Case Report: Refractory macrophage activation syndrome requiring high-dose anakinra, emapalumab, and etoposide therapy in early-onset systemic juvenile idiopathic arthritis associated with adenoviremia
 Elizabeth D. Slaney1*
Elizabeth D. Slaney1*  Renee Modica1,2
Renee Modica1,2  Leandra Woolnough1,2,† Dina Kafisheh1,2 Denise Heather Bell-Brunson1,2,† Melissa Elder1,2,†
Leandra Woolnough1,2,† Dina Kafisheh1,2 Denise Heather Bell-Brunson1,2,† Melissa Elder1,2,†
- 1College of Medicine, University of Florida, Gainesville, FL, United States
- 2Division of Pediatric Allergy, Immunology, and Rheumatology, Department of Pediatrics, University of Florida, Gainesville, FL, United States
Macrophage activation syndrome (MAS) is a life-threatening condition characterized by the excessive stimulation of macrophages and T lymphocytes, provoked by infections, malignancy, and autoimmune or autoinflammatory conditions such as systemic juvenile idiopathic arthritis (sJIA). Clinical signs of sJIA may include high-spiking, quotidian fevers, lymphadenopathy, hepatosplenomegaly, and a salmon-colored migratory, evanescent rash. By contrast, MAS is characterized by unremitting fevers and diffuse, fixed, maculopapular rashes. In addition to hepatosplenomegaly and lymphadenopathy, patients with MAS may also have clinical signs of coagulopathy, as well as cardiac, lung, renal, and central nervous system dysfunction. The empiric treatment for MAS is initially high-dose IV corticosteroids, but usually requires addition of immunomodulators such as tacrolimus or a biologic such as Anakinra to control. The addition of immunotherapies for MAS has improved patient outcomes. We present a 2-year-old male patient with a history of early-onset sJIA, who presented with MAS refractory to corticosteroids and anakinra triggered by adenoviremia that required addition of emapalumab to control. We believe this is the first reported case of a combination of immunosuppressive therapy of emapalumab, etoposide, anakinra, tacrolimus, and corticosteroids used in the successful treatment of infection-induced MAS in early-onset sJIA. Given the lack of treatment guidelines and approved therapies for MAS, alternative strategies should be considered for patients with an intractable course.
1 Introduction
Hemophagocytic Lymphohistiocytosis (HLH) is a “cytokine storm syndrome” characterized by the overproduction of proinflammatory cytokines, mainly interleukin 1β (IL-1β), interleukin 6 (IL-6), interleukin 18 (IL-18), interleukin 33 (IL-33), tumor necrosis factor-α (TNFα), and interferon-γ (IFNγ) by activated immune cells (T lymphocytes and macrophages), which leads to life-threatening systemic inflammation. HLH can be primary (genetic or familial) or secondary (triggered by infection, malignancy, or rheumatologic disease). When secondary HLH (sHLH) is associated with an autoimmune/inflammatory disease, it is known as either macrophage activation syndrome (MAS) or “MAS-HLH,” and is estimated to complicate approximately 10%–13% of pediatric patients with systemic juvenile idiopathic arthritis (sJIA) (1, 2). The incidence of subclinical MAS may be as high as 30%. Several known genetic mutations predispose patients to primary HLH (pHLH), including PRF1, UNC13D, STXBP2, STX11, Rab27a, SH2D1A, and XIAP (3–8).
We report the first case of a child with early-onset sJIA with corticosteroid and anakinra refractory MAS triggered by adenoviremia, who required further dose escalation of anakinra (up to 46 mg/kg/day) as well as the addition of emapalumab (up to 10 mg/kg biweekly) and a course of etoposide for the resolution of his symptoms. We believe this is the first case report of this aggressive combination of immunosuppressive therapy and the highest reported dose of non-continuous intravenous anakinra used in the treatment of MAS in the setting of sJIA.
2 Case description
The patient is a 2-year-old Caucasian male, ex-33-week premature infant with fetal alcohol syndrome who was diagnosed with early onset sJIA at 13 months of age after he presented with persistent quotidian fevers, evanescent rash, lymphadenopathy, and arthritis. Initial treatment included anakinra 100 mg subcutaneously daily. He had five prior hospitalizations for bouts of MAS, requiring treatment with high-dose intravenous (IV) corticosteroids and IV anakinra. His most recent admission, reported here, was prompted by persistent fevers, rash, laboratory values concerning for MAS flare, and chest x-ray suggestive of multifocal pneumonia. His home regimen prior to admission was 8 mg/kg/day of subcutaneous (SC) anakinra and 0.17 mg/kg/day of tacrolimus (with trough level of 7–9). It was initially unclear whether this episode was a continuation of a previous weeks-long MAS flare or constituted a new flare until results of infectious evaluation revealed extreme adenoviremia.
During his admission, which lasted for a total of 105 days, he was initially given pulse IV methylprednisolone (10 mg/kg/day) and was continued on high-dose steroids with inability to taper for weeks. On hospital day (HD) 2, anakinra was increased (25 mg/kg/day IV in divided doses) in the setting of worsening MAS labs. His oral tacrolimus was switched to IV cyclosporine from HD 10–38; however, due to persistently subtherapeutic levels, he was later switched back to oral tacrolimus after developing hypertension. Anakinra 25 mg/kg/day was discontinued on HD 11 to initiate emergent use of low-dose emapalumab 1 mg/kg twice weekly, given worsening MAS labs and fever despite high dose IV anakinra. However, unanticipated issues with IV access delayed his emapalumab infusion and he began to experience spiking fevers as a result. Biweekly emapalumab was added on HD 12 (1 mg/kg) with subsequent weekly dose escalation (3 mg/kg HD 13, 6 mg/kg HD 15, 10 mg/kg HD 19, and 10 mg/kg HD 22 and thereafter). On HD 13, his hyperferritinemia worsened to 67,000 ng/ml, AST/ALT > 1,000 IU/L, LDH >10,000 IU/L, and persistently down-trending platelets, hemoglobin, and leukocyte count. This laboratory trend in conjunction with persistent fevers and worsening respiratory status necessitated increased oxygen supplementation and prompted a transfer to the pediatric intensive care unit (PICU) from HD 13 to 15. Very high dose anakinra was resumed in the PICU (37.5 mg/kg/day IV in divided doses on HD 13). His respiratory deterioration improved after receiving furosemide for interstitial pulmonary edema. He was switched to IV dexamethasone 1.5 mg/kg/day on HD 15. However, owing to worsening MAS labs, his IV anakinra dose was increased further (46 mg/kg/day IV in divided doses from HD 13 to 58). Etoposide (150 mg/m2 weekly for 8 doses) was added on HD 26 per pHLH protocol as recommended by pediatric hematology/oncology based on his severe refractory course despite aggressive treatment for sHLH (9). Of note, he did not have a pathogenic mutation in genes responsible for pHLH. He eventually stabilized after nearly 3 months of this combination therapy plus cidofovir for his adenoviremia, facilitating slow taper of high-dose IV steroids as well as anakinra from 46 mg/kg/day to 2 mg/kg/day over the course of 37 days (HD 59–96). Anakinra was eventually discontinued on HD 101. At discharge (HD 105), he was prescribed prednisolone 2 mg/kg/day, hydrocortisone 5 mg twice daily, emapalumab 10 mg/kg twice weekly, tacrolimus 0.2 mg twice daily (0.03 mg/kg/day) (Figure 1), as well as maintenance cidofovir.
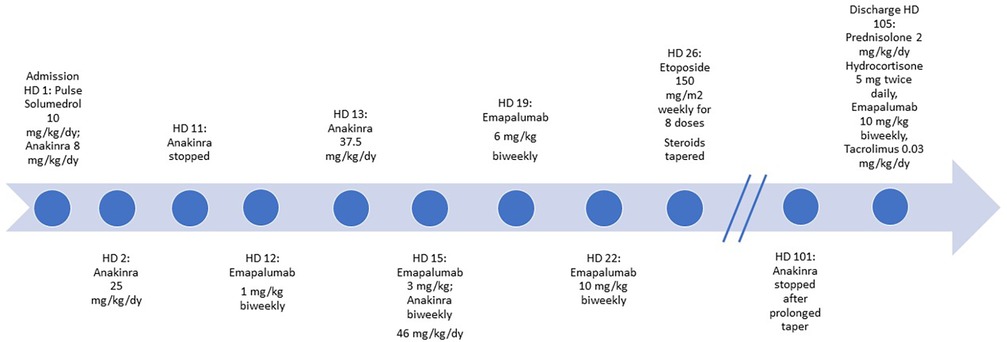
Figure 1. Hospital treatment timeline.
As noted above, evaluation for pHLH was negative by genotyping of blood and bone marrow. Invitae© primary immunodeficiency genetic panel, quantitative immunoglobulins, and lymphocyte subsets were normal and did not indicate an underlying immunodeficiency. Bone marrow biopsies (at initial diagnosis and with current MAS flare) did not demonstrate malignancy and comprehensive infectious evaluation was entirely negative other than adenoviremia (>10,000,000 copies/ml). He was treated with weekly IV cidofovir, which was continued after discharge home, and five doses of IV immunoglobulin G (IVIG) to provide passive immunity. Chest computed tomography (CT) confirmed multifocal pneumonia and excluded interstitial lung disease. He was treated with voriconazole for presumed Aspergillus because of abnormal galactomannan from bronchoalveolar lavage (BAL), which was converted to posaconazole at discharge. However, there remains a debate whether he in fact actually had fungal pneumonia given that only one BAL galactomoannan was abnormal with a negative result on repeat BAL. Repeat BAL universal polymerase chain reaction (PCR) revealed Pneumocystis jiroveci (PJP) despite prophylaxis with nebulized pentamidine, which was treated with atovaquone (Table 1).
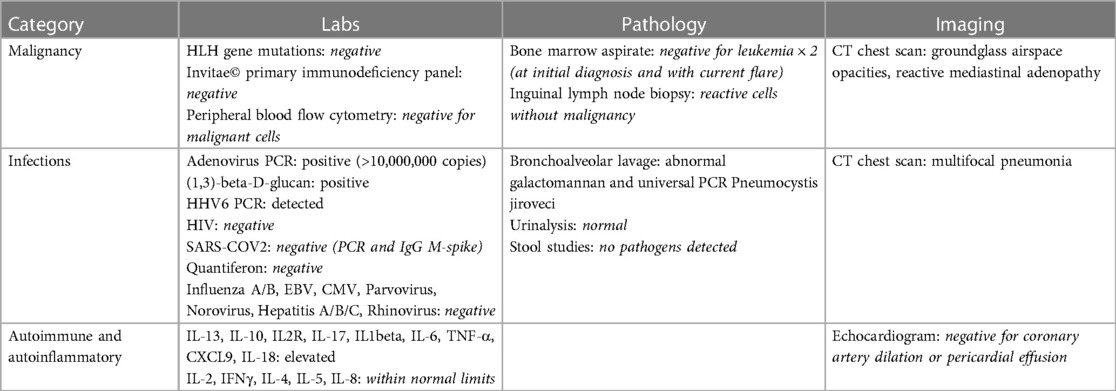
Table 1. Clinical work-up for HLH.
Now 24 months post hospital discharge, he has not had recurrence of MAS off steroids, anakinra, and emapalumab, remaining only on low dose tacrolimus and methotrexate for treatment of his sJIA. He receives monthly inhaled pentamidine for Pneumocystis jirovecii pneumonia (PJP) prophylaxis. He continues to be followed by Pediatric Rheumatology/Immunology for further management.
3 Discussion
MAS is a potentially life-threatening, hyperinflammtory syndrome that may complicate sJIA and lead to other autoimmune disorders resulting in hemophagocytosis primarily involving the bone marrow, liver, and spleen; however, hemophagocytosis can be seen elsewhere such as in the cerebrospinal fluid (CSF) (1). The overlapping clinical features of active sJIA and MAS may delay the diagnosis, and therefore application of the 2016 Classification Criteria for MAS complicating sJIA may be helpful in distinguishing between these conditions (1, 10). The clinical features of MAS include unremitting high fevers, lymphadenopathy, hepatosplenomegaly, maculopapular rash, coagulopathy, renal dysfunction, altered mental status, and may involve other organs as well. Laboratory features include evolving cytopenias, elevated C-reactive protein, precipitously declining erythrocyte sedimentation rate, hypofibrinogenemia, transaminitis, elevated lactate dehydrogenase (LDH), hypertriglyceridemia, hyperferritinemia, and hemophagocytosis on bone marrow biopsy (2). There are no standardized treatment protocols for MAS. Typically, it is managed empirically with high-dose IV corticosteroids, a calcineurin inhibitor, and a biologic. The addition of a variety of novel immunotherapies designed to inhibit specific cytokines involved in the activation of the immune system has improved outcomes for patients with MAS. Anakinra, a recombinant IL-1 receptor antagonist, is efficacious in the treatment of MAS at doses of 1–2 mg/kg/day in children (11–13). More recently, higher doses of anakira have been used in the 4–10 mg/kg/day range.
Although no formal diagnostic criteria or management guidelines exist for treatment of refractory MAS, multiple therapeutic approaches are well described in the literature. Anakinra is not always effective at standard dosing for unremitting MAS in sJIA patients and may require doses >10 mg/kg/day or 2 mg/kg/hour in children with sHLH/MAS. A dose of 2,400 mg/day as a continuous IV infusion was required in a cohort of critically ill adults with MAS (14, 15). Emapalumab, an IFNγ-blocking monoclonal antibody, is effective in the treatment of pHLH, although its use in sHLH/MAS has yet to be approved and is under investigation (16–18). INFγ and IFNγ-related chemokines (CXCL9, CXCL10, CXLC11) levels are notably higher in patients with active sHLH/MAS and are tightly associated with laboratory markers of disease severity (18–20). These studies suggest that IFNγ is a potent driver of sHLH/MAS. Emapalumab has been shown to rapidly neutralize IFNγ and IFNγ-related cytokine levels and has resulted in MAS remission (18, 20–22). Notably, our patient had elevated CXCL9 and IL-18 levels and responded well to emapalumab.
Etoposide, a topoisomerase-II inhibitor, has been employed in the treatment of unrelenting MAS/sHLH (23). Apheresis has been used to manage and induce remission in refractory MAS in patients with sJIA through the removal of proinflammatory cytokines and reduction in peripheral white blood cells (24, 25). Tofacitinib, a Janus kinase inhibitor, which blocks intracellular signaling thereby diminishing the inflammatory cascade, was successfully used in refractory adult-onset Still's disease complicated by MAS, but is now used sparingly (26). The IL-6 inhibitor, tocilizumab, has been used to safely and effectively treat sJIA with refractory MAS (27, 28).
Our patient did not respond to therapeutic doses of cyclosporine or tacrolimus in combination with high-dose steroids (methylprednisolone or dexamethasone) and escalated doses of anakinra. Therefore, emapalumab and etoposide were added for critical worsening of MAS. Our patient's MAS was driven in large part by adenoviremia; however, he did not dramatically respond to initial cidofovir until steroids were tapered. Although plasmapheresis has been used as rescue therapy in refractory MAS, this option was deferred owing to concerns regarding cardiovascular status. Fortunately, he stabilized on this multifaceted regimen and anakinra was discontinued before discharge. He was weaned off emapalumab after 16 months of maintenance treatment. The patient remains on low dose tacrolimus, but steroids were discontinued 4 months after discharge. He was able to discontinue cidofovir after 21 weeks of treatment and his posaconazole prophylaxis for Aspergillus was discontinued after 18 months but he remains on pentamidine prophylaxis. He has not had any flares of MAS since discharge. However, he sustained some infections including norovirus, enteropathogenic E. coli, and Legionella bacteremia treated with azithromycin. He briefly seroconverted positive with low level adenoviremia that responded to an additional dose of cidofovir without recurrence of MAS.
The cytokine storm seen in recalcitrant MAS can cause life-threatening multiorgan failure, in which IFNγ plays a key pathogenic role. Promising, essential, and innovative cytokine-blocking therapeutic approaches that quell the immune dysfunction seen in these hyperinflammatory states are being investigated. Of these therapies, emapalumab, an IFNγ blocking monoclonal antibody, proved efficacious in this patient over steroids, tacrolimus, and anakinra. Given the extreme cytokine cascade in these conditions, high-dose anakinra in combination with other anticytokine and immunosuppressive therapies (such as etoposide) may have a role in treating gravely ill patients who are refractory to standard dosing and/or other therapeutic interventions. Potentially, this combination approach may assist with an expedited laboratory and clinical recovery in critically ill pediatric patients with recalcitrant MAS.
Our patient had no adverse side effects from this treatment regimen other than possible opportunistic pulmonary infections, which were successfully treated. His severe adenoviremia was the precipitating factor in his MAS flare and cidofovir was necessary adjunct therapy. Given the lack of treatment guidelines and approved therapies for refractory MAS, alternative and aggressive management strategies need to be considered for patients with an intractable course.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
ES: Writing – original draft, Writing – review & editing. RM: Writing – original draft, Writing – review & editing. ME: Writing – review & editing. LW: Writing – review & editing. DK: Writing – review & editing. DB: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank our advanced practice registered nurses and administrative staff in the Division of Pediatric Allergy, Immunology, and Rheumatology at the University of Florida, Gainesville, who helped facilitate the care of this complex patient.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. (2019) 10:119. doi: 10.3389/fimmu.2019.00119
2. Ravelli A, Davì S, Minoia F, Martini A, Cron RQ. Macrophage activation syndrome. Hematol Oncol Clin North Am. (2015) 29(5):927–41. doi: 10.1016/j.hoc.2015.06.010
3. Alfaraidi AT, Alqarni AA, Aqeel MT, Albalawi TA, Hejazi AS. Familial hemophagocytic lymphohistiocytosis secondary to PRF1 mutation. Case Rep Hematol. (2021) 2021:7213939. doi: 10.1155/2021/7213939
4. Giri PP, Biswas N, Chakravarty S. Familial hemophagocytic lymphohistiocytosis due to mutation of UNC13D gene. Indian J Hematol Blood Transfus. (2016) 32(Suppl 1):344–6. doi: 10.1007/s12288-014-0494-x
5. Hu X, Liu D, Jiang X, Gao B, Chen C. Identification of a novel nonsense mutation in the UNC13D gene from a patient with hemophagocytic lymphohistiocytosis: a case report. BMC Med Genet. (2018) 19(1):82. doi: 10.1186/s12881-018-0600-2
6. Xinh PT, Chuong HQ, Diem TPH, Nguyen TM, Van ND, Mai Anh NH, et al. Spectrum mutations of PRF1, UNC13D, STX11, and STXBP2 genes in Vietnamese patients with hemophagocytic lymphohistiocytosis. Int J Lab Hematol. (2021) 43(6):1524–30. doi: 10.1111/ijlh.13674
7. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. (2006) 27(1):62–8. doi: 10.1002/humu.20274
8. Yang X, Miyawaki T, Kanegane H. SAP and XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr Int. (2012) 54(4):447–54. doi: 10.1111/j.1442-200X.2012.03683.x
9. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48(2):124–31. doi: 10.1002/pbc.21039
10. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. (2016) 68(3):566–76. doi: 10.1002/art.39332
11. Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2020) 72(2):326–34. doi: 10.1002/art.41103
12. Sönmez HE, Demir S, Bilginer Y, Özen S. Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol. (2018) 37(12):3329–35. doi: 10.1007/s10067-018-4095-1
13. Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. (2020) 2(6):e358–67. doi: 10.1016/S2665-9913(20)30096-5
14. Meneghel A, Martini G, Amigoni A, Pettenazzo A, Padalino M, Zulian F. Case report: life-threatening macrophage activation syndrome with fulminant myocarditis successfully rescued by high dose intravenous anakinra. Front Pediatr. (2021) 8:635080. doi: 10.3389/fped.2020.635080
15. Bami S, Vagrecha A, Soberman D, Badawi M, Cannone D, Lipton JM, et al. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2020) 67(11):e28581. doi: 10.1002/pbc.28581
16. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382(19):1811–22. doi: 10.1056/NEJMoa1911326
17. Garonzi C, Chinello M, Cesaro S. Emapalumab for adult and pediatric patients with hemophagocytic lymphohistiocytosis. Expert Rev Clin Pharmacol. (2021) 14(5):527–34. doi: 10.1080/17512433.2021.1901576
18. De Benedetti F, Grom AA, Brogan PA, Bracaglia C, Pardeo M, Marucci G, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis. (2023) 82(6):857–65. doi: 10.1136/ard-2022-223739
19. Bracaglia C, Marafon DP, Caiello I, de Graaf K, Guilhot F, Ferlin W, et al. High levels of interferon-gamma (IFNg) in macrophage activation syndrome (MAS) and CXCL9 levels as a biomarker for IFNg production in MAS. Pediatric Rheumatology. (2015) 13(Suppl 1):O84. doi: 10.1186/1546-0096-13-S1-O84
20. De Benedetti F, Brogan P, Bracaglia C, Pardeo M, Marucci G, Sacco E, et al. OP0290 emapalumab (anti-interferon-gamma monoclonal antibody) in patients with macrophage activation syndrome (MAS) complicating systemic juvenile idiopathic arthritis (SJIA). Ann Rheum Dis. (2020) 79:180. doi: 10.1136/annrheumdis-2020-eular.3169
21. Chellapandian D, Milojevic D. Case report: Emapalumab for active disease control prior to hematopoietic stem cell transplantation in refractory systemic juvenile idiopathic arthritis complicated by macrophage activation syndrome. Front Pediatr. (2023) 11:1123104. doi: 10.3389/fped.2023.1123104
22. Gabr JB, Liu E, Mian S, Pillittere J, Bonilla E, Banki K, et al. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset still’s disease: case report and review of the literature. Ann Transl Med. (2020) 8(14):887. doi: 10.21037/atm-20-3127
23. Horne A, von Bahr Greenwood T, Chiang SCC, Meeths M, Björklund C, Ekelund M, et al. Efficacy of moderately dosed etoposide in macrophage activation syndrome-hemophagocytic lymphohistiocytosis. J Rheumatol. (2021) 48(10):1596–602. doi: 10.3899/jrheum.200941
24. Kinjo N, Hamada K, Hirayama C, Shimizu M. Role of plasma exchange, leukocytapheresis, and plasma diafiltration in management of refractory macrophage activation syndrome. J Clin Apher. (2018) 33(1):117–20. doi: 10.1002/jca.21570
25. Miyazono A, Abe J, Ogura M, Sato M, Fujimaru T, Kamei K, et al. Successful remission induced by plasma exchange combined with leukocytapheresis against refractory systemic juvenile idiopathic arthritis. Eur J Pediatr. (2014) 173(12):1557–60. doi: 10.1007/s00431-013-2093-5
26. Hu Q, Wang M, Jia J, Teng J, Chi H, Liu T, et al. Tofacitinib in refractory adult-onset still’s disease: 14 cases from a single centre in China. Ann Rheum Dis. (2020) 79(6):842–4. doi: 10.1136/annrheumdis-2019-216699
27. Wu J, Sun L, Tang X, Zheng Q, Guo L, Xu L, et al. Tang X, et al. Effective therapy of tocilizumab on systemic juvenile idiopathic arthritis-associated refractory macrophage activation syndrome. Mod Rheumatol. (2022) 32(6):1114–21. doi: 10.1093/mr/roab119
Keywords: systemic arthritis, macrophage activation syndrome, emapalumab, anakinra, etoposide
Citation: Slaney ED, Modica R, Woolnough L, Kafisheh D, Bell-Brunson DH and Elder M (2024) Case Report: Refractory macrophage activation syndrome requiring high-dose anakinra, emapalumab, and etoposide therapy in early-onset systemic juvenile idiopathic arthritis associated with adenoviremia. Front. Pediatr. 11:1336554. doi: 10.3389/fped.2023.1336554
Received: 10 November 2023; Accepted: 21 December 2023;
Published: 22 January 2024.
Edited by:
Giorgia Martini, University Hospital of Padua, ItalyReviewed by:
Concetta Micalizzi, Giannina Gaslini Institute (IRCCS), ItalyGiorgio Costagliola, Azienda Ospedaliero Universitaria Pisana, Italy
© 2024 Slaney, Modica, Woolnough, Kafisheh, Bell-Brunson and Elder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth D. Slaney edawso5@emory.edu
†ORCID Leandra Woolnough orcid.org/0000-0002-4993-6542 Denise Heather Bell-Brunson orcid.org/0000-0002-7736-6882 Melissa Elder orcid.org/0000-0002-8820-5515