 Shilpi Chaudhary1
Shilpi Chaudhary1 Shuvadeep Ganguly1
Shuvadeep Ganguly1 Jayanth Kumar Palanichamy2
Jayanth Kumar Palanichamy2 Archna Singh2
Archna Singh2 Dibyabhaba Pradhan3
Dibyabhaba Pradhan3 Radhika Bakhshi4
Radhika Bakhshi4 Anita Chopra5
Anita Chopra5 Sameer Bakhshi1*
Sameer Bakhshi1*- 1Department of Medical Oncology, All India Institute of Medical Sciences, New Delhi, India
- 2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi, India
- 3Computational Genomics Centre, Indian Council of Medical Research (ICMR), New Delhi, India
- 4Shaheed Rajguru College of Applied Sciences for Women, University of Delhi, Delhi, India
- 5Department of Laboratory Oncology, All India Institute of Medical Sciences, New Delhi, India
Introduction: Gene expression profile of mitochondrial-related genes is not well deciphered in pediatric acute myeloid leukaemia (AML). We aimed to identify mitochondria-related differentially expressed genes (DEGs) in pediatric AML with their prognostic significance.
Methods: Children with de novo AML were included prospectively between July 2016-December 2019. Transcriptomic profiling was done for a subset of samples, stratified by mtDNA copy number. Top mitochondria-related DEGs were identified and validated by real-time PCR. A prognostic gene signature risk score was formulated using DEGs independently predictive of overall survival (OS) in multivariable analysis. Predictive ability of the risk score was estimated along with external validation in The Tumor Genome Atlas (TCGA) AML dataset.
Results: In 143 children with AML, twenty mitochondria-related DEGs were selected for validation, of which 16 were found to be significantly dysregulated. Upregulation of SDHC (p<0.001), CLIC1 (p=0.013) and downregulation of SLC25A29 (p<0.001) were independently predictive of inferior OS, and included for developing prognostic risk score. The risk score model was independently predictive of survival over and above ELN risk categorization (Harrell’s c-index: 0.675). High-risk patients (risk score above median) had significantly inferior OS (p<0.001) and event free survival (p<0.001); they were associated with poor-risk cytogenetics (p=0.021), ELN intermediate/poor risk group (p=0.016), absence of RUNX1-RUNX1T1 (p=0.027), and not attaining remission (p=0.016). On external validation, the risk score also predicted OS (p=0.019) in TCGA dataset.
Discussion: We identified and validated mitochondria-related DEGs with prognostic impact in pediatric AML and also developed a novel 3-gene based externally validated gene signature predictive of survival.
Introduction
Despite recent advancements, the survival in pediatric acute myeloid leukaemia (AML) continues to remain dismal (1). Various molecular and genetic alterations are frequently used for risk stratification, identification of therapeutic targets as well as predicting disease prognosis in AML (2). Whole genome and transcriptome sequencing have been extensively used in AML to identify potential novel molecular targets and developing prognostic gene signatures to predict survival, relapse and risk stratification (3–5). However, data on potential mitochondrial genes with impact on AML are limited.
Dysregulation of mitochondrial pathways have been implicated in pathogenesis and progression of various malignancies (6). Multiple studies have reported the role of mitochondrial DNA(mtDNA) mutations, metabolic pathways and oxidative phosphorylation, on disease biology and prognosis of AML (7, 8). We have previously reported the relationship of mutations in mtDNA regulatory region with mitochondrial gene expression, and their impact on survival in children with AML (9–11). Considering the impact of mitochondrial pathways in outcome of AML, it is important to explore tumor cell heterogeneity in AML with respect to mitochondrial transcriptome and identify potential therapeutic or prognostic molecular targets.
Recently, we have reported that high mtDNA copy number is associated with poor outcome in paediatric AML and also identified its potential regulation through PGC1A (12). In the current study, among children with AML stratified according to mtDNA copy number, we identified mitochondria-related differentially expressed genes (DEGs) through whole transcriptome sequencing. We further validated the topmost identified mitochondria-related DEGs in a cohort of paediatric AML patients and formulated a prognostic mitochondrial gene signature for predicting survival outcome. We then validated this gene signature in an external cohort of adult AML patients from The Cancer Genome Atlas (TCGA) dataset along with estimation of predictive ability of the developed prognostic gene signature.
Methodology
Study design, patient population, treatment and clinical follow up
This was a prospective observational cohort study that included consecutive de novo paediatric (≤18 years) patients with AML registered from July 2016 to December 2019 at medical oncology outpatient clinic of our cancer centre. The workflow of the study is depicted in Figure 1. Study was ethically approved by institute ethics committee and informed consent was taken from care givers and assent was obtained from all participants (≥8 years). Patients with granulocytic sarcoma without marrow involvement, acute promyelocytic leukaemia (AML M3), and mixed phenotypic acute leukaemia were excluded. Fifty age-matched patients of solid malignancies without marrow involvement were also enrolled as controls. Baseline clinical details, European LeukemiaNet (ELN) risk stratification (2), were recorded and all patients were treated uniformly as per institutional protocol (Methods S1 and S2) (13). Remission status and survival outcomes were noted.
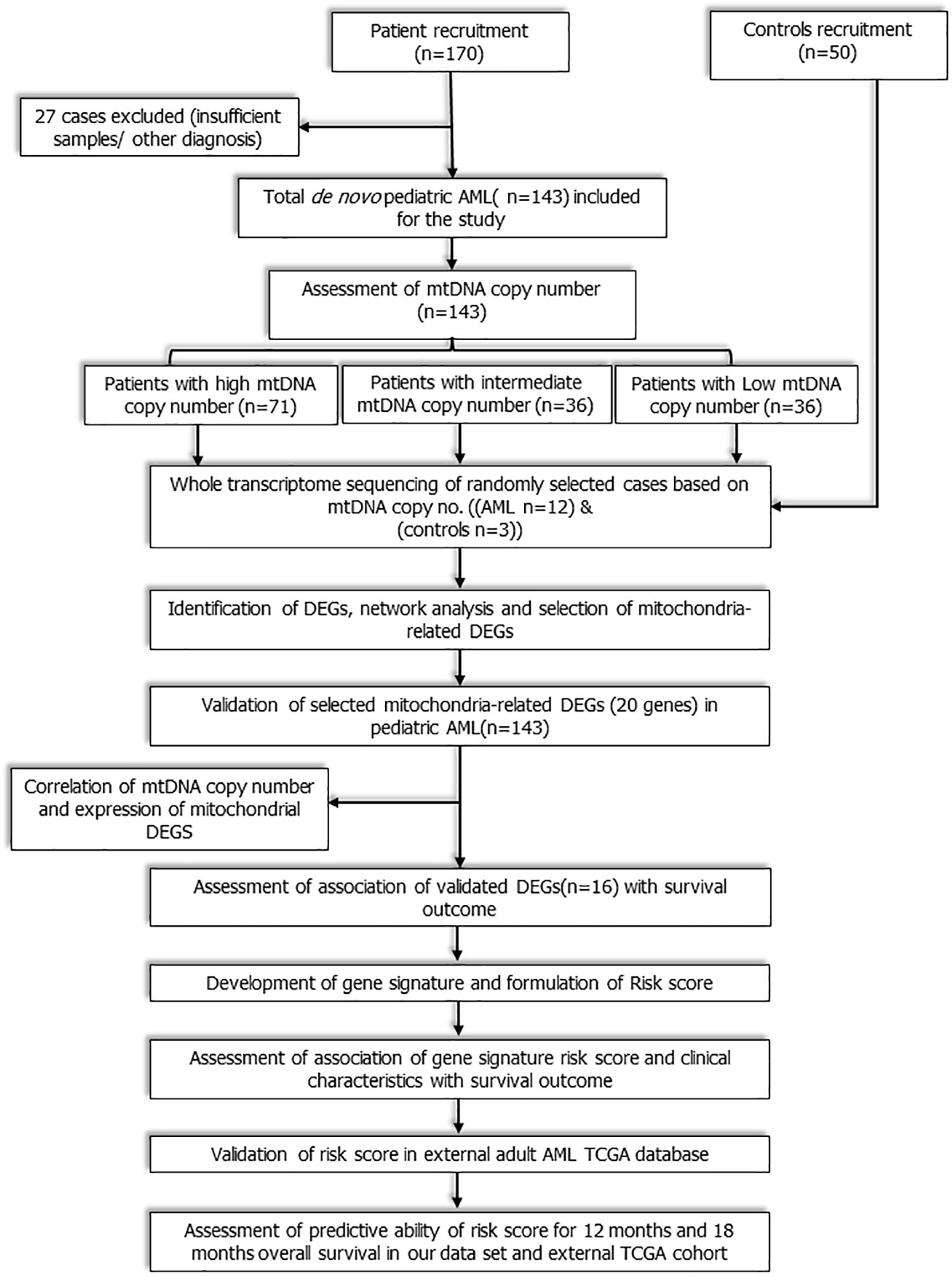
Figure 1 Workflow of the study: Study workflow showing flow of patients from enrolment to RNA sequencing, identification of mitochondria- related DEGs, development and validation of novel 3-gene based Risk score.
Whole transcriptome sequencing, identification of differentially expressed genes and selection of DEG for analysis
Bone marrow mononuclear cells were isolated by Ficoll-Hypaque (Sigma diagnostics, USA) density gradient centrifugation followed by isolation of total RNA and DNA (Methods S3). All the samples were assessed for mtDNA copy number as per previously described protocol (Methods S4) and classified into three separate groups based on relative mtDNA copy number (12). Patients were categorized into: AMLCN_H (mtDNA copy number ≥ 75th percentile), AMLCN_I (mtDNA copy number 50th to 75th percentile) and AMLCN_L (mtDNA copy number< 50th percentile) groups. A subset of samples was randomly selected from each of the three sub-groups and controls with RNA integrity score above 7 and a total of 15 samples (12 patients including 3 from AMLCN_H group, 4 from AMLCN_I group, 5 from AMLCN_L group and 3 controls) were sent for whole transcriptome profiling for the identification of DEGs compared to controls (Methods S5 and S6; Figure S1A).Absolute fold change value ≥ 2 (a ≥ two-fold change in expression, either upregulated or downregulated) and adjusted p value (q ≤ 0.05) threshold compared to controls was considered as differentially expressed genes (DEGs). The sequencing raw data was submitted to NCBI SRA (Sequence Read Archive) and available at PRJNA778747.
Selection and validation of mitochondria-related DEGs
Out of all identified DEGs from transcriptome sequencing, mitochondria-related genes were filtered using Cytoscape compartment mitochondrion score (0 being minimum and 5 being highest) (14). DEGs with topmost mitochondrial compartment score were selected for validation in a cohort of paediatric patients with AML. Along with this, Hub genes as well as maximum interactive genes were identified using CytoHubba and molecular complex detection (MCODE) clustering algorithm respectively (15, 16). The genes of MCODE cluster 1 and Hub genes were assessed for their mitochondrial localization as above and genes in each group with highest mitochondrial compartment score were selected for validation (Methods S7). Based on these selection strategies, a total of 20 mitochondria-related DEGs were selected for validation.
Real time PCR was performed to validate the selected mitochondria-related genes using specific primers (Table S1) and the gene expressions were quantified per previously described protocol (12).
Comparison of validated mitochondria-related DEGs in TCGA data set
For external validation of mitochondrial related DEGs, the RNA-sequencing data (Illumina HiSeq 2000) of TCGA adult AML(LAML) dataset was chosen, which is one of the largest datasets of transcriptomic profile in AML with recorded clinical outcome(https://www.cbioportal.org/study/summary?id=laml_tcga). The adult dataset was specifically chosen to see the impact of prognostic impact of the validated mitochondria-related age group in a different age group as well. The expression of validated DEGs was compared with LAML data set using online available GEPIA2 (Gene Expression Profiling Interactive Analysis) web server (http://gepia2.cancer-pku.cn/#index) (17).
Statistical methods
Prognostic impact of mitochondria-related DEGs and development of mitochondrial gene signature
Statistical analysis was carried out in SPSS (v23, IBM, NY, USA). Descriptive statistics were used to summarize baseline characteristics. Gene expression was reported as median values with interquartile ranges. Gene expression values and clinical continuous variables with non-parametric distribution were compared by Mann Whitney test. Clinical categorical variables were compared by Chi-square test/Fisher’s exact test as applicable. Alpha error was adjusted for multiple comparisons by Bonferroni correction. Kaplan Meier method was used to analyse time to event outcomes. Duration from enrolment to relapse or death due to any cause was considered as event free survival (EFS). Time from enrolment to death due to any cause was defined as overall survival (OS). Survival data was censored till 31st Dec 2020. The follow-up estimation was done by reverse Kaplan Meier method.
Prognostic impact of all validated DEGs on OS of the whole validation cohort was performed by multivariable Cox regression analysis in a forward stepwise manner based on log likelihood change. Validated DEGs with significant (p<0.05) predictive impact on OS in multivariable analysis were included for the prognostic gene signature model. The proportional hazard assumption was assessed by Schoenfeld global test. Internal validation of the multivariable prognostic model was carried out by bootstrapping method (10000 resampling) and genes that did not satisfy bootstrapping validation were excluded. A prognostic risk score was generated using cox regression coefficient Beta (β) values of included genes, of the final multivariable model as below:
The area under the time-dependent receiver operating characteristic (ROC) curve (Timed AUC) for 12-months and 18-months survival was estimated and Harrel’s C-index of the prognostic model was calculated using the R package “survminor” in R (version 4.0.3). Patients were classified into two groups based on their risk score above (High-risk) and below (Low-risk) the median. The survival outcomes of the patients were compared between high-risk score vs low-risk score patients using Kaplan Meier analysis to evaluate the prognostic significance of the gene signature model.
Impact of clinical features and independent prognostic value of the gene signature
The role of demographic and clinical features, including gender, age, haemoglobin, hyperleukocytosis (≥50000/µl), platelet count, presence of chloroma and ELN risk stratification (2)on survival outcome was analysed using the Cox regression. Factors with p<0.1 in univariable analysis were included for multivariable Cox regression in a forward stepwise manner using log likelihood change. Clinico-demographic factors which were significant in multivariable analysis were included in a multivariable Cox regression model along with gene signature risk score to explore the independent predictive value of gene signature. The timed AUC using 12-months survival and 18-months survival as the outcome and Harrel’s C-index of the clinical prognostic model and combined clinical and gene signature prognostic model were compared for identifying the additional prognostic benefits of gene signature over clinical parameters. The impact of mtDNA copy number on survival outcome was also analysed similarly.
External validation of mitochondrial prognostic gene signature in TCGA dataset
The prognostic impact of our gene signature risk score on OS was done in TCGA LAML (n=179) dataset by Cox regression analysis. Patients were similarly sub-grouped into high-risk and low-risk category based on median value of the gene signature; the survival outcomes of the patients were compared between high-risk score vs low-risk score patients using Kaplan Meier analysis and timed AUC of 12-month and 18-month survival was evaluated. Based on available karyotyping data, patients of the TCGA dataset were grouped into poor-risk karyotype and others (including good and intermediate-risk karyotype). The association of risk score with clinical features such as age, sex, and karyotype were also evaluated in TCGA dataset. The karyotype category and mitochondrial gene risk score were assessed for their impact on OS by a multivariable Cox regression model to explore the independent predictive value of the gene signature in the external cohort as well.
Results
Patients’ recruitment and baseline clinical features
Total 170 patients were enrolled, out of which 27 patients (5 patients were AML M3, 4 had granulocytic sarcoma without marrow involvement, and 18 patients had insufficient samples) were excluded. The baseline demographic and clinical characteristics of final 143 patients are summarized in Table S2. Median age was 10 years (range: 0.8-18 years) and 50% of the patients were classified as ELN good risk. Total 104 patients (72.7%) achieved complete remission (CR) after induction therapy. At median follow-up of 36 months (32.67-39.33 months), the median OS was 21.93 months (13.54–30.31months). The clinical characteristics of the TCGA LAML dataset are summarized in Table S3.
Identification of DEGs in paediatric AML based on mtDNA copy number
We identified 898, 769, and 953 significantly dysregulated transcripts in AMLCN_H, AMLCN_I and AMLCN_L groups respectively by whole transcriptome sequencing as represented in volcano plots (Figures S1B–D). Majority of genes were found significantly downregulated in all three groups whereas the number of dysregulated genes were higher in AMLCN_H group compared to other two groups. A total of 351 DEGs (59 upregulated and 292 downregulated) were identified in AMLCN_H. Similarly, AMLCN_I and AMLCN_L groups had 290 (66 upregulated and 224 downregulated) and 332 (47 upregulated and 285 downregulated) DEGs respectively as compared to controls.
Identification of mitochondria-related DEGs, hub genes and selection of genes for validation
Out of all DEGs, 78, 58, and 71 mitochondria-related DEGs were identified in AMLCN_H, AMLCN_I and AMLCN_L groups respectively. Among them, 35 genes were common in all three subgroups, whereas 18, 12 and 14 mitochondria-related DEGs were exclusively present in AMLCN_H, AMLCN_I and AMLCN_L groups respectively (Figure S1E). In AMLCN_H, AMLCN_I and AMLCN_L groups, we identified 17, 18 and 17 hub genes respectively using CytoHubba analysis, of which eight were common among all subgroups (Table S4, Figures S1F–H). Furthermore, using MCODE analysis, clusters with maximum scores were generated and seed gene was determined in the three groups (Figures S1I–K). MMP9 was identified as seed node with maximum MCODE score in both AMLCN_H and AMLCN_I group (Table S5). Based on the mitochondrial compartment score, CytoHubba and MCODE analyses, a total of 20 DEGs were selected for further validation (Table S6). The expression pattern of these selected DEGs in RNA sequencing data were represented in the heatmaps (Figures 2A, C).
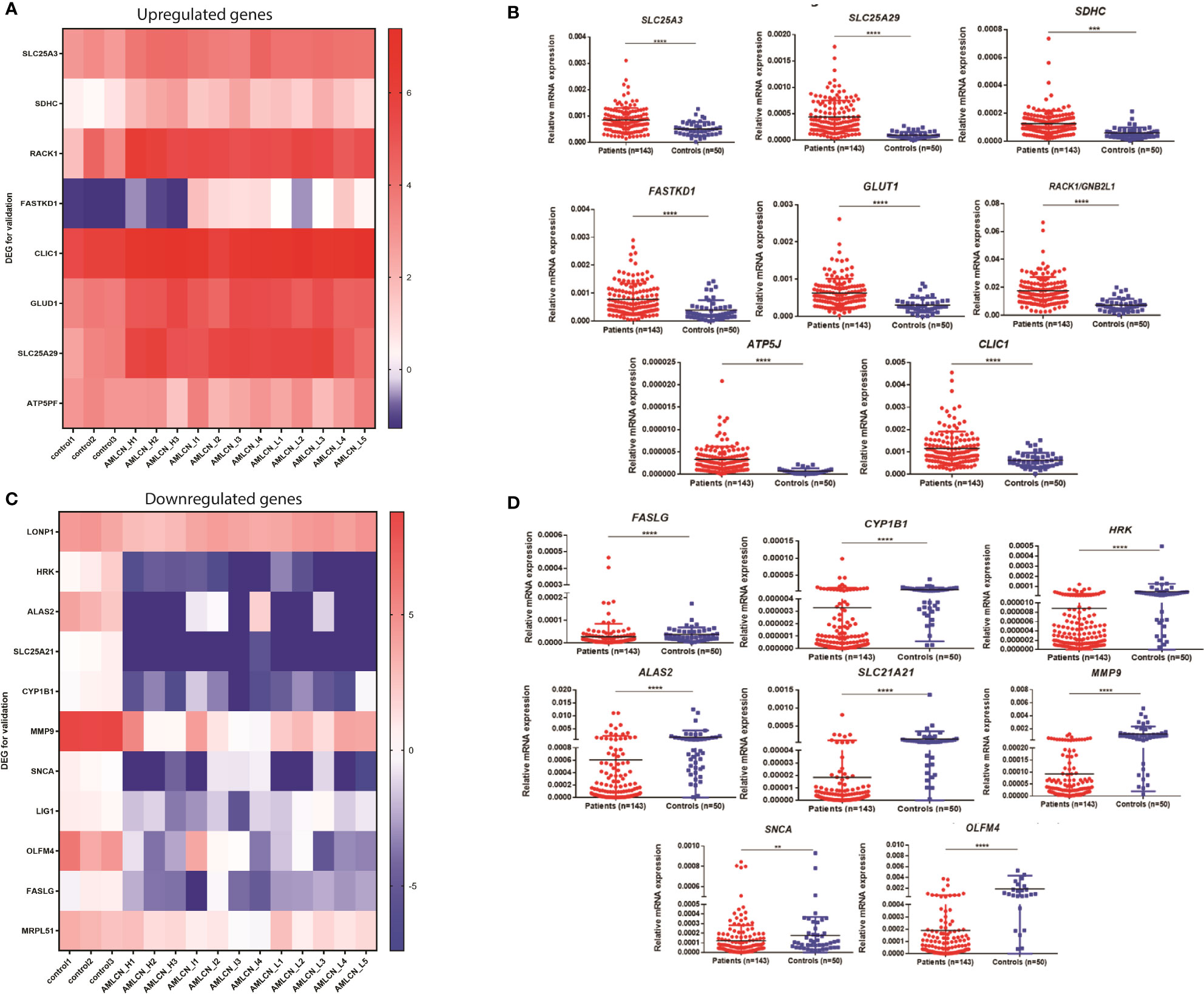
Figure 2 Expression of genes selected for the validation in pediatric AML patients. (A) Heatmap showing expression pattern of upregulated genes selected for validation from RNA sequencing data of pediatric AML patients and controls; (B) Validation of selected upregulated differentially expressed genes (DEGs) in patients as compared to controls. SLC25A3, SLC25A29, SDHC, FASTKD1, GLUD1, RACK1, ATP5J and CLIC1 were significantly upregulated in pediatric AML patients (n=143) compared to controls (n=50). *: P<0.05; **: P< 0.01; ***: P<0.001; ****: P<0.0001; (C) Heatmap showing expression pattern of downregulated genes selected for validation from RNA sequencing data of pediatric AML patients and controls; (D) Validation of selected downregulated differentially expressed genes (DEGs) in patients as compared to controls. FASLG, CYP1B1, HRK, ALAS2, SLC25A21, MMP9, SNCA and OLFM4 were significantly downregulated in pediatric AML patients (n=143) compared to controls (n=50) *: P<0.05; **: P< 0.01; ***: P<0.001; ****: P<0.0001.
Validation of selected DEGs, comparison with TCGA database
In the validation cohort of 143 AML patients, the expression of SLC25A3, SDHC, RACK1/GNB2L1, FASTKD1, ATP5J, CLIC1, GLUD1, and SLC25A29 were found to be significantly upregulated (Figure 2B, Table 1) while FASLG, HRK, ALAS2, SLC25A21, CYP1B1, SNCA, MMP9, and OLFM4 were significantly downregulated (Figure 2D, Table 1) compared to controls. Two selected genes, LIG1 and MRPL51 did not show significant dysregulation while LONP1 had a reverse expression in the validation compared to transcriptomic expression profile. Upon comparison with TCGA dataset of adult AML patients, similar dysregulation was observed for ALAS2, SLC25A21 and SLC25A29 genes while a reverse expression pattern was observed for ATP5J and CLIC1 genes; none of the other genes showed significant dysregulation in the TCGA dataset (Table 1).
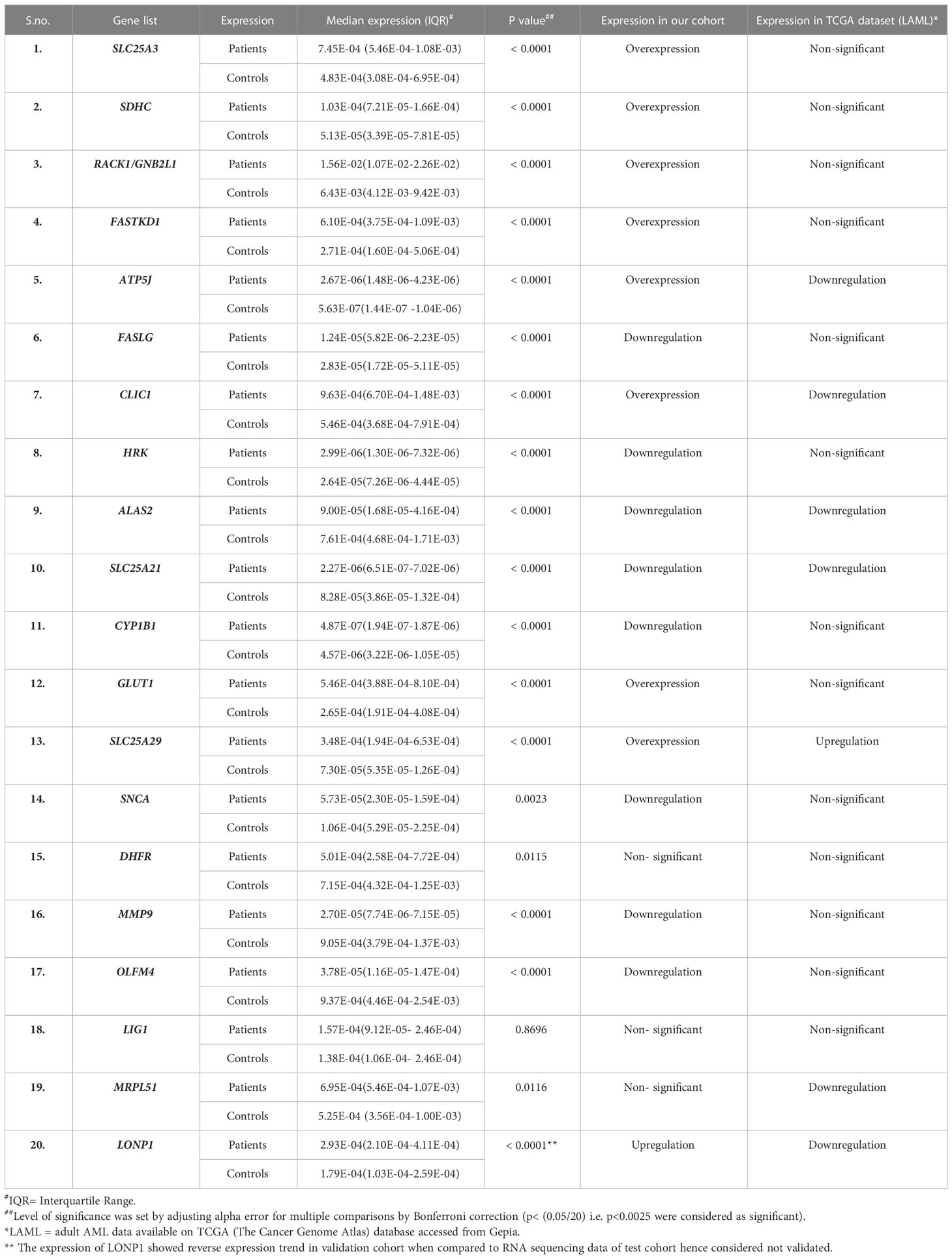
Table 1 Median expression of validated genes in patients (n=143) compared to controls(n=50) and their comparison with TCGA LAML dataset(n=179).
Mitochondria-related DEGs and mtDNA copy number
On univariable analysis, increased mtDNA copy number was significantly associated with poor event free survival (HR= 2.14; 95%CI (1.39-3.29); p=0.001) and overall survival (HR= 2.77; 95% CI (1.70-4.59); p<0.001) (Figures S2A, B). The timed AUC of mtDNA copy number for predicting 12 months and 18 months survival was 0.66 and 0.68 respectively (Figures S2C, D). In patients with increased mtDNA copy number, expression of SLC25A3, SDHC, RACK1/GNB2L1 and FASTKD1, were significantly higher compared to those with low mtDNA copy number (Figures S2E–H). Exclusive elevated expression of these 4 genes were also observed in transcriptome of samples with high/intermediate mtDNA copy number(AMLCN_H/AMLCN_I) compared to low mtDNA copy number (AMLCN_L). On correlation analysis, these 4 genes along with 2 other genes CLIC1 and ATP5J showed significant positive correlation with mtDNA copy number (Table S7).
Predictive ability of expression of validated DEGs on survival outcome and establishment of the prognostic gene signature
On multivariable analysis, upregulated expression of 2 genes, SDHC (HR 1.29; 95% CI (1.14-1.41); p<0.001) andCLIC1(HR 1.20; 95% CI (1.04-1.38); p=0.013), and downregulation of SLC25A29(HR 0.88; 95% CI (0.83-0.93); p<0.001) were found to be independently predictive of worse OS (Table 2) and they were included for the development of a prognostic gene signature model. All these 3 genes (SDHC, CLIC1, SLC25A29) satisfied internal validation by bootstrapping (Table S8), and were finally selected for prognostic model building. Beta coefficient of each of the variables were used for calculation of risk score as follows:
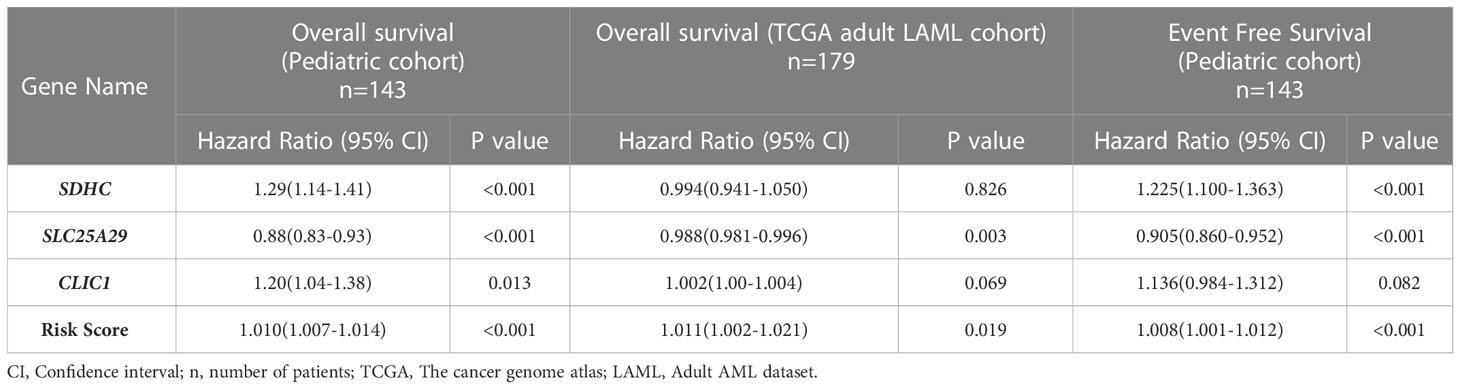
Table 2 Impact of expression of individual genes and overall risk score on overall survival and event free survival of the test cohort (pediatric cohort) and overall survival in validation cohort (TCGA adult LAML cohort).
The formula was used to calculate risk score of all the patients. Risk score median value (10.382) was taken as the cut-off for subgrouping patients into high-risk and low risk group. Patients with high-risk scores (≥10.382) had inferior OS (HR 1.010; 95% CI (1.007-1.014): p<0.001) compared to those with low-risk score (<10.382) (Figure 3A). Harrel’s C-index of the prognostic model was 0.675. The timed AUC of the risk score for 12 months and 18 months survival was 0.747 and 0.736 respectively (Figures 3B, C).
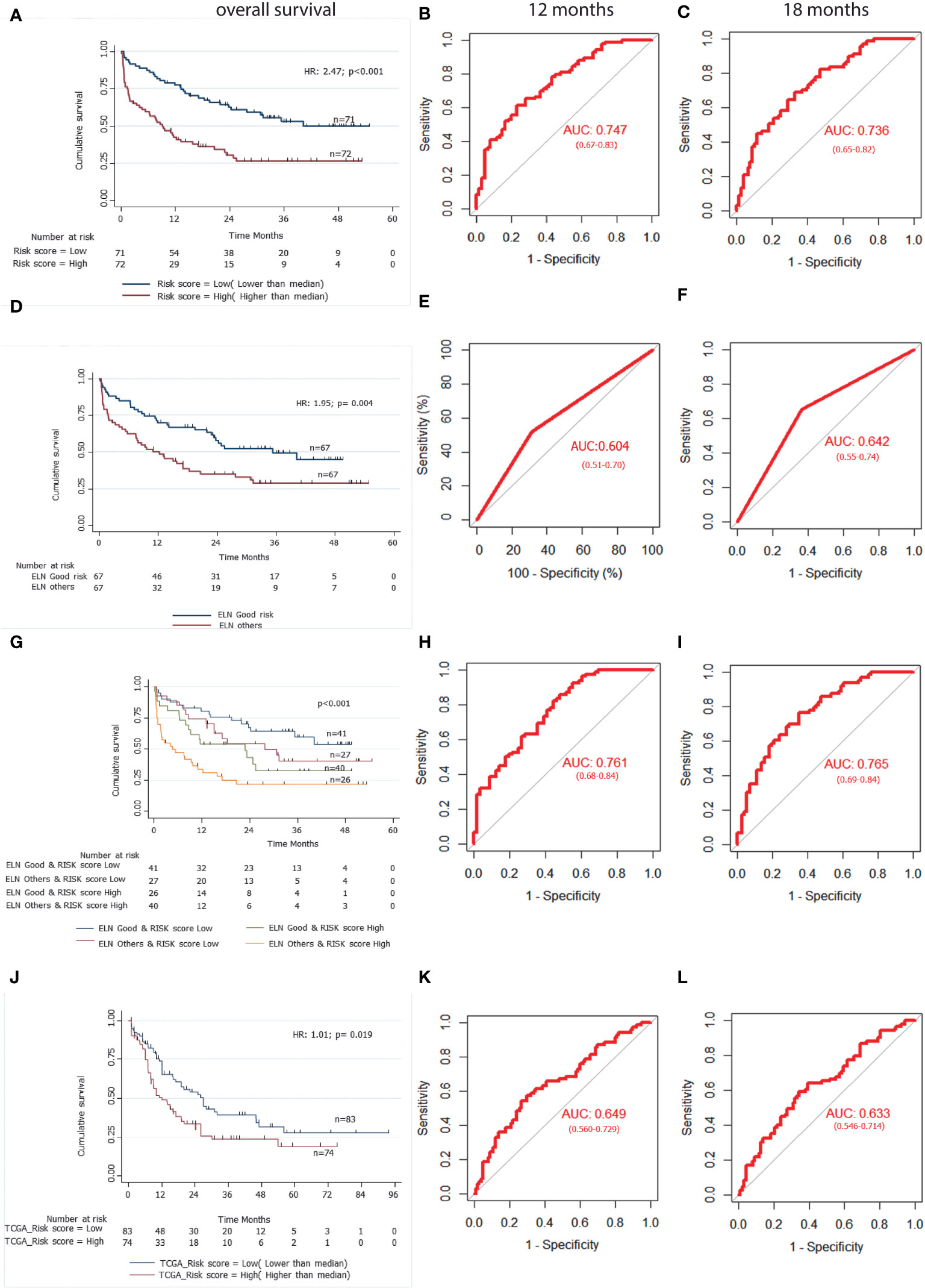
Figure 3 A 3-gene based gene signature stratifies survival in pediatric and adult AML patients along with clinically established European LeukemiaNet (ELN) risk categories. (A) Kaplan Meier estimates of overall survival in pediatric AML patient’s subgroup into high Risk-score and low Risk-score. (B) and (C) AUC curves quantify the ability of our 3-gene based risk score to predict outcome in individual patients (specificity and sensitivity) within the first 12 months(B) and 18 months (C) of treatment initiation respectively. (D) Kaplan Meier estimates of overall survival in pediatric AML patient’s subgroup into ELN good risk and ELN intermediate or poor risk categories. (E) and (F) AUC curves quantify the ability of ELN risk categories to predict outcome in individual patients (specificity and sensitivity) within the first 12 months (E) and 18 months (F) of treatment initiation respectively. (G) Kaplan Meier estimates of overall survival in pediatric AML patient’s subgroup by combining ELN risk categories with our 3 gene-based risk score. (H) and (I) AUC curves quantify the ability of combined model of ELN risk categories and our 3 gene-based risk score to predict outcome in individual patients (specificity and sensitivity) within the first 12 months (H) and 18 months(I) of treatment initiation respectively. (J) Kaplan Meier estimates of overall survival in external adult The Cancer Genome Atlas (TCGA) AML patient’s subgroup into high Risk-score and low Risk-score using our 3 gene-based gene signature model. (K) and (L) AUC curves quantify the ability of our 3-gene based risk score to predict outcome in individual patients of TCGA adult AML datasets (specificity and sensitivity) within the first 12 months (K) and 18 months (L) of treatment initiation respectively. AUC = 1.0 would denote perfect prediction, and AUC = 0.5 would denote no predictive ability.
Association of gene signature-based risk score with event free survival
On multivariable Cox regression analysis, upregulation of SDHC (HR 1.225; 95% CI (1.100-1.363); p<0.001) and downregulation of SLC25A29 (HR 0.905; 95% CI (0.860-0.952); p<0.001) were also predictive of worse EFS. We also found that patients with high-risk score had significantly lower EFS as compared to low-risk score patients (HR 1.008; 95% CI (1.001-1.012); p<0.001) (Table 2). Harrel’s C-index of prognostic model was 0.626. The timed AUC of the risk score for 12 months and 18 months EFS was 0.617 and 0.612 respectively.
Impact of baseline clinical features on survival outcome and association with gene signature model
On univariable Cox regression analysis of clinical variables, ELN intermediate/poor risk and absence of chloroma were significantly associated with inferior OS and only ELN category came out to be an independent prognostic factor in multivariable analysis (Table S9, Figure 3D). Furthermore, on multivariable analysis, both the ELN risk category (p=0.040) and risk score (p<0.001) were found to be independent prognostic factors for OS. We also performed multivariable analysis including mtDNA copy number and observed that all three factors i.e. risk score (p<0.001), ENA risk categories(p=0.012) and mtDNA copy number(p=0.012) were independent prognostic factors for OS (Table S10).
Impact of combined clinical and gene signature model on survival outcome of the cohort
To compare the predictive ability of our gene signature risk score and ELN risk stratification on OS of AML patients, a time dependent AUC was constructed. Harrel’s C-index of the ELN risk stratification was 0.59 and the timed AUC of ELN risk category on 12 months and 18 months survival was 0.60 and0.64 respectively (Figures 3E, F). We combined the ELN risk strategy with our risk score and calculated the predictive ability of the model. The Harrel’s C-index of the model was 0.688 and the timed AUC of combining ELN risk strategy with gene signature risk score for 12 months and 18 months was 0.761 and 0.765 respectively (Figures 3H, I).
Association of gene signature risk score on disease characteristics
We found that a high-risk score was significantly associated with poor risk cytogenetics(p=0.021), absence of RUNX1-RUNX1T1 translocation (p=0.027) and ELN intermediate/poor risk group (p=0.016). Furthermore, the proportion of patients achieving CR was significantly higher in the low-risk group as compared to the high-risk group(p=0.017) (Table 3). On subgroup analysis, it was observed that the mitochondria-related gene signature risk score category was significantly predictive of survival outcome across all clinically relevant subgroups except in those with intermediate/poor-risk karyotype (Figure S3).
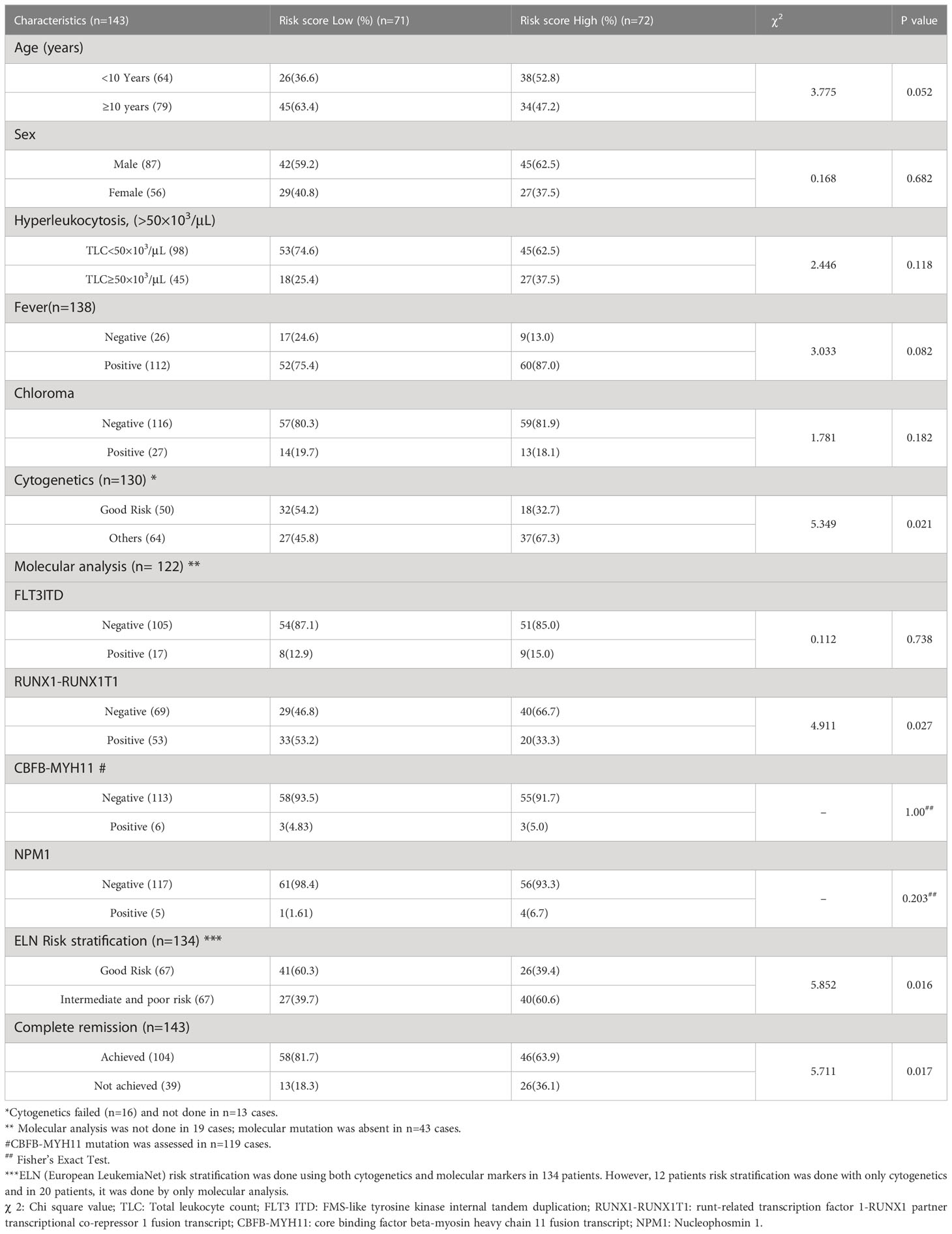
Table 3 Association of 3-gene risk score with clinical and demographic parameters.
Predictive ability of combined gene signature and ELN category model on survival outcome
The predictive ability of gene signature score along with ELN risk stratification on survival outcome of paediatric AML patients was also assessed. Patients with low gene signature score (low risk) belonging to ELN good risk category had significantly better survival outcome (Median OS: Not reached) and predicted 12-months (80% ± 6%), as well as 18-months (75% ± 7%) survival. Similarly, patients with high gene signature score (high risk) belonging to ELN intermediate/poor risk category had significantly inferior outcome (4.67 months (0-3.71)) with 12-months and 18-months predicted survival of 33% ± 8% and 25% ± 7% respectively. On the other hand, patients belonging to other groups (ELN intermediate/poor risk and low-risk; ELN good risk and high-risk score) had intermediate survival outcome (median survival of 27.77-22.90 months respectively) between the two other groups (Figure 3G; Table 4).
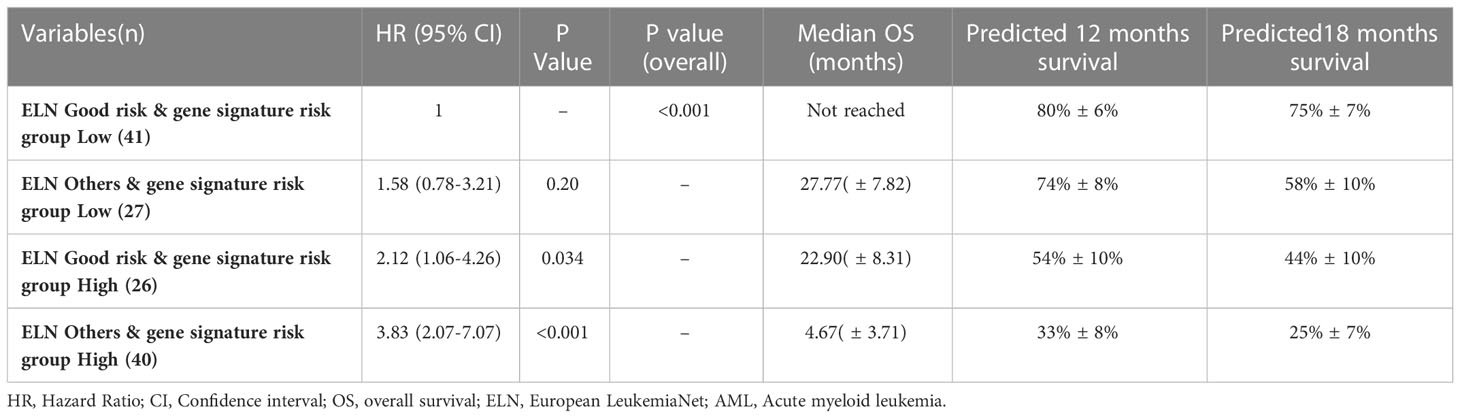
Table 4 Predictive ability of combined gene risk score group and ELN risk category on survival outcome in pediatric AML cohort.
External validation of gene signature risk score in TCGA database
Using our risk calculation model, we calculated the risk score in TCGA dataset (n=179) and similarly, patients were further sub-grouped as high-risk score (higher than median) and low risk score (lower than median) based on the median value (43.434). Kaplan Meier analysis showed that patients with a high-risk score (≥43.434) had inferior OS (HR 1.01;95% CI (1.00-1.02); p<0.019) compared to those with a low-risk score (<43.434) (Figure 3J; Table 2). Along with this, poor risk karyotype patients had worse overall survival (HR 1.89; 95% CI (2.95-1.20); p=0.004) compared to patients with good risk or intermediate risk karyotype (7.03 vs 18.96months). On multivariable analysis karyotype (poor vs good risk/intermediate risk) and risk score were found to be independently predictive of (p=0.002; p=0.025 respectively) for worse OS. The timed AUC of risk score for 12-months and 18-months survival in the TCGA dataset were 0.64 and 0.63 respectively (Figures 3K, L) and Harrel’s C-index of the prognostic model was 0.600. In addition to this, high risk score was also found to be associated with adverse clinical feature of intermediate/poor risk cytogenetics in TCGA dataset as well (Table S11).
Discussion
Mitochondrial adaption is an important phenomenon in leukemic cells and have been shown to impact outcome in patients with AML. The study by Raffel et al. reported that oxidative phosphorylation is an important metabolic alteration which is specific to leukemic stem cells and may be valuable for potential therapeutic targets (18). Similarly, the study by Wu et al. reported that mitochondrial transcription machinery is upregulated in adult AML and confers poor survival outcome (19). Furthermore, using proteomic analysis, a recent study by Jayavelu et al. reported that AML subgroup with high mitochondrial protein expression have shorter remission and poor survival outcomes in adult AML (20). However, there is only limited data on mitochondria-related gene expression profile and its impact on disease outcome of pediatric AML (21, 22).
Our study is the first one to identify and validate mitochondria-related DEGs in paediatric AML along with determining their prognostic significance. In paediatric AML patients, we identified and validated 16 mitochondrial DEGs including 8 upregulated and 8 downregulated genes compared to controls. The dysregulated expression of these genes has been previously reported in the pathogenesis of various malignancies (23–26). However, they have not been studied in paediatric AML. Comparison with LAML dataset of TCGA cohort suggests that the mitochondria-related gene expression profile in paediatric AML is likely distinct. Elevated expression of genes like SLC25A3, FASTKD1, SDHC, ATP5J, which were observed for the first time in our cohort, are involved in mitochondrial energy metabolism (27–29). Genes like FASLG, HRK and SNCA, which were observed to be downregulated, also play role in prevention of mitochondrial damage and apoptosis inhibition in melanoma/medulloblastoma cell lines (30–32). Preliminary data suggests that downregulation of genes like MMP9 and OLFM4, as observed in our cohort, may aid in AML progression (33, 34). The expression of CYP1B1 is reported to be elevated in various malignancies, however, its expression is downregulated in early age leukaemia, as seen in our cohort (35). These findings suggest that the observed mitochondria-related DEGs likely play crucial role in disease progression in paediatric AML, which needs to be studied further mechanistically.
Enhanced mtDNA copy number has been previously reported to be play role on AML initiation, progression as well as predictive of inferior survival outcomes (12, 36). A contrasting finding was recently observed in patients of AML M3 subgroup where elevated mtDNA copy number was predictive of superior survival outcome (37), however, AML M3 subgroup has a distinct disease biology and is not directly comparable with other AML subgroups (38). On the other hand, similar to our previous finding (12), we observed that mtDNA copy number were significantly higher and independently predictive of worse survival outcome in this cohort of pediatric AML patients as well. Furthermore, among the 16 validated mitochondria-related DEGs analysed, we observed that the patients with higher mtDNA copy number had significantly higher expression of SLC25A3, SDHC, RACK1, and FASTKD1 compared to patients with low mtDNA copy number. While, only a small percentage of mitochondrial proteins are coded by the mitochondrial genome, variations in mtDNA may modulate molecular signals through nuclear-mitochondrial crosstalk, which may promote tumorigenesis by upregulating oncogenes (39, 40). This suggests that in paediatric AML, cells with high mtDNA copy number are possibly driven through unique gene expression alterations, influencing disease biology and therapeutic response.
Comprehensive gene expression profiling has been extensively used to identify potential prognostic genes in adult AML; however, dysregulation of mitochondria-related gene expression, especially in children has not been well explored (41–43). Transcriptomic profiling of cytogenetically normal paediatric AML has identified complex genomic rearrangements and/or driver mutations in seemingly normal AML genomes and may even aid risk stratification (44, 45). Cai et al. developed a 3-gene prognostic risk model for children with AML using NCI TARGET dataset, although it was not externally validated (41). Similarly, Duployez et al. and Jiang et al. developed leukaemia stem cell score gene signature and immune checkpoint related gene signature respectively in paediatric AML predictive of survival outcomes (4, 46). The overall comparison of predictive ability of all these available gene signatures with our gene signature model were compiled in the Table S12. None of the above studies evaluated alterations in mitochondrial gene expressions. Mitochondrial gene expression has been evaluated in other malignancies like ovarian cancer, where a mitochondria-related gene signature, consisting of 8 metabolic genes, has been identified with independent prognostic impact (47).
In this study, we identified exclusive mitochondria-related DEGs in paediatric AML and developed a prognostic gene signature including 3 genes (SDHC, CLIC1, and SLC25A29). The gene signature risk score was additionally found to be independently predictive of survival along with established ELN risk stratification with improved predictive ability over clinical risk categorization. The risk score was also found to be associated with poor clinical features of AML like the absence of RUNX1-RUNX1T1 translocation or poor-risk cytogenetics. Hence, the gene signature model is able to categorize the heterogenous molecular landscape of AML into clinically meaningful categories along with identification of adverse disease biology. The developed prognostic score also has the potential to identify high-risk subgroup even among those belonging to ELN good risk and vice-versa allowing better upfront risk stratification and personalized treatment decisions.
TCGA LAML dataset has been extensively used for identifying as well as validating prognostic gene signatures in various AML studies (48, 49). We used the LAML dataset of TCGA for external validation of our gene signature model and observed that the prognostic gene signature score was also independently predictive of survival outcome in a large adult cohort as well with predictive ability over and above known clinical predictors. This suggests that the identified DEGs have a prognostic impact in AML across age group.
Our gene signature included 3 mitochondria-related genes i.e., SDHC, CLIC1, and SLC25A29. SDH mutations lead to decreased activity of SDH with accumulation of succinate and increase in oxidative stress resulting in DNA damage and tumorigenesis (50). In contrast to previous findings, which suggests that the SDH gene is inactivated in solid tumors (51), we observed an increased expression of SDHC gene in AML which was predictive of worse survival. This is likely because, in contrast to solid malignancies, aggressive leukemias like AML depend on cellular oxidative phosphorylation for proliferation which is supported by upregulation of respiratory complex genes (52). Recent study by Erdem et al. has also reported that FLT3-ITD+ AML have high mitochondrial complex II(SDH) activity and inhibition of SDH complex enhance apoptosis of FLT3-ITD+ AML cells in vitro as well as in vivo (53). We also observed that the overall activity of mitochondrial electron transport chain complex II was significantly higher in bone marrow mononuclear cells of pediatric AML patients compared to controls (21). This suggests mitochondrial complex II can be explored as a potential therapeutic target for AML in future studies. Various studies also suggest dysregulation of chloride ion channels such as the CLIC1 gene which plays a role in drug resistance and progression of various malignancies (25, 54). Although, the role of CLIC1 in AML is still unexplored, we observed significant upregulation of CLIC1 in paediatric AML with adverse prognostic impact. The downstream effects of upregulation of CLIC1 on disease biology of AML need to be further deciphered.
In the current study, we observed an upregulation of SLC25A29 in our cohort of paediatric AML patients, which is in line with previous studies where it was found to be significantly elevated in multiple malignancies (26). Similar upregulation was also been observed in adult AML patients of TCGA LAML dataset. However, on survival analysis, downregulation of SLC25A29 was independently predictive of worse OS in our cohort. This finding was consistent even in the external cohort of TCGA LAML dataset, where even though the expression of SLC25A29 was upregulated, a downregulated expression was predictive of worse survival outcomes. This finding was intriguing and the mechanism by which downregulation of SLC25A29 drives a worse survival outcome remains unclear. SLC25A29 is the main arginine transporter in the mitochondrial membrane (55). Aberrant upregulation of SLC25A29 may result in transportation of more arginine into mitochondria, promoting synthesis of metabolites like nitric oxide, polyamines, proline and creatine, which are essential for cell survival and proliferation (56). Mitochondria-derived nitric oxide is known to have a dichotomous role in regulation of cancer progression which is influenced by expression of SLC25A29 likely affecting disease outcome (57). The SLC25 family of genes which encodes for a set of mitochondrial inner membrane carrier proteins, have been identified as a potential biomarker as well as novel therapeutic targets in various malignancies (58). The implications of altered expression of SLC25A29 on disease biology of AML and its assessment as a therapeutic target is an exciting area of further research.
To improve the survival outcomes in AML, advancement in therapies for targeting leukemic cells with heterogenous biology is crucial. We identified 3 gene-based signature, including 3 prognostic genes, which can be explored in future as potential therapeutic targets in AML. SDH inhibitor such as dimethyl malonate has been shown to have effective response in inflammatory disease in vivo (59). Furthermore, several novel SDH inhibitors have been identified using in silico library design which can be potentially utilised in future studies (60). Interestingly, CLIC1 inhibitors has also been explored in glioblastoma cells and found that inhibition of CLIC1 sensitizes glioblastoma stem cells by inhibiting proliferation, migration, invasiveness and self-renewable in vitro and in vivo (61–63). Along with this, using transcriptomic profile of patients with high and low risk score, drug sensitivity assay using FDA approved drugs can be performed to identify drugs precise targeted therapy for patients with high-risk score in future studies (64–66).
Our study has certain limitations. Transcriptomic profile and further validation by RTPCR were done in whole isolated mononuclear cell and not in sorted blasts. However, the gene expression profile as observed in the validation cohort with variable blast percentages using RTPCR remained similar to that observed in transcriptomic profile done in samples with uniform high blast percentage. Initial selection of DEGs were also done from whole RNA sequencing of a limited number of samples, which may lead to a bias in selection, however, external validation of the validated genes confirmed their prognostic impact in an independent cohort.
In conclusion, this is the first study to report a validated set of mitochondria-related DEGs in paediatric AML. We observed that patients with high mtDNA copy number have a unique gene expression pattern possibly affecting disease biology. We developed a 3-gene based mitochondrial gene signature model with ability to predict prognosis in paediatric AML patients over and above established clinical prognostic parameters. The gene signature was also externally validated in a cohort of adult AML patients demonstrating its predictive ability in adult AML as well. Further directions for research include in vitro studies for elucidating the role of prognostic genes in leukemogenesis and their evaluation as potential targets for the treatment of paediatric AML.
Data availability statement
The sequencing data presented in the study are deposited in the Sequence Read Archive (SRA) of the NCBI repository, accession number PRJNA778747.
Ethics statement
The studies involving human participants were reviewed and approved by Institute Ethics Committee, All India Institute of Medical Sciences, New Delhi. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
SC conceptualized the study, conducted the research, performed data analysis and interpreted results and wrote the manuscript. SG analysed data, interpreted results and wrote the manuscript. JP, AS, RB and AC conceptualized the study, provided intellectual inputs, administrative support and edited the manuscript. DP conducted transcriptome data analysis. SB conceptualized the study, provided administrative support, intellectual inputs, interpreted results, wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors acknowledge the funding support from DST-SERB (Department of Science and Technology - Science and Engineering Research Board), Government of India for this work. (Extramural Research grant: EMR/2016/006376 and CRG/2021/001887). The authors also acknowledge the funding support from ICMR (Indian Council of Medical Research), Government of India for this work (ICMR SRF: 2019-6059/CMB/BMS).
Acknowledgments
The authors also acknowledge every member of paediatric oncology team of our center including research staff, nurses and dietician for their exemplary clinical services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1109518/full#supplementary-material
References
1. Lonetti A, Pession A, Masetti R. Targeted therapies for pediatric AML: Gaps and perspective. Front Pediatrics. Front Media S.A (2019) 7:463. doi: 10.3389/fped.2019.00463
2. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129:424–47. American Society of Hematology. doi: 10.1182/blood-2016-08-733196
3. Elsayed AH, Rafiee R, Cao X, Raimondi S, Downing JR, Ribeiro R, et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia (2020) 34(3):735–45. doi: 10.1038/s41375-019-0604-8
4. Duployez N, Marceau-Renaut A, Villenet C, Petit A, Rousseau A, Ng SWK, et al. Evaluation of gene expression signatures predictive of cytogenetic and molecular subtypes of pediatric acute myeloid leukemia. Leukemia (2019) 33(2):348–57. doi: 10.1038/s41375-018-0227-5
5. Wagner S, Vadakekolathu J, Tasian SK, Altmann H, Bornhäuser M, Pockley AG, et al. A parsimonious 3-gene signature predicts clinical outcomes in an acute myeloid leukemia multicohort study. Blood Adv (2019) 3(8):1330–46. doi: 10.1182/bloodadvances.2018030726
6. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol (2015) 11:9–15. Nature Publishing Group. doi: 10.1038/nchembio.1712
7. Panina SB, Baran N, Brasil da Costa FH, Konopleva M, Kirienko NV. A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis (2019) 10(8):1–15. doi: 10.1038/s41419-019-1851-3
8. Al Ageeli E. Alterations of mitochondria and related metabolic pathways in leukemia: A narrative review. Saudi J Med Med Sci (2020) 8(1):3. doi: 10.4103/sjmms.sjmms_112_18
9. Tyagi A, Pramanik R, Bakhshi R, Singh A, Vishnubhatla S, Bakhshi S. Expression of mitochondrial genes predicts survival in pediatric acute myeloid leukemia. Int J Hematol (2019) 110(2):205–12. doi: 10.1007/s12185-019-02666-2
10. Tyagi A, Pramanik R, Vishnubhatla S, Bakhshi R, Bakhshi S. Prognostic impact of mitochondrial DNA d-loop variations in pediatric acute myeloid leukemia. Oncotarget (2019) 10(13):1334–43. doi: 10.18632/oncotarget.26665
11. Sharawat SK, Bakhshi R, Vishnubhatla S, Bakhshi S. Mitochondrial d-loop variations in paediatric acute myeloid leukaemia: A potential prognostic marker. Br J Haematol (2010) 149(3):391–8. doi: 10.1111/j.1365-2141.2010.08084.x
12. Chaudhary S, Ganguly S, Palanichamy JK, Singh A, Bakhshi R, Jain A, et al. PGC1A driven enhanced mitochondrial DNA copy number predicts outcome in pediatric acute myeloid leukemia. Mitochondrion (2021) 58:246–54. doi: 10.1016/j.mito.2021.03.013
13. Arora S, Pushpam D, Tiwari A, Choudhary P, Chopra A, Gupta R, et al. Allogeneic hematopoietic stem cell transplant in pediatric acute myeloid leukemia: Lessons learnt from a tertiary care center in India. Pediatr Transplant (2021) 25(3):e13918. doi: 10.1111/petr.13918
14. Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O’Donoghue SI, Schneider R, et al. COMPARTMENTS: Unification and visualization of protein subcellular localization evidence. Database (2014) 2014. doi: 10.1093/database/bau012
15. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol (2014) 8(4):S11. doi: 10.1186/1752-0509-8-S4-S11
16. Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf (2003) 4:2. doi: 10.1186/1471-2105-4-2
17. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res (2017) 45(W1):W98–102. doi: 10.1093/nar/gkx247
18. Raffel S, Klimmeck D, Falcone M, Demir A, Pouya A, Zeisberger P, et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood (2020) 136(13):1507–19. doi: 10.1182/blood.2019003654
19. Wu S, Fahmy N, Alachkar H. The mitochondrial transcription machinery genes are upregulated in acute myeloid leukemia and associated with poor clinical outcome. Metab Open (2019) 2:100009. doi: 10.1016/j.metop.2019.100009
20. Jayavelu AK, Wolf S, Buettner F, Alexe G, Häupl B, Comoglio F, et al. The proteogenomic subtypes of acute myeloid leukemia. Cancer Cell (2022) 40(3):301–317.e12. doi: 10.1016/j.ccell.2022.02.006
21. Chaudhary S, Ganguly S, Singh A, Palanichamy JK, Chopra A, Bakhshi R, et al. Mitochondrial complex II and V activity is enhanced in pediatric acute myeloid leukemia. Am J Blood Res (2021) 11(5):534–43.
22. Chaudhary S, Ganguly S, Singh A, Palanichamy JK, Bakhshi R, Chopra A, et al. Mitochondrial biogenesis gene POLG correlates with outcome in pediatric acute myeloid leukemia. Leukemia Lymphoma (2022) 63:1005–8. doi: 10.1080/10428194.2021.2010063. Taylor & Francis.
23. Li J-J, Xie D. RACK1, a versatile hub in cancer. Oncogene (2015) 34(15):1890–8. doi: 10.1038/onc.2014.127
24. Oehler VG, Ka YY, Choi YE, Bumgarner RE, Raftery AE, Radich JP. The derivation of diagnostic markers of chronic myeloid leukemia progression from microarray data. Blood. (2009) 114(15):3292–8. doi: 10.1182/blood-2009-03-212969
25. Lee JR, Lee JY, Kim HJ, Hahn MJ, Kang JS, Cho H. The inhibition of chloride intracellular channel 1 enhances Ca2+ and reactive oxygen species signaling in A549 human lung cancer cells. Exp Mol Med (2019) 51(7):1–11. doi: 10.1038/s12276-019-0279-2
26. Zhang H, Wang Q, Gu J, Yin L, Liang S, Wu L, et al. Elevated mitochondrial SLC25A29 in cancer modulates metabolic status by increasing mitochondria-derived nitric oxide. Oncogene. (2018) 37(19):2545–58. doi: 10.1038/s41388-018-0139-x
27. Kolbe HVJ, Costello D, Wong A. Mitochondrial phosphate transport. Large scale isolation and characterization of the phosphate transport protein from beef heart mitochondria. J Biol Chem (1984) 259(14):9115–20.
28. Nazar E, Khatami F, Saffar H, Tavangar SM. The emerging role of succinate dehyrogenase genes (SDHx) in tumorigenesis. Int J Hematol Stem Cell Res (2019) 13(2):72. doi: 10.18502/ijhoscr.v13i2.692
29. Jourdain AA, Popow J, de la Fuente MA, Martinou JC, Anderson P, Simarro M. The FASTK family of proteins: Emerging regulators of mitochondrial RNA biology. Nucleic Acids Res (2017) 45(19):10941–7. doi: 10.1093/nar/gkx772
30. Chen M, Wang W, Ma J, Ye P, Wang K. High glucose induces mitochondrial dysfunction and apoptosis in human retinal pigment epithelium cells via promoting SOCS1 and Fas/FasL signaling. Cytokine (2016) 78:94–102. doi: 10.1016/j.cyto.2015.09.014
31. Li Y, Yu Z, Jiang T, Shao L, Liu Y, Li N, et al. SNCA, a novel biomarker for group 4 medulloblastomas, can inhibit tumor invasion and induce apoptosis. Cancer Sci (2018) 109(4):1263. doi: 10.1111/cas.13515
32. Rizvi F, Heimann T, Herrnreiter A, O’Brien WJ. Mitochondrial dysfunction links ceramide activated HRK expression and cell death. PLoS One (2011) 6(3):e18137. doi: 10.1371/journal.pone.0018137
33. Lin LI, Lin DT, Chang CJ, Lee CY, Tang JL, Tien HF. Marrow matrix metalloproteinases (MMPS) and tissue inhibitors of MMP in acute leukaemia: Potential role of MMP-9 as a surrogate marker to monitor leukaemic status in patients with acute myelogenous leukaemia. Br J Haematol (2002) 117(4):835–41. doi: 10.1046/j.1365-2141.2002.03510.x
34. Liu W, Lee HW, Liu Y, Wang R, Rodgers GP. Olfactomedin 4 is a novel target gene of retinoic acids and 5-aza-2’-deoxycytidine involved in human myeloid leukemia cell growth, differentiation, and apoptosis. Blood (2010) 116(23):4938–47. doi: 10.1182/blood-2009-10-246439
35. Lopes BA, Emerenciano M, Gonçalves BAA, Vieira TM, Rossini A, Pombo-de-Oliveira MS. Polymorphisms in CYP1B1, CYP3A5, GSTT1, and SULT1A1 are associated with early age acute leukemia. PLoS One (2015) 10(5):e0127308. doi: 10.1371/journal.pone.0127308
36. Kang MG, Kim YN, Lee JH, Szardenings M, Baek HJ, Kook H, et al. Clinicopathological implications of mitochondrial genome alterations in pediatric acute myeloid leukemia. Ann Lab Med (2016) 36(2):101–10. doi: 10.3343/alm.2016.36.2.101
37. Pereira-Martins DA, Coelho-Silva JL, Weinhäuser I, Franca-Neto PL, Silveira DR, Ortiz C, et al. Clinical significance of mitochondrial DNA content in acute promyelocytic leukaemia. Br J Haematol (2023) 200(2):170–4. doi: 10.1111/bjh.18510
38. Adès L, Guerci A, Raffoux E, Sanz M, Chevallier P, Lapusan S, et al. Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: The European APL group experience. Blood (2010) 115(9):1690–6. doi: 10.1182/blood-2009-07-233387
39. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell (2008) 134(1):112–23. doi: 10.1016/j.cell.2008.06.016
40. Horan MP, Cooper DN. The emergence of the mitochondrial genome as a partial regulator of nuclear function is providing new insights into the genetic mechanisms underlying age-related complex disease. Hum Genet (2014) 133:435–58. doi: 10.1007/s00439-013-1402-4
41. Cai Z, Wu Y, Zhang F, Wu H. A three-gene signature and clinical outcome in pediatric acute myeloid leukemia. Clin Transl Oncol (2021) 23(4):866–73. doi: 10.1007/s12094-020-02480-x
42. Duployez N, Marceau-Renaut A, Villenet C, Petit A, Rousseau A, Ng SWK, et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia (2019) 33(2):348–57. doi: 10.1038/s41375-018-0227-5
43. Nguyen CH, Glüxam T, Schlerka A, Bauer K, Grandits AM, Hackl H, et al. SOCS2 is part of a highly prognostic 4-gene signature in AML and promotes disease aggressiveness. Sci Rep (2019) 9(1):1–13. doi: 10.1038/s41598-019-45579-0
44. Shiba N, Yoshida K, Hara Y, Yamato G, Shiraishi Y, Matsuo H, et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv (2019) 3(20):3157–69. doi: 10.1182/bloodadvances.2019000404
45. Balgobind BV, van den Heuvel-Eibrink MM, De Menezes RX, Reinhardt D, Hollink IHI, Arentsen-Peters STJCM, et al. Evaluation of gene expression signatures predictive of cytogenetic and molecular subtypes of pediatric acute myeloid leukemia. Haematologica. (2011) 96(2):221–30. doi: 10.3324/haematol.2010.029660
46. Jiang F, Wang XY, Wang MY, Mao Y, Miao XL, Wu CY, et al. An immune checkpoint-related gene signature for predicting survival of pediatric acute myeloid leukemia. J Oncol (2021) 2021:5550116. doi: 10.1155/2021/5550116
47. Wang L, Li X. Identification of an energy metabolism-related gene signature in ovarian cancer prognosis. Oncol Rep (2020) 43(6):1755–70. doi: 10.3892/or.2020.7548
48. Zhang Y, Ma S, Wang M, Shi W, Hu Y. Comprehensive analysis of prognostic markers for acute myeloid leukemia based on four metabolic genes. Front Oncol (2020) 0:1990. doi: 10.3389/fonc.2020.578933
49. Huang R, Liao X, Li Q. Identification and validation of potential prognostic gene biomarkers for predicting survival in patients with acute myeloid leukemia. Onco Targets Ther (2017) 10:5243. doi: 10.2147/OTT.S147717
50. Zhao T, Mu X, You Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget. Impact Journals LLC (2017) 8:53819–28. doi: 10.18632/oncotarget.17734
51. Li J, Liang N, Long X, Zhao J, Yang J, Du X, et al. SDHC-related deficiency of SDH complex activity promotes growth and metastasis of hepatocellular carcinoma via ROS/NFκB signaling. Cancer Lett (2019) 461:44–55. doi: 10.1016/j.canlet.2019.07.001
52. Panina SB, Pei J, Kirienko NV. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab (2021) 9:1–25. doi: 10.1186/s40170-021-00253-w. Springer Science and Business Media LLC.
53. Erdem A, Marin S, Pereira-Martins DA, Geugien M, Cunningham A, Pruis MG, et al. Inhibition of the succinyl dehydrogenase complex in acute myeloid leukemia leads to a lactate-fuelled respiratory metabolic vulnerability. Nat Commun (2022) 13(1):2013. doi: 10.1038/s41467-022-29639-0
54. He YM, Zhang ZL, Liu QY, Xiao YS, Wei L, Xi C, et al. Effect of CLIC1 gene silencing on proliferation, migration, invasion and apoptosis of human gallbladder cancer cells. J Cell Mol Med (2018) 22(5):2569–79. doi: 10.1111/jcmm.13499
55. Porcelli V, Fiermonte G, Longo A, Palmieri F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J Biol Chem (2014) 289(19):13374–84. doi: 10.1074/jbc.M114.547448
56. Keshet R, Erez A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. DMM Dis Models Mech (2018) 11(8):dmm033332. Company of Biologists. doi: 10.1242/dmm.033332
57. Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci (2005) 26:190–5. doi: 10.1016/j.tips.2005.02.005
58. Rochette L, Meloux A, Zeller M, Malka G, Cottin Y, Vergely C. Mitochondrial SLC25 carriers: Novel targets for cancer therapy. Molecules (2020) 25(10):2417. doi: 10.3390/molecules25102417
59. Yang Y, Shao R, Tang L, Li L, Zhu M, Huang J, et al. Succinate dehydrogenase inhibitor dimethyl malonate alleviates LPS/d-galactosamine-induced acute hepatic damage in mice. Innate Immun (2019) 25(8):522–9. doi: 10.1177/1753425919873042
60. Yao TT, Fang SW, Li ZS, Xiao DX, Cheng JL, Ying HZ, et al. Discovery of novel succinate dehydrogenase inhibitors by the integration of in silico library design and pharmacophore mapping. J Agric Food Chem (2017) 65(15):3204–11. doi: 10.1021/acs.jafc.7b00249
61. Barbieri F, Würth R, Pattarozzi A, Verduci I, Mazzola C, Cattaneo MG, et al. Inhibition of chloride intracellular channel 1 (CLIC1) as biguanide class-effect to impair human glioblastoma stem cell viability. Front Pharmacol (2018) 9. doi: 10.3389/fphar.2018.00899
62. Barbieri F, Bosio AG, Pattarozzi A, Tonelli M, Bajetto A, Verduci I, et al. Chloride intracellular channel 1 activity is not required for glioblastoma development but its inhibition dictates glioma stem cell responsivity to novel biguanide derivatives. J Exp Clin Cancer Res (2022) 41(1):1–27. doi: 10.1186/s13046-021-02213-0
63. Wang W, Wan M, Liao D, Peng G, Xu X, Yin W, et al. Identification of potent chloride intracellular channel protein 1 inhibitors from traditional Chinese medicine through structure-based virtual screening and molecular dynamics analysis. BioMed Res Int (2017) 2017:4751780. doi: 10.1155/2017/4751780
64. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature (2018) 562(7728):526–31. doi: 10.1038/s41586-018-0623-z
65. Bottomly D, Long N, Schultz AR, Kurtz SE, Tognon CE, Johnson K, et al. Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell (2022) 40(8):850–864.e9. doi: 10.1016/j.ccell.2022.07.002
Keywords: mitochondria, gene signature, RNA sequencing, child, acute myeloid leukema
Citation: Chaudhary S, Ganguly S, Palanichamy JK, Singh A, Pradhan D, Bakhshi R, Chopra A and Bakhshi S (2023) Mitochondrial gene expression signature predicts prognosis of pediatric acute myeloid leukemia patients. Front. Oncol. 13:1109518. doi: 10.3389/fonc.2023.1109518
Received: 27 November 2022; Accepted: 11 January 2023;
Published: 09 February 2023.
Edited by:
Mario I. Vega, University of California, Los Angeles, United StatesReviewed by:
Diego A. Pereira-Martins, University of Groningen, NetherlandsMario Morales, National Autonomous University of Mexico, Mexico
Copyright © 2023 Chaudhary, Ganguly, Palanichamy, Singh, Pradhan, Bakhshi, Chopra and Bakhshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sameer Bakhshi, sambakh@hotmail.com