 Insoon Chang
Insoon Chang Yi-Ling Loo2
Yi-Ling Loo2 Jin Koo Kim
Jin Koo Kim Paul H. Krebsbach
Paul H. Krebsbach- 1Section of Endodontics, Division of Regenerative and Reconstructive Sciences, School of Dentistry, University of California, Los Angeles, Los Angeles, CA, United States
- 2School of Dentistry, University of California, Los Angeles, Los Angeles, CA, United States
- 3Laboratory of Cancer Biology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 4Oral Immunobiology Unit, National Institutes of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD, United States
- 5Division of Oral and Systemic Health Sciences, School of Dentistry, University of California, Los Angeles, Los Angeles, CA, United States
mEAK-7 (mammalian EAK-7 or MTOR-associated protein, eak-7 homolog), is an evolutionarily conserved lysosomal membrane protein that is highly expressed in several cancer cells. Multiple recent studies have identified mEAK-7 as a positive activator of mTOR (mammalian/mechanistic target of rapamycin) signaling via an alternative mTOR complex, implying that mEAK-7 plays an important role in the promotion of cancer proliferation and migration. In addition, structural analyses investigating interactions between mEAK-7 and V-ATPase, a protein complex responsible for regulating pH homeostasis in cellular compartments, have suggested that mEAK-7 may contribute to V-ATPase-mediated mTORC1 activation. The C-terminal α-helix of mEAK-7 binds to the D and B subunits of the V-ATPase, creating a pincer-like grip around its B subunit. This binding undergoes partial disruption during ATP hydrolysis, potentially enabling other proteins such as mTOR to bind to the α-helix of mEAK-7. mEAK-7 also promotes chemoresistance and radiation resistance by sustaining DNA damage-mediated mTOR signaling through interactions with DNA-PKcs (DNA-dependent protein kinase catalytic subunit). Taken together, these findings indicate that mEAK-7 may be a promising therapeutic target against tumors. However, the precise molecular mechanisms and signal transduction pathways of mEAK-7 in cancer remain largely unknown, motivating the need for further investigation. Here, we summarize the current known roles of mEAK-7 in normal physiology and cancer development by reviewing the latest studies and discuss potential future developments of mEAK-7 in targeted cancer therapy.
Introduction
mEAK-7 (mammalian EAK-7 or MTOR-associated protein, eak-7 homolog), also known through genomics and proteomics studies as KIAA1609 (1, 2), LOC57707 (3), and TLDC1 (TBC/LysM-associated domain-containing 1) (4), is an evolutionarily conserved lysosomal membrane protein that promotes cell proliferation and migration (5). The mEAK-7 protein, first cloned and sequenced from a human fetal brain cDNA library, contains 473 amino acids and is ubiquitously expressed in all human adult and fetal tissues, with highest levels in the brain, kidney, spleen, and ovaries (1). The human ortholog MEAK7 gene (NCBI Gene ID: 57707) is located at chromosome 16q24.1, and an average of 89% of amino acids are conserved in the mEAK-7 protein across eukaryotes (5). In Caenorhabditis elegans, EAK-7 (enhancer-of-akt-1-7), the mEAK-7 homologue in nematodes, nonautonomously controls both dauer arrest and longevity by negatively regulating the nuclear activity of the DAF-16/FoxO transcription factor in parallel to the insulin receptor signaling serine/threonine kinase AKT-1 (6). It has been shown that the mEAK-7 protein is expressed in the hippocampi of mouse brains, and while overexpression of mEAK-7 in N2a (Neuro 2a) cells does not reduce the level of oxidative stress in vitro, expression in mice reduces N2a cell death under oxidative stress conditions through an unknown mechanism (2).
Human mEAK-7 contains two domains: the TLDc (Tre2/Bub2/Cdc16 (TBC), lysine motif (LysM), domain catalytic) domain near the C-terminus and the N-myristoylation motif at the N-terminus (5). The TLDc domain is a highly conserved protein motif which plays an essential role in normal human brain development and is found in a family of five proteins which share protective functions against oxidative stress: NCOA7, TBC1D24, mEAK-7 (TLDC1), OXR1, and C20ORF118 (TLDC2) (2, 7, 8). Computational analysis predicts that mEAK-7 folds into α/β + β sheets to facilitate its enzymatic activity (5). In zebrafish, the crystal structure of the TLDc domain of oxidation resistance protein 2 forms two antiparallel β-sheets sandwiched between two helices and two one-turn helices (9). In humans, the 3-dimensional structure of the TLDc domain is globular and consists of two α-helices at the N-terminus and ten β-strands forming two antiparallel β-sheets (10). The structure of the TLDc domain does not resemble any other enzymes engaged in the protection against reactive oxygen species, and the intricate molecular mechanism underlying its antioxidant attributes remains unknown (11). The N-myristoylation motif anchors proteins to lipid bilayers and endomembrane compartments by irreversibly binding to myristate (12, 13).
Growing evidence indicates that mEAK-7 mediates the activation of the mTOR (mammalian/mechanistic target of rapamycin) pathway through an alternative mTOR complex (5, 14). mTOR regulates many fundamental cell processes including autophagy, apoptosis, cellular growth, proliferation, survival, metabolism, angiogenesis, transcription, and translation. Dysregulation and alterations in mTOR pathways lead to cancer development and advancement (15, 16). The mTOR complex involving mEAK-7 regulates S6K2 and 4E-BP1 in response to insulin, amino acids, and growth factors (5) and has also been shown to interact with V-ATPases (vacular-type adenosine triphosphatases, also known as the H+-ATPase) (8, 17, 18). Recent studies have found that human MEAK7 mRNA is overexpressed in cancers such as hepatocellular carcinoma (19), lymph node-positive breast cancers (20), and non-small-cell lung cancer (14). Here, we present a concise overview of the latest significant discoveries concerning the physiological roles of mEAK-7 and current understanding in the context of carcinogenesis and the advancement of cancer.
mEAK-7 and V-ATPase interaction
Recent findings demonstrate that all five members of the TLDc-containing proteins interact with V-ATPases (11). V-ATPases are conserved protein complexes that regulate pH homeostasis of intracellular compartments and organelles by catalyzing ATP hydrolysis to actively transport protons across biological membranes (21–24). V-ATPase-regulated pH acidification is essential for the normal physiological function of cells. Canonical roles attributed to V-ATPase include endocytic trafficking, protein processing and degradation, plasma membrane functions, and synaptic vesicle loading and coupled transport. Non-canonical functions of V-ATPase include membrane fusion, pH sensing, activation of mTORC1, and scaffolding protein-protein interactions (21). Dysregulated expression, misplacement, and genetic alterations affecting V-ATPases are intricately linked to cancer development and progression, making V-ATPases a central therapeutic target in translational medicine (22). Mammalian V-ATPases are composed of a cytosolic V1 domain (consisting of three copies each of the A, B, G, and E subunits and single copies of C, H, D, and F subunits), responsible for ATP hydrolysis, and a transmembrane V0 subcomplex (containing a single isoform of subunit a, 9 copies of c, and one copy each of d, e, and c’’) embedded in the lipid bilayer that participates in proton translocation (23). The c’ subunit within the V0 domain is only observed in fungi. The A3B3 subcomplex allows rotation of the rotor subcomplex, composed of subunits D, F, d, and the membrane-embedded c-ring (25). Rotation of the c-ring, comprised of nine c subunits and a single c″ subunit, against subunit “a” drives proton translocation through the membrane, and the reversible dissociation of the V1 and V0 regions stops the proton pumping (8, 26). The stationary component, relative to the rotor, has three peripheral stalks each composed of E and G heterodimers, enabling its ‘stator’ function primarily through the binding of C and H subunits of the V1 domain to subunits a of the V0 complex in a cohesive manner (26). In mammalian cells, there are several different isoforms of V-ATPase subunits (27); isoforms of the “a” subunit of the V0 domain contain location-specific information which is essential for directing V-ATPases to their target cellular compartments and creating specific pH values within them (22, 27).
Immunoprecipitation targeting the B subunit of V-ATPase mixed with the lysates from HEK293T cells overexpressing mEAK-7 revealed an interaction between mEAK-7 and the B subunit of V-ATPase (11). In addition, mEAK-7 associates with V-ATPases through its TLDc domain and C-terminal α-helix in rotational State 2 (Figure 1) (8). The C-terminal α-helix binds to the D and B subunits of the V-ATPase and crosslinks the stationary and rotor region of the enzyme through hydrophobic interactions, resulting in the formation of a pincer-like grip around the B subunit of V-ATPase (8, 28). Structural investigations also unveiled interactions between human mEAK7 and human V-ATPase, demonstrating an association between the TLDc domain of mEAK-7 with subunit A, B, and E of V-ATPase and the C-terminal domain of mEAK-7 with subunit D of V-ATPase (17). Another structural analysis complimented this finding by demonstrating that mEAK-7 interacts with subunits A, B, D, and E of V-ATPases in state 2 (18). Collectively, these findings suggest that mEAK-7 is a V-ATPase inhibitor, presumably blocking V1-V0 torque transmission. However, functional studies revealed that mEAK-7 does not affect the enzyme activity of V-ATPases and lysosomal or phagosomal pH in vitro (17, 28).
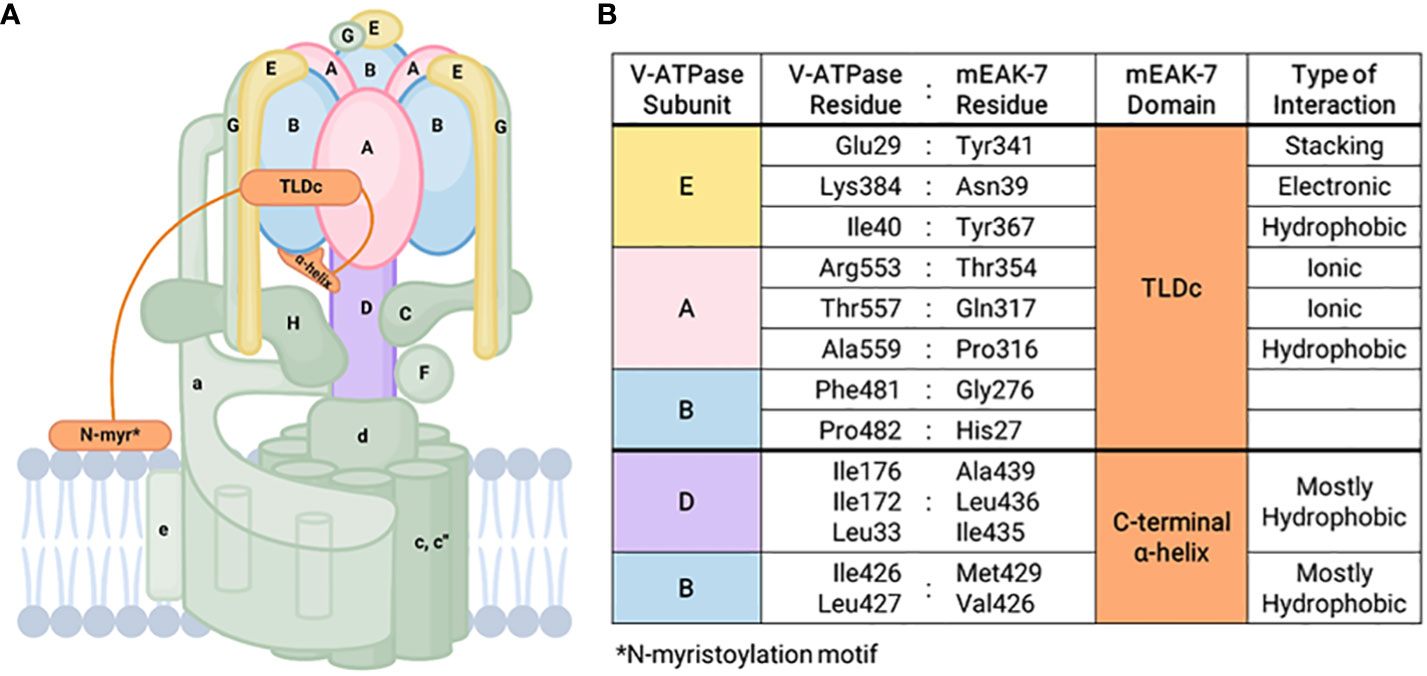
Figure 1 mEAK-7 interaction with V-ATPase. (A) Simplified structural diagram of V-ATPase and its interactions with mEAK-7. The C-terminal α-helix of mEAK-7 was shown to interact with the B and D subunits of V-ATPase, while the TLDc domain of mEAK-7 interacts with the E, A, and B subunits of V-ATPase. The N-myristoylation motif (denoted as “N-myr”) anchors proteins to lipid bilayers and endomembrane compartments by irreversibly attaching to myristate. (B) Specific amino acid interactions between residues in V-ATPase subunits and mEAK-7 domains presented by Tan et al. (8). These indicate potential sites for future targeted drug therapies.
Bafilomycin, a V-ATPase blocker, inhibits lysosomal acidification, thereby preventing the activity of lysosomal proteases and autophagy by interfering with autophagosome-lysosome fusion (29). The overexpression of mEAK-7 also does not affect bafilomycin sensitivity, indicating that the V1 and V0 domains of V-ATPase may remain coupled even when mEAK-7 crosslinks the rotor and stator of V-ATPase. It is also possible that the direct crosslink between mEAK-7 and V-ATPase is interrupted during rotary catalysis (17, 18, 28). CryoEM studies of mEAK-7 and V-ATPase structural interactions showed a clear 3-dimensional structure in rotational State 2 before consumption of ATP, with no area that lacked density (8). However, when ATP was added, a population of particles associated with mEAK-7 in rotational State 2 and the density for the C-terminal α-helix disappeared, indicating that ATP hydrolysis might disrupt mEAK-7 and V-ATPase interactions (8).
mEAK-7, V-ATPase, and the mTOR pathway
mTOR is a conserved serine/threonine kinase in the PI3K (phosphoinositide 3-kinases)-related kinase (PIKK) family and responds to nutrients and growth signals (16). mTOR forms two structurally and functionally distinct complexes known as mTORC1 (mTOR Complex 1) and mTORC2 (15, 16, 30). mTORC1 consists of mTOR, Raptor (regulatory-associated protein of mammalian target of rapamycin), mLST8 (mammalian lethal with Sec13 protein 8, also known as GβL), PRAS40 (proline-rich Akt substrate of 40 kDa), and DEPTOR (DEP domain containing mTOR-interacting protein) (30). mTORC2 consists of mTOR, Rictor (rapamycin-insensitive companion of mTOR), mLST8, DEPTOR, hSin1, and PRR5 (proline-rich protein 5), and predominantly regulates cytoskeletal structure, cellular metabolism, cell survival, and cell response to insulin (30–32). Recently, several studies demonstrated that mTOR activation requires localization to the lysosome surface (33, 34). When amino acids are available, cytosolic mTOR is recruited to the lysosome surface via Rag proteins (34). Upon growth factor stimulation, PI3K triggers the production of PIP3 in the plasma membrane, initiating a cascade to phosphorylate kinase Akt and its activator PDK1 (phosphoinositide-dependent protein kinase 1). This leads to the inhibition of the TSC (Tuberous sclerosis complex) and GAP (GTPase-activating protein) activity, culminating in an enhanced level of GTP-bound Rheb at the lysosome surface, where it directly binds and activates the mTORC1 complex (33–35). The activated mTORC1 has been known to phosphorylate both S6K1 (ribosomal protein S6 kinase) and 4E-BP (the eukaryotic initiation factor 4E binding protein), to promote cellular growth through protein anabolism (16, 36), nucleotide biosynthesis (37), lipogenesis (38), glycolysis (15, 16), and mitochondrial biogenesis (39).
Macrolide V-ATPase inhibitors effectively halt mTORC1 activation, indicating that V-ATPase affects activation of mTOR signaling (40). A small-molecule in vivo activator of autophagy, EN6, has been shown to covalently bind to Cysteine 277 in the ATP6V1A subunit of the lysosomal V-ATPase, decoupling it from RAGs and leading to the inhibition of mTORC1 signaling, enhancement of lysosomal acidification, and activation of autophagy (41). Conversely, mTORC1 has been shown to suppress V-ATPase activity by repressing V1-V0 assembly, enabling mTORC1 to respond promptly to changes in cellular nutrient environments by preventing lysosomal degradation of extracellular proteins (42). It has also been demonstrated that the late endosomal/lysosomal V-ATPase-Ragulator-RAG (Ras-related GTP binding) protein complex is required for mTORC1 activation (43). Upon high nutrient and energy levels, V-ATPase stimulates the GEF (guanine-nucleotide exchange factor) activity of Ragulator, consisting of LAMTOR1 (late endosomal/lysosomal adaptor, MAPK and mTOR activator 1) through LAMTOR5. This promotes the conversion of the GDP-bound form to the GTP-bound form of RAGA/RAGB, increasing the affinity of RAGs for mTORC1 (40, 44, 45). The RAG-bound mTORC1 is then activated after interacting with GTP-bound Rheb on the lysosomal surface (35). These findings highlight the essential role of V-ATPase activity in mTOR signaling and activation.
Previously, we demonstrated that mEAK-7 interacts with mTOR at the lysosome in response to nutrient stimulation and promotes cell proliferation and migration (5). In nematodes, EAK-7 is located in the plasma membrane, and there is some evidence that mEAK-7 can be detected at the plasma membrane in mammalian cells as well (5, 6). Research has shown that mEAK-7 predominantly binds to the lysosomal membrane. This is supported by the strong colocalization between mEAK-7 and LAMP1 (lysosomal-associated membrane protein-1) as well as LAMP2. In contrast, there is minimal to no colocalization observed in the endosome, mitochondria, endoplasmic reticulum, and golgi complex, indicating that mEAK-7’s association is primarily with the lysosomal membrane (5). The knockdown of mEAK-7 in several human cancer cell lines with high endogenous mEAK-7 expression, such as H1975 (non-small cell lung cancer, NSCLC), H1299 cells (NSCLC), and MDA-MB-231 (breast cancer), resulted in decreased lysosomal localization of mTOR, marked attenuation of S6K2 and 4E-BP1 phosphorylation, and diminished cell proliferation and migration. These effects were rescued upon overexpression of mEAK-7, which reactivated mTOR signaling (5). Furthermore, the TLDc domain and C-terminus of mEAK-7 are crucial for facilitating mTOR kinase activity, and although mEAK-7 interacts with mTOR and mLST8, it does not engage with Raptor or Rictor, key constituents of the mTORC1 and mTORC2 complexes (5). Taken together, these findings suggest that mEAK-7 might play a key role in recruiting mTOR to the lysosome and activating mTOR kinase function via an alternative mTOR complex in mammalian cells.
As discussed above, numerous studies have investigated interactions among mEAK-7, V-ATPase, and mTORC1. Although the exact nature of these interactions remains unclear, the discoveries made so far are as follows: 1) The mEAK-7 and mTORC1 have physical and functional interactions; 2) The interaction between mEAK-7 and the V-ATPase is predicted to be inhibitory; however, this interaction could be disrupted by ATP hydrolysis, potentially liberating part of mEAK-7 to serve as a binding site for other molecules. There is speculation that mTORC1 might bind to the released segment of mEAK-7, although empirical evidence to support this interaction remains elusive; and 3) The interaction between V-ATPase and mTORC1 exhibits significant complexity. V-ATPase interactions with Rag proteins are required for mTORC1 activity (40), but inhibition of mTORC1 activates V-ATPase by both increasing its assembly (42) and enhancing synthesis via the TFEB pathway (46). Furthermore, despite the interactions between each pair, currently, there is limited data supporting simultaneous physical or functional interactions amongst all three of the molecules (mEAK-7, mTORC1, and V-ATPase). Also, the precise role mEAK-7 may play in such interactions remains unclear. Recently, structural investigations have unveiled the potential involvement of mEAK-7 in V-ATPase-mediated mTOR signaling (8, 17, 18). While it was initially suggested that the C-terminal α-helix of mEAK-7 could engage with mTOR (5), this interaction through the α-helix seems unfeasible when mEAK-7 is bound to V-ATPase. However, a structural and functional investigation showed that the interaction between mEAK-7 and V-ATPase, hinged on the C-terminal α-helix, undergoes partial disruption during ATP hydrolysis (8, 28). This potentially enables binding of other proteins (47). Alternatively, this mechanism might serve to regulate binding of mEAK-7 to both V-ATPase and other proteins (8). Furthermore, one recent study suggests that mEAK-7 may interfere with V-ATPase-mTOR signaling (17). This validation involved the generation of a stable HCT116 cell line (human colorectal carcinoma) that constitutively expresses the α4 subunit of V-ATPase (HCT-a4), which localizes to the lysosome. Western blot analysis demonstrated that after treatment with bafilomycin A1, the phosphorylation of S6 protein at Ser235/236 was completely abolished. In addition, overexpression of mEAK7 in HC6-a4 cells inhibited the phosphorylation of S6 at Ser235/236 and 4E-BP1 at Ser65, indicating that mEAK7 may interfere with the V-ATPase-mTOR signaling (17). However, the authors of this study explicitly stated that this outcome might be due to using a ‘special class of cells’ with V-ATPase overexpression. Figure 2 illustrates the potential pathways involving mTORC1, V-ATPase, and the alternative mTOR complex. Collectively, these findings reinforce the need for further studies aimed at unraveling the details of mEAK-7-mediated regulation within the context of V-ATPase-mediated mTOR signaling or mEAK-7-mediated mTOR signaling, while also delving into the prospective contributions of mEAK-7 in other pathways, potentially mediated by its interactions involving the α-helix.
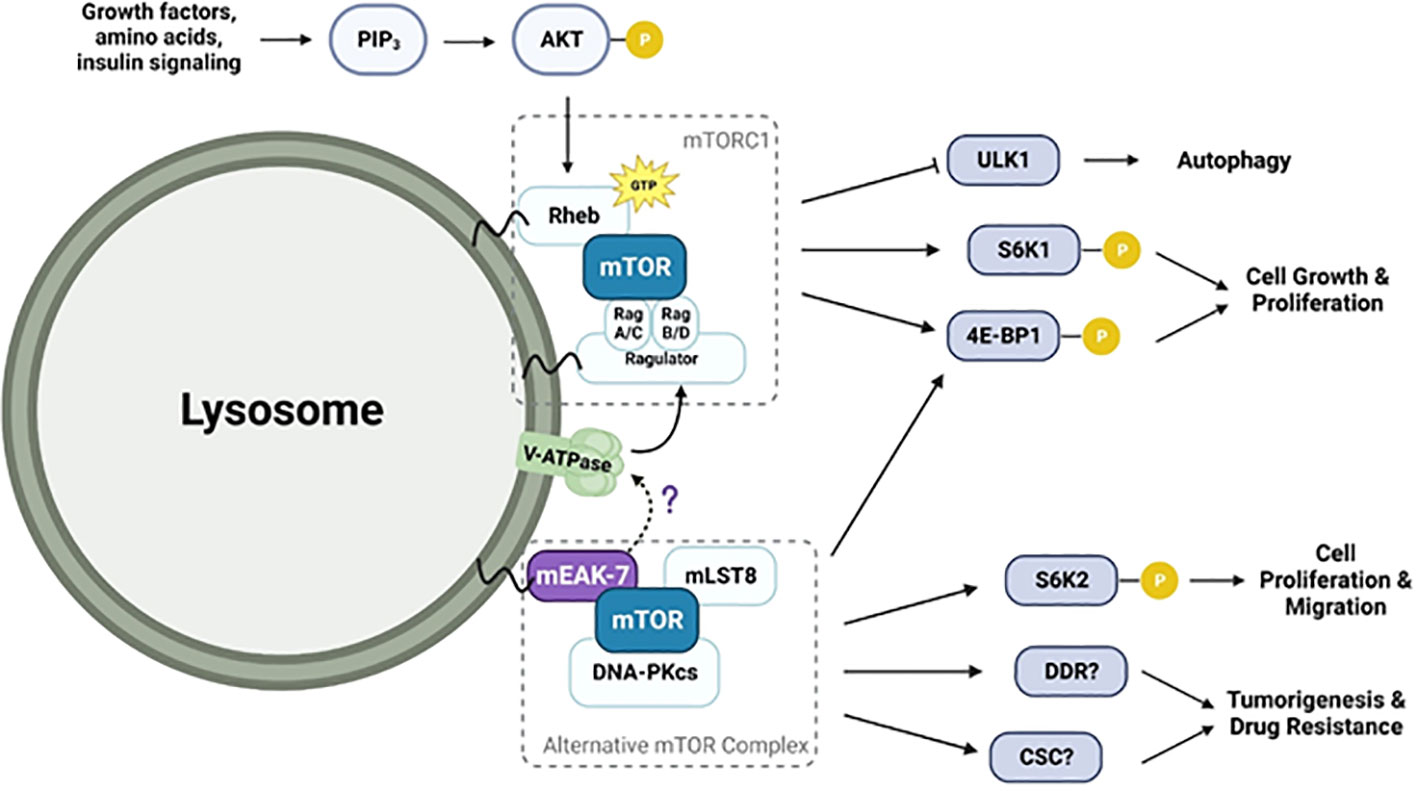
Figure 2 Diagram illustrating the potential mEAK-7-mediated mTOR activation pathway and their interactions with V-ATPase. V-ATPase stimulates GEF activity of Ragulator, promoting conversion of GDP to GTP-bound RAGs and increasing their affinity for mTORC1. RAG-bound mTORC1 is then activated after interacting with GTP-bound Rheb. mEAK-7 interacts with mTOR to form an alternative mTOR complex involving mLST8 and DNA-PKcs. This complex is known to play a role in phosphorylating S6K2 and 4E-BP1, leading to cell growth, proliferation, and migration. Although structural studies have suggested that mEAK-7 interacts with V-ATPase, the precise mechanism is still unknown.
mEAK-7 and cancer
As previously explored, mEAK-7 plays a crucial role in activating the mTOR pathway that regulates cellular growth and migration, maintains physiological hemostasis, and orchestrates metabolism in response to external stimuli (16). The dysregulation of these processes contributes to the development and progression of pathophysiological conditions such as cancer and neurological disorders (5, 14, 15). Heightened mRNA expression levels of mEAK-7 have been observed in several cancer cell lines (5) and there is significant elevation of both mEAK-7 and mTOR signaling in tumor and metastatic lymph nodes of patients diagnosed with NSCLC (14). In two well-established NSCLC cell lines, H1975 and H1299, stem-like CD44+/CD90+ cells, which represented 1% of the total cell population, yielded elevated protein levels of mEAK-7, S6K2, n-cadherin (marker for the epithelial-mesenchymal transition state in cancer stem cells), and phosphorylated S6 and 4E-BP1, indicating activated mTOR signaling with higher invasive potential compared to CD44-/CD90- cells (14). Furthermore, mEAK-7 is necessary for clonogenic potential and spheroid formation of CD44+/CD90+ NSCLS cells and promotion of cisplatin- and radiation resistance through DNA damage-mediated mTOR signaling, in part through interaction with DNA-PKcs (DNA-dependent protein kinase catalytic subunit) (14, 48). Knockdown of mEAK-7 impairs the DNA damage response and enhances proapoptotic Noxa levels and PARP (poly (ADP-ribose) polymerase) cleavage in cancer cells. A bioinformatic search using the Exiqon miRSearch V3.0 algorithm revealed that MicroRNA-1911-3p regulates mEAK-7 translation (49). The study revealed that MicroRNA-1911-3p targets mEAK-7 mRNA at 3’UTR and decreases mEAK-7 protein levels, leading to suppressed mTOR signaling, which was evidenced by significantly decreased mTOR-dependent S6 and 4E-BP1 phosphorylation in NSCLC cell lines. Furthermore, H1299 cells transfected with a MicroRNA-1911-3p mimic significantly decreased mTOR and LAMP2 colocalization, demonstrating that repressed mEAK-7 levels impair co-localization of mTOR to the lysosome and subsequently attenuates NSCLC cell proliferation and migration. Collectively, these studies provide a novel perspective towards developing a cancer treatment targeting mEAK-7-associated mTOR signaling in NSCLC and other malignancies exhibiting elevated mEAK-7 expression.
Conclusion
The lysosome-bound mEAK-7 protein plays an important role in the intricate regulation of cellular proliferation and migration by activating mTOR via an alternative pathway and/or interacting with V-ATPase to modulate V-ATPase-mediated mTOR activation. Structural and functional analyses conducted in both physiological and pathophysiological conditions suggest mEAK-7 could serve as a promising therapeutic target against tumors, particularly non-small-cell lung cancer. However, despite recent findings, the precise molecular mechanisms and signal transduction pathways controlled by mEAK-7 in cancer remain largely unknown, with in vivo investigations yet to be systematically explored. The elucidation of the mEAK-7 protein structure and its interaction with V-ATPases presents a remarkable opportunity for the development of a potent mEAK-7 inhibitor with specific binding characteristics while minimizing adverse effects, paving the way for the generation of mEAK-7-targeted therapy in cancer. This exciting potential not only broadens the current understanding of cancer biology but also highlights the possibility of tailored interventions that could revolutionize treatment strategies.
Author contributions
IC: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing. JP: Writing – review & editing. JN: Writing – review & editing. JK: Writing – review & editing. PK: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institute of Dental and Craniofacial Research (1F30DE026048-01, R01-DE016530, and T32-DE007057); the Stuart & Barbara Padnos Research Award from the Comprehensive Cancer Center at the University of Michigan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nagase T. Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 New cDNA Clones from Brain which code for large proteins in vitro. DNA Res. (2000) 7:271–81. doi: 10.1093/dnares/7.4.271
2. Finelli MJ, Sanchez-Pulido L, Liu KX, Davies KE, Oliver PL. The evolutionarily conserved tre2/bub2/cdc16 (TBC), lysin motif (LysM), domain catalytic (TLDc) domain is neuroprotective against oxidative stress. J Biol Chem. (2016) 291:2751–63. doi: 10.1074/jbc.M115.685222
3. Schröder B, Wrocklage C, Pan C, Jäger R, Kösters B, Schäfer H, et al. Integral and associated lysosomal membrane proteins. Traffic. (2007) 8:1676–86. doi: 10.1111/j.1600-0854.2007.00643.x
4. Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. (2015) 43:D1079–85. doi: 10.1093/nar/gku1071
5. Nguyen JT, Ray C, Fox AL, Mendonça DB, Kim JK, Krebsbach PH. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci Adv. (2018) 4:eaao5838. doi: 10.1126/sciadv.aao5838
6. Alam H, Williams TW, Dumas KJ, Guo C, Yoshina S, Mitani S, et al. EAK-7 controls development and life span by regulating nuclear DAF-16/foxO activity. Cell Metab. (2010) 12:30–41. doi: 10.1016/j.cmet.2010.05.004
7. Finelli MJ, Oliver PL. TLDc proteins: new players in the oxidative stress response and neurological disease. Mamm Genome. (2017) 28:395–406. doi: 10.1007/s00335-017-9706-7
8. Tan YZ, Abbas YM, Wu JZ, Wu D, Keon KA, Hesketh GG, et al. CryoEM of endogenous mammalian V-ATPase interacting with the TLDc protein mEAK-7. Life Sci Alliance. (2022) 5:e202201527. doi: 10.26508/lsa.202201527
9. Blaise M, Alsarraf HMAB, Wong JEMM, Midtgaard SR, Laroche F, Schack L, et al. Crystal structure of the TLDc domain of oxidation resistance protein 2 from zebrafish: Crystal Structure of the TLDc Domain. Proteins Struct Funct Bioinforma. (2012) 80:1694–8. doi: 10.1002/prot.24050
10. Arnaud-Arnould M, Tauziet M, Moncorgé O, Goujon C, Blaise M. Crystal structure of the TLDc domain of human NCOA7-AS. Acta Crystallogr Sect F Struct Biol Commun. (2021) 77:230–7. doi: 10.1107/S2053230X21006853
11. Eaton AF, Brown D, Merkulova M. The evolutionary conserved TLDc domain defines a new class of (H+)V-ATPase interacting proteins. Sci Rep. (2021) 11:22654. doi: 10.1038/s41598-021-01809-y
12. Wang B, Dai T, Sun W, Wei Y, Ren J, Zhang L, et al. Protein N-myristoylation: functions and mechanisms in control of innate immunity. Cell Mol Immunol. (2021) 18:878–88. doi: 10.1038/s41423-021-00663-2
13. Maurer-Stroh S, Eisenhaber B, Eisenhaber F. N-terminal N -myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. J Mol Biol. (2002) 317:523–40. doi: 10.1006/jmbi.2002.5425
14. Nguyen JT, Haidar FS, Fox AL, Ray C, Mendonça DB, Kim JK, et al. mEAK-7 forms an alternative mTOR complex with DNA-PKcs in human cancer. iScience. (2019) 17:190–207. doi: 10.1016/j.sci.2019.06.029
15. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
16. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
17. Wang R, Qin Y, Xie XS, Li X. Molecular basis of mEAK7-mediated human V-ATPase regulation. Nat Commun. (2022) 13:3272. doi: 10.1038/s41467-022-30899-z
18. Wang L, Wu D, Robinson CV, Fu TM. Identification of mEAK-7 as a human V-ATPase regulator via cryo-EM data mining. Proc Natl Acad Sci. (2022) 119:e2203742119. doi: 10.1073/pnsa.2203742119
19. Riou P, Saffroy R, Comoy J, Gross-Goupil M, Thiéry JP, Emile JF, et al. Investigation in liver tissues and cell lines of the transcription of 13 genes mapping to the 16q24 region that are frequently deleted in hepatocellular carcinoma. Clin Cancer Res. (2002) 8:3178–86.
20. Ellsworth RE, Field LA, Love B, Kane JL, Hooke JA, Shriver CD. Differential gene expression in primary breast tumors associated with lymph node metastasis. Int J Breast Cancer. (2011) 2011:1–7. doi: 10.4061/2011/142763
21. Maxson ME, Grinstein S. The vacuolar-type H+-ATPase at a glance – more than a proton pump. J Cell Sci. (2014) 127:4987–93. doi: 10.1242/jcs.158550
22. Chen F, Kang R, Liu J, Tang D. The V-ATPases in cancer and cell death. Cancer Gene Ther. (2022) 29:1529–41. doi: 10.1038/s41417-022-00477-y
23. Pérez-Sayáns M, Somoza-Martín JM, Barros-Angueira F, Rey JMG, García-García A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat Rev. (2009) 35:707–13. doi: 10.1016/j.ctrv.2009.08.003
24. Collins MP, Forgac M. Regulation of V-ATPase assembly in nutrient sensing and function of V-ATPases in breast cancer metastasis. Front Physiol. (2018) 9:902. doi: 10.3389/fphys.2018.00902
25. Vasanthakumar T, Keon KA, Bueler SA, Jaskolka MC, Rubinstein JL. Coordinated conformational changes in the V1 complex during V-ATPase reversible dissociation. Nat Struct Mol Biol. (2022) 29:430–9. doi: 10.1038/s41594-022-00757-z
26. Abbas YM, Wu D, Bueler SA, Robinson CV, Rubinstein JL. Structure of V-ATPase from the mammalian brain. Science. (2020) 367:1240–6. doi: 10.1126/science/aaz2924
27. Hinton A, Sennoune SR, Bond S, Fang M, Reuveni M, Sahagian GG, et al. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J Biol Chem. (2009) 284:16400–8. doi: 10.1074/jbc.M091201200
28. Tan YZ, Abbas YM, Wu JZ, Wu D, Hesketh GG, Bueler SA, et al. CryoEM of endogenous mammalian V-ATPase interacting with the TLDc protein mEAK-7. bioRxiv. (2021). doi: 10.1101/2021.11.03.466369
29. Vinod V, Padmakrishnan CJ, Vijayan B, Gopala S. ‘How can I halt thee?’ The puzzles involved in autophagic inhibition. Pharmacol Res. (2014) 82:1–8. doi: 10.1016/j.phrs.2014.03.005
30. Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. (2021) 101:1371–426. doi: 10.1152/physrev.00026.2020
31. Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. (2009) 34:620–7. doi: 10.1016/j.tibs.2009.09.004
32. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. (2011) 10:2305–16. doi: 10.4161/cc.10.14.16586
34. Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. (2013) 203:563–74. doi: 10.1083/jcb.201306041
35. Yang H, Yu Z, Chen X, Li J, Li N, Cheng J, et al. Structural insights into TSC complex assembly and GAP activity on Rheb. Nat Commun. (2021) 12:339. doi: 10.1038/s41467-020-20522-4
36. Howell JJ, Ricoult SJH, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans. (2013) 41:906–12. doi: 10.1042/BST20130041
37. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. (2013) 339:1323–8. doi: 10.1126/science.1228792
38. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. (2011) 146:408–20. doi: 10.1016/j.cell.2011.06.034
39. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. (2013), 126:1713–9. jcs125773. doi: 10.1242/jcs.125773
40. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H + -ATPase. Science. (2011) 334:678–83. doi: 10.1126/science.1207056
41. Chung CYS, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat Chem Biol. (2019) 15:776–85. doi: 10.1038/s41589-019-0308-4
42. Ratto E, Chowdhury SR, Siefert NS, Schneider M, Wittmann M, Helm D, et al. Direct control of lysosomal catabolic activity by mTORC1 through regulation of V-ATPase assembly. Nat Commun. (2022) 13:4848. doi: 10.1038/s41467-022-32515-6
43. Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, et al. The Lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, Acting as a switch between Catabolism and Anabolism. Cell Metab. (2014) 20:526–40. doi: 10.1016/j.cmet.2014.06.014
44. Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. (2013) 493:679–83. doi: 10.1038/nature11745
45. Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. (2012) 150:1196–208. doi: 10.1016/j.cell.2012.07.032
46. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB: Self-regulation of the lysosome via mTOR and TFEB. EMBO J. (2012) 31:1095–108. doi: 10.1038/emboj.2012.32
47. Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, et al. Mass-spectrometry-based draft of the human proteome. Nature. (2014) 509:582–7. doi: 10.1038/nature13319
48. He Z, Houghton PJ, Williams TM, Shen C. Regulation of DNA duplication by the mTOR signaling pathway. Cell Cycle. (2021) 20:742–51. doi: 10.1080/15384101.2021.1897271
Keywords: mEAK-7, V-ATPase, lysosomal membrane protein, mTOR signaling, cancer
Citation: Chang I, Loo Y-L, Patel J, Nguyen JT, Kim JK and Krebsbach PH (2024) Targeting of lysosomal-bound protein mEAK-7 for cancer therapy. Front. Oncol. 14:1375498. doi: 10.3389/fonc.2024.1375498
Received: 23 January 2024; Accepted: 29 February 2024;
Published: 12 March 2024.
Edited by:
Daniel P. Bezerra, Oswaldo Cruz Foudantion (FIOCRUZ), BrazilReviewed by:
Patricia Marie Kane, Upstate Medical University, United StatesCopyright © 2024 Chang, Loo, Patel, Nguyen, Kim and Krebsbach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul H. Krebsbach, pkrebsbach@dentistry.ucla.edu