y. H., Chang, F. C., Lai, C. F., et al. (2016). DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Invest. 126 (2), 721–731. doi:10.1172/JCI82819
Chen, P. S., Li, Y. P., and Ni, H. F. (2019). Morphology and evaluation of renal fibrosis. Adv. Exp. Med. Biol. 1165, 17–36. doi:10.1007/978-981-13-8871-2_2
Chen, P. Y., Qin, L., Tellides, G., and Simons, M. (2014). Fibroblast growth factor receptor 1 is a key inhibitor of TGFβ signaling in the endothelium. Sci. Signal. 7 (344), ra90. doi:10.1126/scisignal.2005504
Chen, Y. T., Chang, F. C., Wu, C. F., Chou, Y. H., Hsu, H. L., Chiang, W. C., et al. (2011). Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 80 (11), 1170–1181. doi:10.1038/ki.2011.208
Collins, A. J., Foley, R. N., Chavers, B., Gilbertson, D., Herzog, C., Ishani, A., et al. (2014). US renal data system 2012 annual data report. Am. J. Kidney Dis. 63, e1–e476. doi:10.1053/j.ajkd.2012.11.031
Dai, H., Zhang, Y., Yuan, L., Wu, J., Ma, L., and Shi, H. (2016). CTGF mediates high-glucose induced epithelial-mesenchymal transition through activation of beta-catenin in podocytes. Ren. Fail. 38 (10), 1711–1716. doi:10.3109/0886022X.2016.1158069
Dai, H. Y., Ma, L. N., Cao, Y., Chen, X. L., Shi, H., Fan, Y. P., et al. (2017). Protection of CTGF antibody against diabetic nephropathy in mice via reducing glomerular beta-catenin expression and podocyte epithelial-mesenchymal transition. J. Cell. Biochem. 118 (11), 3706–3712. doi:10.1002/jcb.26017
Dai, H. Y., Zheng, M., Lv, L. L., Tang, R. N., Ma, K. L., Liu, D., et al. (2012). The roles of connective tissue growth factor and integrin-linked kinase in high glucose-induced phenotypic alterations of podocytes. J. Cell. Biochem. 113 (1), 293–301. doi:10.1002/jcb.23355
Das, V., Bhattacharya, S., Chikkaputtaiah, C., Hazra, S., and Pal, M. (2019). The basics of epithelial-mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J. Cell. Physiol. 234, 14535–14555. doi:10.1002/jcp.28160
DeFronzo, R. A., Reeves, W. B., and Awad, A. S. (2021). Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 17 (5), 319–334. doi:10.1038/s41581-021-00393-8
Dejana, E., Hirschi, K. K., and Simons, M. (2017). The molecular basis of endothelial cell plasticity. Nat. Commun. 8, 14361. doi:10.1038/ncomms14361
Deng, B., Yang, W., Wang, D., Cheng, L., Bu, L., Rao, J., et al. (2020). Peptide DR8 suppresses epithelial-to-mesenchymal transition via the TGF-β/MAPK signaling pathway in renal fibrosis. Life Sci. 261, 118465. doi:10.1016/j.lfs.2020.118465
Desmouliere, A., Geinoz, A., Gabbiani, F., and Gabbiani, G. (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell. Biol. 122 (1), 103–111. doi:10.1083/jcb.122.1.103
Domingo-Lopez, D. A., Lattanzi, G., Hjs, L., Wallace, E. J., Wylie, R., O'Sullivan, J., et al. (2022). Medical devices, smart drug delivery, wearables and technology for the treatment of Diabetes Mellitus. Adv. Drug Deliv. Rev. 185, 114280. doi:10.1016/j.addr.2022.114280
Dong, Z., Sun, Y., Wei, G., Li, S., and Zhao, Z. (2019). A nucleoside/nucleobase-rich extract from cordyceps sinensis inhibits the epithelial-mesenchymal transition and protects against renal fibrosis in diabetic nephropathy. Molecules 24 (22), E4119. doi:10.3390/molecules24224119
Dong, Z., Sun, Y., Wei, G., Li, S., and Zhao, Z. (2019). Ergosterol ameliorates diabetic nephropathy by attenuating mesangial cell proliferation and extracellular matrix deposition via the TGF-β1/smad2 signaling pathway. Nutrients 11 (2), E483. doi:10.3390/nu11020483
Du, M., Wang, Q., Li, W., Ma, X., Wu, L., Guo, F., et al. (2016). Overexpression of FOXO1 ameliorates the podocyte epithelial-mesenchymal transition induced by high glucose in vitro and in vivo. Biochem. Biophys. Res. Commun. 471 (4), 416–422. doi:10.1016/j.bbrc.2016.02.066
Duffield, J. S. (2014). Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest. 124 (6), 2299–2306. doi:10.1172/JCI72267
Dussaule, J. C., Guerrot, D., Huby, A. C., Chadjichristos, C., Shweke, N., Boffa, J. J., et al. (2011). The role of cell plasticity in progression and reversal of renal fibrosis. Int. J. Exp. Pathol. 92 (3), 151–157. doi:10.1111/j.1365-2613.2011.00760.x
Fan, J. M., Ng, Y. Y., Hill, P. A., Nikolic-Paterson, D. J., Mu, W., Atkins, R. C., et al. (1999). Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56 (4), 1455–1467. doi:10.1046/j.1523-1755.1999.00656.x
Faul, C., Asanuma, K., Yanagida-Asanuma, E., Kim, K., and Mundel, P. (2007). Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell. Biol. 17 (9), 428–437. doi:10.1016/j.tcb.2007.06.006
Feng, J., Hu, S., Liu, K., Sun, G., and Zhang, Y. (2022). The role of MicroRNA in the regulation of tumor epithelial-mesenchymal transition. Cells 11 (13), 1981. doi:10.3390/cells11131981
Fioretto, P., and Mauer, M. (2007). Histopathology of diabetic nephropathy. Semin. Nephrol. 27 (2), 195–207. doi:10.1016/j.semnephrol.2007.01.012
Fu, H., Chu, D., and Geng, X. (2021). Downregulation of miR-17 suppresses TGF-β1-mediated renal fibrosis through targeting Smad7. Mol. Cell. Biochem. 476 (8), 3051–3064. doi:10.1007/s11010-021-04140-2
Fu, J., Lee, K., Chuang, P. Y., Liu, Z. H., and He, J. C. (2015). Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 308 (4), F287–F297. doi:10.1152/ajprenal.00533.2014
Gabbiani, G. (2003). The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200 (4), 500–503. doi:10.1002/path.1427
Gajjala, P. R., Sanati, M., and Jankowski, J. (2015). Cellular and molecular mechanisms of chronic kidney disease with diabetes mellitus and cardiovascular diseases as its comorbidities. Front. Immunol. 6, 340. doi:10.3389/fimmu.2015.00340
Galichon, P., and Hertig, A. (2011). Epithelial to mesenchymal transition as a biomarker in renal fibrosis: Are we ready for the bedside? Fibrogenes. Tissue Repair 4, 11. doi:10.1186/1755-1536-4-11
Galichon, P., Vittoz, N., Xu-Dubois, Y. C., Cornaire, E., Vandermeersch, S., Mesnard, L., et al. (2011). Epithelial phenotypic changes detect cyclosporine in vivo nephrotoxicity at a reversible stage. Transplantation 92 (9), 993–998. doi:10.1097/TP.0b013e31822fa495
Garg, P., and Holzman, L. B. (2012). Podocytes: Gaining a foothold. Exp. Cell. Res. 318 (9), 955–963. doi:10.1016/j.yexcr.2012.02.030
Geevarghese, A., and Herman, I. M. (2014). Pericyte-endothelial crosstalk: Implications and opportunities for advanced cellular therapies. Transl. Res. 163 (4), 296–306. doi:10.1016/j.trsl.2014.01.011
Gerrits, T., Zandbergen, M., Wolterbeek, R., Bruijn, J. A., Baelde, H. J., and Scharpfenecker, M. (2020). Endoglin promotes myofibroblast differentiation and extracellular matrix production in diabetic nephropathy. Int. J. Mol. Sci. 21 (20), E7713. doi:10.3390/ijms21207713
Ghiggeri, G. M., Gigante, M., and Di Donato, A. (2013). Constitutional nephrin deficiency in conditionally immortalized human podocytes induced epithelial-mesenchymal transition, supported by beta-catenin/NF-kappa B activation: A consequence of cell junction impairment? Int. J. Nephrol. 2013, 457490. doi:10.1155/2013/457490
Gonzalez, D. M., and Medici, D. (2014). Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 7 (344), re8. doi:10.1126/scisignal.2005189
Grande, M. T., Sanchez-Laorden, B., Lopez-Blau, C., De Frutos, C. A., Boutet, A., Arevalo, M., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21 (9), 989–997. doi:10.1038/nm.3901
Greka, A., and Mundel, P. (2012). Cell biology and pathology of podocytes. Annu. Rev. Physiol. 74, 299–323. doi:10.1146/annurev-physiol-020911-153238
Gujarati, N. A., Leonardo, A. R., Vasquez, J. M., Guo, Y., Frimpong, B. O., Fozilov, E., et al. (2021). Loss of functional SCO2 attenuates oxidative stress in diabetic kidney disease. Diabetes 71, 142–156. doi:10.2337/db21-0316
Guo, J., Xia, N., Yang, L., Zhou, S., Zhang, Q., Qiao, Y., et al. (2014). GSK-3β and vitamin D receptor are involved in β-catenin and snail signaling in high glucose-induced epithelial-mesenchymal transition of mouse podocytes. Cell. Physiol. biochem. 33 (4), 1087–1096. doi:10.1159/000358678
Gwon, M. G., An, H. J., Kim, J. Y., Kim, W. H., Gu, H., Kim, H. J., et al. (2020). Anti-fibrotic effects of synthetic TGF-β1 and Smad oligodeoxynucleotide on kidney fibrosis in vivo and in vitro through inhibition of both epithelial dedifferentiation and endothelial-mesenchymal transitions. FASEB J. 34 (1), 333–349. doi:10.1096/fj.201901307RR
Hajarnis, S., Yheskel, M., Williams, D., Brefort, T., Glaudemans, B., Debaix, H., et al. (2018). Suppression of microRNA activity in kidney collecting ducts induces partial loss of epithelial phenotype and renal fibrosis. J. Am. Soc. Nephrol. 29 (2), 518–531. doi:10.1681/ASN.2017030334
Hannigan, G., Troussard, A. A., and Dedhar, S. (2005). Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 5 (1), 51–63. doi:10.1038/nrc1524
Hayes, K. L. (2019). Pericytes in type 2 diabetes. Adv. Exp. Med. Biol. 1147, 265–278. doi:10.1007/978-3-030-16908-4_12
Hayward, P., Kalmar, T., and Arias, A. M. (2008). Wnt/Notch signalling and information processing during development. Development 135 (3), 411–424. doi:10.1242/dev.000505
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117 (12), 3810–3820. doi:10.1172/JCI30487
Hills, C. E., and Squires, P. E. (2010). TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 31 (1), 68–74. doi:10.1159/000256659
Hinz, B., Phan, S. H., Thannickal, V. J., Galli, A., Bochaton-Piallat, M. L., and Gabbiani, G. (2007). The myofibroblast - one function, multiple origins. Am. J. Pathol. 170 (6), 1807–1816. doi:10.2353/ajpath.2007.070112
Holderied, A., Romoli, S., Eberhard, J., Konrad, L. A., Devarapu, S. K., Marschner, J. A., et al. (2015). Glomerular parietal epithelial cell activation induces collagen secretion and thickening of Bowman's capsule in diabetes. Lab. Invest. 95 (3), 273–282. doi:10.1038/labinvest.2014.160
Humphreys, B. D., Lin, S. L., Kobayashi, A., Hudson, T. E., Nowlin, B. T., Bonventre, J. V., et al. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176 (1), 85–97. doi:10.2353/ajpath.2010.090517
Humphreys, B. D. (2018). Mechanisms of renal fibrosis. Annu. Rev. Physiol. 80, 309–326. doi:10.1146/annurev-physiol-022516-034227
Hung, P. H., Hsu, Y. C., Chen, T. H., and Lin, C. L. (2021). Recent advances in diabetic kidney diseases: From kidney injury to kidney fibrosis. Int. J. Mol. Sci. 22 (21), 11857. doi:10.3390/ijms222111857
Isono, M., Chen, S., Hong, S. W., Iglesias-de la Cruz, M. C., and Ziyadeh, F. N. (2002). Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem. Biophys. Res. Commun. 296 (5), 1356–1365. doi:10.1016/s0006-291x(02)02084-3
Jourde-Chiche, N., Fakhouri, F., Dou, L., Bellien, J., Burtey, S., Frimat, M., et al. (2019). Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 15 (2), 87–108. doi:10.1038/s41581-018-0098-z
Kalluri, R., and Neilson, E. G. (2003). Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112 (12), 1776–1784. doi:10.1172/JCI20530
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119 (6), 1420–1428. doi:10.1172/JCI39104
Kang, Y. S., Li, Y. J., Dai, C. S., Kiss, L. P., Wu, C. Y., and Liu, Y. H. (2010). Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int. 78 (4), 363–373. doi:10.1038/ki.2010.137
Kaverina, N. V., Eng, D. G., Miner, J. H., Pippin, J. W., and Shankland, S. J. (2020). Parietal epithelial cell differentiation to a podocyte fate in the aged mouse kidney. Aging (Albany NY) 12 (17), 17601–17624. doi:10.18632/aging.103788
Kessler, D., Dethlefsen, S., Haase, I., Plomann, M., Hirche, F., Krieg, T., et al. (2001). Fibroblasts in mechanically stressed collagen lattices assume a "synthetic" phenotype. J. Biol. Chem. 276 (39), 36575–36585. doi:10.1074/jbc.M101602200
Kida, Y., Ieronimakis, N., Schrimpf, C., Reyes, M., and Duffield, J. S. (2013). EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J. Am. Soc. Nephrol. 24 (4), 559–572. doi:10.1681/ASN.2012080871
Kietzmann, L., Guhr, S. S., Meyer, T. N., Ni, L., Sachs, M., Panzer, U., et al. (2015). MicroRNA-193a regulates the transdifferentiation of human parietal epithelial cells toward a podocyte phenotype. J. Am. Soc. Nephrol. 26 (6), 1389–1401. doi:10.1681/ASN.2014020190
Kim, D., You, E., Jeong, J., Ko, P., Kim, J. W., and Rhee, S. (2017). DDR2 controls the epithelial-mesenchymal-transition-related gene expression via c-Myb acetylation upon matrix stiffening. Sci. Rep. 7 (1), 6847. doi:10.1038/s41598-017-07126-7
Kizu, A., Medici, D., and Kalluri, R. (2009). Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am. J. Pathol. 175 (4), 1371–1373. doi:10.2353/ajpath.2009.090698
Koya, D., Haneda, M., Nakagawa, H., Isshiki, K., Sato, H., Maeda, S., et al. (2000). Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 14 (3), 439–447. doi:10.1096/fasebj.14.3.439
Kramann, R., and Humphreys, B. D. (2014). Kidney pericytes: Roles in regeneration and fibrosis. Semin. Nephrol. 34 (4), 374–383. doi:10.1016/j.semnephrol.2014.06.004
Kriz, W., Kaissling, B., and Le Hir, M. (2011). Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Invest. 121 (2), 468–474. doi:10.1172/jci44595
Kuppe, C., Ibrahim, M. M., Kranz, J., Zhang, X., Ziegler, S., Perales-Paton, J., et al. (2021). Decoding myofibroblast origins in human kidney fibrosis. Nature 589 (7841), 281–286. doi:10.1038/s41586-020-2941-1
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 15 (3), 178–196. doi:10.1038/nrm3758
Lan, H. Y. (2012). Transforming growth factor-β/Smad signalling in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 39 (8), 731–738. doi:10.1111/j.1440-1681.2011.05663.x
Lazareth, H., Lenoir, O., and Tharaux, P. L. (2020). Parietal epithelial cells role in repair versus scarring after glomerular injury. Curr. Opin. Nephrol. Hypertens. 29 (3), 293–301. doi:10.1097/MNH.0000000000000600
LeBleu, V. S., Taduri, G., O'Connell, J., Teng, Y., Cooke, V. G., Woda, C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19 (8), 1047–1053. doi:10.1038/nm.3218
Lee, Y. J., and Han, H. J. (2010). Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3β, Snail1, and β-catenin in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 298 (5), F1263–F1275. doi:10.1152/ajprenal.00475.2009
Leveen, P., Pekny, M., Gebre-Medhin, S., Swolin, B., Larsson, E., and Betsholtz, C. (1994). Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes. Dev. 8 (16), 1875–1887. doi:10.1101/gad.8.16.1875
Li, C. W., Xia, W., Huo, L., Lim, S. O., Wu, Y., Hsu, J. L., et al. (2012). Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 72 (5), 1290–1300. doi:10.1158/0008-5472.CAN-11-3123
Li, J., Liu, H., Takagi, S., Nitta, K., Kitada, M., Srivastava, S. P., et al. (2020). Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 5 (6), 129034. doi:10.1172/jci.insight.129034
Li, J., Qu, X., Yao, J., Caruana, G., Ricardo, S. D., Yamamoto, Y., et al. (2010). Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59 (10), 2612–2624. doi:10.2337/db09-1631
Li, J. J., Kwak, S. J., Jung, D. S., Kim, J. J., Yoo, T. H., Ryu, D. R., et al. (2007). Podocyte biology in diabetic nephropathy. Kidney Int. 72 (106), S36–S42. doi:10.1038/sj.ki.5002384
Li, J. P., Liu, H. J., Srivastava, S. P., Hu, Q. Y., Gao, R. F., Li, S. L., et al. (2020). Endothelial FGFR1 (fibroblast growth factor receptor 1) deficiency contributes differential fibrogenic effects in kidney and heart of diabetic mice. Hypertension 76 (6), 1935–1944. doi:10.1161/HYPERTENSIONAHA.120.15587
Li, J. P., Shi, S., Srivastava, S. P., Kitada, M., Nagai, T., Nitta, K., et al. (2017). FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell. Death Dis. 8, e2965. doi:10.1038/cddis.2017.353
Li, L., Chen, L., Zang, J., Tang, X., Liu, Y., Zhang, J., et al. (2015). C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism. 64 (5), 597–610. doi:10.1016/j.metabol.2015.01.014
Li, L., Feng, Y., Zhang, J., Zhang, Q., Ren, J., Sun, C., et al. (2022). Microtubule associated protein 4 phosphorylation-induced epithelial-to-mesenchymal transition of podocyte leads to proteinuria in diabetic nephropathy. Cell. Commun. Signal. 20 (1), 115. doi:10.1186/s12964-022-00883-7
Li, M., Zhou, H., Di, J., Yang, M., and Jia, F. (2019). ILK participates in renal interstitial fibrosis by altering the phenotype of renal tubular epithelial cells via TGF-β1/smad pathway. Eur. Rev. Med. Pharmacol. Sci. 23 (1), 289–296. doi:10.26355/eurrev_201901_16775
Li, Y., Hu, Q., Li, C., Liang, K., Xiang, Y., Hsiao, H., et al. (2019). PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J. Clin. Invest. 129 (3), 1129–1151. doi:10.1172/JCI121987
Li, Y., Kang, Y. S., Dai, C., Kiss, L. P., Wen, X., and Liu, Y. (2008). Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am. J. Pathol. 172 (2), 299–308. doi:10.2353/ajpath.2008.070057
Li, Y., Tan, X., Dai, C., Stolz, D. B., Wang, D., and Liu, Y. (2009). Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J. Am. Soc. Nephrol. 20 (9), 1907–1918. doi:10.1681/ASN.2008090930
Li, Y., Yang, J., Dai, C., Wu, C., and Liu, Y. (2003). Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J. Clin. Invest. 112 (4), 503–516. doi:10.1172/JCI17913
Li, Z., Liu, X., Wang, B., Nie, Y., Wen, J., Wang, Q., et al. (2017). Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology 22 (8), 589–597. doi:10.1111/nep.12831
Liang, G., Song, L., Chen, Z., Qian, Y., Xie, J., Zhao, L., et al. (2018). Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 93 (1), 95–109. doi:10.1016/j.kint.2017.05.013
Lin, S. L., Kisseleva, T., Brenner, D. A., and Duffield, J. S. (2008). Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 173 (6), 1617–1627. doi:10.2353/ajpath.2008.080433
Lin, Y. C., Chang, Y. H., Yang, S. Y., Wu, K. D., and Chu, T. S. (2018). Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 117 (8), 662–675. doi:10.1016/j.jfma.2018.02.007
Liu, B. C., Tang, T. T., Lv, L. L., and Lan, H. Y. (2018). Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 93 (3), 568–579. doi:10.1016/j.kint.2017.09.033
Liu, H., Yan, R., Liang, L., Zhang, H., Xiang, J., Liu, L., et al. (2021). The role of CDX2 in renal tubular lesions during diabetic kidney disease. Aging (Albany NY) 13 (5), 6782–6803. doi:10.18632/aging.202537
Liu, W., Chen, X., Wang, Y., Chen, Y., Chen, S., Gong, W., et al. (2019). Micheliolide ameliorates diabetic kidney disease by inhibiting Mtdh-mediated renal inflammation in type 2 diabetic db/db mice. Pharmacol. Res. 150, 104506. doi:10.1016/j.phrs.2019.104506
Liu, W., Wu, Y., Yu, F., Hu, W., Fang, X., and Hao, W. (2018). The implication of Numb-induced Notch signaling in endothelial-mesenchymal transition of diabetic nephropathy. J. Diabetes Complicat. 32 (10), 889–899. doi:10.1016/j.jdiacomp.2018.06.011
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7 (12), 684–696. doi:10.1038/nrneph.2011.149
Liu, Y. (2010). New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 21 (2), 212–222. doi:10.1681/ASN.2008121226
Loeffler, I., and Wolf, G. (2015). Epithelial-to-Mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells 4 (4), 631–652. doi:10.3390/cells4040631
Lovisa, S., LeBleu, V. S., Tampe, B., Sugimoto, H., Vadnagara, K., Carstens, J. L., et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21 (9), 998–1009. doi:10.1038/nm.3902
Lovisa, S., Zeisberg, M., and Kalluri, R. (2016). Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol. Metab. 27 (10), 681–695. doi:10.1016/j.tem.2016.06.004
Lu, L., Zhong, Z., Gu, J., Nan, K., Zhu, M., and Miao, C. (2021). ets1 associates with KMT5A to participate in high glucose-mediated EndMT via upregulation of PFN2 expression in diabetic nephropathy. Mol. Med. 27 (1), 74. doi:10.1186/s10020-021-00339-7
Lu, Q., Wang, W. W., Zhang, M. Z., Ma, Z. X., Qiu, X. R., Shen, M., et al. (2019). ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 17 (1), 835–846. doi:10.3892/etm.2018.7014
Luna-Antonio, B. I., Rodriguez-Munoz, R., Namorado-Tonix, C., Vergara, P., Segovia, J., and Reyes, J. L. (2017). Gas1 expression in parietal cells of Bowman's capsule in experimental diabetic nephropathy. Histochem. Cell. Biol. 148 (1), 33–47. doi:10.1007/s00418-017-1550-z
Lv, Z., Hu, M., Zhen, J., Lin, J., Wang, Q., and Wang, R. (2013). Rac1/PAK1 signaling promotes epithelial-mesenchymal transition of podocytes in vitro via triggering beta-catenin transcriptional activity under high glucose conditions. Int. J. Biochem. Cell. Biol. 45 (2), 255–264. doi:10.1016/j.biocel.2012.11.003
Maezawa, Y., Takemoto, M., and Yokote, K. (2015). Cell biology of diabetic nephropathy: Roles of endothelial cells, tubulointerstitial cells and podocytes. J. Diabetes Investig. 6 (1), 3–15. doi:10.1111/jdi.12255
Mason, R. M., and Wahab, N. A. (2003). Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 14 (5), 1358–1373. doi:10.1097/01.asn.0000065640.77499.d7
May, C. J., Saleem, M., and Welsh, G. I. (2014). Podocyte dedifferentiation: A specialized process for a specialized cell. Front. Endocrinol. 5, 148. doi:10.3389/fendo.2014.00148
Meran, S., and Steadman, R. (2011). Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 92 (3), 158–167. doi:10.1111/j.1365-2613.2011.00764.x
Mora-Fernandez, C., Dominguez-Pimentel, V., de Fuentes, M. M., Gorriz, J. L., Martinez-Castelao, A., and Navarro-Gonzalez, J. F. (2014). Diabetic kidney disease: From physiology to therapeutics. J. Physiol. 592 (18), 3997–4012. doi:10.1113/jphysiol.2014.272328
Naves, M. A., Requiao-Moura, L. R., Soares, M. F., Silva-Junior, J. A., Mastroianni-Kirsztajn, G., and Teixeira, V. P. (2012). Podocyte Wnt/ß-catenin pathway is activated by integrin-linked kinase in clinical and experimental focal segmental glomerulosclerosis. J. Nephrol. 25 (3), 401–409. doi:10.5301/jn.5000017
Ni, L., Yuan, C., and Wu, X. (2021). The recruitment mechanisms and potential therapeutic targets of podocytes from parietal epithelial cells. J. Transl. Med. 19 (1), 441. doi:10.1186/s12967-021-03101-z
Nishad, R., Meshram, P., Singh, A. K., Reddy, G. B., and Pasupulati, A. K. (2020). Activation of Notch1 signaling in podocytes by glucose-derived AGEs contributes to proteinuria. BMJ Open Diabetes Res. Care 8 (1), e001203. doi:10.1136/bmjdrc-2020-001203
Nishad, R., Mukhi, D., Tahaseen, S. V., Mungamuri, S. K., and Pasupulati, A. K. (2019). Growth hormone induces Notch1 signaling in podocytes and contributes to proteinuria in diabetic nephropathy. J. Biol. Chem. 294 (44), 16109–16122. doi:10.1074/jbc.RA119.008966
Niu, H., Nie, L., Liu, M., Chi, Y., Zhang, T., and Li, Y. (2014). Benazepril affects integrin-linked kinase and smooth muscle alpha-actin expression in diabetic rat glomerulus and cultured mesangial cells. BMC Nephrol. 15, 135. doi:10.1186/1471-2369-15-135
Ohse, T., Chang, A. M., Pippin, J. W., Jarad, G., Hudkins, K. L., Alpers, C. E., et al. (2009). A new function for parietal epithelial cells: A second glomerular barrier. Am. J. Physiol. Ren. Physiol. 297 (6), F1566–F1574. doi:10.1152/ajprenal.00214.2009
Peng, W., Zhou, X., Xu, T., Mao, Y., Zhang, X., Liu, H., et al. (2022). BMP-7 ameliorates partial epithelial-mesenchymal transition by restoring SnoN protein level via Smad1/5 pathway in diabetic kidney disease. Cell. Death Dis. 13 (3), 254. doi:10.1038/s41419-022-04529-x
Pichaiwong, W., Hudkins, K. L., Wietecha, T., Nguyen, T. Q., Tachaudomdach, C., Li, W., et al. (2013). Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J. Am. Soc. Nephrol. 24 (7), 1088–1102. doi:10.1681/ASN.2012050445
Qi, F. H., Cai, P. P., Liu, X., and Si, G. M. (2018). Adenovirus-mediated P311 ameliorates renal fibrosis through inhibition of epithelial-mesenchymal transition via TGF-β1-Smad-ILK pathway in unilateral ureteral obstruction rats. Int. J. Mol. Med. 41 (5), 3015–3023. doi:10.3892/ijmm.2018.3485
Rask-Madsen, C., and King, G. L. (2013). Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell. Metab. 17 (1), 20–33. doi:10.1016/j.cmet.2012.11.012
Sanchez-Duffhues, G., de Vinuesa, A. G., and ten Dijke, P. (2018). Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 247 (3), 492–508. doi:10.1002/dvdy.24589
Schlondorff, D. (1987). The glomerular mesangial cell: An expanding role for a specialized pericyte. FASEB J. 1 (4), 272–281. doi:10.1096/fasebj.1.4.3308611
Schrimpf, C., Xin, C., Campanholle, G., Gill, S. E., Stallcup, W., Lin, S. L., et al. (2012). Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J. Am. Soc. Nephrol. 23 (5), 868–883. doi:10.1681/ASN.2011080851
Schulte, K., Berger, K., Boor, P., Jirak, P., Gelman, I. H., Arkill, K. P., et al. (2014). Origin of parietal podocytes in atubular glomeruli mapped by lineage tracing. J. Am. Soc. Nephrol. 25 (1), 129–141. doi:10.1681/ASN.2013040376
Shankland, S. J., Smeets, B., Pippin, J. W., and Moeller, M. J. (2014). The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 10 (3), 158–173. doi:10.1038/nrneph.2014.1
Shankland, S. J. (2006). The podocyte's response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 69 (12), 2131–2147. doi:10.1038/sj.ki.5000410
Shi, S., Song, L., Yu, H., Feng, S., He, J., Liu, Y., et al. (2020). Knockdown of LncRNA-H19 ameliorates kidney fibrosis in diabetic mice by suppressing miR-29a-mediated EndMT. Front. Pharmacol. 11, 586895. doi:10.3389/fphar.2020.586895
Simonson, M. S. (2007). Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int. 71 (9), 846–854. doi:10.1038/sj.ki.5002180
Song, S., Zhang, R., Cao, W., Fang, G., Yu, Y., Wan, Y., et al. (2019). Foxm1 is a critical driver of TGF-beta-induced EndMT in endothelial cells through Smad2/3 and binds to the Snail promoter. J. Cell. Physiol. 234 (6), 9052–9064. doi:10.1002/jcp.27583
Souma, T., Nezu, M., Nakano, D., Yamazaki, S., Hirano, I., Sekine, H., et al. (2016). Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J. Am. Soc. Nephrol. 27 (2), 428–438. doi:10.1681/ASN.2014121184
Srivastava, S. P., Hedayat, A. F., Kanasaki, K., and Goodwin, J. E. (2019). microRNA crosstalk influences epithelial-to-mesenchymal, endothelial-to-mesenchymal, and macrophage-to-mesenchymal transitions in the kidney. Front. Pharmacol. 10, 904. doi:10.3389/fphar.2019.00904
Srivastava, S. P., Li, J., Takagaki, Y., Kitada, M., Goodwin, J. E., Kanasaki, K., et al. (2021). Endothelial SIRT3 regulates myofibroblast metabolic shifts in diabetic kidneys. iScience 24 (5), 102390. doi:10.1016/j.isci.2021.102390
Stratman, A. N., Malotte, K. M., Mahan, R. D., Davis, M. J., and Davis, G. E. (2009). Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114 (24), 5091–5101. doi:10.1182/blood-2009-05-222364
Su, J., Morgani, S. M., David, C. J., Wang, Q., Er, E. E., Huang, Y. H., et al. (2020). TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577 (7791), 566–571. doi:10.1038/s41586-019-1897-5
Sugahara, M., Pak, W. L. W., Tanaka, T., Tang, S. C. W., and Nangaku, M. (2021). Update on diagnosis, pathophysiology, and management of diabetic kidney disease. Nephrology 26 (6), 491–500. doi:10.1111/nep.13860
Sun, S., Ning, X., Zhang, Y., Lu, Y., Nie, Y., Han, S., et al. (2009). Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 75 (12), 1278–1287. doi:10.1038/ki.2009.62
Sweetwyne, M. T., and Susztak, K. (2013). For better or worse: A niche for notch in parietal epithelial cell activation. Kidney Int. 83 (6), 988–990. doi:10.1038/ki.2013.66
Tharaux, P. L., and Huber, T. B. (2012). How many ways can a podocyte die? Semin. Nephrol. 32 (4), 394–404. doi:10.1016/j.semnephrol.2012.06.011
Thomas, H. Y., and Ford Versypt, A. N. (2022). Pathophysiology of mesangial expansion in diabetic nephropathy: Mesangial structure, glomerular biomechanics, and biochemical signaling and regulation. J. Biol. Eng. 16 (1), 19. doi:10.1186/s13036-022-00299-4
Tian, H., Yang, J., Xie, Z., and Liu, J. (2018). Gliquidone alleviates diabetic nephropathy by inhibiting notch/snail signaling pathway. Cell. Physiol. biochem. 51 (5), 2085–2097. doi:10.1159/000495827
Timmerman, L. A., Grego-Bessa, J., Raya, A., Bertran, E., Perez-Pomares, J. M., Diez, J., et al. (2004). Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes. Dev. 18 (1), 99–115. doi:10.1101/gad.276304
Tonolo, G., and Cherchi, S. (2014). Tubulointerstitial disease in diabetic nephropathy. Int. J. Nephrol. Renov. Dis. 7, 107–115. doi:10.2147/IJNRD.S37883
Umanath, K., and Lewis, J. B. (2018). Update on diabetic nephropathy: Core curriculum 2018. Am. J. Kidney Dis. 71 (6), 884–895. doi:10.1053/j.ajkd.2017.10.026
Urushihara, M., Kondo, S., Kinoshita, Y., Ozaki, N., Jamba, A., Nagai, T., et al. (2020). (Pro)renin receptor promotes crescent formation via the ERK1/2 and Wnt/β-catenin pathways in glomerulonephritis. Am. J. Physiol. Ren. Physiol. 319 (4), F571–F8. doi:10.1152/ajprenal.00250.2020
van Dijk, C. G. M., Nieuweboer, F. E., Pei, J. Y., Xu, Y. J., Burgisser, P., van Mulligen, E., et al. (2015). The complex mural cell: Pericyte function in health and disease. Int. J. Cardiol. 190, 75–89. doi:10.1016/j.ijcard.2015.03.258
Verma, S. K., and Molitoris, B. A. (2015). Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 35 (1), 96–107. doi:10.1016/j.semnephrol.2015.01.010
Vasanth Rao, V. R. A. L. B, Tan, S. H., Candasamy, M., and Bhattamisra, S. K. (2019). Diabetic nephropathy: An update on pathogenesis and drug development. Diabetes Metab. Syndr. 13 (1), 754–762. doi:10.1016/j.dsx.2018.11.054
Wang, D., Dai, C., Li, Y., and Liu, Y. (2011). Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int. 80 (11), 1159–1169. doi:10.1038/ki.2011.255
Wang, E., Wang, L., Ding, R., Zhai, M., Ge, R., Zhou, P., et al. (2020). Astragaloside IV acts through multi-scale mechanisms to effectively reduce diabetic nephropathy. Pharmacol. Res. 157, 104831. doi:10.1016/j.phrs.2020.104831
Wang, W., Sun, W., Cheng, Y., Xu, Z., and Cai, L. (2019). Role of sirtuin-1 in diabetic nephropathy. J. Mol. Med. 97 (3), 291–309. doi:10.1007/s00109-019-01743-7
Wei, J., Deng, X., Li, Y., Li, R., Yang, Z., Li, X., et al. (2021). PP2 ameliorates renal fibrosis by regulating the NF-κB/COX-2 and PPARγ/UCP2 pathway in diabetic mice. Oxid. Med. Cell. Longev. 2021, 7394344. doi:10.1155/2021/7394344
Wu, X., Gao, Y., Xu, L., Dang, W., Yan, H., Zou, D., et al. (2017). Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci. Rep. 7 (1), 9371. doi:10.1038/s41598-017-09907-6
Xiao, K., He, W., Guan, W., Hou, F., Yan, P., Xu, J., et al. (2020). Mesenchymal stem cells reverse EMT process through blocking the activation of NF-κB and Hedgehog pathways in LPS-induced acute lung injury. Cell. Death Dis. 11 (10), 863. doi:10.1038/s41419-020-03034-3
Xie, L., Law, B. K., Chytil, A. M., Brown, K. A., Aakre, M. E., and Moses, H. L. (2004). Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6 (5), 603–610. doi:10.1593/neo.04241
Xie, L., Zhai, R., Chen, T., Gao, C., Xue, R., Wang, N., et al. (2020). Panax Notoginseng ameliorates podocyte EMT by targeting the wnt/β-catenin signaling pathway in STZ-induced diabetic rats. Drug Des. devel. Ther. 14, 527–538. doi:10.2147/DDDT.S235491
Xie, X., Xia, W., Fei, X., Xu, Q., Yang, X., Qiu, D., et al. (2015). Relaxin inhibits high glucose-induced matrix accumulation in human mesangial cells by interfering with TGF-β1 production and mesangial cells phenotypic transition. Biol. Pharm. Bull. 38 (10), 1464–1469. doi:10.1248/bpb.b15-00127
Xu, Z., Jia, K., Wang, H., Gao, F., Zhao, S., Li, F., et al. (2021). METTL14-regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell. Death Dis. 12 (1), 32. doi:10.1038/s41419-020-03312-0
Yamaguchi, Y., Iwano, M., Suzuki, D., Nakatani, K., Kimura, K., Harada, K., et al. (2009). Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am. J. Kidney Dis. 54 (4), 653–664. doi:10.1053/j.ajkd.2009.05.009
Yang, J., and Liu, Y. (2001). Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 159 (4), 1465–1475. doi:10.1016/S0002-9440(10)62533-3
Yang, Y., Ahn, Y. H., Gibbons, D. L., Zang, Y., Lin, W., Thilaganathan, N., et al. (2011). The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J. Clin. Invest. 121 (4), 1373–1385. doi:10.1172/JCI42579
Yuan, Q., Tan, R. J., and Liu, Y. (2019). Myofibroblast in kidney fibrosis: Origin, activation, and regulation. Adv. Exp. Med. Biol. 1165, 253–283. doi:10.1007/978-981-13-8871-2_12
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M., and Kalluri, R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19 (12), 2282–2287. doi:10.1681/ASN.2008050513
Zeisberg, M., and Neilson, E. G. (2009). Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119 (6), 1429–1437. doi:10.1172/JCI36183
Zhang, J., Wang, Y., Gurung, P., Wang, T., Li, L., Zhang, R., et al. (2018). The relationship between the thickness of glomerular basement membrane and renal outcomes in patients with diabetic nephropathy. Acta Diabetol. 55 (7), 669–679. doi:10.1007/s00592-018-1128-9
Zhang, K., Zhang, H., Xiang, H., Liu, J., Liu, Y., Zhang, X., et al. (2013). TGF-β1 induces the dissolution of tight junctions in human renal proximal tubular cells: Role of the RhoA/ROCK signaling pathway. Int. J. Mol. Med. 32 (2), 464–468. doi:10.3892/ijmm.2013.1396
Zhang, L., Shen, Z. Y., Wang, K., Li, W., Shi, J. M., Osoro, E. K., et al. (2019). C-reactive protein exacerbates epithelial-mesenchymal transition through Wnt/β-catenin and ERK signaling in streptozocin-induced diabetic nephropathy. FASEB J. 33 (5), 6551–6563. doi:10.1096/fj.201801865RR
Zhang, M., Liu, M., Xiong, M., Gong, J., and Tan, X. (2012). Schisandra chinensis fruit extract attenuates albuminuria and protects podocyte integrity in a mouse model of streptozotocin-induced diabetic nephropathy. J. Ethnopharmacol. 141 (1), 111–118. doi:10.1016/j.jep.2012.02.007
Zhang, Q., Xiao, X., Zheng, J., Li, M., Yu, M., Ping, F., et al. (2020). Compound danshen dripping pill inhibits retina cell apoptosis in diabetic rats. Front. Physiol. 11, 1501. doi:10.3389/fphys.2018.01501
Zhang, Y., Wang, B., Guo, F., Li, Z., and Qin, G. (2018). Involvement of the TGFβ1- ILK-Akt signaling pathway in the effects of hesperidin in type 2 diabetic nephropathy. Biomed. Pharmacother. 105, 766–772. doi:10.1016/j.biopha.2018.06.036
Zhao, J., Patel, J., Kaur, S., Sim, S. L., Wong, H. Y., Styke, C., et al. (2021). Sox9 and Rbpj differentially regulate endothelial to mesenchymal transition and wound scarring in murine endovascular progenitors. Nat. Commun. 12 (1), 2564. doi:10.1038/s41467-021-22717-9
Zhao, X., Zhang, Y., Li, L., Mann, D., Imig, J. D., Emmett, N., et al. (2011). Glomerular expression of kidney injury molecule-1 and podocytopenia in diabetic glomerulopathy. Am. J. Nephrol. 34 (3), 268–280. doi:10.1159/000330187
Zhao, X. P., Chang, S. Y., Liao, M. C., Lo, C. S., Chenier, I., Luo, H., et al. (2018). Hedgehog interacting protein promotes fibrosis and apoptosis in glomerular endothelial cells in murine diabetes. Sci. Rep. 8 (1), 5958. doi:10.1038/s41598-018-24220-6
Zhao, Y., Qiao, X., Wang, L., Tan, T. K., Zhao, H., Zhang, Y., et al. (2016). Matrix metalloproteinase 9 induces endothelial-mesenchymal transition via Notch activation in human kidney glomerular endothelial cells. BMC Cell. Biol. 17 (1), 21. doi:10.1186/s12860-016-0101-0
Zhao, Y., Yin, Z., Li, H., Fan, J., Yang, S., Chen, C., et al. (2017). MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell. 16 (2), 387–400. doi:10.1111/acel.12563
Zhou, L., and Liu, Y. (2015). Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat. Rev. Nephrol. 11 (9), 535–545. doi:10.1038/nrneph.2015.88
Zhou, L., Xue, H., Yuan, P., Ni, J., Yu, C., Huang, Y., et al. (2010). Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin. Exp. Pharmacol. Physiol. 37 (9), e152–e157. doi:10.1111/j.1440-1681.2010.05421.x
Glossary
DM diabetes mellitus
DN diabetic nephropathy
GFR glomerular filtration rate
ECM extracellular matrix
PECs parietal epithelial cells;
ER endoplasmic reticulum
FN fibronectin
GBM glomerular basement membrane
α-SMA α-smooth muscle actin
PDGFR platelet-derived growth factor receptor
EndoMT endothelial-mesenchymal transition
EMT epithelial-mesenchymal transition
Snail Snail family transcriptional repressor
Twist Twist family
BHLH Transcription Factor
ZEB Zinc finger E-Box binding homeobox
ECs endothelial cells
Cd2-ap CD2-associated protein
ZO-1 zonula occludens-1
MMP9 matrix metalloproteinase 9
FSP1 fibroblast-specific protein-1
HSP47 heat shock protein 47
DDR2 discoidin or domain receptor 2
WT1 Wilms’ tumor 1
PAX8 paired Box 8
ERK extracellular signal-regulated kinase;
MMPs matrix metalloproteinases
TECs tubular epithelial cells
UUO unilateral ureteral obstruction
TGF-β transforming growth factor β
NO nitric oxide
PECAM-1/CD31 platelet endothelial cell adhesion molecule-1
Tie1 tyrosine kinase with immunoglobulin like and EGF like domains 1
Tie2 TEK receptor tyrosine kinase
EPO erythropoietin
Smad SMAD family member
CTGF connective tissue growth factor
SnoN ski-related protein N
Sirt6 Sirtuin 6
RhoA The Ras homolog gene family, member A
MAPK p38 mitogen-activated protein kinase
PI3K phosphoinositide 3-kinases
AKT protein kinase B
ILK integrin-linked kinase
NF-κB nuclear factor kappa B
CRP C-reactive protein
METTL14 methyltransferase Like 14
PTEN phosphatase and tensin homolog
GSK-3β glycogen synthase kinase 3β
Rac1 RAS-related C3 botulinum toxin substrate 1
PAK1 p21-activated kinase 1
CDX2 caudal-type homeobox transcription factor 2
VDR vitamin D receptor
TCF4 transcription Factor 4
Sirt3 mitochondrial deacetylase sirtuin 3
FGFRs fibroblast growth factor receptors
HIF-1α hypoxia inducible factor 1
SGLT2 sodium-glucose cotransporter-2
ROS reactive oxygen species
FGF fibroblast growth factors
NICD notch intracellular domain
AGEs advanced glycation end-products
Numb NUMB endocytic adaptor protein
Glis GLI family zinc finger
Hhip hedgehog-interacting protein
Keywords: diabetic nephropathy, phenotypic transition, fibrosis, EMT, EndoMT, signaling pathway
Citation: Cao Y, Lin J-H, Hammes H-P and Zhang C (2022) Cellular phenotypic transitions in diabetic nephropathy: An update. Front. Pharmacol. 13:1038073. doi: 10.3389/fphar.2022.1038073
Received: 06 September 2022; Accepted: 17 October 2022;
Published: 02 November 2022.
Edited by:
Keizo Kanasaki, Shimane University, JapanReviewed by:
Khadija Banu, Yale University, United StatesSandeep Kumar, Augusta University, United States
Copyright © 2022 Cao, Lin, Hammes and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun Zhang, drzhangchun@hust.edu.cn