 Tomotaka Suzuki1†
Tomotaka Suzuki1† Rui Yokomori2†
Rui Yokomori2† Takaomi Sanda1,2*
Takaomi Sanda1,2* Takaki Kikuchi1Yoshiaki Marumo1Shiori Kinoshita1Tomoko Narita1Ayako Masaki3Asahi Ito1
Takaki Kikuchi1Yoshiaki Marumo1Shiori Kinoshita1Tomoko Narita1Ayako Masaki3Asahi Ito1 Masaki Ri1Shigeru Kusumoto1Hirokazu Komatsu1Hiroshi Inagaki3Shinsuke Iida1
Masaki Ri1Shigeru Kusumoto1Hirokazu Komatsu1Hiroshi Inagaki3Shinsuke Iida1- 1Department of Hematology and Oncology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan
- 2Cancer Science Institute of Singapore, National University of Singapore, Singapore, Singapore
- 3Department of Pathology and Molecular Diagnosis, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan
We report a 69-year-old female who was a human T-cell leukemia virus type 1 carrier and exhibited a unique clinical course of developing three hematological malignancies within a short period: diffuse large B-cell lymphoma (DLBCL), chronic myelomonocytic leukemia (CMMoL), and acute myeloid leukemia (AML). Although the blast cells in AML showed typical morphological and immunophenotypical features of acute promyelocytic leukemia (APL), it did not harbor RARα gene fusion and thus initially diagnosed as APL-like leukemia (APLL). The patient developed heart failure with a fulminant clinical course and died soon after the diagnosis of APLL. Retrospective analysis with whole-genome sequencing detected a chromosomal rearrangement between KMT2A and ACTN4 gene loci both in CMMoL and APLL samples, but not in the DLBCL sample. Therefore, CMMoL and APLL were considered to be derived from the same clone with KMT2A translocation associated with prior immunochemotherapy. However, KMT2A rearrangement is rarely found in CMMoL in general and ACTN4 is also a rare partner of KMT2A translocation. Thus, this case did not follow typical transformational process of CMMoL or KMT2A-rearranged leukemia. Importantly, additional genetic alterations, including NRAS G12 mutation, were found in APLL, but not in CMMoL samples, suggesting that they might contribute to leukemic transformation. This report highlights the diverse effects of KMT2A translocation and NRAS mutation on the transformation of hematological cells as well as the importance of upfront sequencing analysis to detect genetic backgrounds for a better understanding of therapy-related leukemia.
1 Introduction
The development of secondary malignancies is a major clinical problem for cancer survivors who receive intensive chemotherapy for primary malignancies. Treatment with cytotoxic agents, such as topoisomerase II inhibitors (e.g., etoposide and doxorubicin), is known to be the causative factor of therapy-related acute myeloid leukemia (AML), which typically occurs within 1–3 years after the exposure of topoisomerase II inhibitors without passing through myelodysplastic syndrome or myeloproliferative neoplasms. Other myeloid neoplasms, including chronic myelomonocytic leukemia (CMMoL), have also been reported to be therapy-related malignancies.
Herein, we report a female patient who sequentially developed three different types of hematological malignancies (diffuse large B-cell lymphoma [DLBCL], CMMoL, and acute promyelocytic-like leukemia [APLL]) within 3 years. This patient was a human T-cell leukemia virus type 1 (HTLV-1) carrier but did not develop HTLV-1-related disorders. Retrospective analysis with whole-genome sequencing (WGS) of a series of tumor samples detected a chromosomal rearrangement involving the KMT2A gene locus that translocated to ACTN4 gene locus, a rare partner of translocation, which was detected in CMMoL and APLL samples. Although KMT2A-rearrangement has been well known to be associated with therapy-related leukemia, it is rarely found in CMMoL (1). Additionally, although the blast cells in the third malignancy showed all typical morphological and immunophenotypical features of APL, it did not harbor RARα gene fusion. Thus, CMMoL or APLL did not possess typical genetic abnormalities that can confirm the diagnosis and instead followed atypical transformation process likely induced by KMT2A-ACTN4 translocation.
2 Case description
A 69-year-old Japanese woman developed neck lymph node enlargement in November 2017. She was a HTLV-1 carrier but was otherwise healthy previously, without family histories of any hematological malignancies. See the supplemental information for more detailed clinical course and supporting findings (Figures S1-S8). Based on pathological findings of the neck tumor, she was diagnosed with DLBCL of high risk in international prognostic index, and rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHOP) therapy was administered. She ended R-CHOP therapy for up to 7 cycles owing to adverse events, including several episodes of febrile neutropenia and anemia requiring blood transfusion. She achieved a partial metabolic response based on positron emission tomography findings of residual fluorodeoxyglucose accumulation in the neck region at the end of R-CHOP therapy; the disease did not relapse thereafter.
Six months after the last R-CHOP cycle, a second hematological malignancy developed. The patient presented with a high fever that was refractory to antibiotics and lasted for 2 weeks. A peripheral blood sample showed remarkable monocytosis. Bone marrow (BM) samples showed a marked increase in monocytes, ranging from immature to mature stages. She was diagnosed with CMMoL (Table S1), and azacitidine was administered. Monocytosis gradually resolved after three cycles of azacytidine; however, the patient developed cholecystitis and discontinued azacitidine, although cholecystitis was not considered to be related to azacitidine therapy.
Eleven months after the last treatment with azacitidine, a third hematological malignancy developed. The patient presented with pancytopenia. The BM sample showed a marked increase in the number of promyelocytes. Flow cytometry revealed that these cells were positive for CD13, CD33, and MPO and negative for CD34 and HLA-DR, which is a typical immunophenotypical pattern of acute promyelocytic leukemia (APL). Thus, all-trans retinoic acid was administered. However, G-banding analysis of the BM sample showed a normal karyotype, and fluorescence in situ hybridization (FISH) analysis did not show PML-RARA fusion. Hence, we have diagnosed as APLL. However, she developed heart failure with a fulminant clinical course, which might have been caused by Takotsubo cardiomyopathy or myocardial infarction secondary to disseminated intravascular coagulation, and she died soon after the diagnosis. Overview of the clinical course of this patient, along with the possible genetic backgrounds of the series of hematological malignancies, is presented in Figure 1.
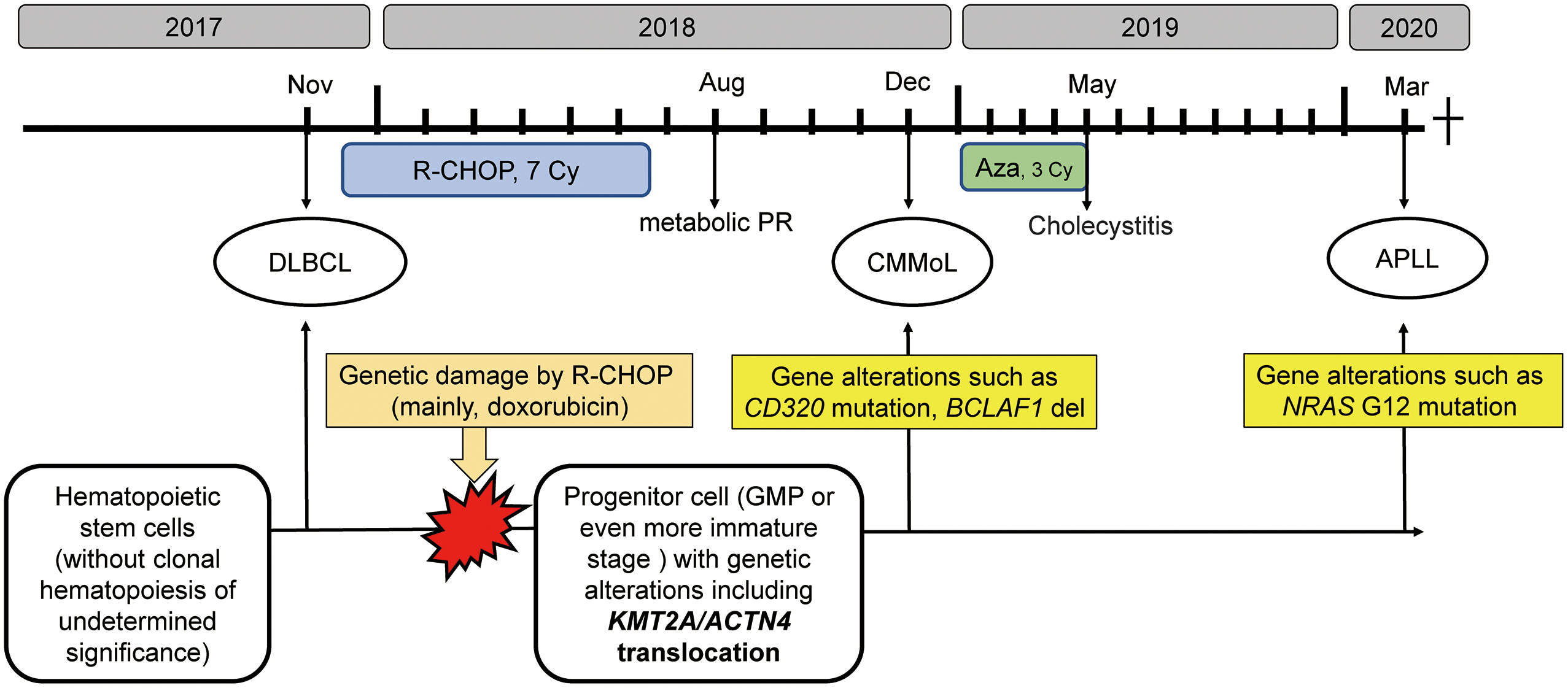
Figure 1 The clinical course along with schematic diagram of possible genetic backgrounds of the series of hematological malignancies The R-CHOP therapy might introduce genetic alterations, such as KMT2A translocation in progenitor cells, from which CMMoL and APLL were derived. In addition, acquiring genetic mutation in NRAS resulted in leukemic translation. R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone; Aza, azacitidine; DLBCL, diffuse large B-cell lymphoma; CMMoL, chronic myelomonocytic leukemia; APLL, acute promyelocytic-like leukemia; PR, partial response; GMP, granulocyte/monocyte progenitors.
3 Diagnostic assessment
Because CMMoL and APLL did not exhibit typical cytogenetic abnormalities, we retrospectively investigated the mutational profiles by WGS analysis using genomic DNAs (gDNAs) from each hematological disease (DLBCL, CMMoL, and APLL) and a somatic control (buccal swab). We detected single nucleotide variants (SNVs) and structural variants (SVs) that are present specifically in tumor samples. We also analyzed SNVs that were found in the buccal swab, but not in the reference human genome, to detect potential germline mutations. See the supplemental methods for detailed information on sample preparation, WGS, and bioinformatic analyses.
WGS analysis of the buccal swab gDNA revealed no notable germline SNVs known to be cancer predisposing factors. Integration of the HTLV-1 genome was not observed in any of the tumor gDNAs. Thus, DLBCL was considered a sporadic case. Importantly, neither SNVs nor SVs were shared between the DLBCL and CMMoL samples (Tables S2, S3), and most of the genetic mutations detected in the CMMoL sample were also found in the APLL sample (Tables 1, S2). No typical genetic mutations associated with clonal hematopoiesis of insignificant potential (CHIP) was identified in any of the samples. This result indicated that CMMoL and APLL cells were derived from the same clone. Notably, WGS analysis was able to detect a chromosomal translocation involving 11q23.3 (KMT2A) and 19q13.2 (ACTN4) loci, both in CMMoL and APLL samples (Figure 2 and Table S3); this is the only SV that was detected commonly in CMMoL and APLL samples. Independent validation by spectral karyotyping (SKY) using Carnoy fluid from CMMoL and APLL samples also supported this finding; translocation of chromosomes 11 and 19 was detected (Figures S5, S8). Hence, according to the fifth edition of the WHO Classification of Haematolymphoid Tumours (2), APLL can be re-diagnosed as “AML with KMT2A rearrangement”.
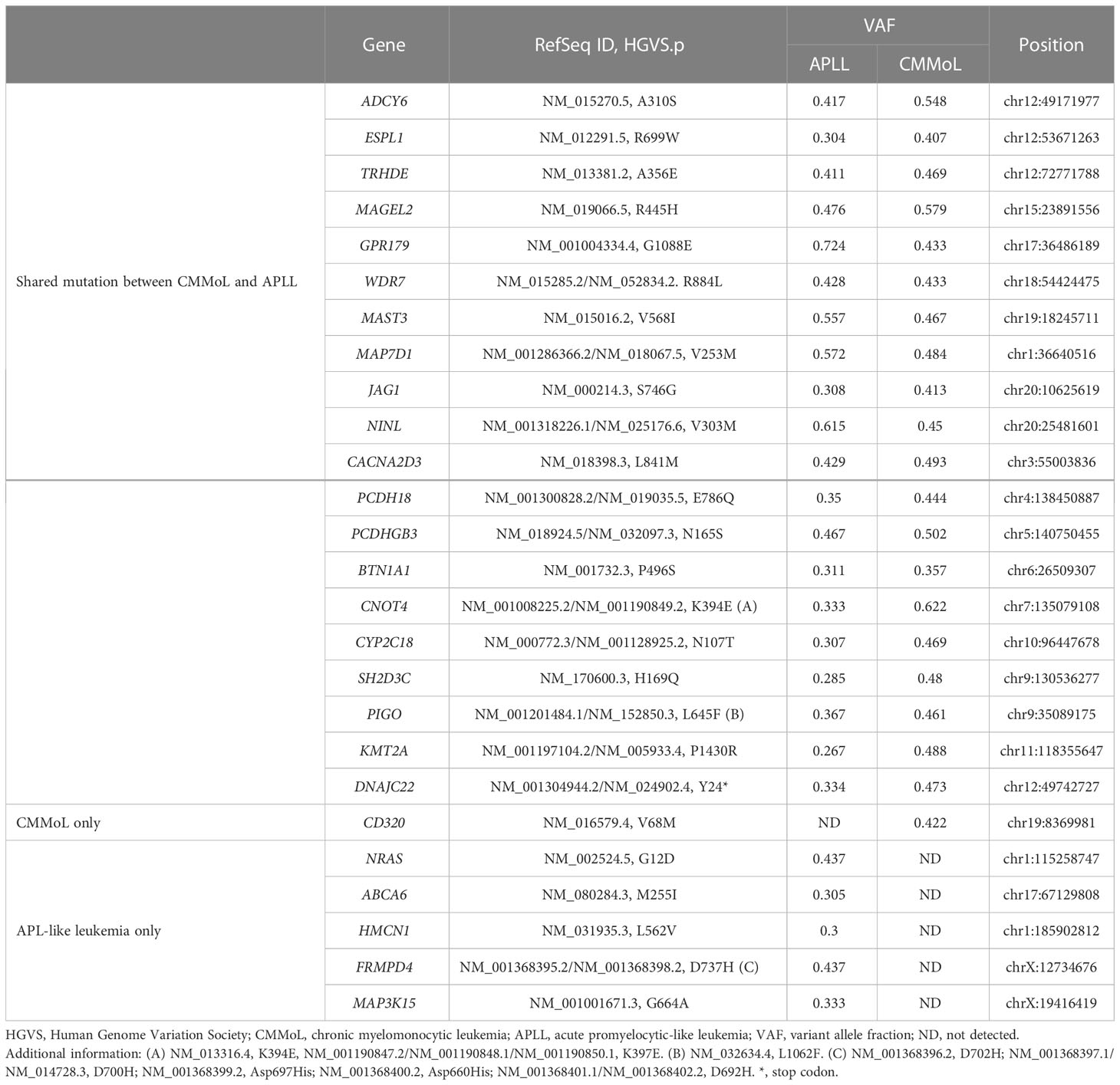
Table 1 Single nucleotide variants detected in CMMoL and/or APL-like leukemia samples.
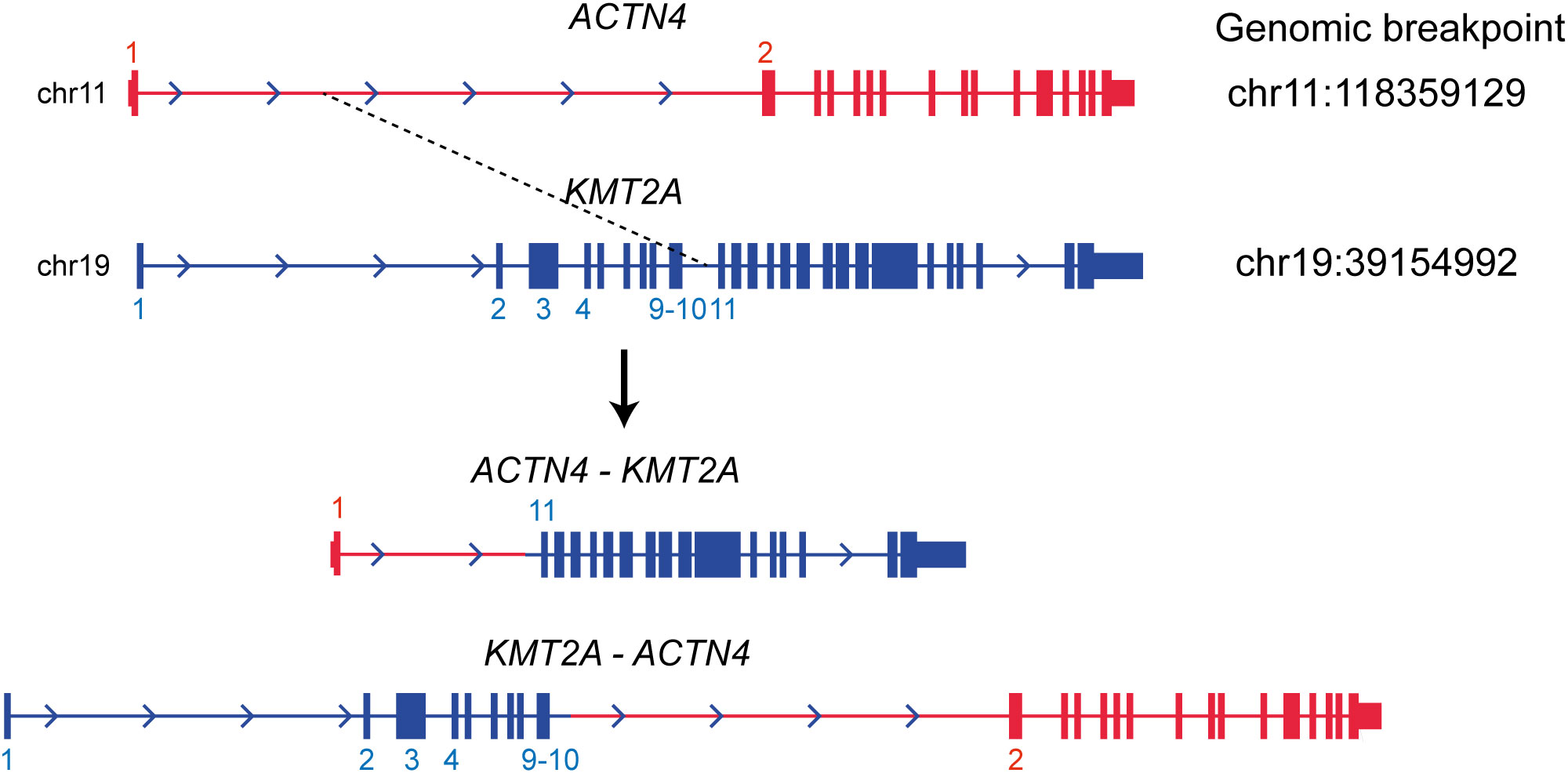
Figure 2 Translocation between KMT2A and ACTN4 genes. It was commonly detected in the CMMoL and APLL samples.
We further selected genetic alterations which were specifically found in either CMMoL or APLL sample. Interestingly, an SNV in the CD320 gene was detected only in the CMMoL sample, whereas several SNVs were detected in only the APLL sample, including NRAS, ABCA6, HMCN1, FRMPD4, and MAP3K15 (Tables 1, S2). The latter abnormalities potentially contributed to the transformation of APLL cells.
4 Discussion
The patient demonstrated a unique clinical course: a series of hematological malignancies (DLBCL, CMMoL, and APLL) occurred within a short period of time. Hence, we hypothesized that these diseases might be related to each other or that the patient might harbor predisposing cancer risks. However, no notable germline mutations or HTLV-1 integration were observed, although HTLV-1 infection might potentially affect anti-tumor immunity. In contrast, our WGS analysis detected KMT2A-ACTN4 translocation, and several other genetic alterations commonly found in CMMoL and APLL samples but not in DLBCL samples. These results suggest that CMMoL and APLL were derived from a common clone that has somatically acquired KMT2A-ACTN4 translocation after immunochemotherapy for DLBCL. APLL clones may have evolved by acquiring additional genetic abnormalities including NRAS G12 mutation.
KMT2A translocation has been reported in patients with therapy-related leukemia, and exposure to topoisomerase-II inhibitors is a risk factor (3). However, KMT2A rearrangement was initially not detected using the conventional method (i.e., karyotyping). Also, because KMT2A rearrangement is rare in CMMoL (1), a targeted approach (e.g., FISH and SKY) or WGS was not considered upfront. Hence, it was originally difficult to suspect or detect KMT2A translocation at the point of diagnosis with CMMoL. Patnaik et al. evaluated genetic and cytogenetic alterations in 492 patients with CMMoL, including 45 therapy-related CMMoLs, using targeted capture sequencing. Although CHIP-associated genes TET2 and ASXL1 were the two most frequent mutations, translocation involving KMT2A was observed in only one patient (1). It is noteworthy that various combinations of somatic mutations involving epigenetic regulation, spliceosome, and signal transduction genes have been associated with CMMoL (2). KMT2A encodes a histone lysine methyltransferase that modifies epigenetic status, which is associated with transcriptional activation (4). Hence, a potential explanation is that a single genetic abnormality, namely, KMT2A translocation, might cause dramatic epigenetic rewiring of the gene expression program that led to the formation of CMMoL.
Identification of the fusion partner of KMT2A, of which >80 genes are known, is also desirable because it provides information for prognosis and disease monitoring (2). In our case, ACTN4, which encodes a ubiquitously expressed actin-bundling protein, was translocated to the KMT2A locus. ACTN4 is a very rare partner of KMT2A translocation; only two cases, therapy-related B-acute lymphoblastic leukemia in a 69-year-old woman and therapy-related MDS in a 5-year-old man, have been reported to date (5). Further investigation is needed to clarify the significance of KMT2A/ACTN4 translocation.
During normal development, monocytes and granulocytes are derived from granulocyte/monocyte progenitors (GMP). Hence, we hypothesized that GMP or even more immature stages that harbor the KMT2A translocation might be the origin of CMMoL and APLL clones. Given that the APLL sample had many other genetic abnormalities, additional abnormalities might be needed for the leukemic transformation of CMMoL to APLL. One such candidate is the NRAS G12 mutation (6). Carr et al. reported that in 48 patients who were initially diagnosed with CMMoL but eventually progressed to AML, whole-exome sequencing analysis of paired CMMoL and AML samples revealed that NRAS was the main gene whose mutation rate increased from CMMoL to leukemic transformation (7). Notably, in AML with KMT2A rearrangements, blast cells usually demonstrate monocytic differentiation (2). However, the blast cells in our case showed typical morphological and immunophenotypical features of APL but did not harbor the RARα fusion. Interestingly, some cases of APLL have possessed KMT2A translocation without RARα translocation (Table 2) (8–11). Therefore, the combination of KMT2A translocation and additional genetic mutations, e.g., NRAS mutations, may contribute to APLL formation.
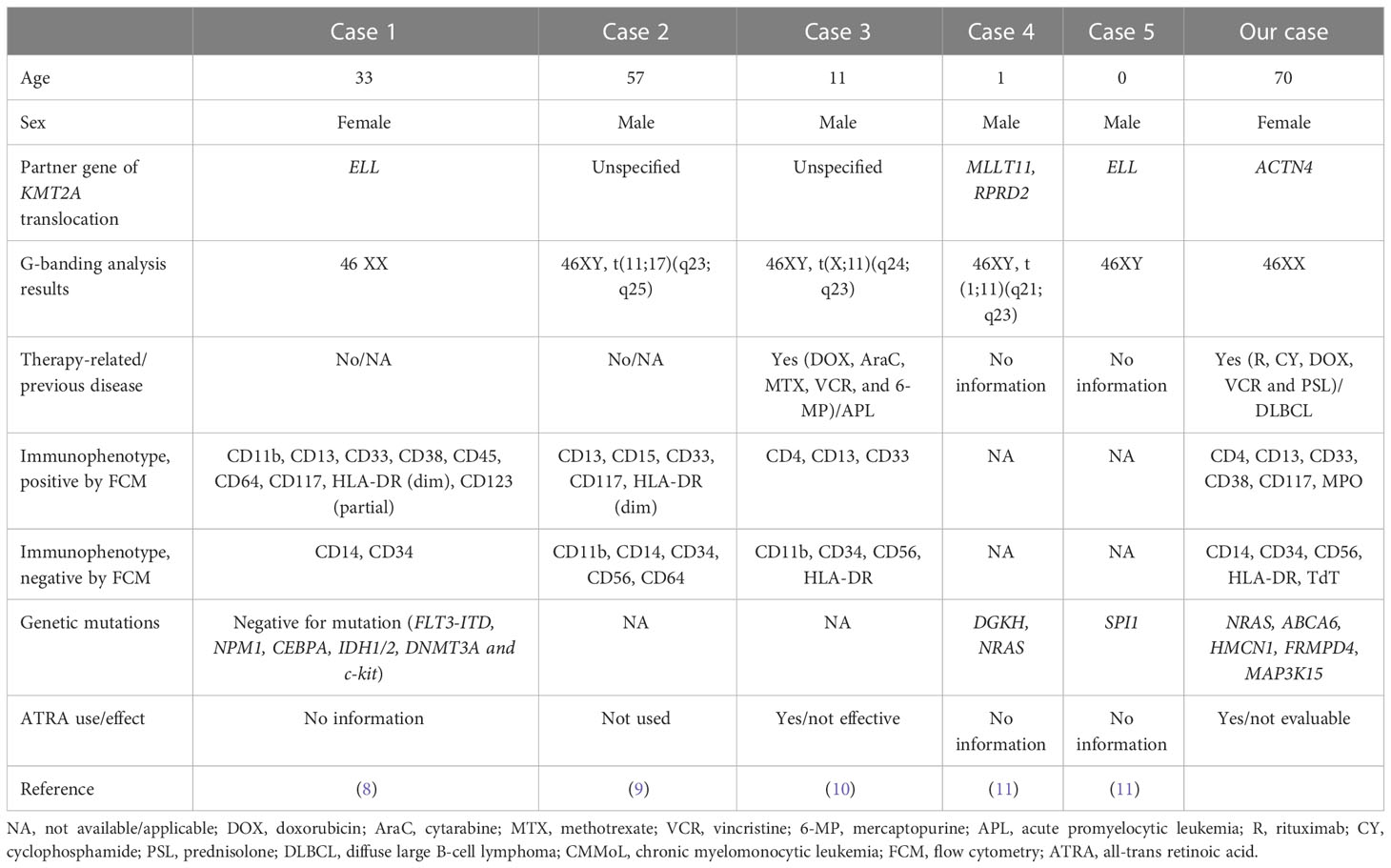
Table 2 Previous reports and our cases of acute promyelocytic-like leukemia with KMT2A translocation.
In conclusion, this report highlights the diverse effects of KMT2A translocation on the transformation of hematological cells and the potential effect of NRAS mutations on the evolution of leukemic clones. Upfront WGS analysis may be considered, especially for suspected cases of secondary hematological malignancies, to unbiasedly detect genetic alterations and understand the transformational process.
5 Patient perspective
The patient received explanation of her medical condition and agreed to receive treatments. The patient hoped to know the reason of developing multiple hematological malignancies.
Data availability statement
The datasets presented in this article are not readily available because of ethical/privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
This study was approved by the institutional review board of Nagoya City University Hospital. Written informed consent was obtained from the patient to use her samples for genetic analyses. This study was conducted in accordance with the principles of the Declaration of Helsinki. The patient provided their written informed consent to participate in this study. Written informed consent was obtained from the individual for the publication of any potentially identifiable images or data included in this article.
Author contributions
TSu, RY and TSa designed the study and wrote the manuscript. TSu extracted the genomic DNA. RY performed the bioinformatic analyses. TSu, RY, and TSa interpreted the sequencing data. TK, YM, SKi, TN, AI, MR, SKu, HK, and SI were responsible for patient medical care. AM and HI were responsible for pathological diagnosis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Japan Society for the Promotion of Science, KAKENHI (18K19960).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1116418/full#supplementary-material
References
1. Patnaik MM, Vallapureddy R, Yalniz FF, Hanson CA, Ketterling RP, Lasho TL, et al. Therapy related-chronic myelomonocytic leukemia (CMML): Molecular, cytogenetic, and clinical distinctions from de novo CMML. Am J Hematol. (2018) 93(1):65–73. doi: 10.1002/ajh.24939
2. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Myeloid and Histiocytic/Dendritic neoplasms. Leukemia (2022) 36(7):1703–19. doi: 10.1038/s41375-022-01613-1
3. Super HJ, McCabe NR, Thirman MJ, Larson RA, Le Beau MM, Pedersen-Bjergaard J, et al. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood (1993) 82(12):3705–11. doi: 10.1182/blood.V82.12.3705.3705
4. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer (2007) 7(11):823–33. doi: 10.1038/nrc2253
5. Yang JJ, Park TS, Lee ST, Seo JY, Oh SH, Cho EH, et al. Molecular characterization and clinical course of MLL-ACTN4 rearrangement in therapy-related hematologic malignancies. Haematologica (2014) 99(4):e49–51. doi: 10.3324/haematol.2013.102798
6. Wang J, Kong G, Liu Y, Du J, Chang YI, Tey SR, et al. Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood (2013) 121(26):5203–7. doi: 10.1182/blood-2012-12-475863
7. Carr RM, Vorobyev D, Lasho T, Marks DL, Tolosa EJ, Vedder A, et al. RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat Commun (2021) 12(1):2901. doi: 10.1038/s41467-021-23186-w
8. Zhang X, Huang X, Xu H, Li J, Yu W. MLL-rearrangement can resemble acute promyelocytic leukemia. Leuk Lymphoma (2019) 60(11):2841–3. doi: 10.1080/10428194.2019.1607328
9. Kang LC, Smith SV, Kaiser-Rogers K, Rao K, Dunphy CH. Two cases of acute myeloid leukemia with t(11;17) associated with varying morphology and immunophenotype: Rearrangement of the MLL gene and a region proximal to the RARalpha gene. Cancer Genet Cytogenetics (2005) 159(2):168–73. doi: 10.1016/j.cancergencyto.2004.10.009
10. Nakata Y, Mori T, Yamazaki T, Suzuki T, Okazaki T, Kurosawa Y, et al. Acute myeloid leukemia with hypergranular cytoplasm accompanied by t(X;11)(q24;q23) and rearrangement of the MLL gene. Leukemia Res (1999) 23(1):85–8. doi: 10.1016/S0145-2126(98)00131-3
Keywords: case report, chronic myelomonocytic leukemia, therapy-related myeloid neoplasm, KMT2A, NRAS
Citation: Suzuki T, Yokomori R, Sanda T, Kikuchi T, Marumo Y, Kinoshita S, Narita T, Masaki A, Ito A, Ri M, Kusumoto S, Komatsu H, Inagaki H and Iida S (2023) Case report: Genomic analysis of a therapy-related chronic myelomonocytic leukemia with KMT2A rearrangement that progressed to acute myeloid leukemia with acute promyelocytic leukemia-like features. Front. Oncol. 13:1116418. doi: 10.3389/fonc.2023.1116418
Received: 05 December 2022; Accepted: 26 January 2023;
Published: 17 February 2023.
Edited by:
Nicola Stefano Fracchiolla, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
Zimu Gong, Houston Methodist Hospital, United StatesGuangsheng He, Nanjing Medical University, China
Copyright © 2023 Suzuki, Yokomori, Sanda, Kikuchi, Marumo, Kinoshita, Narita, Masaki, Ito, Ri, Kusumoto, Komatsu, Inagaki and Iida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takaomi Sanda, tsanda@med.nagoya-cu.ac.jp; takaomi_sanda@nus.edu.sg
†These authors have contributed equally to this work