 Eduard Yakubov1*
Eduard Yakubov1* Sebastian Schmid2
Sebastian Schmid2 Alexander Hammer1,3
Alexander Hammer1,3 Daishi Chen4
Daishi Chen4 Jana Katharina Dahlmanns5Ivana Mitrovic6
Jana Katharina Dahlmanns5Ivana Mitrovic6 Luka Zurabashvili7
Luka Zurabashvili7 Nicolai Savaskan8,9
Nicolai Savaskan8,9 Hans-Herbert Steiner1
Hans-Herbert Steiner1 Marc Dahlmanns5
Marc Dahlmanns5- 1Department of Neurosurgery, Paracelsus Medical University, Nuremberg, Germany
- 2Department of Trauma, Orthopaedics, Plastic and Hand Surgery, University Hospital Augsburg, Augsburg, Germany
- 3Center for Spine and Scoliosis Therapy, Malteser Waldkrankenhaus St. Marien, Erlangen, Germany
- 4Department of Otorhinolaryngology, Shenzhen People's Hospital, Jinan University, Shenzhen, China
- 5Institute for Physiology and Pathophysiology, Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg, Erlangen, Germany
- 6Department of Cardiac Surgery, Bogenhausen Hospital, Munich, Germany
- 7Department of Neurosurgery, Inova Medical Center, Tbilisi, Georgia
- 8Department of Neurosurgery, University Medical School Hospital Universitätsklinikum Erlangen (UKER), Friedrich-Alexander Universität (FAU) Erlangen-Nürnberg, Erlangen, Germany
- 9Department of Public Health Neukölln, District Office Neukölln of Berlin Neukölln, Berlin, Germany
Human malignant brain tumors such as gliomas are devastating due to the induction of cerebral edema and neurodegeneration. A major contributor to glioma-induced neurodegeneration has been identified as glutamate. Glutamate promotes cell growth and proliferation in variety of tumor types. Intriguently, glutamate is also an excitatory neurotransmitter and evokes neuronal cell death at high concentrations. Even though glutamate signaling at the receptor and its downstream effectors has been extensively investigated at the molecular level, there has been little insight into how glutamate enters the tumor microenvironment and impacts on metabolic equilibration until recently. Surprisingly, the 12 transmembrane spanning tranporter xCT (SLC7A11) appeared to be a major player in this process, mediating glutamate secretion and ferroptosis. Also, PPARγ is associated with ferroptosis in neurodegeneration, thereby destroying neurons and causing brain swelling. Although these data are intriguing, tumor-associated edema has so far been quoted as of vasogenic origin. Hence, glutamate and PPARγ biology in the process of glioma-induced brain swelling is conceptually challenging. By inhibiting xCT transporter or AMPA receptors in vivo, brain swelling and peritumoral alterations can be mitigated. This review sheds light on the role of glutamate in brain tumors presenting the conceptual challenge that xCT disruption causes ferroptosis activation in malignant brain tumors. Thus, interfering with glutamate takes center stage in forming the basis of a metabolic equilibration approach.
Introduction
Malignant primary brain tumors account for approximately 30% of all primary brain tumors diagnosed annually in the United States (1). Gliomas are the most common of these and represent one of the leading causes of morbidity and mortality in neurological practice (2). Glioblastomas (also referred to CNS WHO grade 4 glioma) with their median survival time of less than 15 months are considered to be the most malignant brain tumor entity (3). To date, conventional treatment includes surgical resection of the bulk tumor mass, followed by radiotherapy and alkylating agent-based chemotherapy. Even with these advanced therapies developed over the last two decades, survival times have only been extended a few months, and a cure remains elusive. In addition, certain biological properties of glioma make complete surgical resection nearly impossible and radiochemotherapy less effective or ineffective in treating residual glioma cells (4–6). At the cellular level, treatment resistance can be explained by the intra- and intertumoral heterogeneity observed in glioblastomas. Based on genomic and transcriptomic analyses of bulk tumors, glioblastomas can be categorized into four molecular subtypes, namely proneural, neural, classical, and mesenchymal (7, 8). However, a follow-up study revealed that all molecular subtypes coexist within a brain tumor heterogeneously (5). The clinical prognosis remained unaffected except for individuals belonging to the proneural subtype. In glioblastomas with high proportions of alternative subtypes, patients with dominant proneural subtype had poorer survival outcomes. In addition, the existence of glioblastoma stem cells (GSC) also contributes to resistance to adjuvant therapy and promote tumor recurrence (9).
Untreated cases of glioblastoma are commonly associated with perifocal edema resulting from blood-brain barrier disruption. These events can lead to devastating neurological sequelae, such as hemiparesis or cognitive decline (10). Whether the tumor-related edema zone should be resected presents a controversial issue until now, but according to a recent study, surgical resection of the peritumoral edema zone has been found not to carry a greater risk of postoperative complications. It even delayed tumor recurrence than simply removing the contrast-enhancing tumor alone (11).
There has been an association between glioblastoma-induced edema and alterations of tumor-associated genes inside the edema region, in terms of upregulations of e.g. c-myc, ERK, or AKT, and downregulation of tumor-suppressors such as p53 (11). Additional bioinformatic analysis of ‘The Cancer Genome Atlas’ (TCGA) data revealed that occurrence of tumor-related brain edema affects inflammatory gene expression, e.g. by increasing IL-10 levels (12), which in turn was shown to promote glioma cell invasion (13, 14). As a result, the threshold for T cell activation can be raised, and their antitumor activity can be directly suppressed (15). Aside from that, HMOX1-positive myeloid cells are also capable of secreting IL-10, causing T cell dysfunction and immune evasion (16). These recent data strongly suggest that these edema-enriched genes are crucial for gliomagenesis and tumor angiogenesis. The presence of these gene alterations in the edema region is in line with the observation that this area represents the biologically active part of the glioma microenvironment (6).
Brain tissues in the peritumoral area show evidence of oxidative stress, manifested through increased production of reactive oxygen species (ROS) and decreased antioxidant-enzyme defenses such as catalase, glutathione peroxidase, and superoxide dismutase (17, 18). The oxidative stress in the glioma microenvironment is closely related to iron homeostasis, since the balanced amount of intracellular iron governs the oxidation state of phospolipids (19, 20) (Figure 1). An iron overload induces lipid peroxidation and subsequent cell death (21). In a recent study, it was found that GSC can absorb iron from the glioma microenvironment more effectively by upregulating their expression levels of ferritin and transferrin receptor (TfR) 1 (22). The proliferation of glioma cells is facilitated by such TfR overexpression-mediated oxidant accumulation, which inactivates cell cycle regulators and promotes S-phase entry (23).
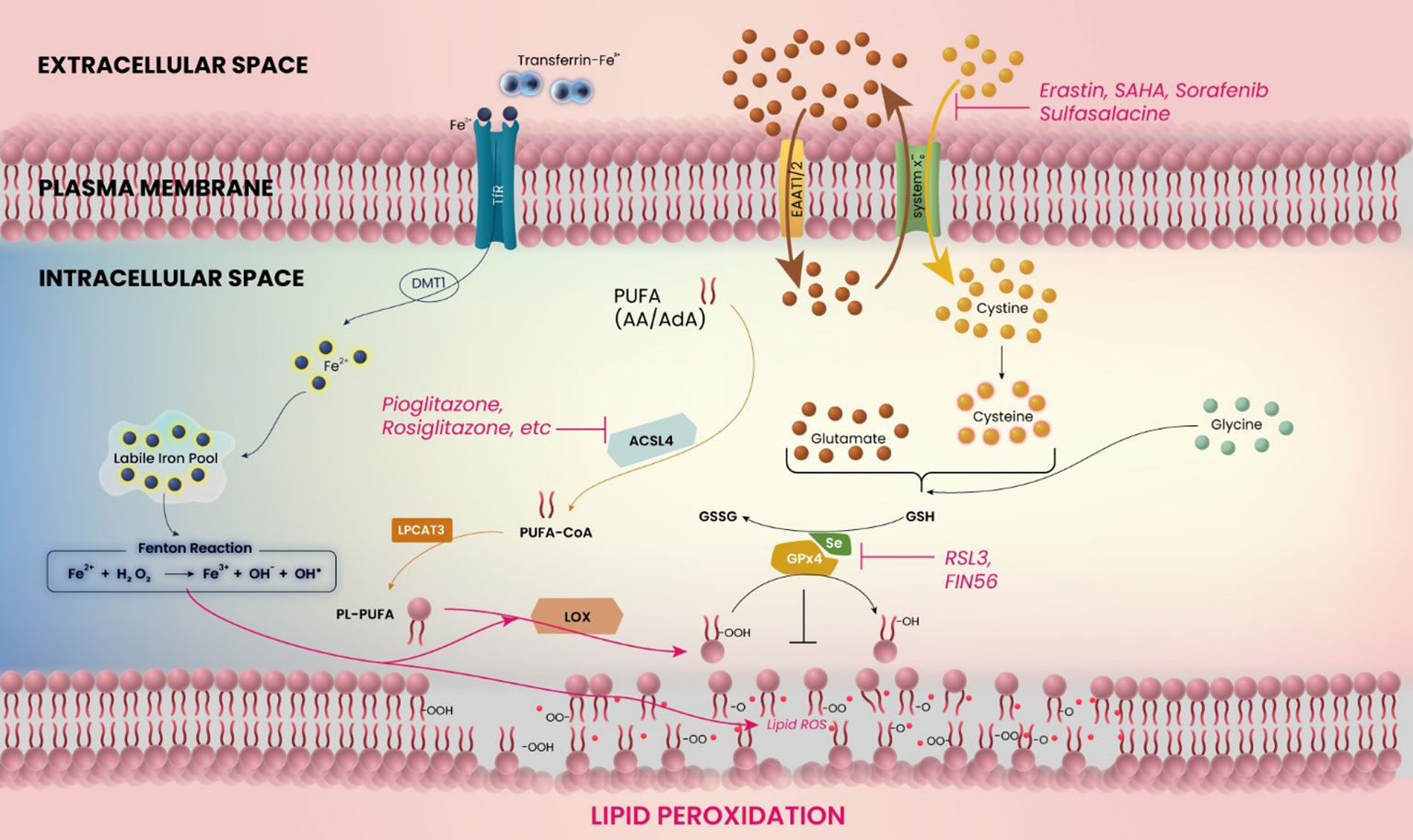
Figure 1 Schematic model for the mechanism of ferroptosis in glioblastoma. The figure shows the related molecules and pathways of ferroptosis. Ferroptosis is induced by inhibition of system xc− or glutathione peroxidase 4 (GPX4), or accumulation of iron (Fe2+) ions. The catabolic enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) must be expressed.
In addition to the direct iron-related mechanism of tumor progression, another relevant upstream mechanism for the development of edema or glutamate-induced excitotoxicity is represented by the glutamate-cystine antiporter system xc− (24, 25) (Figure 1). In glioblastoma cells, increasing cystine import through system xc− drives the production of antioxidant glutathione, whereas the inhibition of cystine import, e.g. by application of system xc− inhibitors, decreases the antioxidative capabilities of (brain) tumor cells (26). Several molecular aspects modulate these antioxidative properties, including the expression of mitoferrin-1, Nrf2, and catalase, among others (27–29). Thus, the tumor itself expresses a variety of molecules that serve to decrease the oxidative stress in its tissue, promoting growth. Interestingly, blocking system xc− allows oxidative agents to accumulate intracellularly, which leads to cell death in the form of ferroptosis (30) (Figure 1). Ferroptosis inhibits malignant brain tumors and tumor-related edema by inducing oxidative stress in tumor cells and through antagonism of the treatment resistance that is strongly displayed by malignant gliomas (31).
In recent years, different pathways have been identified that promote an understanding of the biological actions of malignant tumors and their related cerebral edema. The benefits of current treatment options are still modest, so the investigation of newly identified, tumor- and edema-specific targets could translate quickly into clinical applications, ultimately improving survival rates and quality of life for patients. In this review, we outline recent advances in the treatment of tumor-associated cerebral edema and discuss the overlap between ferroptosis induction in the tumor and the role of ferroptosis adjacent to the edema site.
Origins and relevance of tumor-induced cerebral edema
Aside from rapid growth and diffuse brain infiltration, peritumoral cerebral edema represents a feared hallmark of high-grade gliomas (HGGs, CNS WHO grades 3 and 4) (6, 32). This process involves an increase in fluid content in the surrounding parenchyma. As a result, the volume and, correspondingly, the clinical reflection of the mass-effect of the space-occupying intracranial process rises significantly (33, 34). According to their cause, there are four known types of cerebral edema, namely vasogenic, cytotoxic, interstitial, and osmotic edema (6, 32, 35–37). It cannot be denied that the vasogenic component is the major player in the progression of HGGs. In most cases, disruption of the blood-brain barrier and increased vascular permeability are responsible for the described fluid accumulation (38). This results in impaired oxygen transport, which increases the symptoms elicited by the edema (39). The disturbed fluid discharge increases intracranial pressure (40). This can be compensated in the first line by reduction of the intracerebral volume of blood and liquor (41), but soon this reserve is exhausted and the intracranial pressure rapidly rises. In the final stage, the swelling brain compresses essential brain areas as well as the venous outflow from the brain and thus results in an episode of ischemia that consequently leads to brain death. In fact, the malignancy of primary brain tumors correlates highly with the development of peritumoral edema (6, 17). The modified tumor microenvironment, known as perifocal or perilesional brain swelling, has been traditionally believed to originate primarily from vasogenic mechanisms and is also associated with the region of tumor-related angiogenesis (6, 42). Morphologically, tumor vessels show characteristic features such as altered capillary endothelial with fenestration of hyperplasia (glomeruloid tufts), irregular basal membranes and extravascular spaces, and also convoluted and sinusoidal abnormalities (43). As a consequence of this altered vascular architecture, gliomas accumulate extracellular water in the peritumoral zone while losing blood-brain barrier integrity and permeability selectivity (44). A crucial cellular component found within the tumor microenvironment is the astrocytic glial cell. Astroglial changes such as altered cytoskeletal arrangements, cytoplasmatic processes and filopodia, and altered expression of water channels (i.e. aquaporin-4) were indentified in perifocal areas (45, 46). These astroglial transformations may reflect a desperate attempt to restore the extracellular balance of fluids. In addition to astrocytes, microglia are also present in the tumor microenvironment and may influence the survival of patients (47). Recently, tumor-associated microglia/macrophages have been found to hold an important role in shaping the tumor microenvironment in mice (48). These data are particularly interesting because up to 50% of microglia/macrophages are estimated to constitute the tumor (49). Furthermore, beyond the bulk tumor mass, an analysis of the peritumoral zone has shown that activated microglia accumulate at the tumor and therby constitute a major component of the perifocal area that contributes to tumor-related edema (45). In total, the mechanisms of this perilesional edema and the cellular and molecular composition of the microenvironment are only partially understood, and further studies are required to assess how a cell-type specific intervention may elevate patients’ symptoms.
Based on the mostly vasogenic nature of peritumoral edema (44), it is relevant to identify the various angiogenic factors secreted by HGGs. The most prominent candidates associated with tumor angiogenesis are vascular endothelial growth factor (VEGF) and angiopoietin, as both can stimulate endothelial and perivascular progenitor cell growth as well as tube formation (50). Several mechanisms in which VEGF is involved, lead to an increase in the membrane permeability and thus are responsible for edema formation. Secretion of nitrogen oxide and phosphorylation of occludin are mainly driven by VEGF and result in relaxation of tight junctions (51). Another factor is the hydrostatic edema. This type of edema induces a shift in the liquor drainage due to the bulk tumor mass, creating a subsequent hydrostatic pressure gradient between the ventricle and brain parenchyma. As a result, fluid is forced into the brain tissue (52–54).
Possibilities to broaden the therapeutic toolbox to treat perifocal edema
In clinical settings, the first choice remains the administration of synthetic glucocorticoids (i. e. dexamethasone) and, in rare cases, osmotically active compounds like mannitol (55). These clinical procedures usually reduce the edema rapidly. The underlying mechanisms consist of blocking nitrogen oxide synthase (NOS), accelerating the depletion of bradykinin, and reducing expression of VEGF by tumor cells (56, 57). These procedures stabilize the tight junctions and therefore reduce the efflux of capillary fluid into the brain parenchyma. However, there is also evidence that glucocorticoids may even accelerate the process of tumor progression (58). For instance, glucocorticoids have strong glycolytic effects and enhance fructose 2,6-bisphosphate production - the most potent stimulator of phosphofructokinase 1 - as well as lactate secretion, which may counteract the further action of anticancer drugs (59). Furthermore, frequent adverse reactions become increasingly relevant in long-term treatment with glucocorticoids, and can cause immuno-suppression, reduction in quality of life, and limiting treatment modalities (60).
With this wide variety of undesirable side effects, alternative treatment targets need to be indentified and utilized. Since it has been found that glutamate influx may contribute to cell-swelling (61, 62), therapeutic targeting of glutamate homeostastis-related proteins as system xc- and EAAT1/2 may be potentially beneficial in treatment of tumor-related cerebral edema. In glioma, the decrease in EAAT2 (also known as solute carrier family 1 member 2 (SLC1A2) or glutamate transporter 1 (GLT-1)) correlates with tumor malignancy (63), making the potential involvement of EAAT in tumor-associated diseases much more relevant. For breast and colon cancers beneficial effects of EAAT2 upregulation have already been reported, and the antineoplastic effects have already been well studied (64, 65). Contrary to the system xc-, EAAT2 is poorly expressed by glioblastoma cells (66, 67). It regulates the entry of glutamate into these cells (Figure 1), ultimately decreasing extracellular clearance. Under normal conditions, EAAT2 is predominantly expressed in astrocytes, although detection is also possible in oligodendrocytes and neurons (68, 69). EAATs, in general, are membrane-bound pumps. Up to 90% of extracellular glutamate uptake can be accounted for by these transporters (70, 71), making them the single most important mechanism for a glutamate equilibrium. Upregulation of these receptors, in turn, leads to a substantial shift of extracellular to intracellular glutamate. Their expression can be modified by multiple substances and levels, including signaling pathways through PI3K and NF-κB, as well as EGF, PPARγ and pituitary adenylate cyclase-activating polypeptide (67, 71, 72).
When EAATs get activated, the sodium ion-driven uptake of glutamate also leads to the uptake of water, which may contribute to cytotoxic edema (54). However, observations from liver failure-induced cerebral edema suggest a decrease of EAAT2 accompanied by a concurrent increase in AQP4 expression (73, 74). These actions can elevate extracellular glutamate levels, until reaching neurotoxic amounts of glutamate leading to the activation of NMDA receptors associated with neurotoxicity (75, 76). Mechanistically, in terms of tumor-related edema, its formation can result from a rise in glutamate, as well as leukotrienes and vascular endothelial growth factors, which increase the permeability of the brain vessels surrounding the tumor, that leads to the influx of protein-rich influx in the brains’ white matter (77). In cases of the other forms of edema such as cellular or interstitial edema, the pathophysiological mechanism differs and may involve other consequences, e.g. increased sodium influx.
When AMPA receptor involvement in cerebral edema, tumor unrelated and evoked e.g. by traumatic brain injury, was assessed, it was found that blocking AMPA receptor activity attenuates edema (78–80). Interestingly, in studies using rodent ischemic models, AMPA-R and NMDA-R antagonistic actions have demonstrated promising results. However, despite these positive preclinical findings, clinical trials using AMPA receptor and NMDA receptor antagonists have been unsuccessful (81, 82). Nonetheless, the neuronal microenvironment near the tumor plays an important role for the tumor progression. In breast and prostate carcinoma cells, it was shown that tumor behavior is responsive to modulation of neurotransmitter activity (83), indicating the importance of chemical released by neuronal tissue. In the specific case of brain cancer, it is important to understand how malignant tumor cells and the neuronal cells in the brain communicate with each other in a reciprocal manner. Previous efforts have been made to address these questions. A soluble form of neuroligin-3, a synaptic protein, was able to activate PI3K-mTOR in high-grade glioma, and increased neuroligin-3 expression was negatively associated with patient survival (84). In addition to the dependency on such molecules, it was shown that glioma membrane depolarization drove tumor proliferation (85). In the same study, it was found that glioma provide electrical feedback to neurons in the circuit, thereby regulating their own, activity-driven growth. These data strongly illustrate how the neuronal compartment in the brain in the near vicinity of the tumor is involved in the tumor’s progression.
xCT, microenvironment and tumor-associated cytotoxicity
As another glutamate level-regulating protein, the amino acid transporter xCT (system xc-, SLC7A11) is expressed in various cancers including high-grade gliomas (HGGs). Its specific modulation of tumor microenvironment is revealed to be a hallmark of primary malignant brain tumors. In particular, this modulation influences the tumor-induced neurotoxicity and perifocal edema (32). De- and methylation processes as well as imbalances between the histone deacetylases and acetylases play a critical role in tumor development, whereas the link between these epigenetic regulatory mechanisms and the malignant glioma progression is the transporter system (86).
The inhibition of the cystine/glutamate antiporter xCT leads to a decrease in neurodegeneration, perifocal edema and prolonged survival in vivo. Furthermore, this supports the hypothesis that the formation of edema is, to some extent, influenced by the death of peritumoral cells (24) (Figure 1). Inhibition of xCT primarily disrupts its neurodegenerative and microenvironment-toxifying activity (87). A decrease in glioma cell proliferation was associated with higher concentrations of xCT inhibitors in an L-cystine-dependent manner (24, 88, 89). Glioblastoma cells derived from human patients have been shown to be susceptible to the cytotoxic effects of xCT-inhibitors (90). Additionally, cytotoxic effects of substances such as temozolomide are augmented during sulfasalazine-mediated xCT inhibition (91), and in another study it was found that the glioma-toxic effect of sulfasalazine alone was detectable at concentrations above 200 µM (89). HGGs use xCT to increase glutamate levels and manipulate neuronal glutamate signaling for their own growth advantage, leading to chemotherapeutic resistance and a toxic tumor microenvironment for neurons. Reactive oxygen species (ROS) activate transient receptor potential (TRP) channels with the result of a potentiated glutamate release via the TRP-channels. The system xc- modulates the tumor microenvironment with impact on host cells and the cancer stem cells (92).
Glioma-associated microglia/macrophages (GAMs) are an important cell population component of glioblastoma microenvironment. The increasing glutamate levels cause transcriptional changes in GAMs. These cells respond to extracellular glutamate excess in the glioblastoma microenvironment with increasing expressions of genes related to glutamate transport and metabolism such as GRIA2 (GluA2 or AMPA receptor 2), SLC1A2 (EAAT2), SLC1A3 (EAAT1), decreasing expression of xCT and increasing expression of GLUL (glutamine synthetase) (93).
Regarding the modulation of chemotherapeutic therapy of HGGs, many promising inhibitors and activators of xCT have been detected so far. The key problem of a specific modulation of xCT in gliomas is the ubiquitous expression of xCT also in vital tissue cells which makes it difficult to specifically target expression of xCT in tumor cells, especially because of its essential role in physiology of the CNS (94, 95).
The main cytotoxic tool in countering HGGs is the autophagy-inducing standard chemotherapeutic agent temozolomide. Interestingly, silencing xCT expression in human glioma cells is associated with a higher vulnerability towards temozolomide. However, gliomas with a high xCT expression are more vulnerable towards combinatorial treatment with temozolomide and erastin, a ferroptosis inducing agent (87).
The HDAC-inhibitor SAHA achieves equilibrium with the xCT transporter and is specific to malignant brain tumors, while leaving physiological xCT levels in healthy brain parenchyma unaffected. Consequently, the reduction of extracellular glutamate levels leads to a decrease in neuronal cell death and normalization of the tumor microenvironment. Reducing neurodegeneration results in less damage to the surrounding healthy brain parenchyma (94).
Activating transcription factor 4 (ATF4) is a critical oxido-metabolic regulator that contributes to the malignancy of HGGs by promoting cell proliferation, migration and tumor angiogenesis through the modification of the microenvironment in a potentially harmful way. ATF4 activation is associated with an elevation of xCT levels. The ATF4-induced proliferation is extenuated by xCT inhibition and ferroptosis inducers such as sorafenib and erastin. Moreover, erastin is able to reduce the ATF4-induced angiogenesis. ATF4 and xCT are tightly connected via a xCT-dependent configuring of the vascular architecture (31). Interestingly, ATF4 suppression comes along with an increased temozolomide susceptibility and autophagy in HGGs leading to a migratory stop after temozolomide application. ATF4 activation comes along with a xCT elevation resulting in an elevated temozolomide resistance. Thus, ATF4 can be regarded as a chemo-resistance gene in gliomas being determined by its transcriptional target xCT. Inactivation of ATF4 might be a key strategy to eliminate chemo-resistance in human gliomas (96). These findings open the door to new strategies of pharmacological interventions on tumor-associated genes by epigenetic priming (86).
Involvment of ferroptosis in brain tumor treatment and the implications for associated edema
Ferroptosis is a recently discovered mechanism for cell death, characterized by the accumulation of iron ions and lipid peroxidation during cell death (30, 97) (Figure 1). This can ultimately be caused by ROS accumulation through inhibition of glutathione (GSH) or GSH-dependent selenoprotein glutathione peroxidase 4 (GPx4) (6, 30), with the latter being expressed most abundantly in testes and brains (98). This accumulation of iron ions can then lead to a continuous cycle of lipid oxidation and further iron accumulation. Interestingly, inhibitors of apoptosis, necrosis and autophagy cannot reverse this type of cell death (30).
Blocking system xc-, which displays a strong expression in malignant tumors such as glioblastoma, induces ferroptosis in the tumor cells. As this poses the question if system xc- may be a potential target during chemotherapeutic intervention, it also of particular interest to examine whether ferroptosis is involved in the tumor-associated edema. Recently, it was found that the standard treatment for edema, dexamethasone, sensitizes cells to ferroptosis (99), which would allow to target the tumor and its edema simulatenously by potential medication regimens. In vivo treatment with the ferroptosis inducer sulfasalazine in mice with glioma also reduces the tumor-associated edema (89). In line with this finding, system xc- inhibition by RNA-mediated silencing improves tumor associated-edema in glioma (24). In contrast to this improvement, rats after subarachnoid hemorrhage developed edema that improved after the inhibition of ferroptosis (100), instead of its induction. Thus, the underlying pathology that leads to edema seems to be important for the interventions to be taken in edema, and it may be possible that tumor-related edema appear to be more reliant on the ferroptosis status at the tumor site.
Therefore, it would be valuable to assess other ferroptosis-inducing drugs regarding their ability to modify tumor-related edema. At the moment, four distinct classes of ferroptosis-inducing drugs play a major role. Most important in treatment are class 1 and class 2 inducers (95, 101). However, class 3 (GSH depletion compounds, e. g. acetaminophen) and class 4 (lipid peroxidation inducers, e. g. FINO2) also play significant roles.
Class 1 inducers, such as erastin, primarily target the aforementioned transporter system xc-, ultimately depleting the cells of cystine and glutathione. Interestingly, it has been reported, that especially some glioma cells, that were therapy-resistent to current treatments, are characterized by increased synthesis of polyunsaturated fats (102) – a dependency that can be readily exploited by GPx4 inhibitors (103).
Class 2 inducers, such as Ras-selective lethal 3 (RSL3) and ferroptosis inducer 56 (FIN56), directly target and inhibit GPx4 through/via downstream process (101). This presents another therapeutic angle, as some trials with knockout human cancer cells have shown a partial resistance to erastin, but not RSL3 (104).
However, serious side effects have been reported involving the induction of ferroptosis in cardiomyocytes (105, 106). A probable explanation for this could be the counteraction of the vital, protective, and antioxidant role that GPx4 plays in many cell types (17, 107). Therefore, therapeutic use should be exercised with caution.
As a side note, a number of compounds can protect against ferroptosis-induced tissue damage. These include thiazolidinediones (TZDs), which are a class of PPARγ agonists (108, 109), as well as LOX-inhibitors, DPP4-mediated lipid peroxidation suppression and iron chelators (97).
Ultimately, ferroptosis-inducing agents such as erastin and RSL3 could potentially serve as extension to standard treatments, especially in cancers that seem resistant to current drug regiments. Potential side effects, however, should be taken into consideration.
Role of PPARγ in brain tumors and cerebral edema
Glioma cells are represented by various cellular and molecular alterations that may contribute to their pathological effects and may also represent therapeutical targets. Amongst those altered signaling pathways, glioma cells express lower endogenous levels of peroxisome proliferator-activated receptor gamma (PPARγ) compared to healthy brain tissue (67, 110). PPARγ is a ligand-activated transcription factor that plays an important role in differentiation at a cellular level, as well as glucose and lipid homeostasis. It has been shown to inhibit cellular proliferation and angiogenesis, while promoting differentiation and inducing apoptosis through multiple pathways (111). One of the main obstacles for drugs designated for intracranial effect is the crossing of the blood-brain barrier. This can be easily overcome by PPARγ agonists, as demonstrated for pioglitazone in human glioma xenograft model (112). As shown in Table 1, a wide variety of antineoplastic efficacy could be seen with PPARγ agonists.
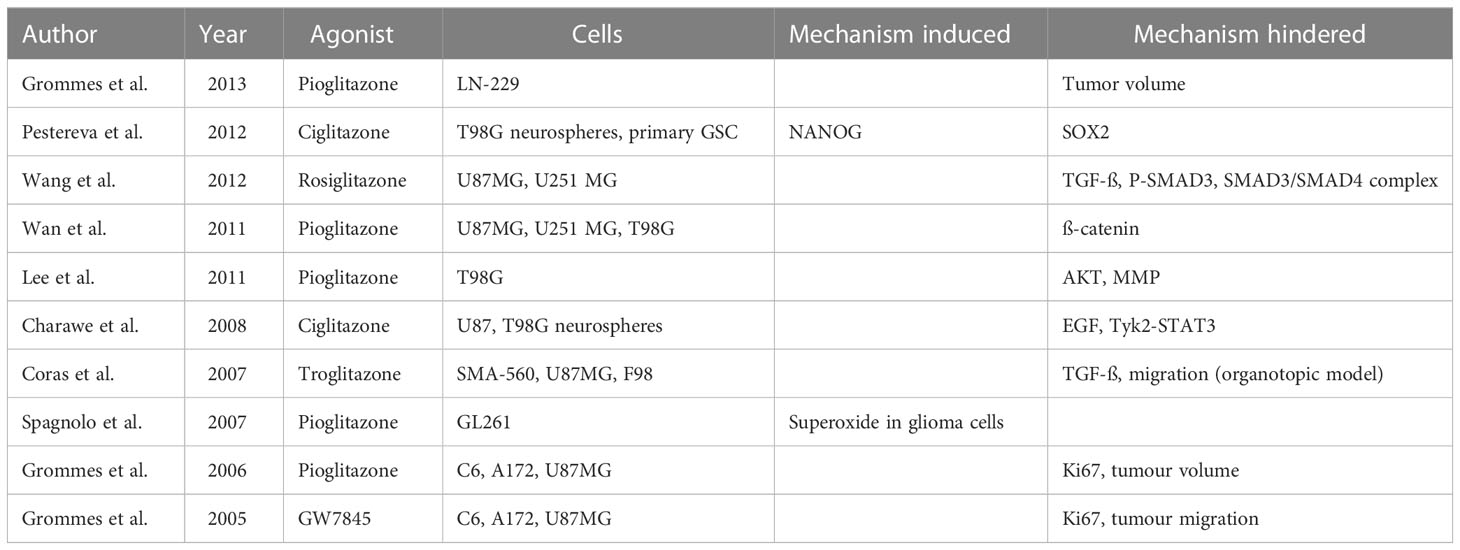
Table 1 Assessment of PPAR gamma agonists for their oncological value.
While some effects in cerebral neoplasms and edema have been reported, the exact relation is not yet fully understood. To illustrate, rosiglitazone has been shown to cause G2/M arrest and apoptosis in certain glioblastoma cell lines (113), furthermore a delay in the age of onset of seizures has been demonstrated in genetically susceptible mice, when utilizing pioglitazone (114). In a clinical trial, an extended median survival of 19 months has also been reported for diabetic patients with glioblastoma who received additional treatment with PPARγ agonists, compared to 6 months of extended survival for patients receiving the standard treatment (115). However, it should be noted that the observed result indicating longer survival for the PPARγ agonist group was not considered statistically significant due to the small sample size used in the study. Currently, classic PPARγ agonists such as pioglitazone are FDA approved primarily as oral antidiabetics but the possibility of generating tissue-specific drugs has been validated (116). At the moment, treatment with first-generation TZDs poses many obstacles, as they hold a wide variety of side effects, limiting their use.
Recent studies have linked PPARγ to ferroptosis. For instance, dendritic cells in the immune system were shown to require PPARγ to undergo ferroptosis in response to RSL3, a finding that has an impact on antitumor immunity (117). The impact of PPARγ is not limited to cancer, since also other pathologies such as diabetic retinopathy were shown to be influenced in their ferroptotic behavior by PPARγ (118). In neuronal tissue, PPARγ-mediated ferroptosis was found to be relevant in the context of traumatic brain injury (119) and in intracerebral hemorrhage (120).
In addition to PPARγ’s role in tumor tissue and ferroptosis that ocurrs within, its importance also expands to edema. For example, in the context of traumatic brain injury PPARγ-modulating substances like pioglitazone and rosiglitazone led to decreases in edema in rodent models (121). Their use is, however, associated with peripheral edema (122). Though rosiglitazone, for example, has been shown to decrease edema following a hemorrhagic event (123), further studies are required to investigate PPARγ specifically in the context of cerebral edema as a result of an adjacent glioma.
According to studies in glioma cell lines and glioma stem cells, PPARγ agonist pioglitazone enhances the functional expression of EAAT2 (124). It suggests that glioblastoma cells at the peritumoral zone may be able to improve glutamate transport, which may lead to alleviation of tumor-related edema. PPARγ agonists have additional effects associated with lipid metabolism and ferroptosis (Figure 1). Polyunsaturated fatty acids (PUFAs) play a crucial role in the process of ferroptosis. To induce lipid peroxidation, the PUFA catabolic enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) must be expressed (109). This enzyme is essential for ferroptosis and responsible for esterifying CoA into PUFAs such as arachidonic acid (AA) and adrenic acid (AdA). By forming Acyl-CoA, PUFAs are activated for fatty acid oxidation. Utilizing pharmaceutical inhibition of ACSL4 with thiazolidinedione ligands, a class of PPARγ agonists such as pioglitazone and rosiglitazone, has demonstrated that PPARγ agonists can suppress ferroptosis sensitivity (109). Based on our current understanding, there is a notable absence of comprehensive and in-depth studies exploring the molecular mechanisms underlying the regulation of PPARγ and ferroptosis in glioblastoma. Further investigations on the role of PPARγ in glioma microenvironment and ferroptosis are required.
Conclusion
In this review, we propose a fresh view on metabolic homeostasis in context of glioma-induced neurodegeneration and peritumoral edema. We discussed the glutamate signaling cascade and glutamate-EAAT-xCT axis with a special focus on ferroptosis. Brain tumors display a deranged microenvironment with metabolic changes. We discussed xCT and PPARγ as therapeutic targets addressing brain swelling and metabolic homeostasis. We provide supporting evidence for the conceptual challenge that xCT disruption causes ferroptosis activation in malignant brain tumors. We raised the potential involvement of PPARγ agonists in the context of glioblastoma and tumor-related edema.
Author contributions
EY and MD designed the concept, structure and content of the review. EY and MD wrote the manuscript with input from SS, AH, DC, JKD, IM, LZ, H-HS, and NS. EY, SS and MD prepared all tables and figures. All authors listed provided critical revisions to the article. All authors contributed to the article and agreed to submit the manuscript in its current state.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23:iii1–iii105. doi: 10.1093/neuonc/noab200
3. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
4. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, Mcguire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U.S.A. (2012) 109:3041–6. doi: 10.1073/pnas.1114033109
5. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344:1396–401. doi: 10.1126/science.1254257
6. Yakubov E, Eibl T, Hammer A, Holtmannspotter M, Savaskan N, Steiner HH. Therapeutic potential of selenium in glioblastoma. Front Neurosci (2021) 15:666679. doi: 10.3389/fnins.2021.666679
7. Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PloS One (2009) 4:e7752. doi: 10.1371/journal.pone.0007752
8. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
9. Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer (2020) 6:223–35. doi: 10.1016/j.trecan.2020.01.009
10. Esquenazi Y, Lo VP, Lee K. Critical care management of cerebral edema in brain tumors. J Intensive Care Med (2017) 32:15–24. doi: 10.1177/0885066615619618
11. Qin X, Liu R, Akter F, Qin L, Xie Q, Li Y, et al. Peri-tumoral brain edema associated with glioblastoma correlates with tumor recurrence. J Cancer (2021) 12:2073–82. doi: 10.7150/jca.53198
12. Wu Y, Peng Z, Wang H, Xiang W. Identifying the hub genes of glioma peritumoral brain edema using bioinformatical methods. Brain Sci (2022) 12:805. doi: 10.3390/brainsci12060805
13. Ghoochani A, Schwarz MA, Yakubov E, Engelhorn T, Doerfler A, Buchfelder M, et al. MIF-CD74 signaling impedes microglial M1 polarization and facilitates brain tumorigenesis. Oncogene (2016) 35:6246–61. doi: 10.1038/onc.2016.160
14. Zhang Z, Huang X, Li J, Fan H, Yang F, Zhang R, et al. Interleukin 10 promotes growth and invasion of glioma cells by up-regulating KPNA 2 in vitro. J Cancer Res Ther (2019) 15:927–32. doi: 10.4103/jcrt.JCRT_284_19
15. Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front Oncol (2015) 5:153. doi: 10.3389/fonc.2015.00153
16. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kuckelhaus J, et al. T-Cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun (2022) 13:925. doi: 10.1038/s41467-022-28523-1
17. Yakubov E. Der einfluss des zerebralen selenspiegels auf die progression maligner hirntumoren. (2019) (Dissertation. Erlangen: Friedrich-Alexander University of Erlangen-Nürnberg).
18. Obukhova L, Kopytova T, Murach E, Shchelchkova N, Kontorshchikova C, Medyanik I, et al. Glutathione and its metabolic enzymes in gliomal tumor tissue and the peritumoral zone at different degrees of anaplasia. Curr Issues Mol Biol (2022) 44:6439–49. doi: 10.3390/cimb44120439
19. Liu S, Dong L, Shi W, Zheng Z, Liu Z, Meng L, et al. Potential targets and treatments affect oxidative stress in gliomas: an overview of molecular mechanisms. Front Pharmacol (2022) 13:921070. doi: 10.3389/fphar.2022.921070
20. Zhao J, Wang Y, Tao L, Chen L. Iron transporters and ferroptosis in malignant brain tumors. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.861834
21. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ (2016) 23:369–79. doi: 10.1038/cdd.2015.158
22. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell (2015) 28:441–55. doi: 10.1016/j.ccell.2015.09.002
23. Chirasani SR, Markovic DS, Synowitz M, Eichler SA, Wisniewski P, Kaminska B, et al. Transferrin-receptor-mediated iron accumulation controls proliferation and glutamate release in glioma cells. J Mol Med (Berl) (2009) 87:153–67. doi: 10.1007/s00109-008-0414-3
24. Savaskan NE, Heckel A, Hahnen E, Engelhorn T, Doerfler A, Ganslandt O, et al. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med (2008) 14:629–32. doi: 10.1038/nm1772
25. Dahlmanns M, Dahlmanns JK, Savaskan N, Steiner HH, Yakubov E. Glial glutamate transporter-mediated plasticity: system xc–/xCT/SLC7A11 and EAAT1/2 in brain diseases. Front Biosci (Landmark Ed) (2023) 28(3):57. doi: 10.31083/j.fbl2803057
26. Jyotsana N, Ta KT, Delgiorno KE. The role of Cystine/Glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.858462
27. Flor S, Oliva CR, Ali MY, Coleman KL, Greenlee JD, Jones KA, et al. Catalase overexpression drives an aggressive phenotype in glioblastoma. Antioxidants (Basel) (2021) 10:1988. doi: 10.3390/antiox10121988
28. Awuah WA, Toufik AR, Yarlagadda R, Mikhailova T, Mehta A, Huang H, et al. Exploring the role of Nrf2 signaling in glioblastoma multiforme. Discovery Oncol (2022) 13:94. doi: 10.1007/s12672-022-00556-4
29. Ali MY, Griguer CE, Flor S, Oliva CR. Mitoferrin-1 promotes proliferation and abrogates protein oxidation via the glutathione pathway in glioblastoma. Antioxidants (Basel) (2023) 12:349. doi: 10.3390/antiox12020349
30. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
31. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene (2017) 36:5593–608. doi: 10.1038/onc.2017.146
32. Savaskan NE, Eyupoglu IY. xCT modulation in gliomas: relevance to energy metabolism and tumor microenvironment normalization. Ann Anat (2010) 192:309–13. doi: 10.1016/j.aanat.2010.07.003
33. Silbergeld DL, Rostomily RC, Alvord EC Jr. The cause of death in patients with glioblastoma is multifactorial: clinical factors and autopsy findings in 117 cases of supratentorial glioblastoma in adults. J Neurooncol (1991) 10:179–85. doi: 10.1007/BF00146880
34. Roth P, Pace A, Le Rhun E, Weller M, Ay C, Cohen-Jonathan Moyal E, et al. Neurological and vascular complications of primary and secondary brain tumours: EANO-ESMO clinical practice guidelines for prophylaxis, diagnosis, treatment and follow-up. Ann Oncol (2021) 32:171–82. Office@Eano.Eu, E.E.B.E.A., and Clinicalguidelines@Esmo.Org, E.G.C.E.A. doi: 10.1016/j.annonc.2020.11.003
35. Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS. Molecular mechanisms of brain tumor edema. Neuroscience (2004) 129:1011–20. doi: 10.1016/j.neuroscience.2004.05.044
36. Lawrence SE, Cummings EA, Gaboury I, Daneman D. Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J Pediatr (2005) 146:688–92. doi: 10.1016/j.jpeds.2004.12.041
37. Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol (2010) 28:1963–72. doi: 10.1200/JCO.2009.26.3541
38. Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol (2004) 16:593–600. doi: 10.1097/01.cco.0000142076.52721.b3
39. Seano G, Nia HT, Emblem KE, Datta M, Ren J, Krishnan S, et al. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat BioMed Eng (2019) 3:230–45. doi: 10.1038/s41551-018-0334-7
40. Liotta EM. Management of cerebral edema, brain compression, and intracranial pressure. Continuum (Minneap Minn) (2021) 27:1172–200. doi: 10.1212/CON.0000000000000988
41. Marmarou A, Takagi H, Shulman K. Biomechanics of brain edema and effects on local cerebral blood flow. Adv Neurol (1980) 28:345–58.
42. Fan Z, Sehm T, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan NE. Dexamethasone alleviates tumor-associated brain damage and angiogenesis. PloS One (2014) 9:e93264. doi: 10.1371/journal.pone.0093264
43. Stummer W. Mechanisms of tumor-related brain edema. Neurosurg Focus (2007) 22:E8. doi: 10.3171/foc.2007.22.5.9
44. Cenciarini M, Valentino M, Belia S, Sforna L, Rosa P, Ronchetti S, et al. Dexamethasone in glioblastoma multiforme therapy: mechanisms and controversies. Front Mol Neurosci (2019) 12. doi: 10.3389/fnmol.2019.00065
45. Engelhorn T, Savaskan NE, Schwarz MA, Kreutzer J, Meyer EP, Hahnen E, et al. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci (2009) 100:1856–62. doi: 10.1111/j.1349-7006.2009.01259.x
46. Lan YL, Wang X, Lou JC, Ma XC, Zhang B. The potential roles of aquaporin 4 in malignant gliomas. Oncotarget (2017) 8:32345–55. doi: 10.18632/oncotarget.16017
47. Dai X, Ye L, Li H, Dong X, Tian H, Gao P, et al. Crosstalk between microglia and neural stem cells influences the relapse of glioblastoma in GBM immunological microenvironment. Clin Immunol (2023) 251:109333. doi: 10.1016/j.clim.2023.109333
48. Chipman ME, Wang Z, Sun D, Pedraza AM, Bale TA, Parada LF. Tumor progression is independent of tumor-associated macrophages in cell lineage-based mouse models of glioblastoma. Proc Natl Acad Sci U.S.A. (2023) 120:e2222084120. doi: 10.1073/pnas.2222084120
49. Arrieta VA, Najem H, Petrosyan E, Lee-Chang C, Chen P, Sonabend AM, et al. The eclectic nature of glioma-infiltrating macrophages and microglia. Int J Mol Sci (2021) 22:13382. doi: 10.3390/ijms222413382
50. Takakura N, Kidoya H. Maturation of blood vessels by haematopoietic stem cells and progenitor cells: involvement of apelin/APJ and angiopoietin/Tie2 interactions in vessel caliber size regulation. Thromb Haemost (2009) 101:999–1005. doi: 10.1160/TH08-06-0358
51. Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol (2015) 51:966–79. doi: 10.1007/s12035-014-8752-3
53. Filippidis AS, Carozza RB, Rekate HL. Aquaporins in brain edema and neuropathological conditions. Int J Mol Sci (2016) 18:647–666. doi: 10.3390/ijms18010055
54. Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab (2016) 36:513–38. doi: 10.1177/0271678X15617172
55. Cook AM, Morgan Jones G, Hawryluk GWJ, Mailloux P, Mclaughlin D, Papangelou A, et al. Guidelines for the acute treatment of cerebral edema in neurocritical care patients. Neurocrit Care (2020) 32:647–66. doi: 10.1007/s12028-020-00959-7
56. Murayi R, Chittiboina P. Glucocorticoids in the management of peritumoral brain edema: a review of molecular mechanisms. Childs Nerv Syst (2016) 32:2293–302. doi: 10.1007/s00381-016-3240-x
57. Naro GR, Noverati N, Craig T. The role of C1-esterase inhibitors in the management of vasogenic edema in glioblastoma. Case Rep Med (2020) 2020:7981609. doi: 10.1155/2020/7981609
58. Kostopoulou ON, Mohammad AA, Bartek J Jr., Winter J, Jung M, Stragliotto G, et al. Glucocorticoids promote a glioma stem cell-like phenotype and resistance to chemotherapy in human glioblastoma primary cells: biological and prognostic significance. Int J Cancer (2018) 142:1266–76. doi: 10.1002/ijc.31132
59. Bartrons R, Simon-Molas H, Rodríguez-García A, Castaño E, Navarro-Sabaté À., Manzano A, et al. Fructose 2,6-bisphosphate in cancer cell metabolism. Front Oncol (2018) 8:331. doi: 10.3389/fonc.2018.00331
60. Oray M, Abu Samra K, Ebrahimiadib N, Meese H, Foster CS. Long-term side effects of glucocorticoids. Expert Opin Drug Saf (2016) 15:457–65. doi: 10.1517/14740338.2016.1140743
61. Bender AS, Schousboe A, Reichelt W, Norenberg MD. Ionic mechanisms in glutamate-induced astrocyte swelling: role of k+ influx. J Neurosci Res (1998) 52:307–21. doi: 10.1002/(SICI)1097-4547(19980501)52:3<307::AID-JNR7>3.0.CO;2-H
62. Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, Blomstrand F, et al. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem Int (2000) 37:317–29. doi: 10.1016/S0197-0186(00)00033-4
63. Robert SM, Sontheimer H. Glutamate transporters in the biology of malignant gliomas. Cell Mol Life Sci (2014) 71:1839–54. doi: 10.1007/s00018-013-1521-z
64. Sarraf P, Mueller E, Jones D, King FJ, Deangelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med (1998) 4:1046–52. doi: 10.1038/2030
65. Elstner E, Williamson EA, Zang C, Fritz J, Heber D, Fenner M, et al. Novel therapeutic approach: ligands for PPARgamma and retinoid receptors induce apoptosis in bcl-2-positive human breast cancer cells. Breast Cancer Res Treat (2002) 74:155–65. doi: 10.1023/A:1016114026769
66. Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res (1999) 59:4383–91.
67. Ching J, Amiridis S, Stylli SS, Bjorksten AR, Kountouri N, Zheng T, et al. The peroxisome proliferator activated receptor gamma agonist pioglitazone increases functional expression of the glutamate transporter excitatory amino acid transporter 2 (EAAT2) in human glioblastoma cells. Oncotarget (2015) 6:21301–14. doi: 10.18632/oncotarget.4019
68. Maragakis NJ, Dietrich J, Wong V, Xue H, Mayer-Proschel M, Rao MS, et al. Glutamate transporter expression and function in human glial progenitors. Glia (2004) 45:133–43. doi: 10.1002/glia.10310
69. Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int (2007) 51:333–55. doi: 10.1016/j.neuint.2007.03.012
70. Seal RP, Amara SG. Excitatory amino acid transporters: a family in flux. Annu Rev Pharmacol Toxicol (1999) 39:431–56. doi: 10.1146/annurev.pharmtox.39.1.431
71. Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia (2000) 32:1–14. doi: 10.1002/1098-1136(200010)32:1<1::AID-GLIA10>3.0.CO;2-W
72. Bunch L, Erichsen MN, Jensen AA. Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets (2009) 13:719–31. doi: 10.1517/14728220902926127
73. Desjardins P, Belanger M, Butterworth RF. Alterations in expression of genes coding for key astrocytic proteins in acute liver failure. J Neurosci Res (2001) 66:967–71. doi: 10.1002/jnr.10045
74. Thumburu KK, Dhiman RK, Vasishta RK, Chakraborti A, Butterworth RF, Beauchesne E, et al. Expression of astrocytic genes coding for proteins implicated in neural excitation and brain edema is altered after acute liver failure. J Neurochem (2014) 128:617–27. doi: 10.1111/jnc.12511
75. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch (2010) 460:525–42. doi: 10.1007/s00424-010-0809-1
76. Li V, Wang YT. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: implications for neuroprotective therapeutics for stroke. Neural Regener Res (2016) 11:1752–3. doi: 10.4103/1673-5374.194713
77. Senger DR, Van De Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, et al. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev (1993) 12:303–24. doi: 10.1007/BF00665960
78. Westergren I, Johansson BB. NBQX, an AMPA antagonist, reduces glutamate-mediated brain edema. Brain Res (1992) 573:324–6. doi: 10.1016/0006-8993(92)90781-4
79. Atsumi T, Hoshino S, Furukawa T, Kobayashi S, Asakura T, Takahashi M, et al. The glutamate AMPA receptor antagonist, YM872, attenuates regional cerebral edema and IgG immunoreactivity following experimental brain injury in rats. Acta Neurochir Suppl (2003) 86:305–7. doi: 10.1007/978-3-7091-0651-8_66
80. Chen T, Liu WB, Qian X, Xie KL, Wang YH. The AMPAR antagonist perampanel protects the neurovascular unit against traumatic injury via regulating Sirt3. CNS Neurosci Ther (2021) 27:134–44. doi: 10.1111/cns.13580
81. Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science (1990) 247:571–4. doi: 10.1126/science.2154034
82. Shen H, Chen GJ, Harvey BK, Bickford PC, Wang Y. Inosine reduces ischemic brain injury in rats. Stroke (2005) 36:654–9. doi: 10.1161/01.STR.0000155747.15679.04
83. Lang K, Drell TLT, Lindecke A, Niggemann B, Kaltschmidt C, Zaenker KS, et al. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int J Cancer (2004) 112:231–8. doi: 10.1002/ijc.20410
84. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell (2015) 161:803–16. doi: 10.1016/j.cell.2015.04.012
85. Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, et al. Electrical and synaptic integration of glioma into neural circuits. Nature (2019) 573:539–45. doi: 10.1038/s41586-019-1563-y
86. Eyupoglu IY, Savaskan NE. Epigenetics in brain tumors: HDACs take center stage. Curr Neuropharmacol (2016) 14:48–54. doi: 10.2174/1570159X13666151030162457
87. Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyupoglu IY, Savaskan NE. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget (2016) 7:74630–47. doi: 10.18632/oncotarget.11858
88. Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci (2005) 25:7101–10. doi: 10.1523/JNEUROSCI.5258-04.2005
89. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget (2016) 7:36021–33. doi: 10.18632/oncotarget.8651
90. Patel D, Kharkar PS, Gandhi NS, Kaur E, Dutt S, Nandave M. Novel analogs of sulfasalazine as system x(c)(-) antiporter inhibitors: insights from the molecular modeling studies. Drug Dev Res (2019) 80:758–77. doi: 10.1002/ddr.21557
91. Ignarro RS, Facchini G, Vieira AS, De Melo DR, Lopes-Cendes I, Castilho RF, et al. Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol Cell Biochem (2016) 418:167–78. doi: 10.1007/s11010-016-2742-x
92. Savaskan NE, Fan Z, Broggini T, Buchfelder M, Eyupoglu IY. Neurodegeneration and the brain tumor microenvironment. [corrected]. Curr Neuropharmacol (2015) 13:258–65. doi: 10.2174/1570159X13666150122224158
93. Choi J, Stradmann-Bellinghausen B, Yakubov E, Savaskan NE, Regnier-Vigouroux A. Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol Ther (2015) 16:1205–13. doi: 10.1080/15384047.2015.1056406
94. Wolf IM, Fan Z, Rauh M, Seufert S, Hore N, Buchfelder M, et al. Histone deacetylases inhibition by SAHA/Vorinostat normalizes the glioma microenvironment via xCT equilibration. Sci Rep (2014) 4:6226. doi: 10.1038/srep06226
95. Dahlmanns M, Yakubov E, Dahlmanns JK. Genetic profiles of ferroptosis in malignant brain tumors and off-target effects of ferroptosis induction. Front Oncol (2021) 11:783067. doi: 10.3389/fonc.2021.783067
96. Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget (2017) 8:51164–76. doi: 10.18632/oncotarget.17737
97. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
98. Liang H, Yoo SE, Na R, Walter CA, Richardson A, Ran Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem (2009) 284:30836–44. doi: 10.1074/jbc.M109.032839
99. Von Massenhausen A, Zamora Gonzalez N, Maremonti F, Belavgeni A, Tonnus W, Meyer C, et al. Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci Adv (2022) 8:eabl8920. doi: 10.1126/sciadv.abl8920
100. Li Y, Liu Y, Wu P, Tian Y, Liu B, Wang J, et al. Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage In vitro and In vivo via reduction of lipid peroxidation. Cell Mol Neurobiol (2021) 41:263–78. doi: 10.1007/s10571-020-00850-1
101. Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
102. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
103. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature (2017) 551:247–50. doi: 10.1038/nature24297
104. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U.S.A. (2016) 113:E4966–4975. doi: 10.1073/pnas.1603244113
105. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol (2018) 314:H659–68. doi: 10.1152/ajpheart.00452.2017
106. Tang M, Huang Z, Luo X, Liu M, Wang L, Qi Z, et al. Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radic Biol Med (2019) 134:445–57. doi: 10.1016/j.freeradbiomed.2019.01.052
107. Yakubov E, Buchfelder M, Eyupoglu IY, Savaskan NE. Selenium action in neuro-oncology. Biol Trace Elem Res (2014) 161:246–54. doi: 10.1007/s12011-014-0111-8
108. Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat acyl-CoA synthetases 1, 4, and 5. selective inhibition by triacsin c and thiazolidinediones. J Biol Chem (2001) 276:24667–73. doi: 10.1074/jbc.M010793200
109. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
110. Lee MW, Kim DS, Kim HR, Kim HJ, Yang JM, Ryu S, et al. Cell death is induced by ciglitazone, a peroxisome proliferator-activated receptor gamma (PPARgamma) agonist, independently of PPARgamma in human glioma cells. Biochem Biophys Res Commun (2012) 417:552–7. doi: 10.1016/j.bbrc.2011.12.001
111. Tatenhorst L, Hahnen E, Heneka MT. Peroxisome proliferator-activated receptors (PPARs) as potential inducers of antineoplastic effects in CNS tumors. PPAR Res (2008) 2008:204514. doi: 10.1155/2008/204514
112. Grommes C, Karlo JC, Caprariello A, Blankenship D, Dechant A, Landreth GE. The PPARgamma agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemother Pharmacol (2013) 71:929–36. doi: 10.1007/s00280-013-2084-2
113. Morosetti R, Servidei T, Mirabella M, Rutella S, Mangiola A, Maira G, et al. The PPARgamma ligands PGJ2 and rosiglitazone show a differential ability to inhibit proliferation and to induce apoptosis and differentiation of human glioblastoma cell lines. Int J Oncol (2004) 25:493–502. doi: 10.3892/ijo.25.2.493
114. Okada K, Yamashita U, Tsuji S. Ameliorative effect of pioglitazone on seizure responses in genetically epilepsy-susceptible EL mice. Brain Res (2006) 1102:175–8. doi: 10.1016/j.brainres.2006.04.108
115. Grommes C, Conway DS, Alshekhlee A, Barnholtz-Sloan JS. Inverse association of PPARgamma agonists use and high grade glioma development. J Neurooncol (2010) 100:233–9. doi: 10.1007/s11060-010-0185-x
116. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19:557–66. doi: 10.1038/nm.3159
117. Han L, Bai L, Qu C, Dai E, Liu J, Kang R, et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun (2021) 576:33–9. doi: 10.1016/j.bbrc.2021.08.082
118. Fan X, Xu M, Ren Q, Fan Y, Liu B, Chen J, et al. Downregulation of fatty acid binding protein 4 alleviates lipid peroxidation and oxidative stress in diabetic retinopathy by regulating peroxisome proliferator-activated receptor γ-mediated ferroptosis. Bioengineered (2022) 13:10540–51. doi: 10.1080/21655979.2022.2062533
119. Liang H, Tang T, Huang H, Li T, Gao C, Han Y, et al. Peroxisome proliferator-activated receptor-γ ameliorates neuronal ferroptosis after traumatic brain injury in mice by inhibiting cyclooxygenase-2. Exp Neurol (2022) 354:114100. doi: 10.1016/j.expneurol.2022.114100
120. Duan C, Jiao D, Wang H, Wu Q, Men W, Yan H, et al. Activation of the PPARγ prevents ferroptosis-induced neuronal loss in response to intracerebral hemorrhage through synergistic actions with the Nrf2. Front Pharmacol (2022) 13:869300. doi: 10.3389/fphar.2022.869300
121. Halstead MR, Geocadin RG. The medical management of cerebral edema: past, present, and future therapies. Neurotherapeutics (2019) 16:1133–48. doi: 10.1007/s13311-019-00779-4
122. Blazer-Yost BL. PPARgamma agonists: blood pressure and edema. PPAR Res (2010) 2010:785369. doi: 10.1155/2010/785369
123. Gu C, Wang Y, Li J, Chen J, Yan F, Wu C, et al. Rosiglitazone attenuates early brain injury after experimental subarachnoid hemorrhage in rats. Brain Res (2015) 1624:199–207. doi: 10.1016/j.brainres.2015.07.025
Keywords: glioblastoma, glutamate, peritumoral edema, SLC7A11, ferroptosis
Citation: Yakubov E, Schmid S, Hammer A, Chen D, Dahlmanns JK, Mitrovic I, Zurabashvili L, Savaskan N, Steiner H-H and Dahlmanns M (2023) Ferroptosis and PPAR-gamma in the limelight of brain tumors and edema. Front. Oncol. 13:1176038. doi: 10.3389/fonc.2023.1176038
Received: 28 February 2023; Accepted: 04 July 2023;
Published: 24 July 2023.
Edited by:
Hailiang Tang, Fudan University, ChinaReviewed by:
Kevin Joseph, University of Freiburg Medical Center, GermanyPeter Canoll, Columbia University, United States
Copyright © 2023 Yakubov, Schmid, Hammer, Chen, Dahlmanns, Mitrovic, Zurabashvili, Savaskan, Steiner and Dahlmanns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduard Yakubov, edmond-ariel@hotmail.de