 Binbin Wang1,2†
Binbin Wang1,2† Qiongfeng Wu1†
Qiongfeng Wu1† Jie Liao1†
Jie Liao1† Shaoshao Zhang1Huixia Liu3
Shaoshao Zhang1Huixia Liu3 Cui Yang3Qian Dong1Ning Zhao1Zhengrong Huang3Kefang Guo4*
Cui Yang3Qian Dong1Ning Zhao1Zhengrong Huang3Kefang Guo4* Yimei Du1*
Yimei Du1*- 1Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Critical Care Medicine, Zhongnan Hospital of Wuhan University, Wuhan, China
- 3Department of Cardiology, The First Affiliated Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China
- 4Department of Anesthesiology, Zhongshan Hospital, Fudan University, Shanghai, China
Ca2+ entry via the transient receptor potential vanilloid 4 (TRPV4) channel contributes to Ca2+ overload and triggers many pathophysiological conditions, including myocardial ischemia/reperfusion (I/R) injury. Propofol, a widely used intravenous anesthetic, attenuates myocardial I/R injury. However, the mechanism of propofol remains to be examined. The present study aims to test the hypothesis that propofol attenuates myocardial I/R injury through the suppression of TRPV4. We used a murine ex vivo model of myocardial I/R and in vitro cultured myocytes subjected to hypoxia/reoxygenation (H/R). Propofol or TRPV4 antagonist, HC-067047, attenuates myocardial I/R injury in isolated hearts. In addition, propofol, HC-067047, or TRPV4-siRNA attenuates H/R-induced intracellular Ca2+ concentration ([Ca2+]i) increase and cell viability reduction. On the contrary, TRPV4 agonist GSK1016790A exacerbates both ex vivo and in vitro myocardial injury. Pretreatment with propofol reverses the myocardial injury and intracellular Ca2+ overload induced by GSK1016790A at least in vitro. However, neither the combination of propofol and HC-067047 nor applying propofol to cells transfected with TRPV4-siRNA creates additional protective effects. In addition, propofol dose-dependently inhibits TRPV4-mediated Ca2+ entry induced by GSK1016790A and 4α-PDD. Propofol attenuates myocardial I/R injury partially through the suppression of TRPV4 channel and the subsequent inhibition of intracellular Ca2+ overload.
Introduction
Propofol (2, 6-diisopropylphenol), an intravenous anesthetic frequently used during surgery, has been proved to dose-dependently attenuate myocardial ischemia/reperfusion (I/R) and hypoxia/reoxygenation (H/R) injury (Xia et al., 2006; Sun et al., 2017; Zhao et al., 2019). The potential mechanisms include the reduction of reactive oxygen species (ROS) (Xia et al., 2003), the inhibition of Ca2+ overload (Kim et al., 2008), the suppression of Ca2+ entry channels (L-type Ca2+ channel, T-Type Ca2+ channel, and Na+/Ca2+ exchange) (Luk et al., 1995; Kojima et al., 2015), and the reduction of apoptosis (Liu et al., 2017).
An increased level of intracellular Ca2+ is detrimental to cardiomyocytes and results in myocardial cell death during I/R injury (Garcia-Dorado et al., 2012). One of our recent studies has shown that Ca2+ entry via the transient receptor potential vanilloid 4 (TRPV4) channel in cardiomyocytes plays a critical role in mediating Ca2+ overload and ROS release during the process of myocardial I/R injury (Wu et al., 2017). Similar to the cardioprotective effects of propofol, TRPV4 blockage reduces infarct size and troponin T (TnT) and improves in vivo heart function (Dong et al., 2017). TRPV4 protein levels have also been found to increase in the brain I/R model (Jie et al., 2016), while TRPV4 selective antagonist HC-067047 (Everaerts et al., 2010) attenuates I/R-induced brain injury (Jie et al., 2015). In addition, excessive Ca2+ influx through TRPV4 induced by TRPV4 agonists GSK1016790A and 4α-PDD leads to the apoptosis of retinal ganglion cells and neuronal death in the hippocampus (Ryskamp et al., 2011; Jie et al., 2016). Based on these findings, TRPV4 channel is a promising target in the treatment of I/R-induced myocardial injury (Jones et al., 2019; Wu et al., 2019). Moreover, propofol has been found to directly interact with TRP channels, including TRPC5 and TRPA1 (Bahnasi et al., 2008; Ton et al., 2017). However, there is no direct evidence showing that propofol inhibits TRPV4-mediated Ca2+ entry. Therefore, in this study, we hypothesize that propofol activates protective mechanisms through the suppression of TRPV4 channel and the subsequent inhibition of intracellular Ca2+ overload in ex vivo isolated hearts under I/R and in vitro cell models under H/R.
We first investigated the dose-dependent protective effects of propofol against reperfusion-induced myocardial injury. We subsequently confirmed the involvement of TRPV4 channel in reperfusion-induced myocardial injury using pharmacological approaches. Furthermore, we observed the effects of propofol on TRPV4 agonist-induced myocardial I/R injury and examined the cardioprotective effects of propofol in TRPV4-siRNA transfected cells. Finally, we analyzed the effects of propofol on TRPV4-mediated Ca2+ influx and intracellular Ca2+ concentration ([Ca2+]i) in HEK 293 cells, H9C2 cells, and adult rat ventricle myocytes (ARVMs).
Materials and Methods
Animals
Male C57BL/6 mice were purchased from Vital River Laboratories, Beijing, China, and Sprague-Dawley rats were purchased from the Experimental Animal Center, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. All animals were kept in the Experimental Animal Center of Tongji Medical College. This study was carried out in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978). The protocol was approved by the the Animal Research Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology. All experiments were conducted on age and body weight-matched animal groups.
Pharmacologic Agents
The propofol we used on isolated mice hearts (10 mg/ml, Fresenius Kabi GmbH, Graz, Austria) was dissolved in an intralipid suspension including soybean oil (10%), egg phosphatide (1.2%), and glycerol (2.25%). An equivalent volume of intralipid (INTRA, 20%, Sigma Aldrich, St. Louis, MO, USA) was used as a vehicle control. In cell experiments, we applied pure propofol monomers (TCI Shanghai, Shanghai, China), which were dissolved in dimethyl sulfoxide (DMSO). Propofol dosage (25, 50, 100 μM ex vivo; 12.5, 25, 50, 100 μM in vitro) was based on the volume previous studies applied (Xia et al., 2006; Sun et al., 2017). TRPV4 agonists GSK1016790A (20 nM ex vivo and 300 nM in vitro) and 4α-PDD (3 μΜ in vitro) and TRPV4 antagonist HC-067047 (0.1 μM ex vivo and 1 μM in vitro) were all from Sigma Aldrich and were dissolved in DMSO. The percentage of DMSO in the final solution was less than 0.01%.
Langendorff Perfusion and Experimental Protocol
Mice (22–28 g) were anesthetized with 2.5% avertin 0.02 ml/g and heparinized with sodium heparin 125 IU intraperitoneally. Avertin was prepared by mixing 5 g of tribromoethanol (Sigma-Aldrich, St. Louis, MO, USA), 5 ml of tert-amyl alcohol (Sigma-Aldrich, St. Louis, MO, USA), and 195 ml of normal saline solution. After thoracotomy, mice hearts were quickly excised and mounted for retrograde perfusion using a Krebs-Henseleit buffer. The buffer (in mM: NaCl 118, KCl 4.7, NaH2PO4 1.2, MgSO4 1.2, NaHCO3 25, glucose 15, CaCl2 2.5, EDTA 0.5, pH = 7.4) was gassed with 95% O2 and 5% CO2 at 37°C, and its constant flow rate was 2–4 ml/min. A latex water-filled balloon fixed to a pressure transducer was inserted through the mitral valve into the left ventricle to constantly monitor heart rate (HR), left ventricular end diastolic pressure (LVEDP), left ventricular systolic pressure (LVSP), left ventricle developed pressure (LVDP = LVSP-LVEDP), rate pressure product (RPP = LVDP*HR), as well as the maximum increase/decrease rate of left ventricular pressure (± dP/dt max). LVEDP was adjusted to approximately 5 mmHg before the start of the experiment by adjusting the intraventricular balloon volume. Selection criteria include: HR ≥ 300 beats/min, LVDP ≥ 60 mmHg after 15 min of equilibration, maintenance sinus rhythm, and no prolonged arrhythmias (Sutherland et al., 2003).
In the experimental protocol, we divided the mice hearts into 14 treatment groups (Figure 1): INTRA, POP 25, POP 50, POP 100, DMSO, GSK, HC, INTRA + DMSO, INTRA + GSK, POP 50 + DMSO, POP 50 + GSK, POP 25 + DMSO, POP 25 + HC, and INTRA + HC. I/R was achieved by 30 min of global ischemia (zero flow) and then 60 min of reperfusion.
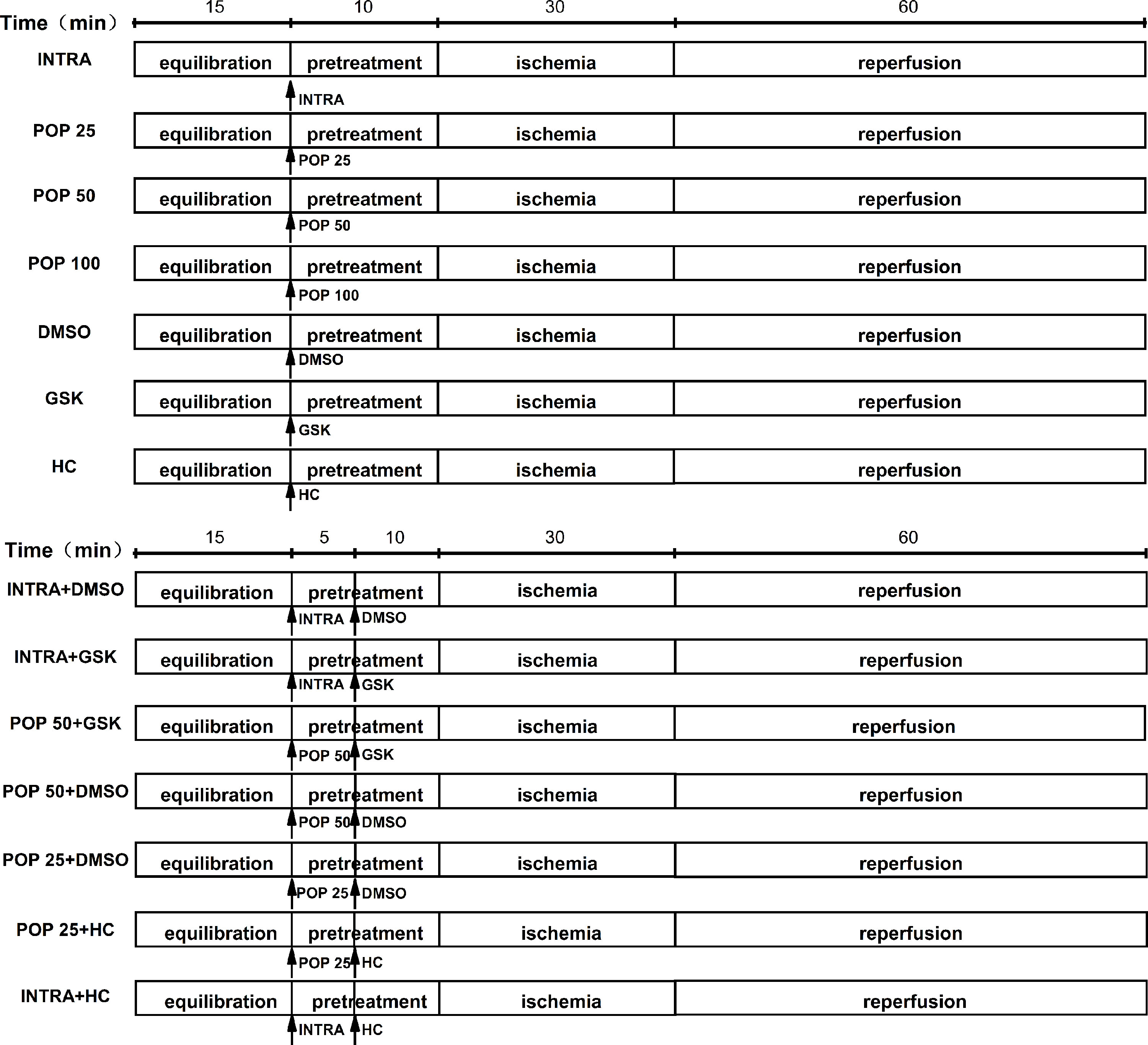
Figure 1 The protocol for myocardial I/R injury in isolated mice hearts.
Lactate Dehydrogenase Release
Lactate dehydrogenase (LDH) activity was assessed by an assay kit (Nanjing Jiancheng Biochemistry Co., Nanjing, China) according to the manufacturer’s instructions. Coronary effluent samples were collected 0, 5, 10, 15, 30, 45, and 60 min after reperfusion (Rossello et al., 2016).
Myocardial Infarct Size
Myocardial infarction size was measured by 2,3,5-triphenyltetrazolium chloride (Sigma Aldrich, St. Louis, MO, USA) staining. At the end of reperfusion, the left ventricle was cut into 1 mm-thick transverse slices from the apex to the base. The size of 2,3,5-triphenyltetrazolium chloride -negative areas (infarct area, white) was calculated using the planimetry function in Image-Pro Plus v 6.0 (Media Cybernetics, MD, USA). The myocardial infarct size is presented as the percentage of infarct area size in the total left ventricle.
Cell Culture and Treatment
As previously described, adult rat ventricle myocytes (ARVMs) were isolated by enzymatic dissociation from the left ventricles of Sprague-Dawley rats. ARVMs were incubated in a medium comprised of 5% fetal bovine serum (Hangzhou Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou, China), 10 mM 2,3-butanedione monoxime, and M199 (Gibco, Grand Island, NY, USA) at 37°C in 5% CO2 for 24 h before experiments. H9C2 cells were purchased from ATCC (Rockefeller, MD, USA) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 15% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere with 95% air and 5% CO2. Cells were treated with TRPV4 agonists GSK1016790A (300 nM), 4α-PDD (3 μM), or TRPV4 antagonist HC-067047 (1 μM) at the onset of hypoxia, while propofol (12.5, 20, 50, 100 μM) was applied 30 min earlier.
H9C2 cells with > 50% sub-confluency were transfected with TRPV4-siRNA (5-GGAGCTGAACAAGAACTCA-3, RIBOBIO, Guangzhou, China) using Lipofectamine 3000 (Invitrogen). A scrambled non-silencing siRNA (5-TTCTCCGAACGTGTCACGTdTdT-3, RIBOBIO) was used as a negative control. The knockdown efficiency of TRPV4-siRNA is shown in the Supplementary Figure S1.
HEK-293T cells were purchased from ATCC and maintained in the same conditions as H9C2 cells. HEK-293T cells were transiently transfected with hTRPV4/pCDNA 4.0 (generously provided by Dr. Ke-wei Wang, Peking University School of Pharmaceutical Sciences, Qingdao University School of Pharmacy, China) using the Lipofectamine 3000 method as described previously (Fu et al., 2013).
H/R Model
Before hypoxia, the culture medium was changed to a fetal bovine serum-free DMEM. Hypoxia was achieved by placing the cells in a controlled hypoxic plastic chamber (HiTech Photelectricity Biotechnology Co., LTD., Guangzhou, China) containing 95% N2 and 5% CO2 for 6 h. Subsequently, the medium was replaced by a normal medium and the cells were incubated in 95% air and 5% CO2 for a 6 h reoxygenation.
[Ca2+]i Measurement
[Ca2+]i was measured according to previously described procedures (Wu et al., 2017). Cells were loaded with 2 μM Fluo-4 AM for 30 min. Florescence was captured with the Enspire Multimode Plate Reader (PerkinElmer, Boston, MA, USA). Relative changes in Ca2+ influx stimulated by GSK1016790A (300 nM) or 4α-PDD (3 μM) are presented as (F/F0) or fold changes (∆F/F0), respectively. F represents fluorescence intensity, F0 represents the average fluorescence intensity before GSK1016790A or 4α-PDD stimulation, and ΔF represents the mean fluorescence intensity at the steady state after drug stimulation minus F0. Ionomycin (1 μM, Sigma, St. Louis, MO, USA) was set as a positive control.
Cell Viability Measurement
Cell viability was measured using the Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Kyushu, Japan) as previously described (Fu et al., 2015; Wu et al., 2017). Optical density at the 450-nm wavelength was measured using a microplate reader (DG5033A, Nanjing, China).
Statistical Analysis
All values are presented as mean ± SD. Data was analyzed by a two-tailed t test (data on Ca2+ influx in H9C2 and ARVMs) or a one-way ANOVA followed by a Bonferroni method analysis (data on ex vivo and in vitro of I/R and H/R). Statistical analysis was performed using SigmaStat3.5 (Systat Software Inc, San Jose, California, USA). The dose-response curve was fitted using the Hill equation in Origin 9 (OriginLab Corporation, Northampton, MA, USA). IC50 represents the propofol dosage that produces 50% of the maximal inhibitory effect. Only when p < 0.05 was the difference considered statistically significant.
Results
As shown in the Supplementary Tables S1 and S2, all groups showed identical measured values (HR, LVDP, ± dP/dt max, and RPP) after 15 min of equilibration as well as before global ischemia. Original tracings of the left ventricle pressure for each experiment are shown in Supplementary Figures S2–S5.
Propofol Concentration-Dependently Attenuates Myocardial I/R Injury in Isolated Mice Hearts
We first examined the protective effects of propofol against myocardial I/R injury in isolated hearts. Figures 2 (A–D) and Supplementary Figures S2 (A–D) show that propofol (25, 50, 100 μM) promoted the recovery of LVDP, +dP/dt max, -dP/dt max, and RPP in a concentration-dependent manner. In addition, the hearts pretreated with 50 μM propofol recovered best from INTRA (LVDP increases from 55.1 ± 2.7% to 91.8 ± 2.8%, +dP/dt max from 74.9 ± 2.9% to 98.4 ± 3.6%, -dP/dt max from 77.2 ± 3.0% to 93.3 ± 2.6%, and RPP from 49.6 ± 2.6% to 80.2 ± 1.9%). As shown in Figure 2E, propofol reduced the peak value and AUC of LDH release in a dose-dependent manner. Similarly, propofol dose-dependently reduced the infarct size of the left ventricle, and 50 μM propofol had the best protective effects (from 34.5 ± 3.1% to 10.5 ± 2.9%, Figure 2F).
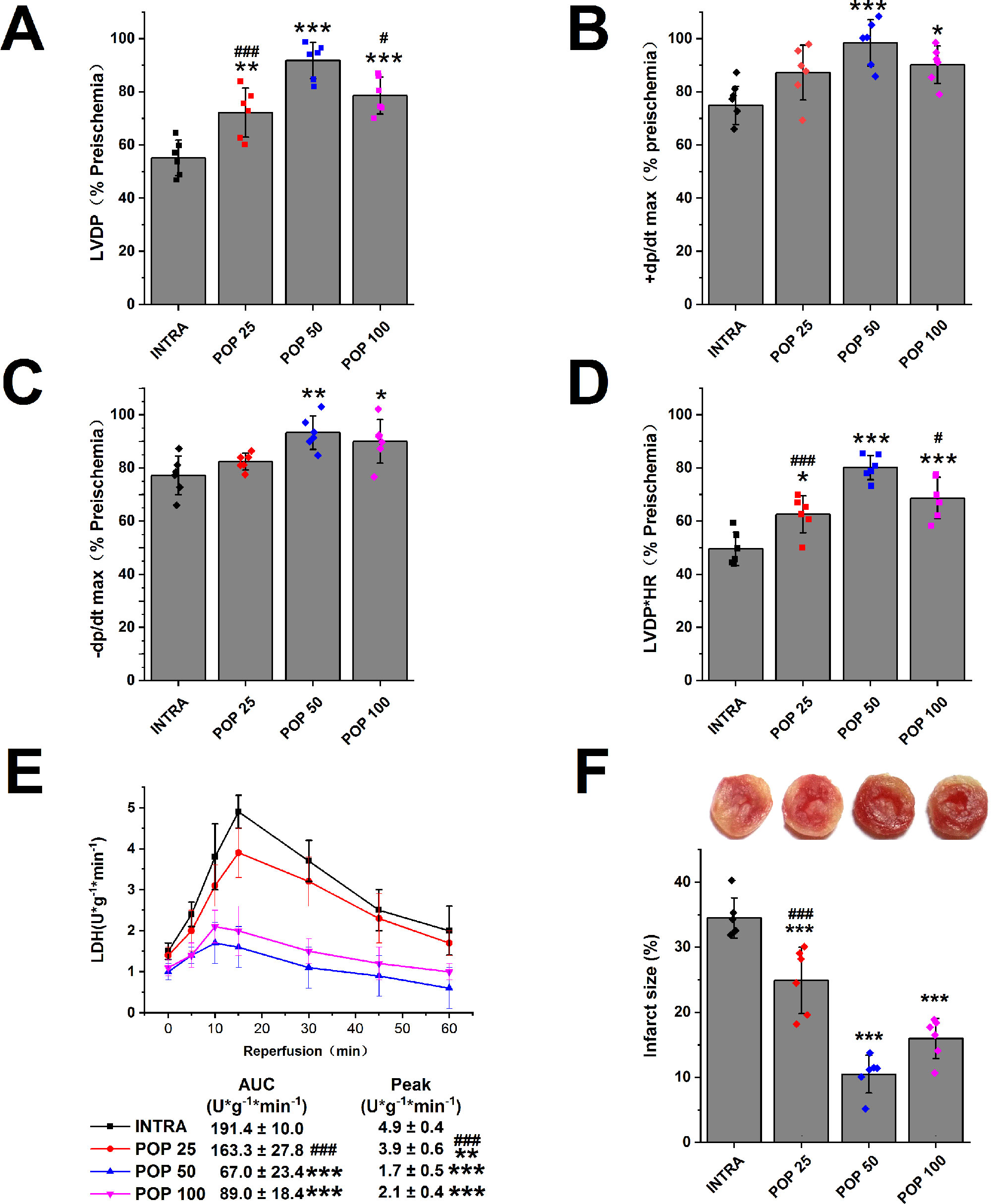
Figure 2 The dose-dependent effect of propofol on myocardial I/R injury. The recovery of LVDP (A), +dP/dt max (B), -dP/dt max (C), and RPP (D) 60 minutes after reperfusion displayed as the percentage of respective preischemia values. (E) The amount of LDH release in coronary effluent at different times after reperfusion. (F) Representative images of TTC stained LV slices. Infarct size is quantified. Values are presented as mean ± SD, n = 6 for all groups, a one-way ANOVA followed by Bonferroni test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. INTRA. #p < 0.05, ###p < 0.001 vs. POP 50.
TRPV4 Agonists/Antagonists Aggravate/Attenuate Myocardial I/R Injury in Isolated Mice Hearts
We evaluated the role of TRPV4 channel in isolated hearts after I/R injury. In comparison with DMSO treatment, TRPV4 agonist GSK1016790A impaired the recovery of LVDP, +dP/dt max, -dP/dt max, and RPP (LVDP decreases from 53.3 ± 8.5% to 29.4 ± 10.9%, +dP/dt max from 77.7 ± 8.1% to 52.3 ± 8.0%, -dP/dt max from 78.4 ± 5.2% to 60.4 ± 7.2%, and RPP from 56.1 ± 5.5% to 28.7 ± 8.9%), while TRPV4 antagonist HC-067047 promoted the recovery of the above indexes, as shown in Figures 3A–D and Supplementary Figures S3A–D. Similarly, TRPV4 antagonist HC-067047 reduced LDH release and infarct size, while TRPV4 agonist GSK1016790A had completely opposite effects (Figures 3E, F). Consistent with previous in vivo studies (Dong et al., 2017; Wu et al., 2017), these results confirmed that TRPV4 channel plays a pathogenic role in myocardial I/R injury.
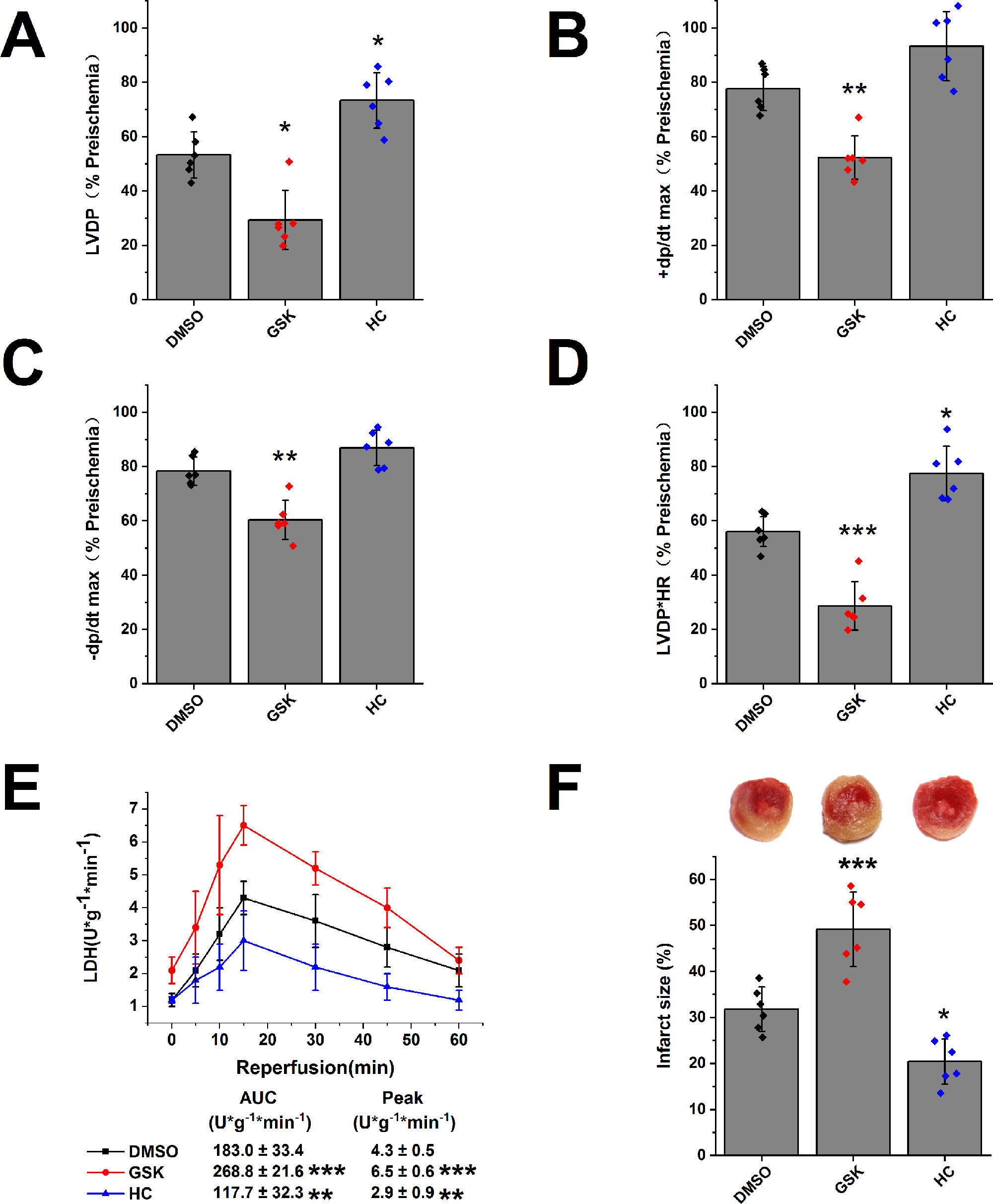
Figure 3 The effect of TRPV4 agonist GSK1016790A and TRPV4 antagonist HC-067047 on myocardial I/R injury. The recovery of LVDP (A), +dP/dt max (B), -dP/dt max (C), and RPP (D) 60 minutes after reperfusion displayed as the percentage of respective preischemia values. (E) The amount of LDH release in the coronary effluent at different times after reperfusion. (F) Representative images of TTC stained LV slices. Infarct size is quantified. Values are presented as mean ± SD, n = 6 for all groups, a one-way ANOVA followed by Bonferroni test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. DMSO.
Effects of the Combination of Propofol and TRPV4 Agonists/Antagonists on Myocardial I/R Injury in Isolated Mice Hearts
Next, we investigated the effect of propofol on TRPV4 agonist-induced myocardial I/R injury in isolated mice hearts. As shown in Figure 4 and Supplementary Figure S4, compared with INTRA+ DMSO, treatment with TRPV4 agonist GSK1016790A induced the detrimental effects. These effects, however, can be reduced by applying a combination of GSK1016790A and propofol at 50 μM. POP + DMSO had better recovery rates of cardiac function (LVDP, +dp/dt max, and RRR) and less release of LDH as well as infarct size compared with POP + GSK. It appears that propofol (50 μM) cannot completely reverse the detrimental effects induced by GSK1016790A in isolated mice hearts. We further investigated the effect of the combination of propofol and TRPV4 antagonists on myocardial I/R injury in isolated mice hearts. To leave sufficient room for HC-067047 to play an additive protective effect, we reduced the concentration of propofol to 25 μM. As shown in Figures 5 and 5S, combining TRPV4 antagonist HC-067047 and propofol produced no additional protective effect compared to propofol or HC-067047 alone. These results suggest that propofol attenuates myocardial I/R injury at least partially via the suppression of TRPV4 channel.
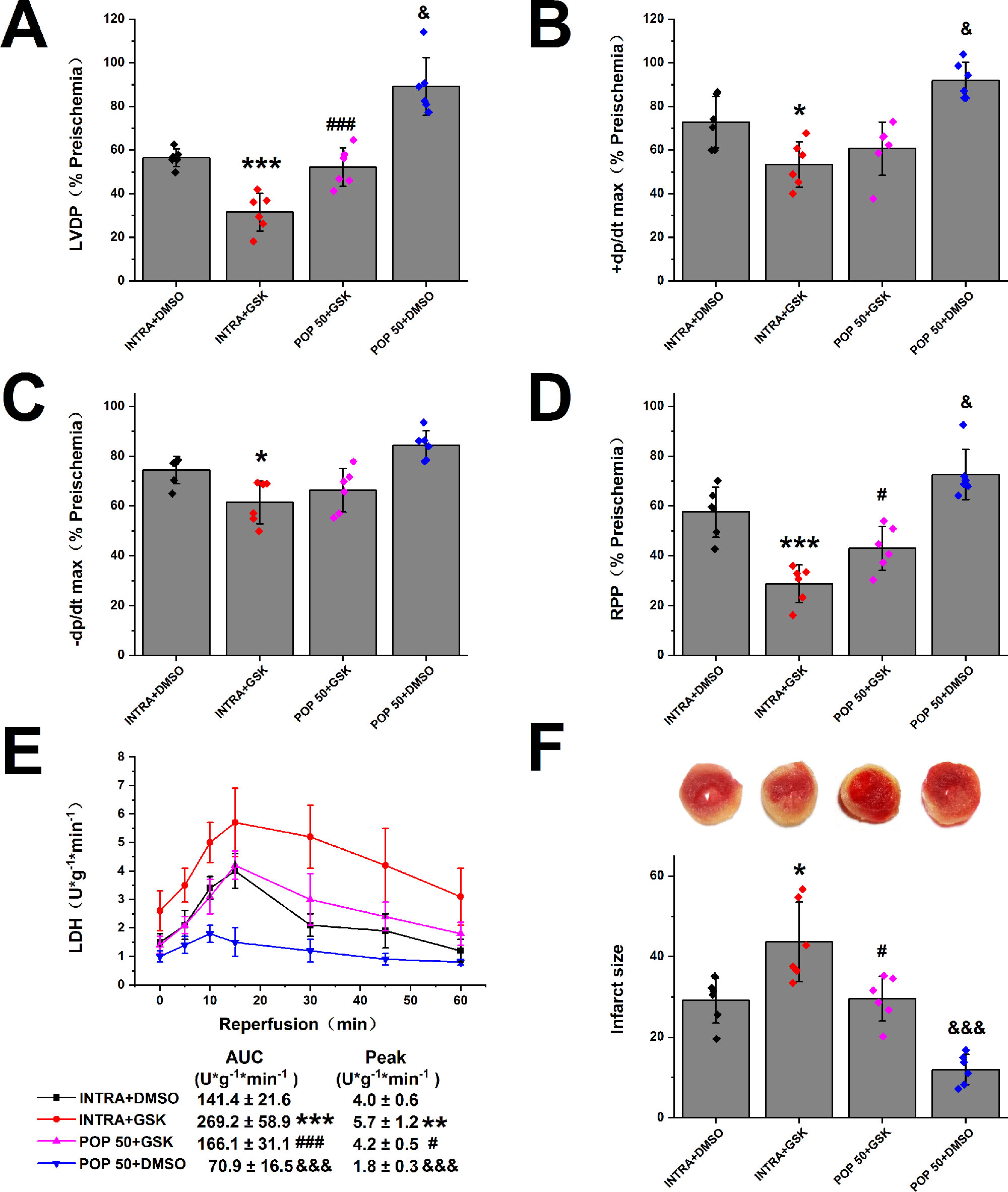
Figure 4 The effect of propofol on TRPV4 agonist-induced myocardial I/R injury. Propofol or intralipid is given 5 minutes before applying TRPV4 agonist or DMSO. The recovery of LVDP (A), +dP/dt max (B), -dP/dt max (C), and RPP (D) 60 minutes after reperfusion displayed as the percentage of respective preischemia values. (E) The amount of LDH release in the coronary effluent at different times after reperfusion. (F) Representative images of TTC stained LV slices. Infarct size is quantified. Values are presented as mean ± SD, n = 6 for all groups, a one-way ANOVA followed by a Bonferroni test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. INTRA + DMSO. #p < 0.05, ###p < 0.001 vs. INTRA + GSK. &p < 0.05, &&&p < 0001 vs. POP50 + GSK.
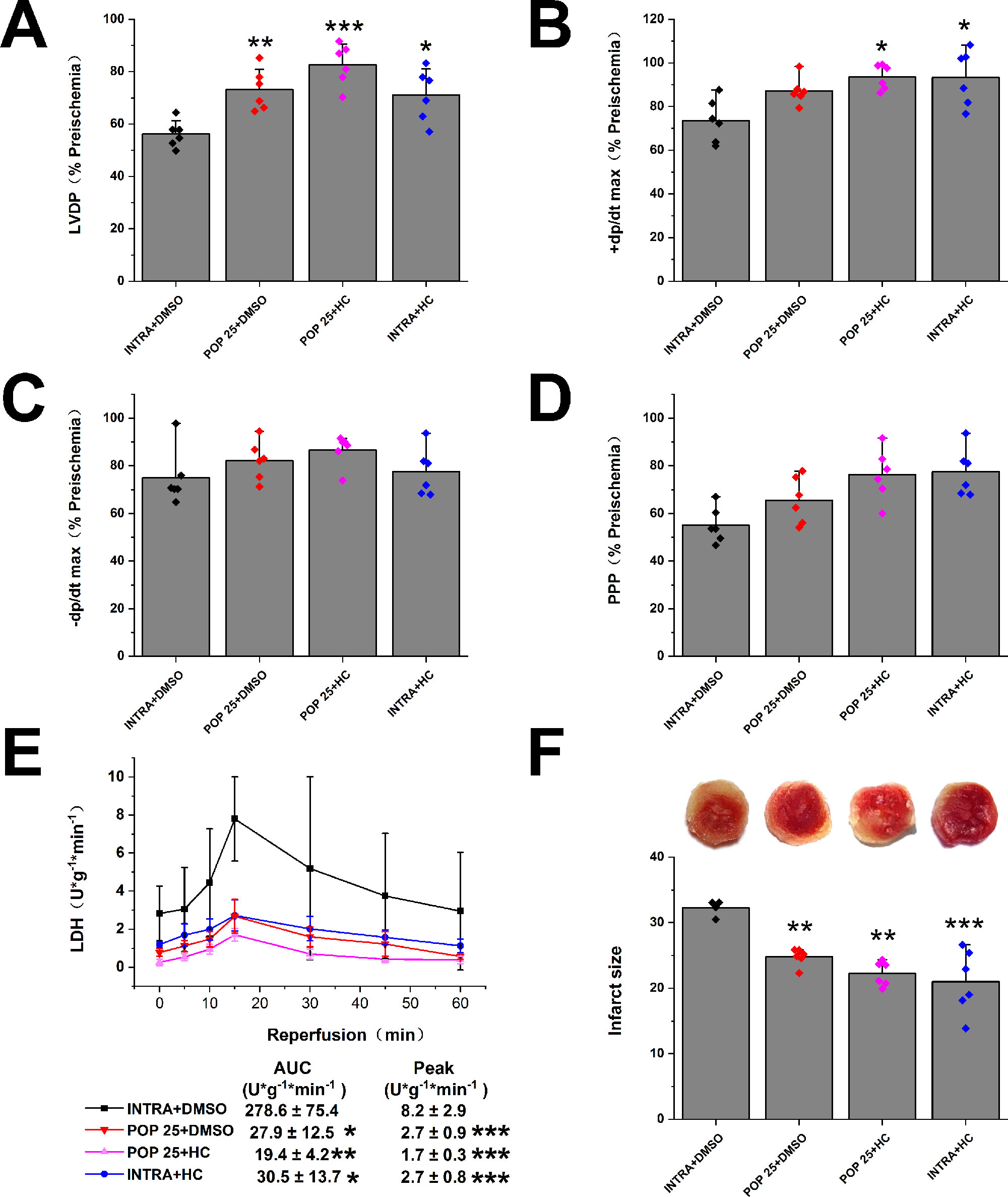
Figure 5 The effect of the combination of propofol and TRPV4 antagonist on myocardial I/R injury. Propofol or intralipid is given 5 minutes before applying TRPV4 antagonist or DMSO. The recovery of LVDP (A), +dP/dt max (B), -dP/dt max (C), and RPP (D) 60 minutes after reperfusion displayed as the percentage of respective preischemia values. (E) The amount of LDH release in the coronary effluent at different times after reperfusion. (F) Representative images of TTC stained LV slices. Infarct size is quantified. Values are presented as mean ± SD, n = 6 for all groups, a one-way ANOVA followed by a Bonferroni test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. INTRA + DMSO.
Propofol Attenuates H/R-Induced Intracellular Ca2+ Overload and Cell Injury in Cardiomyocytes Via the Suppression of TRPV4 Channel
As shown in Figures 6A, B, propofol has no obvious effect on the [Ca2+]i or cell viability in H9C2 cells under normoxic conditions ([Ca2+]i: 172.3 ± 47.4 nM vs. 149.1 ± 51.2 nM, cell viability: 100 ± 9.8% vs. 95.6 ± 5.4%). However, pretreatment with propofol (25, 50 and 100 μΜ) dose-dependently reduced the [Ca2+]i (from 464.0 ± 94.1 nM to 383.9 ± 97.8 nM, 245.8 ± 80.8 nM, and 196.9 ± 38.6 nM, respectively) and increased the cell viability in H9C2 cells subjected to H/R (from 65.9 ± 9.6% to 72.0 ± 9.3%, 80.9 ± 9.4%, and 82.6 ± 7.0%, respectively). Moreover, propofol significantly inhibited the enhancement effects of TRPV4 agonist GSK1016790A on H/R-induced intracellular Ca2+ overload and cell injury ([Ca2+]i: 238.0 ± 79.2 nM vs. 712.4 ± 216.0 nM, cell viability: 79.8 ± 10.0% vs. 41.8 ± 6.3% , Figures 6C, D). TRPV4-siRNA protected cells from H/R injury ([Ca2+]i: 311.9 ± 68.3 nM vs. 500.2 ± 178.1 nM, cell viability: 81.4 ± 6.8% vs. 64.3 ± 9.0%, Figures 6E, F), but this effect was not further enhanced when 50 μΜ propofol was applied to H9C2 cells transfected with TRPV4-siRNA ([Ca2+]i: 244.1 ± 52.3 nM vs. 311.9 ± 68.3 nM, cell viability: 85.9 ± 6.4% vs. 81.4 ± 6.8%). This indicates that propofol has no further cardioprotective effect in H9C2 cells transfected with TRPV4-siRNA. Therefore, we could infer that propofol attenuates H/R-induced intracellular Ca2+ overload and cell injury in H9C2 cells at least partially via the suppression of TRPV4 channel.
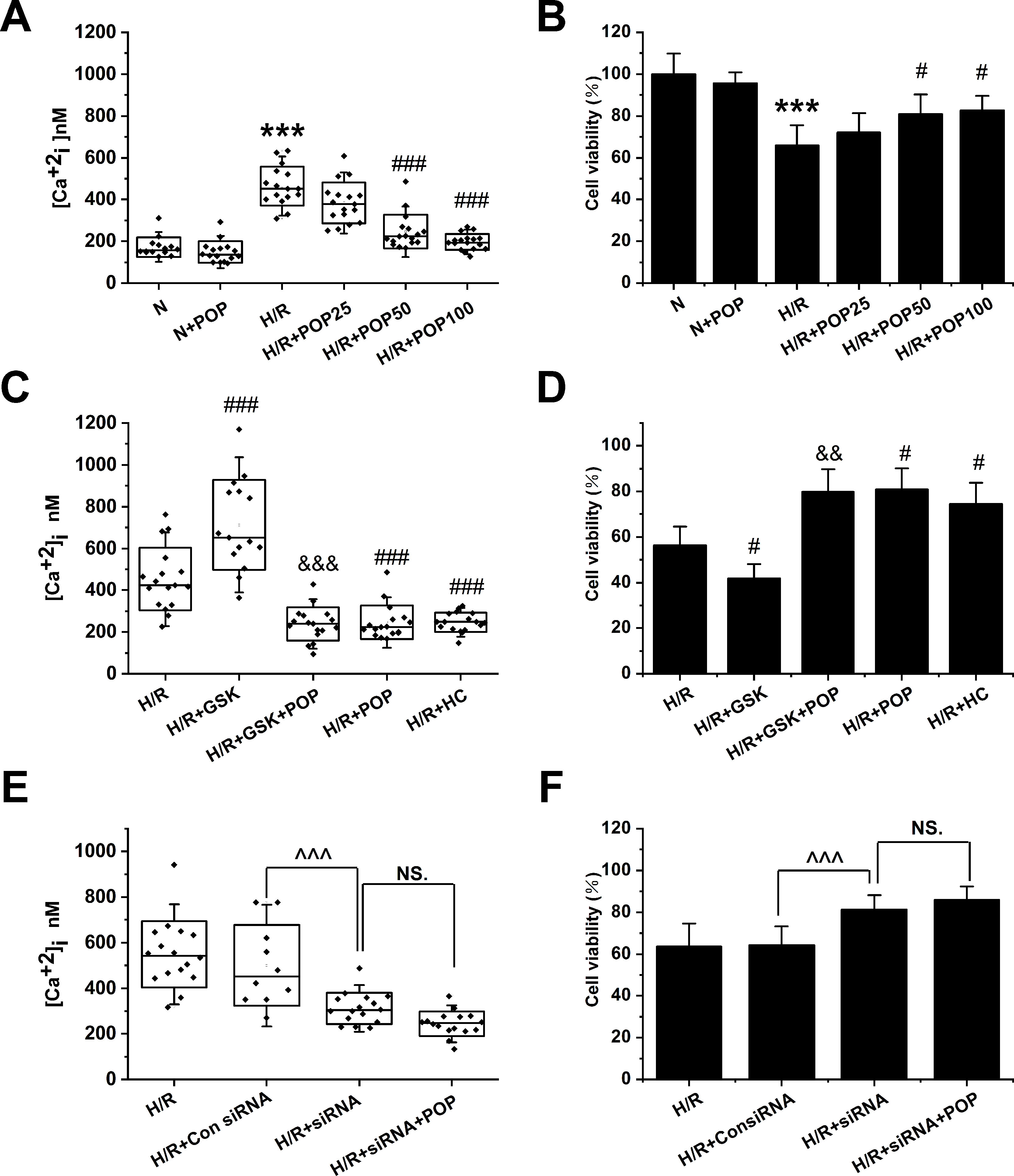
Figure 6 The effect of the combination of propofol and TRPV4 agonist/TRPV4-siRNA on the [Ca2+]i and cell viability in H9C2 cells subjected to H/R. Pretreatment with propofol dose-dependently attenuates [Ca2+]i (A, n = 14 in N group, n = 16 in N + POP and H/R groups, n = 17 in H/R + POP 25, H/R + POP 50, and H/R + POP 100 groups) and increases cell viability (B, n = 6 for all groups). (C, n = 17 in H/R, H/R + GSK + POP,H/R + POP, H/R + HC, and H/R + HC + POP groups, n = 15 in H/R + GSK group) and (D, n = 6 for all groups) show the effects of propofol in the presence of TRPV4 agonist GSK1016790A. (E, n = 16 in H/R, H/R + siRNA, and H/R + siRNA + POP groups, n = 10 in H/R + Con siRNA and H/R + POP groups) and (F, n = 6 for all groups) show the protective effects of TRPV4-siRNA combined with propofol. Values are presented as mean ± SD, a one-way ANOVA followed by a Bonferroni test, ***p < 0.001 vs. N. #p < 0.05, ###p < 0.001 vs. H/R. &&p < 0.01, &&&p < 0.001 vs. H/R+GSK. ^^^p < 0.001 vs. HR+Con siRNA, NS. means no statistical significance.
We also tested whether propofol had similar effects on ARVMs. ARVMs were isolated from adult rat hearts and were subjected to H/R after 1 day of incubation. Figure 7 shows H/R-induced [Ca2+]i increase (from 88.8 ± 20.0 nM to 278.5 ± 47.8 nM, p < 0.001 vs. N) and cell death expressed by the LDH release (from 53.8 ± 8.2 U/L to 113.5 ± 18.5U/L). Pretreatment with 50 μΜ propofol significantly suppressed the above effects ([Ca2+]i = 140.4 ± 38.1 nM, LDH = 75.1 ± 8.7 U/L). Similar to the effects in H9C2 cells, propofol reversed the intracellular Ca2+ overload and cell death induced by TRPV4 agonist GSK1016790A ([Ca2+] i: 156.4 ± 30.7 nM vs. 630.9 ± 144.6 nM, LDH: 131.4 ± 20.1 U/L vs. 220.8 ± 41.7 U/L).
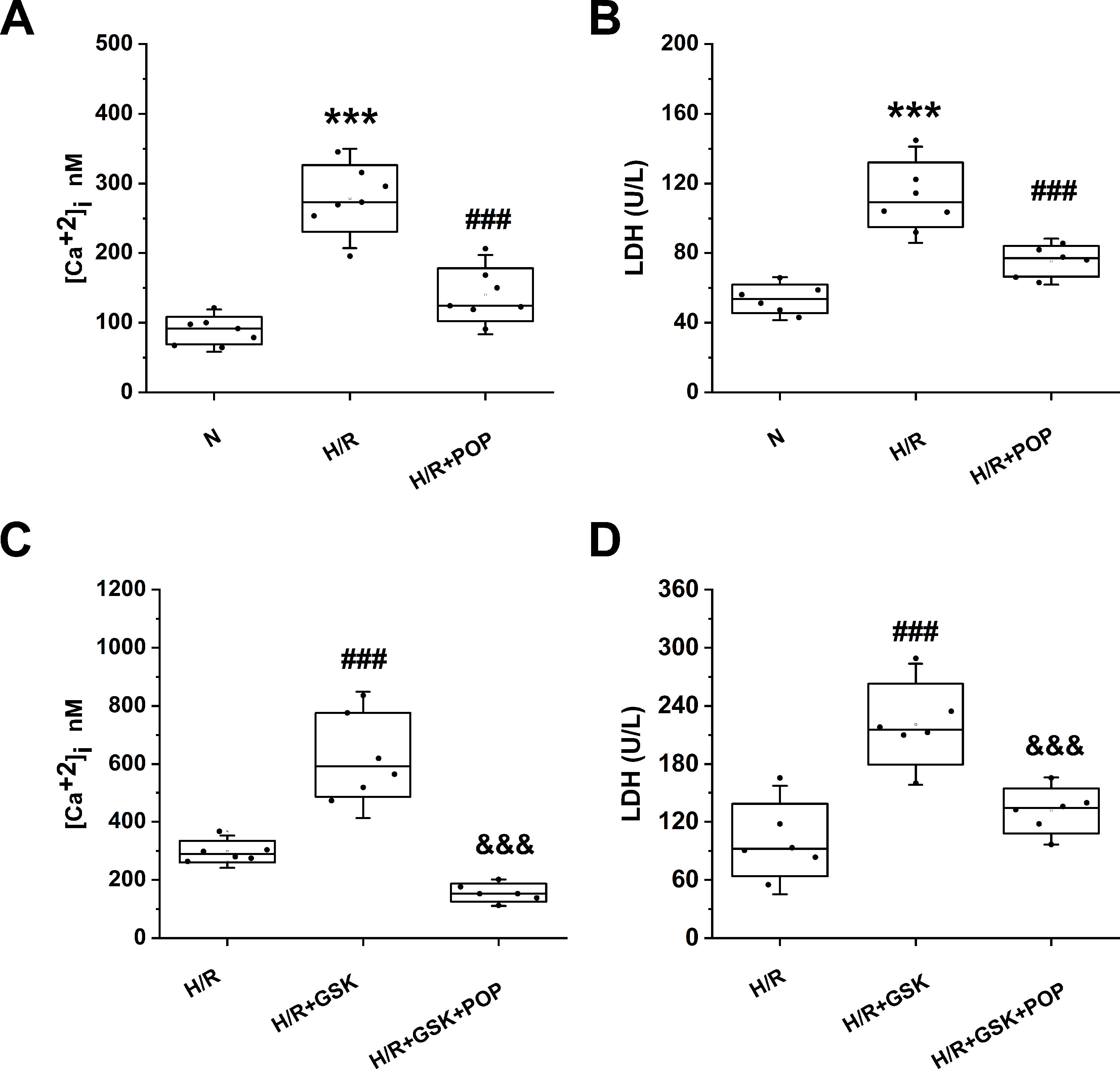
Figure 7 The effect propofol on TRPV4 agonist-induced [Ca2+]i and the LDH release in ARVMs subjected to H/R. Pretreatment with 50 µM propofol attenuates [Ca2+]i (A) and LDH release (B), (C) and (D) show the effects of propofol in the presence of TRPV4 agonist GSK1016790A. Values are presented as mean ± SD, n = 6 for all groups, a one-way ANOVA followed by a Bonferroni test, ***p < 0.001 vs. N. ###p< 0.001 vs. H/R. &&&p < 0.001 vs. H/R+GSK.
Propofol Suppresses TRPV4 Channel Function in a Concentration-Dependent Manner in 293T Cells Transfected With hTRPV4
To test the hypothesis that propofol inhibits TRPV4 channel function, we measured the Ca2+ influx induced by specific TRPV4 agonists GSK1016790A and 4α-PDD in HEK-293T cells transfected with hTRPV4. GSK1016790A and 4α-PDD both induced significant Ca2+ influx, which is reduced by pretreatment with propofol (12.5, 25, 50, 100 µM, Figures 8A, B). The quantitative analysis of the relative changes (∆F/F0) in Ca2+ influx is displayed in Figures 8 C, D. The concentration response data fitted by the Hill equation shows a propofol IC50 of 39.1 ± 4.5 μΜ under GSK1016790A and 45.3 ± 6.9 μΜ under 4α-PDD (Figures 8E, F). Our results proved that propofol suppresses the function of TRPV4 channel in a concentration-dependent manner.
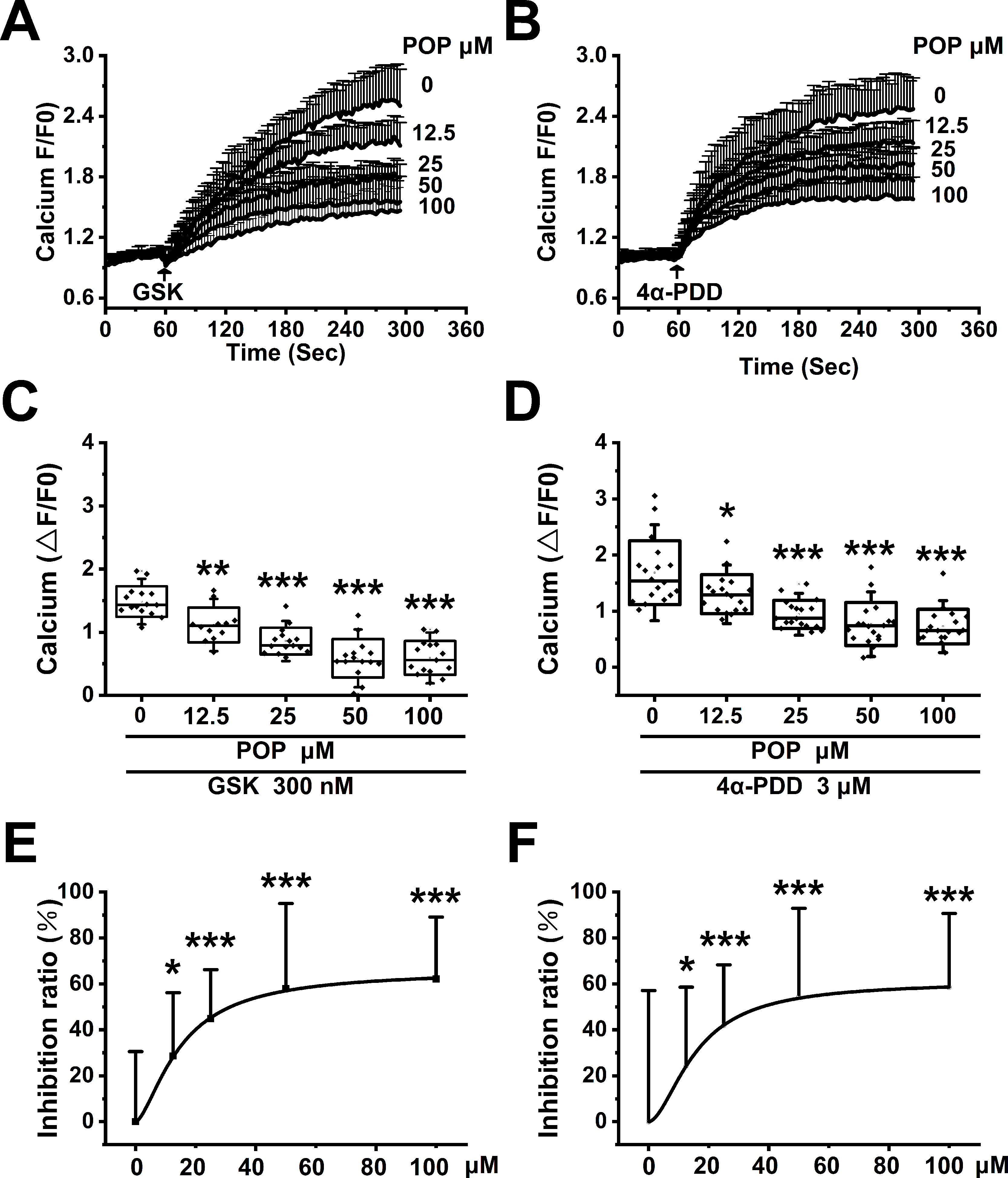
Figure 8 Propofol dose-dependently suppresses the Ca2+ influx induced by TRPV4 agonists in HEK-293T cells transfected with hTRPV4. The above graphs show intracellular Ca2+ level change induced by 300 nM GSK1016790A (A) and 3 μM 4α-PDD (B). Arrows indicate when GSK1016790A and 4α-PDD are applied. Quantitative analyses of the relative changes in Ca2+ influx induced by GSK1016790A (C) and 4α-PDD (D) (∆F/F0). The dose-response curve fitted by the Hill function showing the inhibition of Ca2+ influx induced by GSK1016790A (E) and 4α-PDD (F). Values are presented as mean ± SD, GSK1016790A: n = 15 in the absence of POP, n = 12 in the presence of propofol at 12.5 µM, n = 15 in the presence of propofol at 25, 50 and 100 µM, 4α-PDD: n = 18 for all groups, a one-way ANOVA followed by a Bonferroni test, *p < 0.05, **p < 0.01, ***p < 0.001 vs. without propofol.
Propofol Suppresses TRPV4 Channel Function in Cardiomyocytes
Confirming our previous observation (Wu et al., 2017) H9C2 and ARVMs show obvious Ca2+ influx after stimulation with 300 nM GSK1016790A (Figures 9A, D). Similar to our results from 293T cells transfected with hTRPV4, pretreatment with 50 μM propofol significantly reduced Ca2+ influx induced by GSK1016790A, but had no effect on Ca2+ influx induced by Ionomycin in H9C2 or ARVMs (Figures 9 B, E). The quantitative analysis of the relative changes (∆F/F0) in Ca2+ influx in cardiomyocytes is presented in Figures 9 C, F, and Ionomycin was set as a positive control. This demonstrates that propofol suppresses the function of TRPV4 channel in cardiomyocytes.
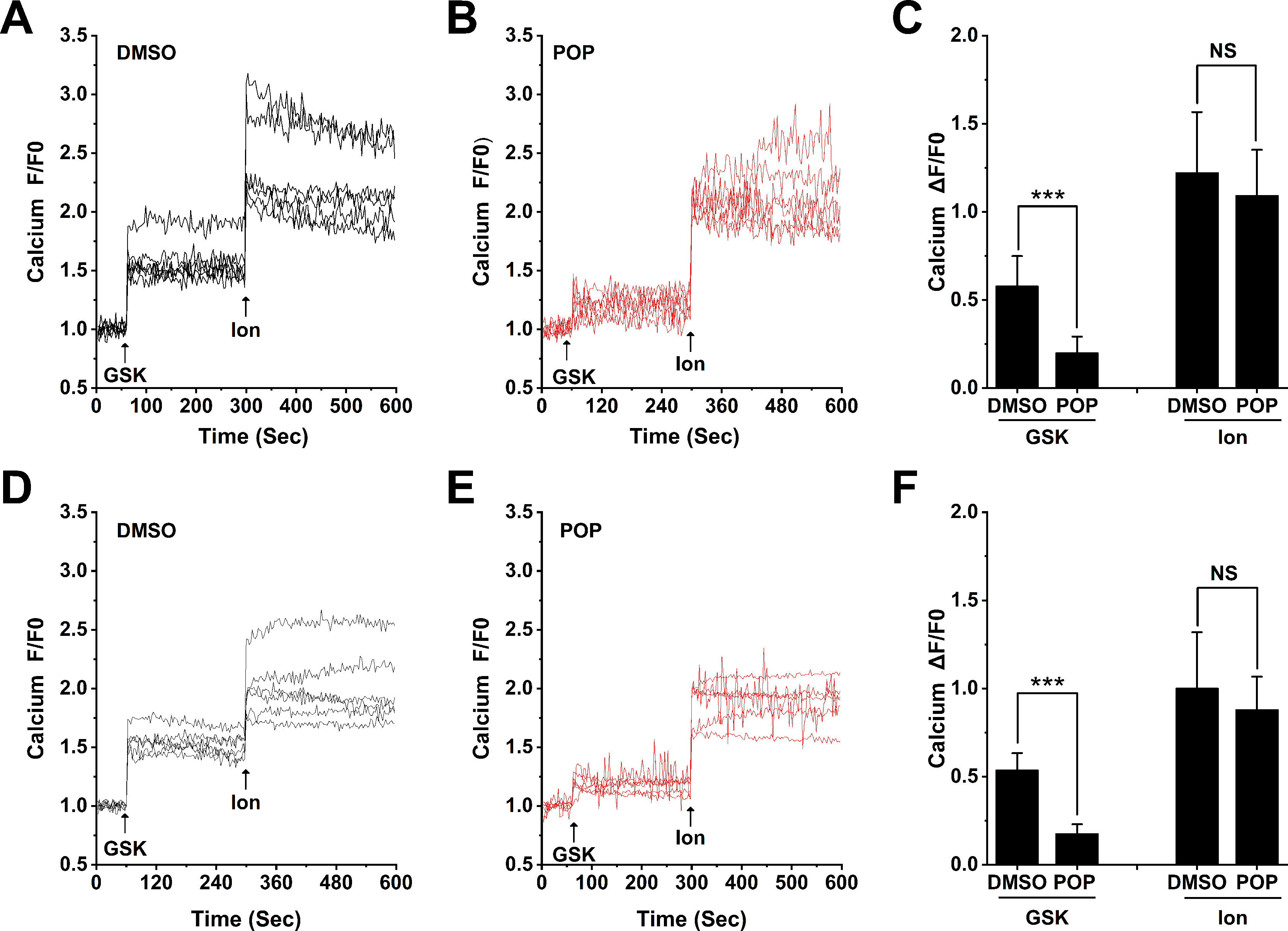
Figure 9 Propofol suppresses the Ca2+ influx induced by TRPV4 agonists in cardiomyocytes. Original time-lapse Ca2+ influx traces and quantification of GSK1016790A -induced [Ca2+]i response in H9C2 cells (A-C) and ARVMs (D-F). Arrows indicate when GSK1016790A (300 nM) and Ionomycin (1 μM) are applied. Cells are pretreated with 50 μM propofol for 30 minutes. Ionomycin is set as a positive control. Values are presented as mean ± SD, n = 6 for all groups, a two-tailed t test, ***p < 0.001 vs. DMSO, NS. means no statistical significance
Discussion
Earlier studies have demonstrated that propofol protects the heart against I/R injury through inhibiting intracellular Ca2+ overload (Kim et al., 2008). However, the specific mechanisms are not yet clear. Our previous research has shown that TRPV4 channel functional expression significantly increases in both the in vivo and in vitro model of myocardial I/R, resulting in intracellular Ca2+ overload and myocardial injury (Dong et al., 2017; Wu et al., 2017). This study is the first to prove that propofol attenuates myocardial I/R injury at least partially through the suppression of TRPV4 channel and the subsequent inhibition of intracellular Ca2+ overload.
We first confirmed the previous finding that propofol protects isolated hearts from I/R injury in a concentration-dependent manner. However, our results showed that 50 μM propofol had the best protective effects on mice, while Shao et al. and Ko et al. found that 100 μM has the best results on rat (Ko et al., 1997; Shao et al., 2008). The reason might be that the concentration of 100 μM is too high for mice and that high dosage propofol suppresses heart contraction by inhibiting Ca2+ influx as well as Ca2+-induced excitation-contraction coupling (Li et al., 1997; Han et al., 2016). Therefore, a concentration of 25 or 50 μM was chosen in the following experiment.
Using the Langendorff model, we found that 0.1 μM TRPV4 antagonist HC-067047 significantly promoted the recovery of LVDP, +dP/dt max, -dP/dt max, and RPP, and reduced LDH release as well as infarct size. On the contrary, 20 nM TRPV4 agonist GSK1016790A exacerbated myocardial I/R injury. These results demonstrated that TRPV4 channel activation contributed to myocardial I/R injury in ex vivo isolated mice hearts, which is consistent with the results of our previous in vivo and in vitro studies (Dong et al., 2017; Wu et al., 2017). We further investigated the effects of the combination propofol and TRPV4 agonist/antagonist on myocardial I/R injury. Our results show that the detrimental effects induced by GSK1016790A were reversed by pretreatment with propofol in vitro but not in ex vivo isolated hearts. TRPV4 activation during I/R is likely mediated by an endogenous mechanism that is less vigorous than GSK1067790A. Moreover, the exogenous GSK1067790A and the endogenous TRPV4 activators, e.g. EETs, may activate TRPV4 via different mechanisms. TRPV4 activation by I/R alone could be reduced by propofol. Furthermore, the combination of propofol and TRPV4 antagonist HC-067047 did not create additional protective effects ex vivo. Similar results were also found in H9C2 transfected with TRPV4-siRNA. This suggested that propofol reduced myocardial I/R injury at least partially via the suppression of TRPV4 channel.
Ca2+ is vital for heart contraction. However, after myocardial I/R, intracellular Ca2+ overload impairs left ventricular mechanical function (Steenbergen et al., 1993; Meissner and Morgan, 1995). A number of pathways have been found to mediate cardiomyocytes Ca2+ overload, including the Na+/Ca2+ exchanger, Ca2+-ATPase, L-type Ca2+ channel, sarcoplasmic reticulum Ca2+ release channel, and TRPV4 channel (Takahashi et al., 1994; Garcia-Dorado et al., 2012; Wu et al., 2017; Jones et al., 2019). One of our previous studies shows that the activation of TRPV4 channel contributes to H/R-induced intracellular Ca2+ overload in cardiomyocytes (Wu et al., 2017). In this study, we also measured the [Ca2+] i in H9C2 cells and ARVMs subjected to H/R. Pretreatment with propofol inhibited H/R-induced [Ca2+]i increase and also prevented the effect of TRPV4 agonist GSK1016790A on [Ca2+]i. Furthermore, we found no obvious difference in [Ca2+]i between H/R + POP, H/R + TRPV4 siRNA, and H/R + TRPV4 siRNA + POP groups. Therefore, we assume that propofol attenuates H/R-induced intracellular Ca2+ overload through the suppression of TRPV4 channel. In addition, many previous studies have already demonstrated that the blockage of TRPV4 channel is a promising target for I/R therapy (Jie et al., 2015; Jie et al., 2016; Dong et al., 2017; Jones et al., 2019; Wu et al., 2019).
In order to investigate whether propofol could inhibit TRPV4-mediared effects, we recorded the Ca2+ signals in H9C2 cells, ARVMs, and HEK-293T cells transfected with hTRPV4. Our results showed that propofol suppressed the Ca2+ influx induced by TRPV4 agonists GSK1016790A and 4α-PDD in a concentration-dependent manner. The IC50 is 39.1 ± 4.5 μΜ and 45.3 ± 6.9 μΜ, respectively, both of which were close to the concentration levels of 35 μΜ in clinical applications (Mathur et al., 1999), indicating that propofol can suppress TRPV4 channel to induce cardioprotective effects under I/R.
Propofol has also been reported to reduce Ca2+ entry via TRPC5 (Bahnasi et al., 2008) with an estimated IC50 value of 84.5 μΜ in transfected HEK293 cells. TRPC5 is present in the heart and is upregulated in the cardiac myocyte hypertrophy model and human heart failure conditions (Lau et al., 2016). Therefore, more research is required to determine whether propofol-induced cardiac protection acts through a TRPC5-dependent mechanism. Propofol, as a commonly used anesthetic in clinical practice, is recognized as a safe and effective drug. However, unwanted side effects with propofol use have been documented and include hypotension, postoperative pain, and propofol-related infusion syndrome, a rare but seriously life-threatening complication (Mirrakhimov et al., 2015). Activation of TRPA1 and TRPV1 has been demonstrated to contribute to propofol-induced vasodilation and pain (Nishimoto et al., 2015; Sinharoy et al., 2017). Interestingly, the concentrations of propofol that induced Ca2+ influx via TRPA1 (EC50 = 65.4 μM) and TRPV1 (90 μM) are similar to those that affect TRPV4. Moreover, TRPV4 is also abundant in smooth muscle and endothelial cells of blood vessels (Randhawa and Jaggi, 2015). TRPV4 activation involves in shear stress-induced vasodilation as well as the damage of the vascular barrier. Whether TRPV4 inhibition involved in propofol-induced vasoreactivity or propofol-related infusion syndrome requires further research.
Our study has a few limitations. First, ex vivo or in vitro findings might be different from in vivo results. Second, although we discovered that propofol suppresses TRPV4 channel, which has yielded new insights into how propofol induces cardioprotective effects, the binding sites of propofol and TRPV4 channel are still unclear. This requires further research that applies site-directed mutagenesis methods, gene delivery techniques, and advanced computational technologies. In addition, although common in other studies, the knockdown efficiency (50%) of TRPV4-siRNA in the present study is low (Adapala et al., 2013; Suresh et al., 2015).
In conclusion, we conducted a series of ex vivo and in vitro experiments and demonstrated that propofol directly inhibited Ca2+ entry via TRPV4 channel, which is a newfound mechanism whereby propofol reduces intracellular Ca2+ overload and subsequently attenuates myocardial I/R injury. Due to the widespread expression and numerous effects of TRPV4 channel, the findings of this research are also relevant to other organ systems, including the respiratory, digestive, and urinary systems (Randhawa and Jaggi, 2015). Recent evidence has demonstrated that TRPV4 channel activation is related to pulmonary edema, gastrointestinal disorders, and bladder dysfunction. Moreover, a recent review article has highlighted the importance of TRPV4 channel in the pathogenesis of various diseases (White et al., 2016). Therefore, we think the use of propofol during the perioperative period is potentially beneficial to the prevention of TRPV4 channel-related diseases. However, this is beyond the scope of this study and requires further research. In addition, future research will need to examine whether other general anesthetics, such as volatile anesthetics or etomidate, also suppress TRPV4 channel to produce cardioprotective effects.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
All procedures concerning animal use were performed in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and approved by Tongji Medical College Committee on Animal Care.
Author Contributions
BW, QW, NZ, ZH, KG, and YD contributed conception and design of the study; BW, QW, JL, SZ, HL, CY, QD, and NZ organized the database; BW, QW, and JL performed the statistical analysis; BW, QW, and JL wrote the first draft of the manuscript; ZH, KG, and YD wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This research was funded by the National Nature Science Foundation of China, grant number 81470421 (YD), 81770328 (YD), and 81700332 (NZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Prof. Kewei Wang (Peking University School of Pharmaceutical Sciences, Qingdao University School of Pharmacy, China) for hTRPV4 plasmid. YD thanks Mr. Li Xiao for his consistent support during this research and Ms. Jenny Xiao (The University of Hong Kong, Hong Kong, China) for providing linguistic assistance.
Supplementary Material
The Supplementary Material for this article can be found online At: https://www.frontiersin.org/articles/10.3389/fphar.2019.01150/full#supplementary-material
References
Adapala, R. K., Thoppil, R. J., Luther, D. J., Paruchuri, S., Meszaros, J. G., Chilian, W. M., et al. (2013). TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell Cardiol. 54, 45–52. doi: 10.1016/j.yjmcc.2012.10.016
Bahnasi, Y. M., Wright, H. M., Milligan, C. J., Dedman, A. M., Zeng, F., Hopkins, P.M., et al. (2008). Modulation of TRPC5 cation channels by halothane, chloroform and propofol. Br. J. Pharmacol. 153, 1505–1512. doi: 10.1038/sj.bjp.0707689
Dong, Q., Li, J., Wu, Q. F., Zhao, N., Qian, C., Ding, D., et al. (2017). Blockage of transient receptor potential vanilloid 4 alleviates myocardial ischemia/reperfusion injury in mice. Sci. Rep. 7, 42678. doi: 10.1038/srep42678
Everaerts, W., Zhen, X., Ghosh, D., Vriens, J., Gevaert, T., Gilbert, J.P., et al. (2010). Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc. Natl. Acad. Sci. U. S. A. 107, 19084–19089. doi: 10.1073/pnas.1005333107
Fu, X., Du, L., Zhao, N., Dong, Q., Liao, Y., Du, Y. (2013). 18B-Glycyrrhetinic acid potently inhibits Kv1.3 potassium channels and T cell activation in human Jurkat T cells. J. Ethnopharmacol. 148, 647–654. doi: 10.1016/j.jep.2013.05.022
Fu, X. X., Zhao, N., Dong, Q., Du, L. L., Chen, X. J., Wu, Q. F., et al. (2015). Interleukin-17A contributes to the development of post-operative atrial fibrillation by regulating inflammation and fibrosis in rats with sterile pericarditis. Int. J. Mol. Med. 36, 83–92. doi: 10.3892/ijmm.2015.2204
Garcia-Dorado, D., Ruiz-Meana, M., Inserte, J., Rodriguez-Sinovas, A., Piper, H. M. (2012). Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 94, 168–180. doi: 10.1093/cvr/cvs116
Han, L., Fuqua, S., Li, Q., Zhu, L., Hao, X., Li, A., et al. (2016). Propofol-induced inhibition of catecholamine release is reversed by maintaining calcium influx. Anesthesiology 124, 878–884. doi: 10.1097/ALN.0000000000001015
Jie, P., Hong, Z., Tian, Y., Li, Y., Lin, L., Zhou, L., et al. (2015). Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 6, e1775. doi: 10.1038/cddis.2015.146
Jie, P., Lu, Z., Hong, Z., Li, L., Zhou, L., Li, Y., et al. (2016). Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Mol. Neurobiol. 53, 8–17. doi: 10.1007/s12035-014-8992-2
Jie, P., Tian, Y., Hong, Z., Li, L., Zhou, L., Chen, L., et al. (2015). Blockage of transient receptor potential vanilloid 4 inhibits brain edema in middle cerebral artery occlusion mice. Front. Cell Neurosci. 9, 141. doi: 10.3389/fncel.2015.00141
Jones, J. L., Peana, D., Veteto, A. B., Lambert, M. D., Nourian, Z., Karasseva, N. G., et al. (2019). TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc. Res. 115, 46–56. doi: 10.1093/cvr/cvy156
Kim, H. S., Chang, W. C., Hwang, K. C., Choi, I. G., Park, W. K. (2008). Effect of propofol on calcium homeostasis in hypoxia-reoxygenated neonatal rat cardiomyocytes. Eur. J. Pharmacol. 594, 139–145. doi: 10.1016/j.ejphar.2008.07.027
Ko, S. H., Yu, C. W., Lee, S. K., Choe, H., Chung, M. J., Kwak, Y. G., et al. (1997). Propofol attenuates ischemia-reperfusion injury in the isolated rat heart. Anesth. Analg. 85, 719–724. doi: 10.1213/00000539-199710000-00002
Kojima, A., Ito, Y., Kitagawa, H., Matsuura, H. (2015). Ionic mechanisms underlying the negative chronotropic action of propofol on sinoatrial node automaticity in guinea pig heart. Br. J. Pharmacol. 172, 799–814. doi: 10.1111/bph.12936
Lau, O. C., Shen, B., Wong, C. O., Tjong, Y. W., Lo, C. Y., Wang, H. C., et al. (2016). TRPC5 channels participate in pressure-sensing in aortic baroreceptors. Nat Commun 7, 11947. doi: 10.1038/ncomms11947
Li, Y. C., Ridefelt, P., Wiklund, L., Bjerneroth, G. (1997). Propofol induces a lowering of free cytosolic calcium in myocardial cells. Acta Anaesthesiol. Scand. 41, 633–638. doi: 10.1111/j.1399-6576.1997.tb04756.x
Liu, X. R., Cao, L., Li, T., Chen, L. L., Yu, Y. Y., Huang, W. J., et al. (2017). Propofol attenuates H2O2-induced oxidative stress and apoptosis via the mitochondria- and ER-medicated pathways in neonatal rat cardiomyocytes. Apoptosis 22, 639–646. doi: 10.1007/s10495-017-1349-3
Luk, H. N., Yu, C. C., Lin, C. I., Yang, C. Y. (1995). Electropharmacological actions of propofol on calcium current in guinea-pig ventricular myocytes. J. Electrocardiol. 28, 332–333. doi: 10.1016/S0022-0736(05)80052-4
Mathur, S., Farhangkhgoee, P., Karmazyn, M. (1999). Cardioprotective effects of propofol and sevoflurane in ischemic and reperfused rat hearts: Role of K(ATP) channels and interaction with the sodium-hydrogen exchange inhibitor HOE 642 (cariporide). Anesthesiology 91, 1349–1360. doi: 10.1097/00000542-199911000-00027
Meissner, A., Morgan, J. P. (1995). Contractile dysfunction and abnormal Ca2+ modulation during postischemic reperfusion in rat heart. Am. J. Physiol. 268, H100–H111. doi: 10.1152/ajpheart.1995.268.1.H100
Mirrakhimov, A. E., Voore, P., Halytskyy, O., Khan, M., Ali, A. M. (2015). Propofol infusion syndrome in adults: A clinical update. Crit. Care Res. Pract. 2015, 260385. doi: 10.1155/2015/260385
Nishimoto, R., Kashio, M., Tominaga, M. (2015). Propofol-induced pain sensation involves multiple mechanisms in sensory neurons. Pflugers Arch. 467, 2011–2020. doi: 10.1007/s00424-014-1620-1
Randhawa, P. K., Jaggi, A. S. (2015). TRPV4 channels: Physiological and pathological role in cardiovascular system. Basic Res. Cardiol. 110, 54. doi: 10.1007/s00395-015-0512-7
Rossello, X., Hall, A. R., Bell, R. M., Yellon, D. M. (2016). Characterization of the langendorff perfused isolated mouse heart model of global ischemia-reperfusion injury: Impact of ischemia and reperfusion length on infarct size and LDH release. J. Cardiovasc. Pharmacol. Ther. 21, 286–295. doi: 10.1177/1074248415604462
Ryskamp, D. A., Witkovsky, P., Barabas, P., Huang, W., Koehler, C., Akimov, N. P., et al. (2011). The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 31, 7089–7101. doi: 10.1523/JNEUROSCI.0359-11.2011
Shao, H., Li, J., Zhou, Y., Ge, Z., Fan, J., Shao, Z., et al. (2008). Dose-dependent protective effect of propofol against mitochondrial dysfunction in ischaemic/reperfused rat heart: Role of cardiolipin. Br. J. Pharmacol. 153, 1641–1649. doi: 10.1038/bjp.2008.45
Sinharoy, P., Bratz, I. N., Sinha, S., Showalter, L. E., Andrei, S. R., Damron, D. S. (2017). TRPA1 and TRPV1 contribute to propofol-mediated antagonism of U46619-induced constriction in murine coronary arteries. PLoS One 12, e180106. doi: 10.1371/journal.pone.0180106
Steenbergen, C., Fralix, T. A., Murphy, E. (1993). Role of increased cytosolic free calcium concentration in myocardial ischemic injury. Basic Res. Cardiol. 88, 456–470. doi: 10.1007/BF00795412
Sun, H. J., Lu, Y., Wang, H. W., Zhang, H., Wang, S. R., Xu, W. Y., et al. (2017). Activation of endocannabinoid receptor 2 as a mechanism of propofol pretreatment-induced cardioprotection against ischemia-reperfusion injury in rats. Oxid. Med. Cell Longev. 2017, 2186383. doi: 10.1155/2017/2186383
Suresh, K., Servinsky, L., Reyes, J., Baksh, S., Undem, C., Caterina, M., et al. (2015). Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell Mol. Physiol. 309, L1467–L1477. doi: 10.1152/ajplung.00275.2015
Sutherland, F. J., Shattock, M. J., Baker, K. E., Hearse, D. J. (2003). Mouse isolated perfused heart: Characteristics and cautions. Clin. Exp. Pharmacol. Physiol. 30, 867–878. doi: 10.1046/j.1440-1681.2003.03925.x
Takahashi, H., Puttick, R. M., Terrar, D. A. (1994). The effects of propofol and enflurane on single calcium channel currents of guinea-pig isolated ventricular myocytes. Br. J. Pharmacol. 111, 1147–1153. doi: 10.1111/j.1476-5381.1994.tb14865.x
Ton, H. T., Phan, T. X., Abramyan, A. M., Shi, L., Ahern, G. P. (2017). Identification of a putative binding site critical for general anesthetic activation of TRPA1. Proc. Natl. Acad. Sci. U. S. A. 114, 3762–3767. doi: 10.1073/pnas.1618144114
White, J. P., Cibelli, M., Urban, L., Nilius, B., McGeown, J. G., Nagy, I. (2016). TRPV4: Molecular conductor of a diverse orchestra. Physiol. Rev. 96, 911–973. doi: 10.1152/physrev.00016.2015
Wu, Q., Lu, K., Zhao, Z., Wang, B., Liu, H., Zhang, S., et al. (2019). Blockade of transient receptor potential vanilloid 4 enhances antioxidation after myocardial ischemia/reperfusion. Oxid. Med. Cell Longev. 2019, 7283683. doi: 10.1155/2019/7283683
Wu, Q. F., Qian, C., Zhao, N., Dong, Q., Li, J., Wang, B. B., et al. (2017). Activation of transient receptor potential vanilloid 4 involves in hypoxia/reoxygenation injury in cardiomyocytes. Cell Death Dis. 8, e2828. doi: 10.1038/cddis.2017.227
Xia, Z., Godin, D. V., Ansley, D. M. (2003). Propofol enhances ischemic tolerance of middle-aged rat hearts: Effects on 15-F(2t)-isoprostane formation and tissue antioxidant capacity. Cardiovasc. Res. 59, 113–121. doi: 10.1016/S0008-6363(03)00351-1
Xia, Z., Huang, Z., Ansley, D. M. (2006). Large-dose propofol during cardiopulmonary bypass decreases biochemical markers of myocardial injury in coronary surgery patients: A comparison with isoflurane. Anesth. Analg. 103, 527–532. doi: 10.1213/01.ane.0000230612.29452.a6
Keywords: propofol, TRPV4, cardioprotection, ischemia-reperfusion injury, calcium
Citation: Wang B, Wu Q, Liao J, Zhang S, Liu H, Yang C, Dong Q, Zhao N, Huang Z, Guo K and Du Y (2019) Propofol Induces Cardioprotection Against Ischemia-Reperfusion Injury via Suppression of Transient Receptor Potential Vanilloid 4 Channel. Front. Pharmacol. 10:1150. doi: 10.3389/fphar.2019.01150
Received: 21 April 2019; Accepted: 06 September 2019;
Published: 04 October 2019.
Edited by:
Issy Laher, University of British Columbia, CanadaReviewed by:
James S. K. Sham, Johns Hopkins University, United StatesTim Murphy, University of New South Wales, Australia
Copyright © 2019 Wang, Wu, Liao, Zhang, Liu, Yang, Dong, Zhao, Huang, Guo and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kefang Guo, drguokefang@foxmail.com; Yimei Du, yimeidu@mail.hust.edu.cn
†These authors have contributed equally to this work