 Zuheng Liu1,2†
Zuheng Liu1,2† Haiping Tian1,2,3†Jinghai Hua1,2Wanqiang Cai1,2Yujia Bai1,2Qiong Zhan1,2Wenyan Lai1,2
Haiping Tian1,2,3†Jinghai Hua1,2Wanqiang Cai1,2Yujia Bai1,2Qiong Zhan1,2Wenyan Lai1,2 Qingchun Zeng1,2Hao Ren2,4*
Qingchun Zeng1,2Hao Ren2,4* Dingli Xu1,2*
Dingli Xu1,2*- 1State Key Laboratory of Organ Failure Research, Department of Cardiology, Nanfang Hospital, Southern Medical University, Guangzhou, China
- 2Key Laboratory for Organ Failure Research, Ministry of Education of the People’s Republic of China, Guangzhou, China
- 3Department of Cardiology, The Affiliated Hospital, Inner Mongolia Medical University, Hohhot, China
- 4Department of Rheumatology, Nanfang Hospital, Southern Medical University, Guangzhou, China
Chromosomal maintenance 1 (CRM1) inhibitors display antihypertrophic effects and control protein trafficking between the nucleus and the cytoplasm. PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1alpha) is a type of transcriptional coactivator that predominantly resides in the nucleus and is downregulated during heart failure. NT-PGC-1α is an alternative splicing variant of PGC-1α that is primarily distributed in the cytoplasm. We hypothesized that the use of a CRM1 inhibitor could shuttle NT-PGC-1α into the nucleus and activate PGC-1α target genes to potentially improve cardiac function in a mouse model of myocardial infarction (MI). We showed that PGC-1α and NT-PGC-1α were decreased in MI-induced heart failure mice. Phenylephrine and angiotensin II were applied to induce hypertrophy in neonatal rat ventricular myocytes (NRVMs). The antihypertrophic effects of the CRM1-inhibitor Selinexor was verified through profiling the expression of β-MHC and through visualizing the cell cross-sectional area. NRVMs were transfected with adenovirus-NT-PGC-1α or adenovirus-NLS (nucleus localization sequence)-NT-PGC-1α and then exposed to Selinexor. Confocal microscopy was then used to observe the shuttling of NT-PGC-1α. After NT-PGC-1α was shuttled into the nucleus, there was increased expression of its related genes, including PPAR-α, Tfam, ERR-γ, CPT1b, PDK4, and Nrf2. The effects of Selinexor on post-MI C57BL/6j mice were determined by echocardiography and qPCR. We found that Selinexor showed antihypertrophic effects but did not influence the ejection fraction of MI-mice. Interestingly, the antihypertrophic effects of Selinexor might be independent of NT-PGC-1α transportation.
Introduction
Rates of heart failure (HF) are rising at an alarming rate throughout the world (Martinez-Gonzalez and Ruiz-Canela, 2015). HF is commonly accompanied by cardiac hypertrophy (Warren et al., 2017). Previous studies suggest that chromosomal maintenance 1 (CRM1) (also called exportin-1) inhibitors exhibit antihypertrophic effects by influencing β-MHC and HDACs (Monovich et al., 2009; Chahine et al., 2015). Additional studies on NT-PGC-1α, which is an alternative splicing variant of PGC-1α, have revealed that CRM1 inhibitors can lead to a relative increase in the nuclear distribution of NT-PGC-1α (Chang et al., 2010). Thus, the regulation of NT-PGC-1α by CRM1 inhibitors might represent a novel mechanism of their anti-cardiac-hypertrophy effect.
NT-PGC-1α is a powerful regulator of fatty acid oxidation (FAO) in adipose tissue (Zhang et al., 2009; Jun et al., 2014; Kim et al., 2016; Liu et al., 2018). In recent years, a large amount of experimental evidence has demonstrated that HF is associated with metabolic dysfunction, which is accompanied by the down regulation of PGC-1α, a key factor in controlling mitochondrial energy metabolism (Arany et al., 2006; Schilling and Kelly, 2011; Arumugam et al., 2016). It is generally accepted that the dysregulation of mitochondrial energy metabolism aggravates HF (Chang et al., 2016; Parihar and Parihar, 2017). Thus, NT-PGC-1α might be important in the progression and pathogenesis of HF. NT-PGC-1α is predominantly distributed in the cytoplasm, and the PGC1 families fulfill most of their function through the coactivation of transcription factors in the nucleus (Zhang et al., 2009).
Although the subcellular distributions of NT-PGC-1α and PGC-1α are different, NT-PGC-1α is able to partially compensate for the function of PGC-1α, and this capability seems to be even more robust in FAO (Jun et al., 2014). Furthermore, cAMP (cyclic adenosine monophosphate) analogs can trigger both the shuffling of NT-PGC-1α into the nucleus and the activation of PGC-1α (Zhang et al., 2009). Therefore, it is plausible that NT-PGC-1α fulfills the role of PGC-1α by entering the nucleus when energy demands are increased. Importantly, the target genes of the PGC1 families are not limited to genes involved in FAO but include others, such as FOXO, ERRs, and NRFs, which are favorable in cardiovascular diseases (Vega et al., 2000; Riehle and Abel, 2012; Martin et al., 2014). Thus, we hypothesized that transporting NT-PGC-1α into the nucleus would likely lead to the activation of targets of PGC-1α, which might be beneficial to cardiac function. As a result, CRM1 inhibitors might be promising candidates for this strategy.
Materials and Methods
Experimental Animals
The 8- to 10-week-old male C57BL/6J mice and neonatal Sprague-Dawley rats that were used in this study were obtained from the laboratory of animal center of Southern Medical University. All mice were housed in cages and bred in a temperature-controlled room that was maintained at 22–26°C on a 12 h light–dark cycle with standard feed and water. This study was approved by the Southern Medical University Review Board and the animals used in this study comply with the Guide for the Care and Use of Laboratory Animals (NIH, 8th Edition, 2011).
Models of Myocardial Infarction
The mice used in this study were anesthetized with a mixture of xylazine (5 mg/kg) and ketamine (100 mg/kg) delivered by intraperitoneal injection. Once the mouse was anesthetized, the trachea was intubated to provide mechanical ventilation (inspiration/expiration ratio: 1:3, 120 strokes/min), the left thorax of the mouse was opened, and a left coronary artery ligation was performed to induce a myocardial infarction. Successful ligation was confirmed by ST-segment elevation as measured by an electrocardiogram. The sham-operated mice were subjected to the same treatment without ligation. Three days after the operation, Selinexor (10 mg/kg) or DMSO (20 μl) mixed with 0.2 ml distilled water were given by gavage to MI-mice every 3 days. After 30 postoperative days, the mice were sacrificed using an overdose of anesthetic. Their hearts were extracted and soaked in liquid nitrogen, then stored in an −80°C freezer for future use or extracted for HE and immunohistochemical staining.
Echocardiography
Echocardiography was performed on anesthetized (2% isoflurane) mice using a VEVO2100 system (Visual Sonic, North American), which has been previously described (Liu et al., 2017). The LV end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD) were measured using the M-mode.
Isolation and Culture of Neonatal Rat Ventricular Cardiomyocytes (NRVMs)
One- to three-day-old Sprague-Dawley rats were sacrificed via 2% isoflurane inhalation and cervical dislocation. The hearts were then removed, dissected, and enzymatically digested with 0.2% pancreatin overnight. The cells were then isolated by magnetic stirring with collagenase II (1 mg/mL) in a sterile glass vial. After 90 min of differential adhesion, the isolated cells were plated in a culture dish containing 0.1 mM 5-bromo-2′-deoxyuridine (BrdU, Sigma) and 10% fetal bovine serum (FBS, Gibco) to inhibit fibroblast proliferation. After 48 h, spontaneously contracting neonatal rat ventricular myocytes (NRVMs) were treated with either 10 μM phenylephrine (PE, Selleck) for 72 h, 1 μM angiotensin II (AngII, Abcam) for 24 h or 50 nM Selinexor (Selleck) for 4 h in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco; Thermo Fisher) containing penicillin and streptomycin (100:1) (Gibco; Thermo Fisher). Phalloidin staining was performed by first fixing the cells with 4% paraformaldehyde for 10 min, then washing with phosphate buffered saline containing 0.1% Triton X-100, and finally staining with FITC-phalloidin (Actin-tracker green) (Beyotime, China) for 60 min and Hoechst 33258 (Thermo Fisher) for 15 min. Adenovirus-infected cells were stained Hoechst 33258 (Thermo Fisher) for 15 min after being treated with 50 nM Selinexor (Selleck) for 4 h. The results were visualized by confocal microscopy. Cell viability was assessed using the Cell Counting Kit-8 (CCK8) according to the manufacturer’s guidelines and previous reports (Lin et al., 2016). The CCK8 assay was also used to test the toxicity of Selinexor on the NRVMs.
Infection of Adenovirus in NRVMs
The adenoviruses, including mCherry-NT-PGC-1α and mCherry-NLS-NT-PGC-1α, were purchased from the OBiO Technology Corp. Ltd., China. Nuclear localization sequence (NLS) is an amino acid sequence that controls protein import into the cell nucleus. A multiplicity of infection (MOI) of 100 was used for the adenovirus to induce the overexpression of NT-PGC-1α in the NRVMs. Four hours after the initial infection, an equal volume of fresh growth medium was added to the culture. The cells were then incubated for 24 h to allow the virus to achieve its maximum effect.
Real-Time PCR Assays
Total RNA was extracted by RNAiso Plus (Takara). We used 1 μg of total RNA per reaction for the first-strand cDNA synthesis using RT primers (Takara). Quantitative real-time PCR assays were performed using cDNA in a 10 μL reaction volume (SYBR Green PCR kit; Takara) on an Applied LightCycler 480 system (Roche). Gene expression levels were measured using the ΔΔCt method with normalization to β-actin. The primers used in this study were obtained from Sangon (primer sequences were listed in Tables 1, 2).

Table 1. Primer sequences.

Table 2. Primer sequences.
Western Blot and Co-immunoprecipitation
Proteins were collected from the cultured NRVMs and murine hearts by a mixture of RIPA buffer (Beyotime Biotechnology, Shanghai, China) with protease inhibitor (Sigma, United States) (1:100) and quantified by BCA assay (Thermo Fisher). The primary antibodies used included Anti-β-MHC (1:1000, Abcam, United States), Anti-N-terminal NT-PGC-1α (1:1000, Abcam, United States), anti-CRM1 (1:500, Abcam, United States) and anti-β-actin (1:1000, Bioss, Beijing, China). The secondary antibody used was a goat anti-rabbit IgG-HRP (1:5000, Santa Cruz, United States). Immunoreactive bands were detected by the Pierce ECL Substrate (Pierce Biotechnology, Rockford, IL, United States) and Gene Gnome Imaging System (Syngene Bio Imaging) and quantified with the NIH ImageJ software package. The protein A/G agarose beads were purchased from Santa Cruz Biotechnology, and the CO-IP protocol was carried out according to the manufactures’ guidelines.
Statistical Analyses
Quantitative data were displayed as the mean ± SEM (standard error of mean). A normal distribution test was performed to determine if parametric or non-parametric tests should be used. Comparisons between the two experimental groups were based on a two-tailed t-test while comparisons of the parameters across more than three groups were analyzed by ANOVA followed by a Dunnett’s T3 for post hoc multiple comparisons. For all analyses, differences were considered to be statistically significant at a value of P < 0.05.
Results
Downregulation of PGC-1α and NT-PGC-1α in Mice With MI-Induced Heart Failure
Previous studies have thoroughly described alterations in cardiac metabolic substrates during HF. Here, we used a model of HF that was induced by MI. Four weeks after the operation, the myocardial expression of PGC-1α and NT-PGC-1α were significantly decreased (compared to sham-operated mice; n = 5; P < 0.05), as determined by Western blot (Figures 1A,B). The representative photographs of the immunohistochemical staining are shown in Figures 1C,D.
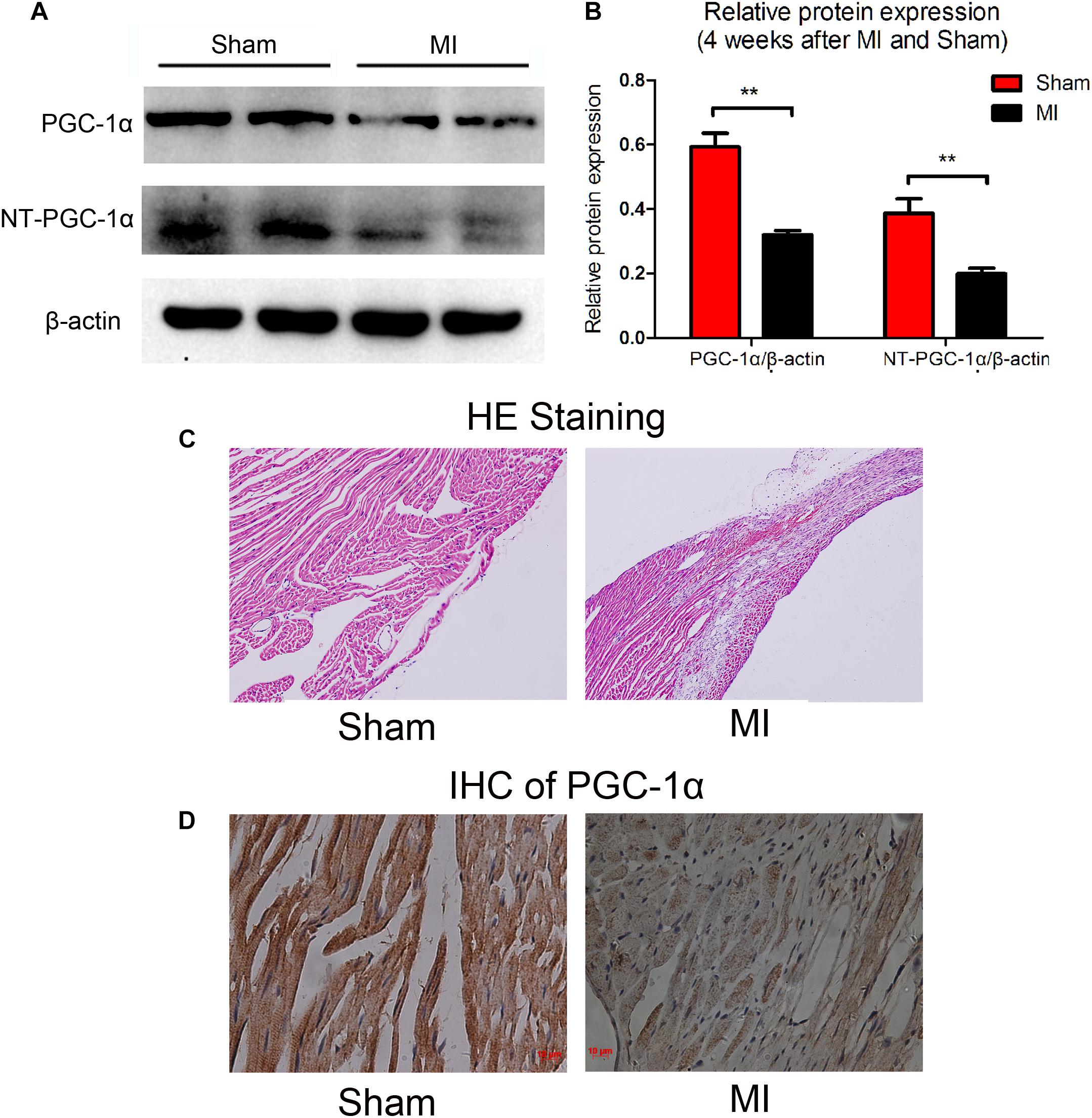
Figure 1. Decreased levels of PGC-1α and NT-PGC-1α in myocardial infarction mice. A representative western blot (A) and relative quantification to β-actin (B) of PGC-1α and NT-PGC-1α in mice subjected to a sham operation or MI. ∗∗p < 0.01 compared to the sham group, n = 4–5 in each group (t-test). The representative photomicrographs of HE staining (C) and IHC of total PGC-1α (D) in sham and MI mice.
Antihypertrophic Effects of the CRM1-Inhibitor Selinexor
We aimed to determine an appropriate concentration of the CRM1-inhibitor Selinexor for NRVMs. Cells were exposed to different concentrations of Selinexor for a range of durations and NRVM viability was assayed using the CCK8 cell viability assay. We discovered that cells exposed to less than 600 nM Selinexor for 4 h did not show any significant differences compared to controls (Figures 2A–D). However, a prolonged reaction time with Selinexor led to decreased OD values. Thus, we applied 50 nM Selinexor to stimulate the NRVMs for 4 h. The antihypertrophic effect of CRM1 inhibitors was demonstrated in previous studies (Harrison et al., 2004; Monovich et al., 2009; Chahine et al., 2015). Here, we verified that Selinexor can restrict PE- and AngII-induced hypertrophy in NRVMs and visualized the cell’s cross-sectional areas using Phalloidin staining and confocal microscopy (Figure 3A). The area of the NRVMs is displayed using a micrometer scale (μm2). As expected, the control group was smaller compared to the PE group (1,148.89 ± 73.85 μm2 vs. 2,756.683 ± 333.48 μm2, p < 0.01) and the AngII group (1,148.89 ± 73.85 μm2 vs. 1,861.60 ± 243.38 μm2, p < 0.05), while the PE group had a larger cell cross-sectional area than the PE+Selinexor group (2,756.683 ± 333.48 μm2 vs. 1,818.56 ± 209.08 μm2, p < 0.05). Similarly, the AngII group had a larger cell cross area compared to the AngII + Selinexor group (1,861.60 ± 243.38 vs. 1,247.71 ± 113.65, p < 0.05) (Figure 3B). Further investigation showed that Selinexor can inhibit the expression of β-MHC that is induced by PE (PE vs. PE+Selinexor: 0.01637 ± 0.00239 vs. 0.00973 ± 0.00047, p < 0.05) (Figures 3C,D). These results show that the CRM1-inhibitor Selinexor, which displays oral activity, can restrict cardiac hypertrophy in vitro.
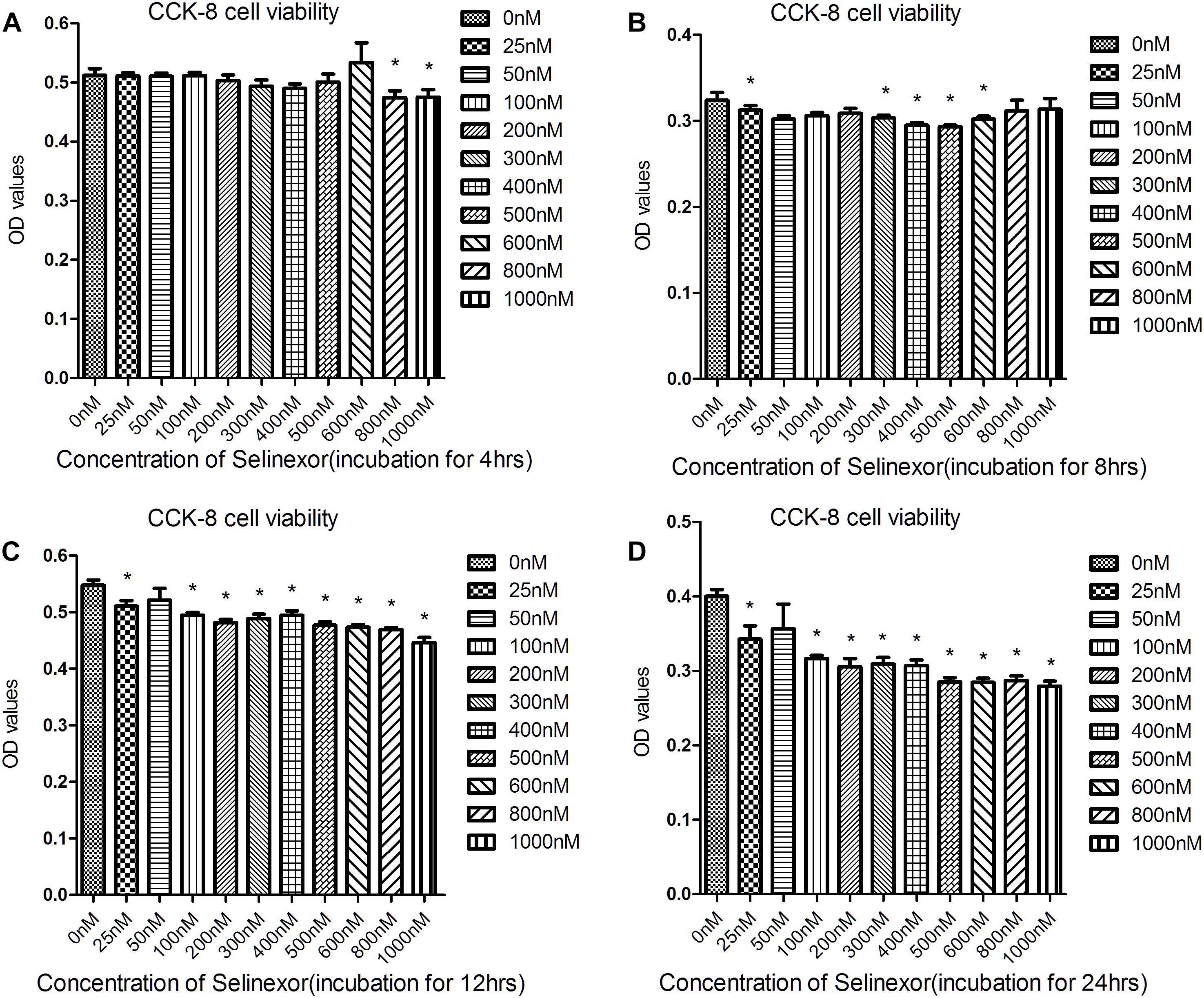
Figure 2. Results of the Cell Counting Kit-8 assay to determine cell viability after exposure to Selinexor. NRVMs were exposed to different concentrations of Selinexor and incubated for (A) 4 h, (B) 8 h, (C) 12 h, and (D) 24 h, respectively. *P < 0.05, compared to the corresponding control group.
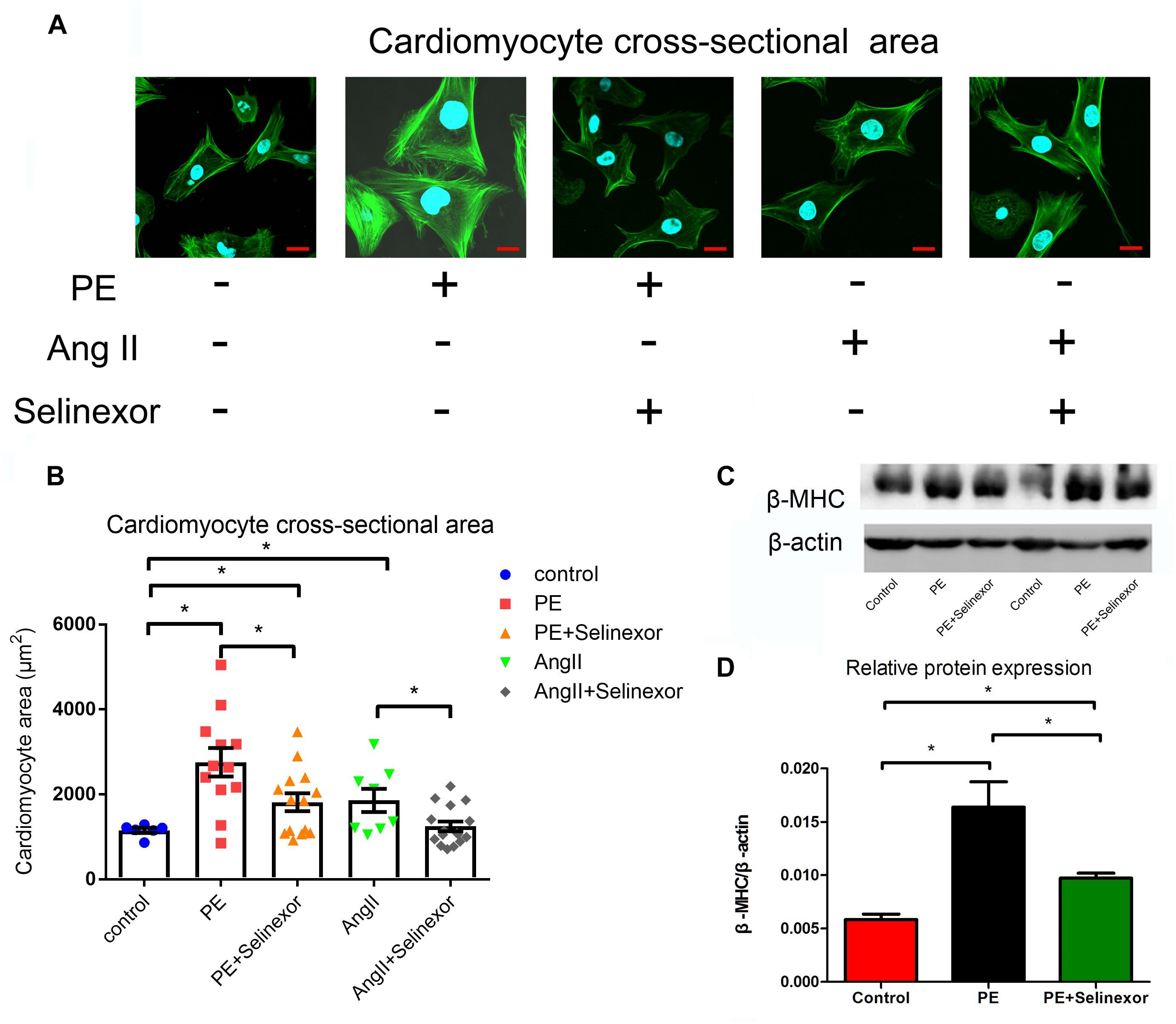
Figure 3. Effects of the CRM1-inhibitor Selinexor on cardiac hypertrophy. (A) Representative photomicrographs of the actin-tracker green stain in NRVMs that are exposed to PE, AngII and Selinexor, and (B) their relative cross-sectional areas. (C,D) The expression of β-MHC in cells that were stimulated by PE and Selinexor as detected by western blot. *P < 0.05, compared to the corresponding control group (n = 4).
Regulation of NT-PGC-1α Distribution by CRM1 Inhibitor and NLS (Nucleus Localization Sequence)
Neonatal rat ventricular myocytes were transfected with adenovirus-mCherry-NT-PGC-1α and adenovirus-mCherry-NLS-NT-PGC-1α to investigate the role of CRM1 inhibitors in the regulation of NT-PGC-1α. Following the infection, the cells were then treated with 50 nM Selinexor. After stimulation, the cells were stained with Hoechst 33258 and visualized with confocal microscopy. We determined that Selinexor and NLS can increase the nuclear density of mCherry, and the nucleus/cytoplasm mean densities were also measured. Comparisons between the AdV-NT-PGC-1α and AdV-NT-PGC-1α+Selinexor groups showed significant differences (0.48 ± 0.01 vs. 0.93 ± 0.03, respectively, p < 0.001); the AdV-NLS-NT-PGC-1α group had lower mean density than the AdV-NLS-NT-PGC-1α+Selinexor group (1.26 ± 0.09 vs. 0.61 ± 0.04, respectively, p < 0.001), while comparisons between the AdV-NT-PGC-1α and Adv-NLS-NT-PGC-1α group showed significant differences (0.48 ± 0.01 vs. 0.61 ± 0.04, respectively, p < 0.05) (Figures 4A,B). Furthermore, we discovered that CRM1 interacted with NT-PGC-1α and PGC-1α in the NRVMs through the use of Co-IP (Figure 4E). These findings suggest that the trafficking of NT-PGC-1α between the nucleus and cytoplasm are likely dependent on the interaction between CRM1 and its corresponding protein.
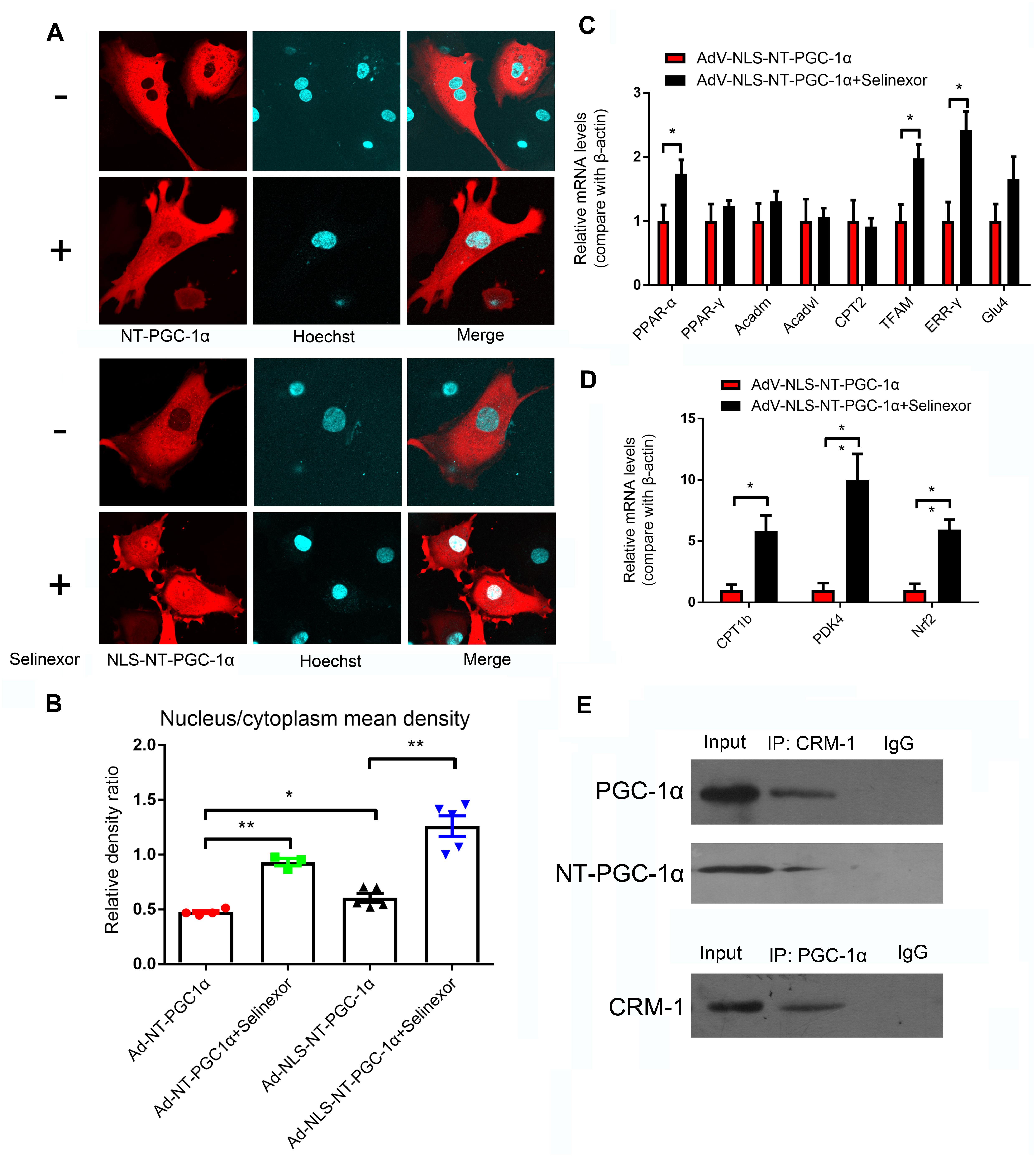
Figure 4. Regulation of the distribution of NT-PGC-1α by the CRM1 inhibitor and relative mRNA expression after NT-PGC-1α shuttling into the nucleus. (A) The influence of the CRM1 inhibitor Selinexor and the NLS (nuclear localization sequence) on the subcellular distribution of NT-PGC-1α, and (B) the mean density ratio of the nucleus/cytoplasm. *P < 0.05, **P < 0.05 compared to the control group, n = 3–5 in each group. (C,D) The relative mRNA expression after NT-PGC-1α is shuttled into the nucleus by Selinexor and NLS in NRVMs that are overexpressing NLS-NT-PGC-1α. *P < 0.05, **P < 0.01 compared to the corresponding group with NLS-NT-PGC-1α overexpression, n = 6–9 in each group. (E) Co-IP of PGC-1α, NT-PGC-1α, and CRM1.
Relative mRNA Expression After Shuttling NT-PGC-1α Into the Nucleus
We discovered that Selinexor and NLS could dramatically increase the nuclear distribution of NT-PGC-1α. Thus, we explored relative mRNA expression after the shuttling of NT-PGC-1α into the nucleus. We discovered that the NRVMs that were transfected with AdV-NLS-NT-PGC-1α and exposed to Selinexor could increase their relative amount of mRNA expression compared to cells that were transfected with AdV-NLS-NT-PGC-1α (Figures 4C,D). These genes include PPAR-α (1.00 ± 0.25 vs. 1.74 ± 0.21, p < 0.05); Tfam (1.00 ± 0.26 vs. 1.98 ± 0.22, p < 0.05); ERR-γ (1.00 ± 0.30 vs. 2.42 ± 0.29, p < 0.01); CPT1b (1.00 ± 0.01 vs. 5.82 ± 1.28, p < 0.05); PDK4 (1.00 ± 0.58 vs. 10.00 ± 2.12, p < 0.05); and Nrf2 (1.00 ± 0.52 vs. 5.96 ± 0.78, p < 0.001). These results suggest that NT-PGC-1α might function in nuclear transcription, and could partially compensate for the function of full-length PGC-1α. Additionally, most of the genes mentioned above have beneficial cardiovascular effects as reported by previous studies.
The Influence of Selinexor on Cardiac Function and mRNA Expression in MI-Induced Heart Failure Mice
If CRM1 inhibitors exhibit cardiovascular protective effects, they might be due to their antihypertrophic effects and the shuttling of NT-PGC-1α into the nucleus. To test this hypothesis, we gavaged Selinexor to C57BL/6j mice that then underwent coronary artery ligation. If Selinexor could ameliorate energy metabolism in vivo, then it would likely enhance the ejection fraction (EF) or reduce the left ventricular end-systolic diameter (LVESD) in mice. We performed an echocardiogram on these mice at 1 week and 4 weeks post-myocardial infraction but the results were not significant between the Selinexor and control groups (Figures 5A,B and Supplementary Table S1). However, we found that the Selinexor treatment reduced HW/BW (the ratio of heart weight and body weight) and HW/TL (the ratio of heart weight and tibia length) in the MI-mice (Figures 5C,D), which is in line with the cellular findings. Furthermore, there were no significant differences in the relative mRNA levels of PGC-1α between these groups, except for SOD3 (Figure 6). These findings indicate the potential effects of Selinexor in heart remodeling.
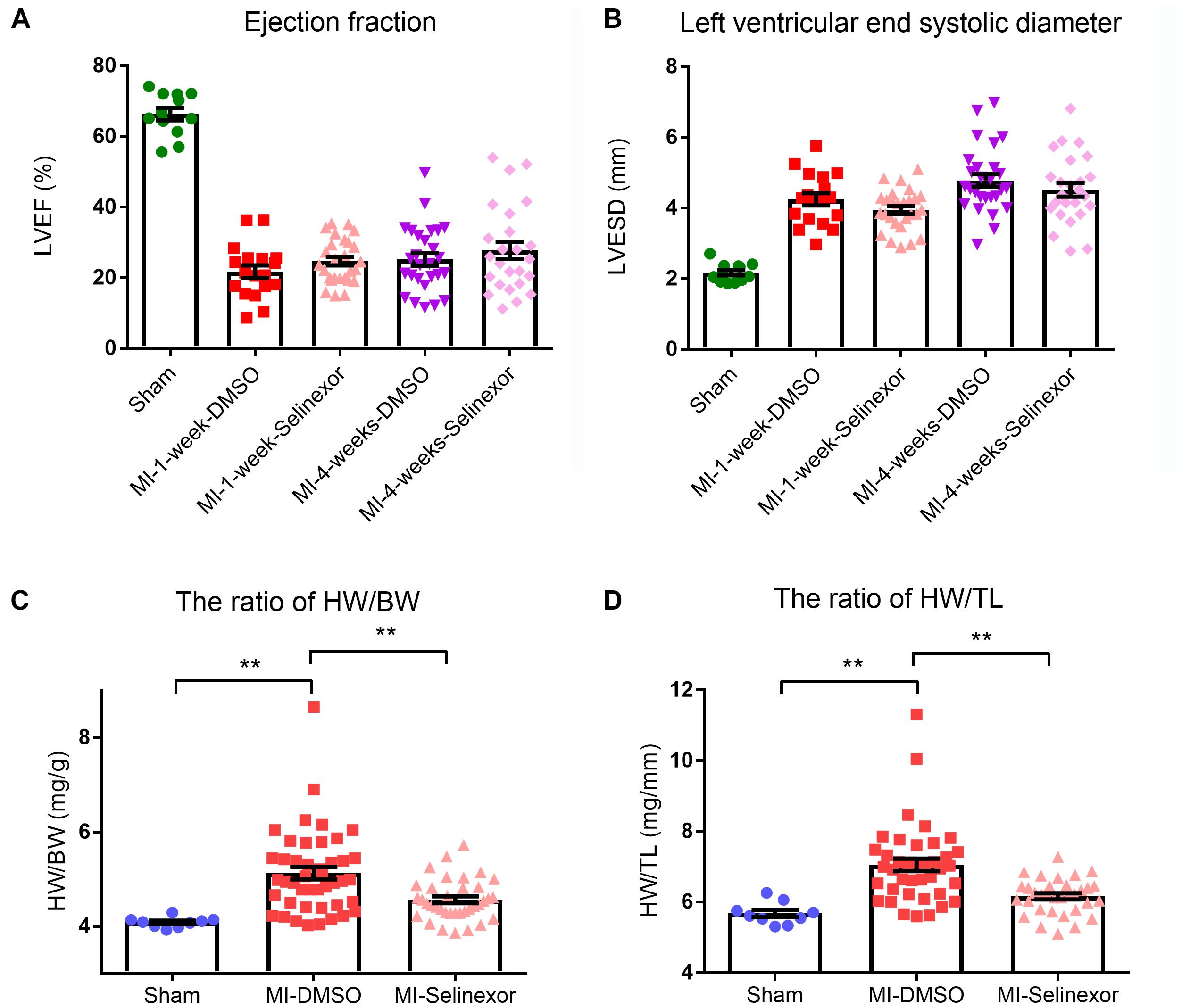
Figure 5. Echocardiography results and HW/BW and HW/TL ratios in MI-mice treated with Selinexor. (A) The ejection fraction (B) in MI-mice treated with Selinexor or vehicle post-myocardial infraction was detected by echocardiogram at 1 week and at 4 weeks (n = 12, 18, 27, 27, and 25 in sham mice, MI-1-week-DMSO, MI-1-week-Selinexor, MI-4-week-DMSO, MI-4-week-Selinexor, respectively). The ratio of (C) HW/BW and (D) HW/TL in MI-mice gavaged with Selinexor (**P < 0.01 compared to the corresponding group).
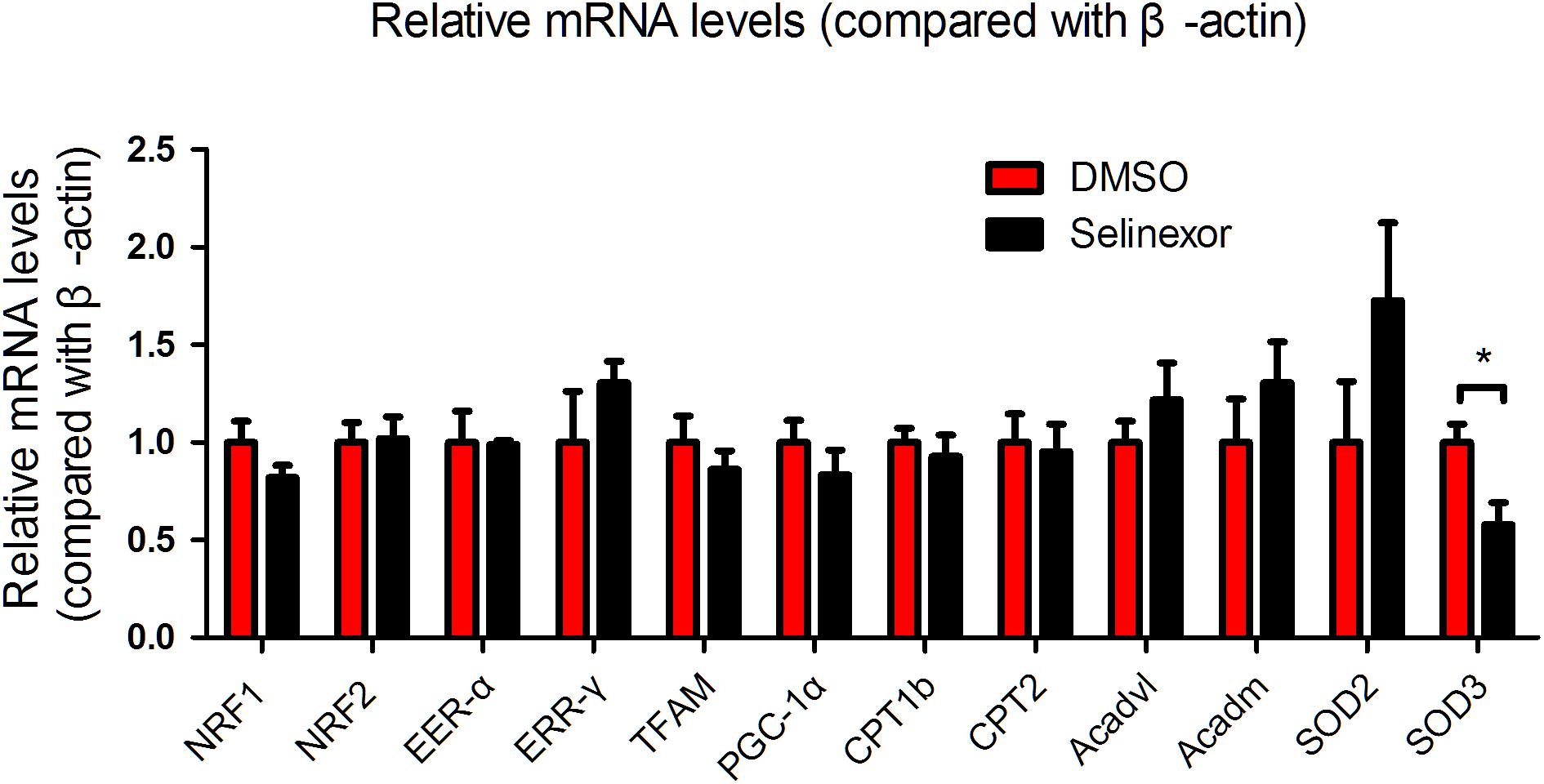
Figure 6. mRNA expression in MI-mice treated with Selinexor. Cardiac mRNA expression in WT mice treated with Selinexor and vehicle post-MI at 1 month (n = 5 in each group, *P < 0.05 compared to the corresponding group).
Discussion
In the present study, we suggest that the antihypertrophic effects of the CRM1 inhibitor Selinexor might be independent of NT-PGC-1α. We found that PGC-1α and NT-PGC-1α were decreased in the MI-induced HF mouse model, which supports the role of energy metabolism defects in HF (Warren et al., 2017). Although the mechanisms of PGC-1α have been fully elucidated, there are relatively few studies that have investigated the role of NT-PGC-1α in the heart (Liu et al., 2018). Additionally, the trafficking of NT-PGC-1α between the nucleus and the cytoplasm by CRM1 inhibitors in previous studies was only demonstrated in other cell lines (Chang et al., 2010). Therefore, CRM1 might be a promising target in HF due to its antihypertrophic effects and its regulation of NT-PGC-1α.
We detected the antihypertrophic effects of a CRM1 inhibitor that have been confirmed in previous studies (Harrison et al., 2004; Monovich et al., 2009; Chahine et al., 2015). Our results suggest that the CRM1 inhibitor Selinexor can alleviate PE- and AngII-induced cardiac-hypertrophy and inhibit the expression of β-MHC induced by PE. Thus, it is plausible that the antihypertrophic effects of Selinexor are achieved by trafficking NT-PGC-1α between the nucleus and the cytoplasm.
To explore the role of nuclear NT-PGC-1α, we utilized an adenovirus that expresses a NLS-fused protein to infect NRVMs and treated the NRVMs with Selinexor. We found that cells exposed to Selinexor slightly increase their nuclear concentration of NT-PGC-1α, but cells that express NLS-NT-PGC-1α and are exposed to Selinexor dramatically increase their nuclear content of NT-PGC-1α. These results agree with previous studies that showed CRM1 inhibitors can lead to the retention of nuclear NT-PGC-1α levels (Chang et al., 2010; Shen et al., 2012). Zhang et al. (2009) suggested that NT-PGC-1α lacked the structure of the NLS in its C terminus and that, under the influence of the nuclear exporting sequence (NES), NT-PGC-1α is predominantly distributed in cytoplasm. The CRM1 protein affects protein trafficking between the nucleus and the cytoplasm (Lu et al., 2015), which is a reported mechanism in the regulation of NT-PGC-1α subcellular distribution (Chang et al., 2010; Shen et al., 2012). Chang et al. (2010) reported that the CRM1-inhibitor leptomycin B (LMB) resulted in NT-PGC-1α being retained in the nucleus and increase its nuclear concentration.
In this study, we confirmed the interaction between CRM1 and both NT-PGC-1α and PGC-1α by immunoprecipitation. Furthermore, we showed that the expression of mRNAs involved in mitochondrial function are increased after NT-PGC-1α is shuttled into the nucleus. Collectively, these data indicate a possible mechanism of a CRM1 inhibitor in the heart, by which the retention of NT-PGC-1α in the nucleus is able to respond to cellular energy demands.
However, in vivo applications of Selinexor did not improve LV ejection fractions, had no effect on the expression of genes involved in energy metabolism, and even reduced the expression of SOD3, which indicates that it might be involved with other mechanisms and could be associated with toxicity. Nevertheless, Selinexor reduced the ratios of HW/BW and HW/TL, which likely fulfills an antihypertrophic role that is independent of NT-PGC-1α shuttling.
There are some plausible explanations for the in vivo results. First, the CRM1 inhibitor is unable to dramatically increase the levels of nuclear NT-PGC-1α without a fused NLS, and mice do not have the fused NLS in NT-PGC-1α. Second, myocardial infraction is accompanied by severe oxidative stress, which might dampen the expression of NT-PGC-1α. This is supported by previous reports of the inhibition of NT-PGC-1α by H2O2-treated medium (Choi et al., 2014). Third, high doses of Selinexor might induce toxicity, which would restrict higher doses in mice. Although we failed to identify any significant effects of the CRM1 inhibitor on cardiac energy metabolism, the shuttling of NT-PGC-1α into the nucleus remains a possible method to ameliorate metabolic dysfunction (Chang and Ha, 2017). Additionally, recent studies show that cytoplasmic NT-PGC-1α interacts with some mitochondrial proteins (Choi et al., 2013; Chang and Ha, 2017), which indicates its bilateral effects in both the nucleus and the cytoplasm. Further studies are required to determine if the retention of NT-PGC-1α in the nucleus is associated with any additional impairments.
Conclusion
Taken together, our findings indicate that Selinexor enhances the expression of several metabolic genes in the presence of NT-PGC-1α overexpression when using an in vitro model and that its antihypertrophic effects might be independent of NT-PGC-1α shuttling.
Ethics Statement
This study was carried out in accordance with the recommendations of the guidelines of the Animal Care Committee of Nanfang Hospital, Southern Medical University. The protocol was approved by the Animal Care Committee of Nanfang Hospital.
Author Contributions
ZL, HT, JH, WC, YB, QcZ, QoZ, WL, HR, and DX made a substantial contribution to the conception or design of the work or the acquisition, analysis, or interpretation of data for the work; approved the final version to be published; and also agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. ZL, HT, HR, and DX drafted the work or revised it critically for important intellectual content.
Funding
The present study was supported by the National Natural Science Foundation of China (81670367 and 81270320) and partially supported by The National Key Research and Development Program of China (2017YFC1308304).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00465/full#supplementary-material
TABLE S1 | The echocardiography of MI-mice.
References
Arany, Z., Novikov, M., Chin, S., Ma, Y., Rosenzweig, A., and Spiegelman, B. M. (2006). Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. U.S.A. 103, 10086–10091. doi: 10.1073/pnas.0603615103
Arumugam, S., Sreedhar, R., Thandavarayan, R. A., Karuppagounder, V., and Watanabe, K. (2016). Targeting fatty acid metabolism in heart failure: is it a suitable therapeutic approach? Drug Discov. Today 21, 1003–1008. doi: 10.1016/j.drudis.2016.02.010
Chahine, M. N., Mioulane, M., Sikkel, M. B., O’Gara, P., Dos, R. C., Pierce, G. N., et al. (2015). Nuclear pore rearrangements and nuclear trafficking in cardiomyocytes from rat and human failing hearts. Cardiovasc. Res. 105, 31–43. doi: 10.1093/cvr/cvu218
Chang, H. C., Shapiro, J. S., and Ardehali, H. (2016). Getting to the “Heart” of cardiac disease by decreasing mitochondrial iron. Circ. Res. 119, 1164–1166. doi: 10.1161/CIRCRESAHA.116.309746
Chang, J. S., and Ha, K. (2017). An unexpected role for the transcriptional coactivator isoform NT-PGC-1alpha in the regulation of mitochondrial respiration in brown adipocytes. J. Biol. Chem. 292, 9958–9966. doi: 10.1074/jbc.M117.778373
Chang, J. S., Huypens, P., Zhang, Y., Black, C., Kralli, A., and Gettys, T. W. (2010). Regulation of NT-PGC-1alpha subcellular localization and function by protein kinase A-dependent modulation of nuclear export by CRM1. J. Biol. Chem. 285, 18039–18050. doi: 10.1074/jbc.M109.083121
Choi, J., Batchu, V. V., Schubert, M., Castellani, R. J., and Russell, J. W. (2013). A novel PGC-1alpha isoform in brain localizes to mitochondria and associates with PINK1 and VDAC. Biochem. Biophys. Res. Commun. 435, 671–677. doi: 10.1016/j.bbrc.2013.05.041
Choi, J., Ravipati, A., Nimmagadda, V., Schubert, M., Castellani, R. J., and Russell, J. W. (2014). Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 18, 41–48. doi: 10.1016/j.mito.2014.09.005
Harrison, B. C., Roberts, C. R., Hood, D. B., Sweeney, M., Gould, J. M., Bush, E. W., et al. (2004). The CRM1 nuclear export receptor controls pathological cardiac gene expression. Mol. Cell. Biol. 24, 10636–10649. doi: 10.1128/MCB.24.24.10636-10649.2004
Jun, H. J., Joshi, Y., Patil, Y., Noland, R. C., and Chang, J. S. (2014). NT-PGC-1alpha activation attenuates high-fat diet-induced obesity by enhancing brown fat thermogenesis and adipose tissue oxidative metabolism. Diabetes 63, 3615–3625. doi: 10.2337/db13-1837
Kim, J., Fernand, V. E., Henagan, T. M., Shin, J., Huypens, P., Newman, S., et al. (2016). Regulation of brown and white adipocyte transcriptome by the transcriptional coactivator NT-PGC-1alpha. PLoS One 11:e0159990. doi: 10.1371/journal.pone.0159990
Lin, H., Shen, L., Zhang, X., Xie, J., Hao, H., Zhang, Y., et al. (2016). HMGB1-RAGE axis makes no contribution to cardiac remodeling induced by pressure-overload. PLoS One 11:e0158514. doi: 10.1371/journal.pone.0158514
Liu, Z., Hua, J., Cai, W., Zhan, Q., Lai, W., Zeng, Q., et al. (2018). N-terminal truncated peroxisome proliferator activated receptor gamma coactivator1alpha alleviates phenylephrine induced mitochondrial dysfunction and decreases lipid droplet accumulation in neonatal rat cardiomyocytes. Mol. Med. Rep. 18, 2142–2152. doi: 10.3892/mmr.2018.9158
Liu, Z., Liu, H. Y., Zhou, H., Zhan, Q., Lai, W., Zeng, Q., et al. (2017). Moderate-intensity exercise affects gut microbiome composition and influences cardiac function in myocardial infarction mice. Front. Microbiol. 8:1687. doi: 10.3389/fmicb.2017.01687
Lu, C., Figueroa, J. A., Liu, Z., Konala, V., Aulakh, A., Verma, R., et al. (2015). Nuclear export as a novel therapeutic target: the crm1 connection. Curr. Cancer Drug Targets 15, 575–592. doi: 10.2174/156800961507150828223554
Martin, O. J., Lai, L., Soundarapandian, M. M., Leone, T. C., Zorzano, A., Keller, M. P., et al. (2014). A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 114, 626–636. doi: 10.1161/CIRCRESAHA.114.302562
Martinez-Gonzalez, M. A., and Ruiz-Canela, M. (2015). Preventing heart failure: sweetened beverages and healthy lifestyles. Heart 101, 1935–1937. doi: 10.1136/heartjnl-2015-308426
Monovich, L., Koch, K. A., Burgis, R., Osimboni, E., Mann, T., Wall, D., et al. (2009). Suppression of HDAC nuclear export and cardiomyocyte hypertrophy by novel irreversible inhibitors of CRM1. Biochim. Biophys. Acta 1789, 422–431. doi: 10.1016/j.bbagrm.2009.04.001
Parihar, P., and Parihar, M. S. (2017). Metabolic enzymes dysregulation in heart failure: the prospective therapy. Heart Fail. Rev. 22, 109–121. doi: 10.1007/s10741-016-9588-x
Riehle, C., and Abel, E. D. (2012). PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 22, 98–105. doi: 10.1016/j.tcm.2012.07.003
Schilling, J., and Kelly, D. P. (2011). The PGC-1 cascade as a therapeutic target for heart failure. J. Mol. Cell. Cardiol. 51, 578–583. doi: 10.1016/j.yjmcc.2010.09.021
Shen, T., Liu, Y., and Schneider, M. F. (2012). Localization and regulation of the N terminal splice variant of PGC-1alpha in adult skeletal muscle fibers. J. Biomed. Biotechnol. 2012:989263. doi: 10.1155/2012/989263
Vega, R. B., Huss, J. M., and Kelly, D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20, 1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000
Warren, J. S., Oka, S. I., Zablocki, D., and Sadoshima, J. (2017). Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: from metabolomics to epigenetics. Am. J. Physiol. Heart Circ. Physiol. 313, H584–H596. doi: 10.1152/ajpheart.00103.2017
Keywords: CRM1, NT-PGC-1α, myocardial infarction, antihypertrophy, nuclear distribution, cardiac function
Citation: Liu Z, Tian H, Hua J, Cai W, Bai Y, Zhan Q, Lai W, Zeng Q, Ren H and Xu D (2019) A CRM1 Inhibitor Alleviates Cardiac Hypertrophy and Increases the Nuclear Distribution of NT-PGC-1α in NRVMs. Front. Pharmacol. 10:465. doi: 10.3389/fphar.2019.00465
Received: 25 November 2018; Accepted: 12 April 2019;
Published: 07 May 2019.
Edited by:
Nicolau Beckmann, Novartis Institutes for BioMedical Research, SwitzerlandReviewed by:
Hangang Yu, West Virginia University, United StatesTracey Gaspari, Monash University, Australia
Copyright © 2019 Liu, Tian, Hua, Cai, Bai, Zhan, Lai, Zeng, Ren and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Ren, renhao67@aliyun.com Dingli Xu, dinglixu@fimmu.com
†These authors have contributed equally to this work