 Daniel D. Shapiro
Daniel D. Shapiro Maria Virumbrales-Muñoz
Maria Virumbrales-Muñoz David J. Beebe3,5
David J. Beebe3,5 E. Jason Abel
E. Jason Abel- 1Department of Urology, University of Wisconsin School of Medicine and Public Health, Madison, WI, United States
- 2Division of Urology, William S. Middleton Memorial Veterans Hospital, Madison, WI, United States
- 3Department of Pathology and Laboratory Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI, United States
- 4Department of Cell and Regenerative Biology, University of Wisconsin School of Medicine and Public Health, Madison, WI, United States
- 5Department of Biomedical Engineering, University of Wisconsin – Madison, Madison, WI, United States
Modeling renal cell carcinoma is critical to investigating tumor biology and therapeutic mechanisms. Multiple systems have been developed to represent critical components of the tumor and its surrounding microenvironment. Prominent in vitro models include traditional cell cultures, 3D organoid models, and microphysiological devices. In vivo models consist of murine patient derived xenografts or genetically engineered mice. Each system has unique advantages as well as limitations and researchers must thoroughly understand each model to properly investigate research questions. This review addresses common model systems for renal cell carcinoma and critically evaluates their performance and ability to measure tumor characteristics.
Introduction
Preclinical models are necessary for the discovery of molecular pathways that regulate tumor growth and to identify tumor vulnerabilities for potential therapeutic targets. Ideal models faithfully represent key characteristics of human tumors and response to drug therapies. Models should also be easily maintained and rapidly established to allow for efficient, cost-effective investigations. Additionally, models should ideally capture key components of the tumor microenvironment (TME), which is the dynamic space around the tumor, containing multiple cell types, growth and paracrine factors, and structural components (1–3). Hallmark components of the TME include immune cells, stromal cells, blood vessels and extracellular matrix (4). These components of the TME contribute to a tumor’s potential to grow or regress, metastasize, or remain localized (1–5). A deeper understanding of the TME has opened potential targets for new therapies (6, 7).
Modeling renal cell carcinoma (RCC) and the associated TME is challenging but necessary as RCC is a major cause of cancer globally. Renal cell carcinoma affects over 400,000 individuals globally per year (8). Up to one third of patients will present with or develop metastatic disease, which is almost universally fatal (9). Prognosis greatly depends on stage at diagnosis, with metastatic disease only demonstrating a 12% 5-year survival rate (8, 10). Typically, standard of care therapy for RCC consists of surgery or ablative therapy for localized disease and systemic therapy for metastatic disease (11–13). Commonly targeted pathways in RCC include the hypoxia inducible factor (HIF) and mTOR pathways, and more recently immune checkpoint blockade (14). Tyrosine kinase inhibitors and mTOR inhibitors, as well as VEGF blockade have previously been used to treat metastatic RCC until the more recent development of immune checkpoint blockade (anti-PD1, anti-PDL1 and anti-CTLA4 antibodies), which has improved disease-free and overall survival (14). Renal cell carcinoma tumors can be divided into multiple subtypes that range from indolent to aggressive and differ in their response to therapeutics. The tumors can grow large and are spatially heterogenous, consisting of multiple populations of subclones harboring unique genetic mutations in different regions of the tumor (15, 16). Additionally, RCC tumors can modulate the host immune response, which has become the target for modern systemic immune-based therapies (17). No single preclinical model can capture the complex tumor microenvironment or the subsequent host response. Thus, researchers employ multiple systems to capture and control unique components of RCC tumor biology to answer specific questions. The focus of this review will be to discuss commonly used preclinical models for RCC mechanistic and therapeutic investigations.
Renal Cell Carcinoma Genetic Hallmarks
Selecting biologically relevant preclinical models for research requires a thorough understanding of the molecular hallmarks of RCC. Renal cell carcinoma arises from epithelial cells of the nephron. RCC is a heterogenous disease (15) and can be classified into distinct histologic subtypes of which clear cell RCC (ccRCC) is the most common, accounting for about 70-80% kidney cancers (9, 18–20). Other common subtypes include papillary type 1, papillary type 2, and chromophobe RCC; however, a number of other unique subtypes exist (21). Clear cell renal cell carcinoma is defined by loss of the VHL gene expression (22–24). Studies have demonstrated that the most frequent arm-level events involve loss of chromosome 3p (91% of samples) (16, 22). Loss of this allele includes loss of VHL as well as additional tumor suppressor genes in close proximity to VHL including PBRM1, BAP1, and SETD2 (22, 25, 26). These four genes become the targets for subsequent inactivating mutations on the second chromosomal copy (22, 25). While second-hit loss of function mutations in VHL are present in over 90% of ccRCC, complete loss of VHL expression alone is insufficient to produce ccRCC in humans as well as mice (27, 28). The most frequently mutated genes in ccRCC besides VHL are PBRM1 (~45%), SETD2 (10-15%), and BAP1 (10-15%) (22, 29, 30). Other common mutations found in the TCGA cohort include MTOR, PIK3CA, KDM5C, TP53 and PTEN (22, 29). Using a multi-region sampling approach, the TRACERx consortium evaluated the evolutionary trajectories of ccRCC (25). This study demonstrated that loss of chromosome 3p is the initiating driver event, which typically occurs in childhood or adolescence (25). The second critical event is the inactivation of the second VHL allele which may occur decades after the initial 3p loss. Additional driver mutations in PBRM1, SETD2, and BAP1 may occur, and the number of driver events is significantly associated with tumor stage, grade, and presence of necrosis (Figure 1) (16, 25). VHL inactivation leads to loss of VHL protein, which is a critical component of the E3 ubiquitin ligase complex that functions to ubiquitinate hypoxia-inducible transcription factors in the presence of oxygen, leading to proteasomal degradation (23). Loss of the VHL protein expression causes stabilization of the hypoxia inducible factors (HIF-1α and HIF-2α). Unimpeded HIF activation leads to expression of target genes regulating angiogenesis, glycolysis, and apoptosis (23). The next most commonly modified genes, including PBRM1, SETD2, and BAP1, are all chromatin modifiers and play roles in chromatin maintenance (32). Mutations in BAP1 have correlated with poor survival in ccRCC and PBRM1 mutations have been associated with a more indolent phenotype (18). Additional cross-platform molecular analyses demonstrated a correlation between worsened prognosis in patients with ccRCC harboring mutations causing metabolic shifts involving increased reliance on the pentose phosphate shunt, decreased AMPK, decreased Krebs cycle activity, increased glutamine transport and fatty acid production (33). This metabolic shift is similar to the Warburg metabolic phenotype of increased glycolysis and decreased AMPK, glutamine-dependent lipogenesis (22). In general, primary RCC tumors can demonstrate profound intratumoral genomic heterogeneity (15), and multi-region sequencing of primary tumors have demonstrated that up to seven biopsies are required to detect at least 75% of the many subclonal driver events (16).
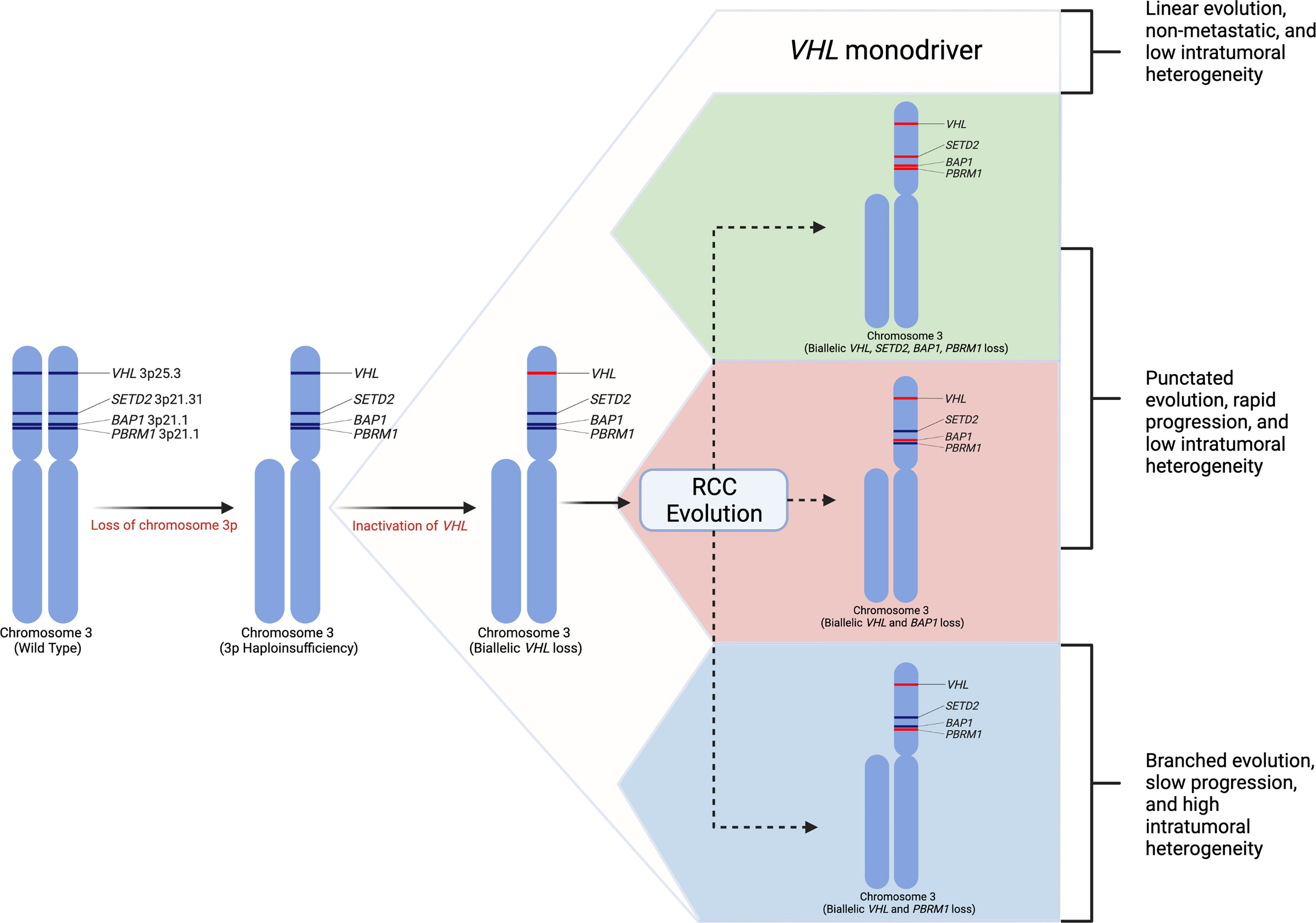
Figure 1 Renal cell carcinoma genetic evolution. This model of renal cell carcinoma evolution demonstrates chromosome 3p loss as the initial “first-hit” event. Subsequent biallelic loss of VHL occurs as a second hit. Tumor cells then may undergo multiple different pathways or “branches” of evolution based on mutations in other driver genes such as SETD2, PBRM1, and BAP1. These distinct evolutionary branches create different tumor phenotypes (31).
Loss of chromosome 9p and 14q appear to be hallmark genomic alterations in ccRCC metastatic competence (31). Additionally, distinct genomic evolutionary subtypes have been identified. The order and timing of driver events leads to distinct metastatic potential. There appear to be two primary modes of metastatic dissemination. Primary tumors with decreased intratumoral heterogeneity and high genomic instability acquire metastatic ability early in tumor clonal evolution leading to rapid progression (31). Among these tumors, subtypes exist including tumors in which metastatic competence is driven by BAP1 inactivation, tumors that maintain VHL wild-type expression, or tumors that contain multiple clonal drivers (31). Conversely, primary tumors with high intratumoral heterogeneity with or without high genomic instability gradually acquire metastatic capacity causing attenuated metastatic progression. These subtypes include PBRM1 followed by PI3K inactivation, PBRM1 followed by SETD2 inactivation, and PBRM1 followed by somatic copy number alterations (31). Genomic characterization of metastatic sites among these subtypes demonstrated more homogenous cells with fewer somatic driver alterations (31). Tumors with high intratumoral heterogeneity and low genomic instability typically demonstrate an initial oligometastatic pattern with gradually increasing metastatic load over time (31).
Beyond ccRCC, additional subtypes of RCC continue to be described with their own unique genetic hallmarks. Fumarate hydratase (FH) deficient RCC is characterized by mutations in the FH gene at 1q42. Patients often present with a hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndromic findings including cutaneous leiomyomas and uterine leiomyomas in females in addition to type 2 papillary RCC tumors. These tumors have reportedly responded to first-line bevacizumab/erlotinib (34). Succinate dehydrogenase (SD) deficient RCC is due to inactivation of one of the genes within the SDH family, often as germline mutations. In addition to kidney tumors, patients may develop pheochromocytomas, GIST and pituitary adenomas (34). MiT family translocation RCC (tRCC) account for around 50% of pediatric RCC and are caused by gene fusions of TFE3 or TFEB. The most common translocation involves Xp11 leading to activation of TFE3 (34). Other rare renal tumors with distinct genetic characterizations include ALK rearrangement-associated RCC (ALK-RCC), TCEB1 renal cell carcinoma, renal medullary carcinoma, collecting duct carcinoma, tubulocystic RCC, clear cell papillary RCC, acquired cystic disease-associated RCC, and mucinous tubular spindle cell carcinoma (34).
Overall, the understanding of these molecular mechanisms has come from work with models of renal cell carcinoma including cell lines, organoids, and murine models. These models can help to recapitulate the molecular mechanisms of human RCC. We will provide a review of the main preclinical models utilized when studying renal cell carcinoma.
Renal Cell Carcinoma Cell Lines
Cell lines provide a low-cost research tool to understand the molecular mechanisms of RCC. Ideal cell lines will faithfully recapitulate defining tumor characteristics such as the critical genomic changes that drive RCC tumor phenotypes (35). Additionally, ideal cell lines will respond to new therapies (e.g., new drug therapies) in a way that predicts outcomes in humans. The first established cell lines for RCC included 786-O, ACHN, CAKI-1, A498, RXF393, UO-31, TK-10, and SN12C (36). These were established as part of the US National Cancer Institute 60 cell line anticancer drug screen effort (36, 37). There are a substantial number of additional RCC cell lines that are derived from various RCC subtypes including lines derived from both primary renal tumors as well as metastatic sites (38). In addition to the commonly used and commercially available immortalized cell lines, more recent efforts have been made to create individual patient derived cell lines that maintain the characteristics of the parental tumor, including parental driver mutations and allele frequency (39). These cell lines may allow for development of personalized drug screening and discovery (39). Each cell line harbors a unique genetic profile which is critical to understand prior to interpreting genetic and cellular events during in vitro studies (Table 1) (38). We will review some of the most frequently used cell lines in RCC research.
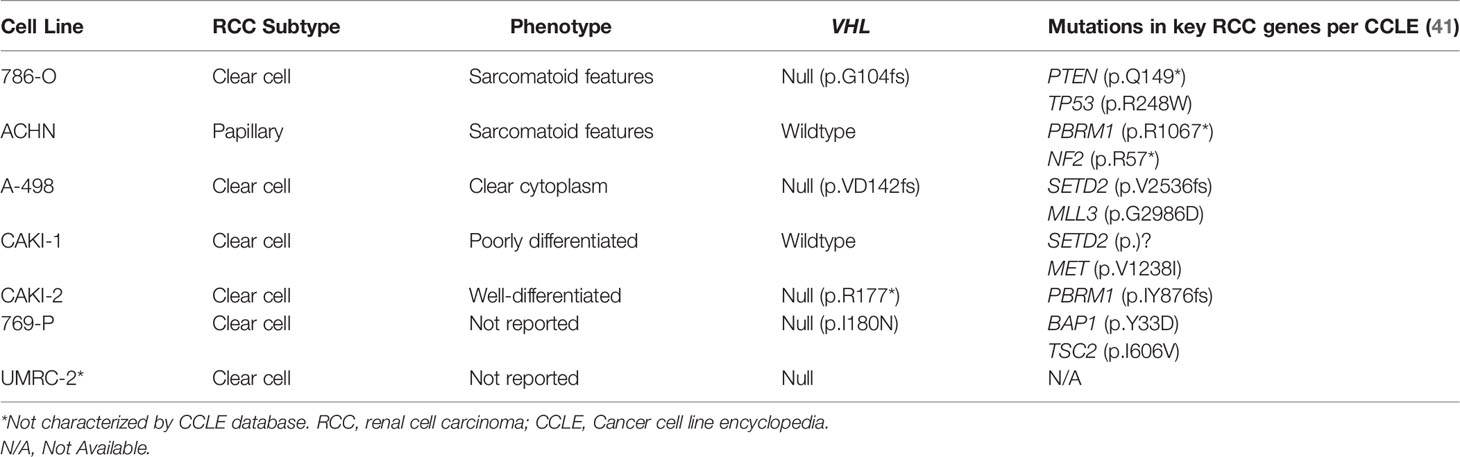
The ACHN cell line was derived from a malignant pleural effusion patient sample and appears to be possibly derived form a papillary RCC subtype. This cell line harbors a MET mutation consistent with papillary RCC (38). Additionally, an analysis of copy number alterations reveal that ACHN cells cluster with papillary tumors when compared to TCGA renal tumor copy number alterations (40). The ACHN cell line is the third most commonly cited cell line in the literature, despite being derived from papillary RCC cells (36, 40). When investigating key RCC mutations, ACHN cells were found to harbor no mutations in genes including VHL, PBRM1, SETD2, BAP1, and TP53 (40). The ACHN line was used by Thomas et al. to demonstrated that after VHL knockdown, ACHN cells became sensitive to mTOR inhibition, providing the support for the clinical use of mTOR inhibitors for RCC treatment (42). Histologically, xenografts derived from ACHN cells tend to show a poorly differentiated carcinoma with aggressive features (40). Despite its genomic correlation with papillary RCC, ACHN tends to show sarcomatoid differentiation rather than papillary architecture in xenografts (40). Given its likely papillary origins, investigators should use caution if using this cell line in an attempt to recapitulate clear cell RCC diseases.
The 786-O cell line is one of the most frequently used cell lines for RCC specific research (36, 38). 786-O cells are characterized by their loss of function mutations in VHL genes leading to clear cell RCC phenotypes (36, 40, 43). These cells demonstrate increased expression of HIF-2α and VEGF proteins. This cell line has been used in numerous studies including initial studies demonstrating the function of the VHL/HIF pathway (24, 36, 44). In a study by Iliopoulos et al, restoration of pVHL in 786-O cells effectively suppressed tumor growth in nude mice (24). Kondo et al. demonstrated that the tumor suppression gained by pVHL can be overridden by a HIF variant that escapes pVHL control. This effectively demonstrated HIF as a target of pVHL and that unsuppressed activation of HIF promotes tumorigenesis (24, 44). Additionally, 786-O cells were used to demonstrate that inhibition of HIF-2α with shRNA suppressed tumor growth in 786-O cell derived xenograft models (44). About 2 decades later, belzutifan, a small molecule HIF-2α inhibitor would receive FDA approval for treatment of patients with VHL disease (45). While 786-O appears to have been derived from ccRCC, the cell line has undergone significant dedifferentiation in vitro, both genomically and histologically. It demonstrates a sarcomatoid appearance in xenograft studies and the cells do not display a clear cytoplasm characteristic of typical ccRCC histology in humans (36, 40).
Another frequently cited cell line is A-498. This cell line contains p.VD142fs frameshift deletion of VHL, suggesting its ccRCC origin (36, 41). This cell line has been classified according to the prognostic expression based subtype ccB, which indicates a more aggressive phenotype, along with other RCC cell lines 786-O, CAKI-1, and OS-RC.2 (40). RCC tumors have previously been classified according to the ccA (“clear cell A”) and ccB prognostic classification system, which are defined according to a highly predictive gene set developed using 48 RCC tumors and subsequently validated using 177 RCC tumors (46). ccA tumors display increased angiogenesis and hypoxic signaling while ccB tumors display decreased hypoxic gene expression, elevated epithelial-to-mesenchymal signaling and more aggressive behavior (46). While both 786-O and A-498 appear to derive from a ccRCC origin, 786-O tends to harbor a greater number of genetic alterations than A-498. Additionally, while 786-O demonstrates poorly differentiated cells histologically, A-498 cell derived xenografts demonstrate more typical histologic findings of ccRCC including nests of malignant cells with clear cytoplasm (40). The A-498 cell line has been used to demonstrate the upregulation of the mTOR signaling pathway in RCC and demonstrated that cell growth could be inhibited by the mTOR inhibitor, rapamycin (47).
Overall, multiple different cell lines derived from RCC tumors exist from which researchers can choose. These cell lines contain a diverse genetic makeup and display variable phenotypes in vitro and when engrafted in vivo to make xenografts. As noted in Table 1, VHL is wild-type in 3 of the 6 most commonly reported cell lines, yet VHL is known to be mutated in over 90% of ccRCC tumors found in patients. This creates a divergent molecular phenotype among cell lines compared to tumors found in the majority of patients. Some studies have investigated generation of ccRCC patient derived cell lines to search for therapeutic vulnerabilities at an individual level (48). This can be challenging as patient derived cell lines are frequently contaminated by normal epithelial cells leading to poor efficiency (48). It is critical for the researcher to select the appropriate cell line based on the research question being investigated and select one that most closely resembles the tumor molecular response and phenotype in vivo. Cell cultures have other significant limitations. Traditional cell cultures grow in only 2 dimensions compared to the 3-dimensional tumor architecture. Cell lines typically become de-differentiated and pick up mutations after serial passages in vitro.
Organoid Models
New model techniques have been developed to overcome some of the limitations found using traditional cell cultures. It is known that the structural phenotype of the tumor influences cellular interactions and subsequent gene expression (35, 49). Thus, 2D cell cultures are limited by their lack of normal tissue architecture. Organoids are 3D cell cultures that attempt to mimic normal and tumor tissue structure in vitro (50, 51). These 3D models imitate environmental pressures in culture such as nutrient gradients in which the inner portion of the tumors receive less nutrients due to devascularization, and also more realistically model how drug therapies reach different portions of the tumors (51, 52). These qualities allow researchers to study tumor drug-sensitivity in addition to other mechanistic and genomic studies (51). Organoid cultures have been utilized in many cancers including breast (53), prostate (54), pancreas (55), colorectal cancer (56), as well as in RCC.
A study by Grassi et al. established normal kidney and renal cell carcinoma organoids. Using portions of excised RCC tumors, the tissue was homogenized both mechanically and enzymatically. The cell suspension was first plated in a stem cell enriching medium and after an incubation period, the cells were transferred to Matrigel containing organoid specific medium to promote organoid growth (50). Tumor organoid colonies were noted to establish at a lower rate than benign kidney derived organoids. Additionally, tumor organoids notably lost their structure after serial passages (50). Sequencing studies demonstrated preserved mutational patterns among organoids when compared to the primary tumor sample, indicating organoids may make excellent patient specific models for personalized drug susceptibility testing (50). It should be noted that Matrigel is frequently used for organoid culture but is a poorly characterized medium derived from mouse sarcoma cells and may alter normal cellular function (57). New methods are being developed to grow organoids without Matrigel (57).
Organoids also provide a platform for incorporating other components of the tumor microenvironment beyond sole tumor cells. Renal cell carcinoma has seen recent treatment advances based on immune activating therapies (58–66). One difficulty in the modern era of immunotherapy for advanced clear cell RCC is developing a model system that incorporates both tumor cells and the surrounding tumor immune microenvironment (TME). These models are critical to discovering potential biomarkers to guide immunotherapy (66, 67). To recapitulate the TME, Neal et al. developed air-liquid interface organoids which preserve primary tumor epithelium en bloc with endogenous immune and non-immune stromal components (68). This organoid model allows for the study of immunotherapy effects in vitro, and this model demonstrated a functional tumor infiltrating lymphocyte response to PD-1/PD-L1 checkpoint blockade (68). While these models better incorporate components of the TME, they cannot measure the effects of the peripheral immune system, which are also critical for anti-tumor immunity (68). Using the same air-liquid interface system, Esser et al. generated patient derived organoids from 42 RCC tumors (69), which better represented the primary tumors as demonstrated by IHC and RNA sequencing analysis (69). Additionally, they used 10 organoids and treated them with either cabozantinib or nivolumab, and they noted different responses of each organoid to either of these therapies. Response to nivolumab was dependent on a higher CD8+ T-cell presence in the organoid model (69). The authors demonstrate that patient derived organoids appear to be a suitable tool for therapeutic testing even with immunotherapeutic agents (69).
Overall, organoids provide advantages over traditional 2D cell cultures in that they more accurately reflect the 3-dimensional tissue architecture and even can mimic properties of the TME including nutrient gradients and incorporating immune or stromal cells. They are, however, more labor and time intensive, and can be limited by higher costs than 2-dimensional cell lines. Additionally, similar to 2D cultures, organoids can develop genetic alteration differences from the primary tumor as the organoids are serially passaged and subjected to in vitro selection pressures (70). Regardless, these preclinical models provide a rational design for testing personalized targeted therapeutic regimens and warrant further prospective investigations.
Microfluidic (Microphysiological) Models
The last few years have seen an explosion in microfluidic tumor models, often called microphysiological models (71). Microfluidic models are housed in microscale in vitro devices that allow for the manipulation of fluids at microliter volumes to control microenvironmental conditions, create biochemical gradients and allow for work with extremely small quantities of samples (72, 73). Of note, microfluidic devices are especially useful for recreating different compartments and geometries to generate physiologically-relevant structures and generate organized co-cultures for in vitro studies (Figure 2).
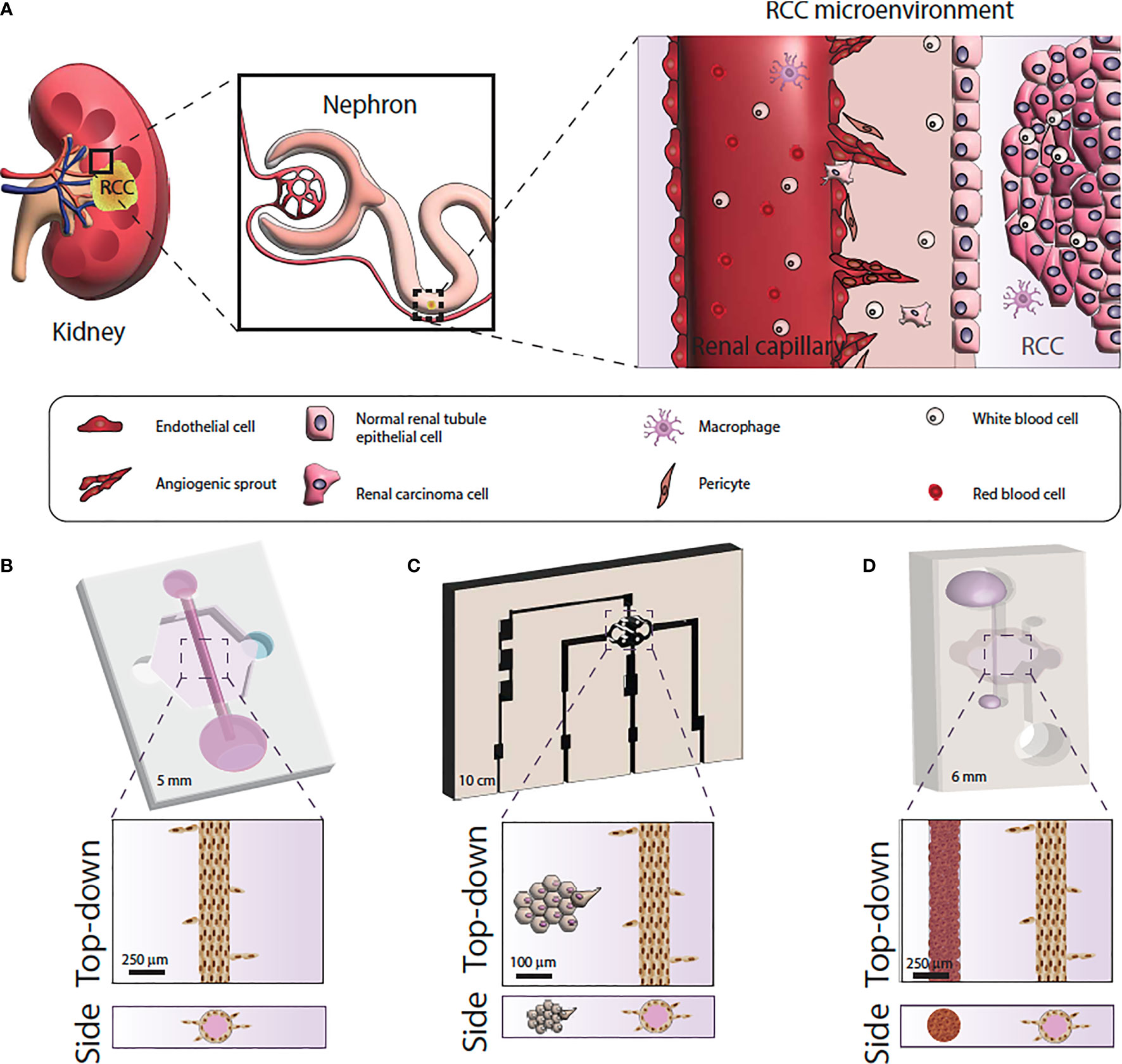
Figure 2 Microfluidic model systems used for renal cell carcinoma research. (A) Schematic of the RCC tumor microenvironment. (B) Microfluidic model of RCC response using primary normal and tumor-associated endothelial vessels. (C) Microfluidic model of RCC angiogenesis using primary epithelial-derived spheroids and Human Umbilical Cord Endothelial cell vessel models. (D) Microfluidic of RCC development and angiogenesis using RCC cell lines or primary epithelial cells and iPSC-derived endothelial cell vessels.
Although the field of microfluidics started to develop in the early 2000s, only recently have microfluidic models come of age and gained acceptance as in vitro tools (74). Therefore, microfluidic models of normal kidney function (or kidney-on-a-chip) have been reported since the early 2010s (75). These models initially consisted of 2D cultures of kidney cell lines where fluid flow was applied to observe the effects of this mechanical stimulus on kidney cell behavior (e.g., changes in morphology, inflammation) (76). However, overtime these models have evolved to include biologically derived matrices, relevant tubular geometries to recreate the renal tubule, and a nearby blood vessel model to mimic a nephron more closely (77). To date, these studies used models to investigate normal kidney function and nephrotoxicity (78–80).
However, microfluidic models are particularly relevant in RCC research. Renal cell carcinoma produces significant angiogenic signals leading to neovascularity formation. Tumor-associated vessels differ from normal vessel in organization, angiogenic sprouting and vessel permeability (81). Traditional in vitro models have been unable to recapitulate tumor-induced angiogenesis, but microfluidics have demonstrated promise in modeling tumor neovascularity (82) and been used to test drugs that target angiogenesis (81).
Despite the great potential of microfluidic modeling in RCC research, few microfluidic RCC models exist. Researchers have mimicked the 3-dimensional vascular microenvironment of RCC by placing tumor cell clusters around engineered human vessels that are subjected to continuous flow (83). Using this design, one study demonstrated that ccRCC cells could stimulate angiogenesis due to the tumor cell upregulation of angiogenic factors, and create what the investigators termed “ccRCC-on-a-chip” (83). Another study used patient-derived normal and tumor-associated endothelial cells to create microfluidic tubular vessel models from multiple patients (81). The microfluidic device demonstrated higher permeability in vessel models built from tumor-associated endothelial cells, recapitulating the phenotypic differences between normal and tumor associated microvasculature. The researchers were able to test anti-angiogenic drugs using the microfluidic devices which caused decreased vessel sprouting and restored tumor vessel permeability (81). A recent study used cell lines and induced pluripotent stem cell-derived endothelial cells to recapitulate RCC development and angiogenic response. Specifically, the authors recapitulated RCC hallmarks like hypoxia, glycolic metabolism, and sprouting angiogenesis, and then tested the model response to a common anti-angiogenic drug. The authors also demonstrated the same model using patient-derived RCC cells instead of cell lines with equivalent results (84).
Microfluidic modeling presents multiple advantages for in vitro research, such as inclusion of many cell types, integration of mechanical cues (e.g., extracellular matrices, perfusion of media) and real-time monitoring capabilities. The high degree of control in microfluidic devices makes these models ideally suited for anti-cancer drug testing studies and investigations into mechanisms of pathology. These types of physiological investigations are more difficult using in vivo models in which the tumor cannot be easily separated into certain components (such as only investigating the tumor cell interaction with the surrounding microvasculature) (72). Existing models are limited by only incorporating 1-2 cell types, thereby failing to include the complex interactions between multiple cell types such as tumor, stromal, and immune cells in RCC (85, 86). Thus, existing RCC research using these devices has typically focused on a single physiologic process within the tumor environment, but there is great potential for more complex RCC modeling using microfluidics (72, 87).
Xenograft Models of Renal Cell Carcinoma
While 2- and 3-D cultures provide an efficient, low-cost way to study cellular mechanisms and potential responses to drug therapies, these models are ultimately limited by their inability to capture the heterogenous tumor microenvironment in vivo including tumor interactions with the surrounding stromal and inflammatory cells. One method to overcome this limitation is to utilize mouse models bearing RCC tumors. These tumors can be generated from a variety of sources. One method involves the xenotransplantation of established RCC cell lines (also known as cell derived xenografts). Another option includes transplantation of tumor tissue taken directly from patient tumor samples into immunodeficient mice to allow growth of the tumors. These tumors are known as “patient derived xenograft” models or PDX models. Newer advances in genetic engineering have allowed for the creation of genetically engineered mouse models (GEMMs), which are discussed below. GEMMs are advantageous because they can mimic the genetic composition, cellular interactions, and therapeutic response of human tumors (88). Cancer cell line transplantation into mice is a commonly used method allowing for rapid preclinical drug testing (88), however these models often poorly predict clinical response in humans. PDX models more faithfully recapitulate the genetic intratumoral heterogeneity and can allow for a personalized medicine approach to drug screening (88, 89). These tumor models are limited by low engraftment rates and must be performed in an immunocompromised mouse, making immunotherapy testing impossible. This is a significant limitation in the current immunotherapy dominant era for advanced RCC treatment (88–90).
Multiple studies have demonstrated the feasibility of developing patient derived xenograft models of RCC. Grisanzio et al. developed a panel of PDX models in immunodeficient mice (91). The authors implanted 20 patient-derived RCC tumors orthotopically (under the renal capsule) using clear cell, papillary and oncocytoma tumors. They demonstrated a high engraftment rate at 95% and found PDX tumors maintained the same genetic characteristics of the original tumors (91). They also noted two cases that developed locally invasive disease and one that developed distant metastases (91). Sivanand et al. established PDX models to investigate molecularly targeted therapies (92). They found engraftment rates were higher for tumors derived from metastatic sites compared to primary kidney tumors (80% versus 14%, respectively) (92). Additional studies have supported this finding, demonstrating higher engraftment rates derived from metastatic sites compared to primary tumors (93). In a large-scale study of PDX model development, Lang et al. implanted 336 RCC tumors of various histologic subtypes into nude mice, with a stable engraftment rate (i.e., greater than 3 successful passages) of 8.9% (94). The authors found higher stable engraftment rates with increasing stage and grade of primary tumors, as well as among tumors with sarcomatoid features. Importantly, the PDX models maintained their genetic characteristics after serial passages. The models also showed significant variation after exposure to different targeted therapies including sunitinib, sorafenib and everolimus, similar to tumor responses in humans (94). Stable engraftment of PDX tumors has shown prognostic value and been associated with worse survival in humans, suggesting a more aggressive tumor phenotype when tumors are able to stably engraft in mice (92, 95, 96). Sivanand et al. additionally evaluated PDX tumor response to targeted therapies sunitinib and sirolimus. Both therapies inhibited PDX tumor growth similar to responses in human tumors (92). They further tested the investigational drug dovitinib, which inhibits VEGFR1-3, PDGFRβ, and FGFR1-3. Dovitinib inhibited PDX tumor growth to a greater extent than either sunitinib or sirolimus, and was well tolerated by the mice (92). This study demonstrated the applicability of using PDX models as a model of human tumor clinical outcomes and responses to targeted therapies, which do not require an active immune system to work. Dong et al. importantly demonstrated that PDX models more accurately represented responses to sunitinib than cell lines derived from the same primary tumor, suggesting that the in vivo models of RCC more faithfully recapitulate human tumor responses than the patient derived cell lines (93).
More recent developments in PDX models include creating a “humanized” mouse model. Humanized mouse models are created by transplantation of a human tumor graft into an immunodeficient mouse and simulating the human immune system by simultaneously transplanting human peripheral blood or tumor infiltrating lymphocytes into the mouse model. This is advantageous because it allows for testing immunotherapeutics within PDX models, which traditionally lack components of the immune system (97). Another method for generating humanized models involves the transplantation of human hematopoietic stem cells (HSCs), allowing for more complete restoration of all hematopoietic cells. These models are limited by the availability of HSCs able to be obtained from cancer patients (98). While humanized PDX models allow for better evaluation of immune therapies, they still are limited by the lack of incorporation of all cell types, robust or reliable immune response, and the frequent development of graft versus host disease in the mouse (98). PDX models have been used as a robust platform for rapid preclinical drug testing and many centers continue to maintain large tumorgraft colonies derived from patient tumor samples (99, 100). These will continue to be widely used as preclinical models for RCC-based studies.
Genetically Engineered Mouse Models
Genetically engineered mouse models (GEMMs) provide a method to study tumorigenesis and metastasis as well as therapeutic testing similar to PDX models. GEMMs are particularly attractive for oncology research due to the relative ease the mouse genome can be manipulated to inactivate tumor suppressors or activate oncogenes (101, 102). These models can be used in a variety of ways for translational research. GEMMs can be used to test novel drug targets, evaluate therapeutic response as well as mechanisms of resistance, and may be used for immunotherapeutic research as these models typically maintain an intact immune system (88). Current GEMMs allow for the conditional somatic inactivation of tumor suppressor genes or activation of oncogenes allowing for tissue specific tumorigenesis (88, 103). Mechanisms to achieve this often utilize Cre-loxP system. Cre-recombinase is a DNA recombinase derived from bacteriophage that causes recombination at 34 base-pair recognition sites called loxP (104). Genes flanked by the loxP recombination site will be deleted after Cre-recombinase activation is achieved such as through cellular delivery of Cre-recombinase by adenovirus (88). Somatic mutations can also be induced at specific times by utilizing Cre-ERT fusion proteins (88). The estrogen receptor binding domain is fused to Cre-recombinase, and administration of tamoxifen to the mouse causes the expression of Cre-recombinase leading to excision of loxP flanked genes (88). These systems allow for immunocompetent mouse models which produce de novo tumors that mimic human tumors.
While valuable, they can be labor intensive, expensive, slow to develop tumors, and require a thorough understanding of the differences between some of the RCC driver gene locations in humans and mice (Figure 3) (88). Recent advances have led to the development of autochthonous RCC models (27, 105–109). Initial efforts to generate GEMMs of ccRCC focused on inactivation of Vhl in mice, since biallelic inactivation of VHL is present in over 90% of sporadic ccRCC in humans (28, 110–112). Interestingly, multiple groups attempted to generate ccRCC tumors in mice through either embryonic or conditional knockout of Vhl in mice without successful development of tumors (105, 108). This led to the realization that additional mutations are necessary to generate ccRCC tumors in humans and mice (105, 108). In humans, the VHL gene is located next to other tumor suppressors (e.g., PBRM1, BAP1 and SETD2) on chromosome 3p. One explanation for the lack of development of ccRCC in mice by sole knockdown of Vhl is that the mouse Vhl gene is located on chromosome 6 while Pbrm1 and Bap1 are located on chromosome 14 and Setd2 is located on chromosome 9 (Figure 3). Thus, loss of Vhl in mice still leaves functional Pbrm1, Bap1, and Setd2 genes, while loss of chromosome 3p, which is the typical first-hit event in humans, often causes loss of PBRM1, BAP1 and, SETD2, predisposing humans to additional second-hit inactivating mutations in these genes and subsequent tumor development (107).
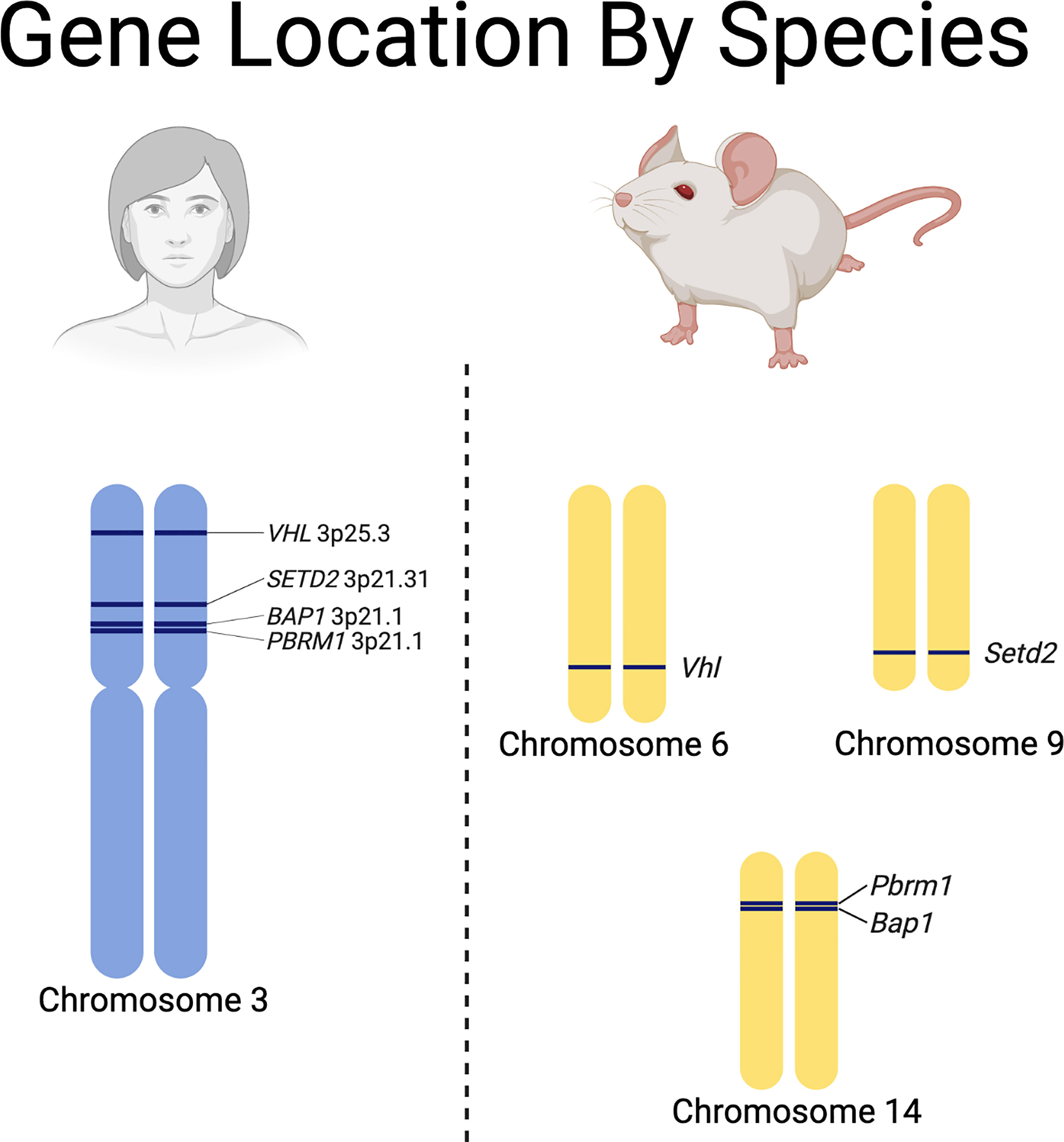
Figure 3 Species specific differences in the chromosomal locations of renal cell carcinoma driver genes.
Harlander et al. demonstrated that mutations in mouse Vhl combined with Trp53 and Rb1 in renal epithelial cells successfully generated ccRCC tumors in mice (27). To do this, the authors utilized the inducible renal epithelial cell specific Ksp1.3-CreERT2 homozygous deletion of the loxP-flanked alleles of Rb1, Vhl, and Trp53. These deletions were induced by exposing mice to tamoxifen. Eighty-two percent of the triple deletion mice developed renal tumors with histologic and transcriptional similarities to human ccRCC (27). The authors further treated mice with sunitinib and demonstrated, similar to humans, heterogenous responses to the drug. After exposure to sunitinib, 32% of tumors progressed, 16% of tumors regressed and 53% of the tumors remained stable in size (27). While VHL, TP53, and RB1 are not common coincident mutations in ccRCC, the investigators speculate that this background mimics the copy number alterations of p53 pathway regulators and cell cycle control enzymes typically found in human ccRCC tumors (27). This model permits the evolution of genetically distinct ccRCC in each mouse (27).
Gu et al. developed a GEMM using a Pax8-Cre deletion of Vhl along with Bap1 and Pbrm1 (107). Deletion of Vhl and Bap1 led to expected changes in effector pathways including upregulation of Hif-1 and Hif-2 as well as increased ubiquitinated histone H2A protein which is normally deubiquitinated by BAP1 (107). Creation of mice that were either deficient in Bap1 or Pbrm1 along with Vhl developed multiple cystic and solid lesions in the kidneys similar to human ccRCC (107). Also similar to humans, Bap1 deficient tumors tended to be higher grade compared to Pbrm1 deficient tumors which were slower to develop, more homogenous and lower grade (29, 107). Bailey et al. developed papillary RCC GEMMs through renal tubular cell specific activation of MYC (109). When the authors combined MYC activation with Vhl and Cdkn2a deletion, kidney tumors more closely resembled ccRCC (109). Clear cell RCC frequently contains focal gains of 8q24 which harbors the MYC gene, as well as losses of 9p21 which harbors CDKN2A (22, 109, 113). This model was unique in that it demonstrated metastatic capability, with 1/3 of the mice developing liver metastases. This model is limited in that only about 6% of human ccRCC tumors have VHL and CDKN2A inactivation with MYC overexpression (109).
These studies demonstrate the utility of GEMMs in recapitulating human RCC and the ability to use these models to study mechanisms of tumorigenesis (Table 2). These models, however, require a thorough understanding of the human and mouse genome and gene-expression differences, which often explain their phenotypic differences between mouse and human pathology (114, 115).

Table 2 Genetically engineered mouse models used for kidney cancer research.
Conclusion
RCC models that incorporate a reliable representation of tumor genomic and phenotypic behavior in addition to the surrounding TME and host response are lacking. Strengths of the individual systems must be balanced against each system’s unique limitations (Figure 4). While cell lines have been the backbone of oncology research in understanding cellular mechanisms, these systems are limited by divergent genetic changes in vitro compared to human tumors. RCC cell lines frequently respond differently to drug therapy and lack a surrounding cellular microenvironment, which is important for RCC tumor biology. Organoids provide a more realistic tumor physical structure and may be developed with components of the surrounding TME, but still lack many of the complex components and interactions of the tumor environment. PDX models provide a more biologically relevant model which may be particularly useful to test drug therapies on individual tumors for a personalized medicine approach. These models, however, often require an immunocompromised mouse which limits the ability to study therapeutics targeting the immune response. Development of humanized models continues to expand in an effort to try and overcome these limitations. GEMMs provide an opportunity for the study of de novo tumor development and investigations into clonal evolution and metastases. These models feature immunocompetent hosts providing a more relevant platform for immunotherapeutic testing. All models require specific expertise and experience to effectively utilize. As each model continues to develop, further discoveries into the driver events of RCC will create new priorities for model development. The insights gained will allow for new diagnostic and therapeutic approaches as well as individualized approaches to RCC management.
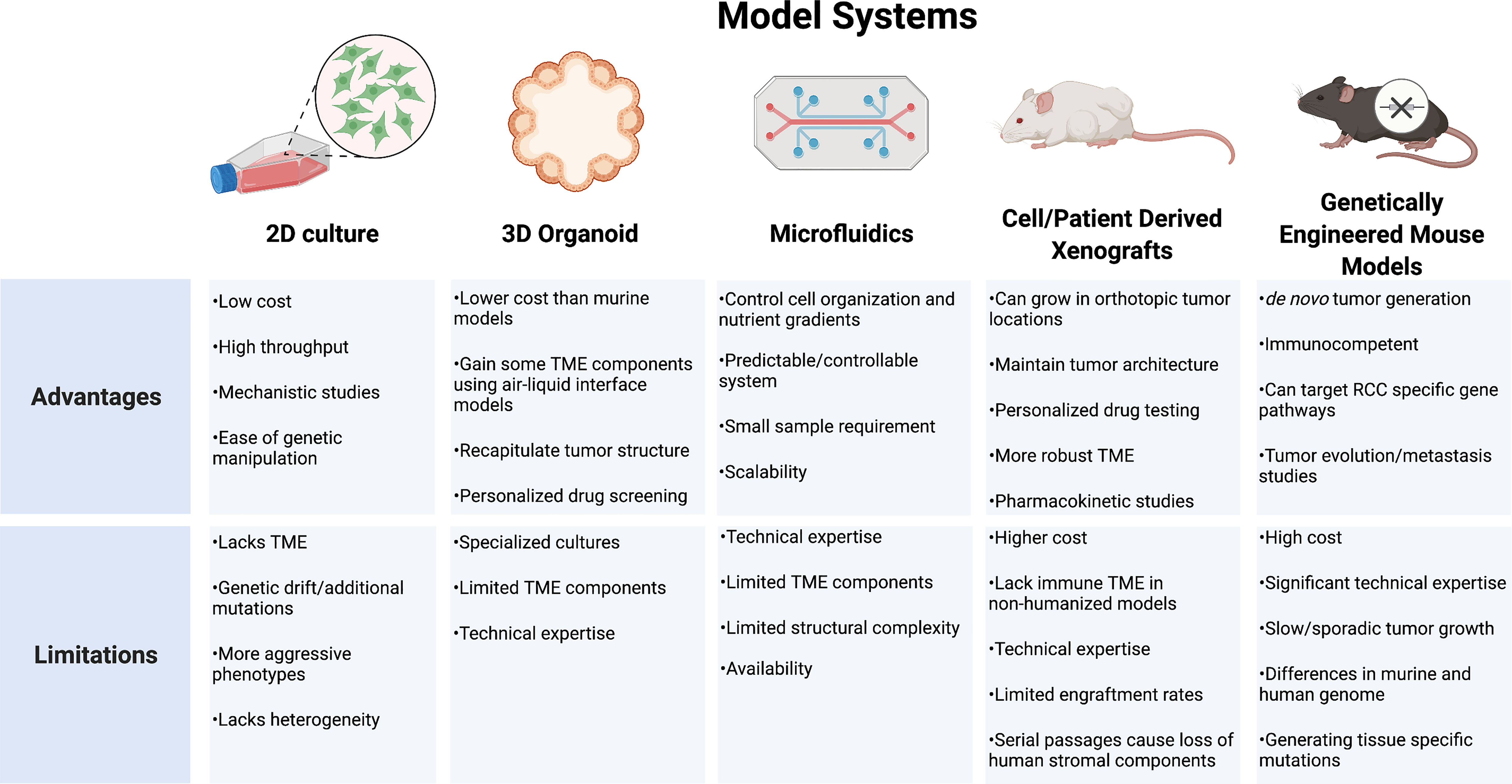
Figure 4 Model systems used for renal cell carcinoma research. TME, tumor microenvironment; RCC, renal cell carcinoma.
Author Contributions
All authors contributed to the conceptualization, writing, editing, figure and table development. All authors contributed to the article and approved the submitted version.
Conflict of Interest
DB holds equity in Bellbrook Labs LLC, Tasso Inc., Turba LLC, Salus Discovery LLC, Stacks to the Future LLC, Lynx Biosciences Inc., Flambeau Diagnostics, and Onexio Biosystems.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures generated with biorender.com.
References
1. Devarasetty M, Forsythe SD, Shelkey E, Soker S. In Vitro Modeling of the Tumor Microenvironment in Tumor Organoids. Tissue Eng regenerative Med (2020) 17(6):759–71. doi: 10.1007/s13770-020-00258-4
2. Balkwill FR, Capasso M, Hagemann T. The Tumor Microenvironment at a Glance. J Cell Sci (2012) 125(23):5591–96. doi: 10.1242/jcs.116392
3. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell communication Signaling CCS (2020) 18(1):59. doi: 10.1186/s12964-020-0530-4
4. Anderson NM, Simon MC. The Tumor Microenvironment. Curr Biol CB (2020) 30(16):R921–5. doi: 10.1016/j.cub.2020.06.081
5. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
6. Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell (2020) 78(6):1019–33. doi: 10.1016/j.molcel.2020.05.034
7. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discovery (2021) 11(4):933–59. doi: 10.1158/2159-8290.CD-20-1808
8. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
9. Jonasch E, Gao J, Rathmell WK. Renal Cell Carcinoma. BMJ (Clinical Res ed) (2014) 349:g4797. doi: 10.1136/bmj.g4797
10. Padala SA, Barsouk A, Thandra KC, Saginala K, Mohammed A, Vakiti A, et al. Epidemiology of Renal Cell Carcinoma. World J Oncol (2020) 11(3):79–87. doi: 10.14740/wjon1279
11. Campbell SC, Clark PE, Chang SS, Karam JA, Souter L, Uzzo RG. Renal Mass and Localized Renal Cancer: Evaluation, Management, and Follow-Up: AUA Guideline: Part I. J Urol (2021) 206(2):199–208. doi: 10.1097/JU.0000000000001911
12. Campbell SC, Uzzo RG, Karam JA, Chang SS, Clark PE, Souter L. Renal Mass and Localized Renal Cancer: Evaluation, Management, and Follow-Up: AUA Guideline: Part II. J Urol (2021) 206(2):209–18. doi: 10.1097/JU.0000000000001912
13. Motzer RJ, Jonasch E, Agarwal N, Alva A, Baine M, Beckermann K, et al. Kidney Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Network (2022) 20(1):71–90. doi: 10.6004/jnccn.2022.0001
14. Choueiri TK, Motzer RJ. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N Engl J Med (2017) 376(4):354–66. doi: 10.1056/NEJMra1601333
15. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New Engl J Med (2012) 366(10):883–92. doi: 10.1056/NEJMoa1113205
16. Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell (2018) 173(3):595–610.e11. doi: 10.1016/j.cell.2018.03.043
17. Xu W, Atkins MB, McDermott DF. Checkpoint Inhibitor Immunotherapy in Kidney Cancer. Nat Rev Urol (2020) 17:137–50. doi: 10.1038/s41585-020-0282-3
18. Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep (2018) 23(1):313–26.e5. doi: 10.1016/j.celrep.2018.03.075
19. Beksac AT, Paulucci DJ, Blum KA, Yadav SS, Sfakianos JP, Badani KK. Heterogeneity in Renal Cell Carcinoma. Urologic Oncology: Semin Original Investigations (2017) 35(8):507–15. doi: 10.1016/j.urolonc.2017.05.006
20. Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, et al. Understanding Pathologic Variants of Renal Cell Carcinoma: Distilling Therapeutic Opportunities From Biologic Complexity. Eur Urol (2015) 67(1):85–97. doi: 10.1016/j.eururo.2014.04.029
21. Warren AY, Harrison D. WHO/ISUP Classification, Grading and Pathological Staging of Renal Cell Carcinoma: Standards and Controversies. World J Urol (2018) 36(12):1913–26. doi: 10.1007/s00345-018-2447-8
22. Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Robertson AG, et al. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature (2013) 499(7456):43–9. doi: 10.1038/nature12222
23. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal Cell Carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi: 10.1038/nrdp.2017.9
24. Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumour Suppression by the Human Von Hippel-Lindau Gene Product. Nat Med (1995) 1(8):822–6. doi: 10.1038/nm0895-822
25. Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O'Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell (2018) 173(3):611–23.e17. doi: 10.1016/j.cell.2018.02.020
26. Chiang YC, Park IY, Terzo EA, Tripathi DN, Mason FM, Fahey CC, et al. SETD2 Haploinsufficiency for Microtubule Methylation Is an Early Driver of Genomic Instability in Renal Cell Carcinoma. Cancer Res (2018) 78(12):canres.3460.2017. doi: 10.1158/0008-5472.can-17-3460
27. Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, et al. Combined Mutation in Vhl, Trp53 and Rb1 Causes Clear Cell Renal Cell Carcinoma in Mice. Nat Med (2017) 23(7):869–77. doi: 10.1038/nm.4343
28. Kleymenova E, Everitt JI, Pluta L, Portis M, Gnarra JR, Walker CL. Susceptibility to Vascular Neoplasms But No Increased Susceptibility to Renal Carcinogenesis in Vhl Knockout Mice. Carcinogenesis (2004) 25(3):309–15. doi: 10.1093/carcin/bgh017
29. Brugarolas J. Molecular Genetics of Clear-Cell Renal Cell Carcinoma. J Clin Oncol Off J Am Soc Clin Oncol (2014) 32(18):1968–76. doi: 10.1200/JCO.2012.45.2003
30. Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, Wang S, et al. BAP1 Loss Defines a New Class of Renal Cell Carcinoma. Nat Genet (2012) 44(7):751–9. doi: 10.1038/ng.2323
31. Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell (2018) 173(3):581–94.e12. doi: 10.1016/j.cell.2018.03.057
32. Haake SM, Weyandt JD, Rathmell WK. Insights Into the Genetic Basis of the Renal Cell Carcinomas From The Cancer Genome Atlas. Mol Cancer Res MCR (2016) 14(7):589–98. doi: 10.1158/1541-7786.MCR-16-0115
33. Wettersten HI, Aboud OA, Lara PN, Weiss RH. Metabolic Reprogramming in Clear Cell Renal Cell Carcinoma. Nat Rev Nephrol (2017) 13(7):410–9. doi: 10.1038/nrneph.2017.59
34. Cimadamore A, Cheng L, Scarpelli M, Massari F, Mollica V, Santoni M, et al. Towards a New WHO Classification of Renal Cell Tumor: What the Clinician Needs to Know—a Narrative Review. Trans Andrology Urol (2020) 10(3):1506520–1520. doi: 10.21037/tau-20-1150
35. Klinghammer K, Walther W, Hoffmann J. Choosing Wisely - Preclinical Test Models in the Era of Precision Medicine. Cancer Treat Rev (2017) 55:36–45. doi: 10.1016/j.ctrv.2017.02.009
36. Wolf MM, Rathmell WK, Beckermann KE. Modeling Clear Cell Renal Cell Carcinoma and Therapeutic Implications. Oncogene (2020) 39(17):3413–26. doi: 10.1038/s41388-020-1234-3
37. Shoemaker RH. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat Rev Cancer (2006) 6(10):813–23. doi: 10.1038/nrc1951
38. Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM. Choosing the Right Cell Line for Renal Cell Cancer Research. Mol Cancer (2016) 15(1):83. doi: 10.1186/s12943-016-0565-8
39. Borodovsky A, McQuiston TJ, Stetson D, Ahmed A, Whitston D, Zhang J, et al. Generation of Stable PDX Derived Cell Lines Using Conditional Reprogramming. Mol Cancer (2017) 16(1):177. doi: 10.1186/s12943-017-0745-1
40. Sinha R, Winer AG, Chevinsky M, Jakubowski C, Chen Y-B, Dong Y, et al. Analysis of Renal Cancer Cell Lines From Two Major Resources Enables Genomics-Guided Cell Line Selection. Nat Commun (2017) 8(1):15165. doi: 10.1038/ncomms15165
41. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature (2012) 483(7391):603–7. doi: 10.1038/nature11003
42. Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-Inducible Factor Determines Sensitivity to Inhibitors of mTOR in Kidney Cancer. Nat Med (2006) 12(1):122–7. doi: 10.1038/nm1337
43. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res (2018) 47(Database issue):gky1015. doi: 10.1093/nar/gky1015
44. Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is Necessary for Tumor Suppression by the Von Hippel-Lindau Protein. Cancer Cell (2002) 1(3):237–46. doi: 10.1016/s1535-6108;(>02;)>00043-0
45. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for Renal Cell Carcinoma in Von Hippel–Lindau Disease. New Engl J Med (2021) 385(22):2036–46. doi: 10.1056/nejmoa2103425
46. Ghatalia P, Rathmell WK. Systematic Review: ClearCode 34 – A Validated Prognostic Signature in Clear Cell Renal Cell Carcinoma (ccRCC). Kidney Cancer (2018) 2(1):23–9. doi: 10.3233/kca-170021
47. Robb VA, Karbowniczek M, Klein-Szanto AJ, Henske EP. Activation of the mTOR Signaling Pathway in Renal Clear Cell Carcinoma. J Urol (2007) 177(1):346–52. doi: 10.1016/j.juro.2006.08.076
48. Lobo NC, Gedye C, Apostoli AJ, Brown KR, Paterson J, Stickle N, et al. Efficient Generation of Patient-Matched Malignant and Normal Primary Cell Cultures From Clear Cell Renal Cell Carcinoma Patients: Clinically Relevant Models for Research and Personalized Medicine. BMC Cancer (2016) 16(1):485. doi: 10.1186/s12885-016-2539-z
49. Schmeichel KL, Bissell MJ. Modeling Tissue-Specific Signaling and Organ Function in Three Dimensions. J Cell Sci (2003) 116(12):2377–88. doi: 10.1242/jcs.00503
50. Grassi L, Alfonsi R, Francescangeli F, Signore M, Angelis MLD, Addario A, et al. Organoids as a New Model for Improving Regenerative Medicine and Cancer Personalized Therapy in Renal Diseases. Cell Death Dis (2019) 10(3):201. doi: 10.1038/s41419-019-1453-0
51. Clevers H. Modeling Development and Disease With Organoids. Cell (2016) 165(7):1586–97. doi: 10.1016/j.cell.2016.05.082
52. Rodenhizer D, Gaude E, Cojocari D, Mahadevan R, Frezza C, Wouters BG, et al. A Three-Dimensional Engineered Tumour for Spatial Snapshot Analysis of Cell Metabolism and Phenotype in Hypoxic Gradients. Nat Materials (2016) 15(2):227–34. doi: 10.1038/nmat4482
53. Sachs N, Jd L, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell (2018) 172(1-2):373–86.e10. doi: 10.1016/j.cell.2017.11.010
54. Gao D, Vela I, Sboner A, Iaquinta Phillip J, Karthaus Wouter R, Gopalan A, et al. Organoid Cultures Derived From Patients With Advanced Prostate Cancer. Cell (2014) 159(1):176–87. doi: 10.1016/j.cell.2014.08.016
55. Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal Pancreatic Cancer Modeling and Drug Screening Using Human Pluripotent Stem Cell– and Patient-Derived Tumor Organoids. Nat Med (2015) 21(11):1364–71. doi: 10.1038/nm.3973
56. Boehnke K, Iversen PW, Schumacher D, Lallena MJ, Haro R, Amat J, et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. Slas Discovery (2016) 21(9):931–41. doi: 10.1177/1087057116650965
57. Kozlowski MT, Crook CJ, Ku HT. Towards Organoid Culture Without Matrigel. Commun Biol (2021) 4(1):1387. doi: 10.1038/s42003-021-02910-8
58. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab Plus Ipilimumab Versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
59. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab Plus Axitinib Versus Sunitinib for Advanced Renal-Cell Carcinoma. New Engl J Med (2019) 380(12):1103–15. doi: 10.1056/nejmoa1816047
60. Santoni M, Massari F, Nunno VD, Conti A, Cimadamore A, Scarpelli M, et al. Immunotherapy in Renal Cell Carcinoma: Latest Evidence and Clinical Implications. Drugs Context (2018) 7:212528. doi: 10.7573/dic.212528
61. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
62. Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab Plus Bevacizumab Versus Sunitinib in Patients With Previously Untreated Metastatic Renal Cell Carcinoma (IMmotion151): A Multicentre, Open-Label, Phase 3, Randomised Controlled Trial. Lancet (2019) 393(10189):2404–15. doi: 10.1016/s0140-6736;(>19;)>30723-8
63. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab Plus Axitinib Versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med (2019) 380(12):1116–27. doi: 10.1056/NEJMoa1816714
64. Choueiri TK, Tomczak P, Park SH, Venugopal B, Ferguson T, Chang Y-H, et al. Adjuvant Pembrolizumab After Nephrectomy in Renal-Cell Carcinoma. New Engl J Med (2021) 385(8):683–94. doi: 10.1056/nejmoa2106391
65. Klaassen Z, Satkunasivam R, Wallis CJD. Immune Checkpoint Blockade Plus Axitinib for Renal-Cell Carcinoma. New Engl J Med (2019) 380(26):2581–2. doi: 10.1056/nejmc1905518
66. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination With Bevacizumab Versus Sunitinib in Renal Cell Carcinoma. Nat Med (2018) 24(6):749–57. doi: 10.1038/s41591-018-0053-3
67. Lopez-Beltran A, Henriques V, Cimadamore A, Santoni M, Cheng L, Gevaert T, et al. The Identification of Immunological Biomarkers in Kidney Cancers. Front Oncol (2018) 8:456:. doi: 10.3389/fonc.2018.00456
68. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell (2018) 175(7):1972–88.e16. doi: 10.1016/j.cell.2018.11.021
69. Esser LK, Branchi V, Leonardelli S, Pelusi N, Simon AG, Klümper N, et al. Cultivation of Clear Cell Renal Cell Carcinoma Patient-Derived Organoids in an Air-Liquid Interface System as a Tool for Studying Individualized Therapy. Front Oncol (2020) 10:1775:. doi: 10.3389/fonc.2020.01775
70. Bolck HA, Corrò C, Kahraman A, Teichman AV, Toussaint NC, Kuipers J, et al. Tracing Clonal Dynamics Reveals That Two- and Three-Dimensional Patient-Derived Cell Models Capture Tumor Heterogeneity of Clear Cell Renal Cell Carcinoma. Eur Urol Focus (2021) 7(1):152–62. doi: 10.1016/j.euf.2019.06.009
71. Wang YI, Carmona C, Hickman JJ, Shuler ML. Multiorgan Microphysiological Systems for Drug Development: Strategies, Advances, and Challenges. Advanced Healthcare Materials (2018) 7(2):1701000. doi: 10.1002/adhm.201701000
72. Ayuso JM, Park KY, Virumbrales-Muñoz M, Beebe DJ. Toward Improved In Vitro Models of Human Cancer. APL bioengineering (2021) 5(1):010902. doi: 10.1063/5.0026857
73. Atencia J, Beebe DJ. Controlled Microfluidic Interfaces. Nature (2005) 437(7059):648–55. doi: 10.1038/nature04163
74. Virumbrales-Muñoz M, Ayuso JM. From Microfluidics to Microphysiological Systems: Past, Present, and Future. Organs-on-a-Chip (2022) 4:100015. doi: 10.1016/j.ooc.2022.100015
75. Zanetti F. Organ-On-a-Chip . Neuchâtel, Switzerland: PMI R&D, Philip Morris Products S.A. (2020), pp. 233–53. doi: 10.1016/b978-0-12-817202-5.00007-3
76. Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh KY, et al. Human Kidney Proximal Tubule-on-a-Chip for Drug Transport and Nephrotoxicity Assessment. Integr Biol quantitative Biosci nano to macro (2013) 5(9):1119–29. doi: 10.1039/c3ib40049b
77. Mu X, Zheng W, Xiao L, Zhang W, Jiang X. Engineering a 3D Vascular Network in Hydrogel for Mimicking a Nephron. Lab chip (2013) 13(8):1612–18. doi: 10.1039/c3lc41342j
78. Adler M, Ramm S, Hafner M, Muhlich JL, Gottwald EM, Weber E, et al. A Quantitative Approach to Screen for Nephrotoxic Compounds In Vitro. J Am Soc Nephrol JASN (2016) 27(4):1015–28. doi: 10.1681/ASN.2015010060
79. Lee J, Kim S. Kidney-On-a-Chip: A New Technology for Predicting Drug Efficacy, Interactions, and Drug-Induced Nephrotoxicity. Curr Drug Metab (2018) 19(7):577–83. doi: 10.2174/1389200219666180309101844
80. Jansen J, Fedecostante M, Wilmer MJ, Peters JG, Kreuser UM, van den Broek PH, et al. Bioengineered Kidney Tubules Efficiently Excrete Uremic Toxins. Sci Rep (2016) 6:26715. doi: 10.1038/srep26715
81. Virumbrales-Muñoz M, Chen J, Ayuso J, Lee M, Abel EJ, Beebe DJ. Organotypic Primary Blood Vessel Models of Clear Cell Renal Cell Carcinoma for Single-Patient Clinical Trials. Lab Chip (2020) 20(23):4420–32. doi: 10.1039/d0lc00252f
82. Virumbrales-Muñoz M, Ayuso JM, Gong MM, Humayun M, Livingston MK, Lugo-Cintrón KM, et al. Microfluidic Lumen-Based Systems for Advancing Tubular Organ Modeling. Chem Soc Rev (2020) 49:6402–42. doi: 10.1039/d0cs00705f
83. Miller CP, Tsuchida C, Zheng Y, Himmelfarb J, Akilesh S. A 3d Human Renal Cell Carcinoma-On-a-Chip for the Study of Tumor Angiogenesis. Neoplasia (New York NY) (2018) 20(6):610–20. doi: 10.1016/j.neo.2018.02.011
84. Virumbrales-Muñoz M, Ayuso JM, Loken JR, Denecke KM, Rehman S, Skala MC, et al. Microphysiological Model of the Renal Cell Carcinoma to Inform Anti-Angiogenic Therapy. Biomaterials (2022) 283:121454. doi: 10.1016/j.biomaterials.2022.121454
85. Lyu Z, Park J, Kim KM, Jin HJ, Wu H, Rajadas J, et al. A Neurovascular-Unit-on-a-Chip for the Evaluation of the Restorative Potential of Stem Cell Therapies for Ischaemic Stroke. Nat Biomed Eng (2021) 5(8):847–63. doi: 10.1038/s41551-021-00744-7
86. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, et al. On-Chip Recapitulation of Clinical Bone-Marrow Toxicities and Patient-Specific Pathophysiology. Nat Biomed Eng (2020) 4(4):394–406. doi: 10.1038/s41551-019-0495-z
87. Ma Y-HV, Middleton K, You L. Sun Y. A Review of Microfluidic Approaches for Investigating Cancer Extravasation During Metastasis. Microsystems Nanoengineering (2018) 4(1):17104. doi: 10.1038/micronano.2017.104
88. Kersten K, Visser KED, Miltenburg MHV, Jonkers J. Genetically Engineered Mouse Models in Oncology Research and Cancer Medicine. EMBO Mol Med (2017) 9(2):137–53. doi: 10.15252/emmm.201606857
89. Okada S, Vaeteewoottacharn K, Kariya R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells (2019) 8(8):889. doi: 10.3390/cells8080889
90. Invrea F, Rovito R, Torchiaro E, Petti C, Isella C, Medico E. Patient-Derived Xenografts (PDXs) as Model Systems for Human Cancer. Curr Opin Biotechnol (2020) 63:151–6. doi: 10.1016/j.copbio.2020.01.003
91. Grisanzio C, Seeley A, Chang M, Collins M, Napoli AD, Cheng SC, et al. Orthotopic Xenografts of RCC Retain Histological, Immunophenotypic and Genetic Features of Tumours in Patients. J Pathol (2011) 225(2):212–21. doi: 10.1002/path.2929
92. Sivanand S, Peña-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, et al. A Validated Tumorgraft Model Reveals Activity of Dovitinib Against Renal Cell Carcinoma. Sci Trans Med (2012) 4(137):137ra75–ra75. doi: 10.1126/scitranslmed.3003643
93. Dong Y, Manley BJ, Becerra MF, Redzematovic A, Casuscelli J, Tennenbaum DM, et al. Tumor Xenografts of Human Clear Cell Renal Cell Carcinoma But Not Corresponding Cell Lines Recapitulate Clinical Response to Sunitinib: Feasibility of Using Biopsy Samples. Eur Urol Focus (2017) 3(6):590–8. doi: 10.1016/j.euf.2016.08.005
94. Lang H, Béraud C, Bethry A, Danilin S, Lindner V, Coquard C, et al. Establishment of a Large Panel of Patient-Derived Preclinical Models of Human Renal Cell Carcinoma. Oncotarget (2016) 7(37):59336–59. doi: 10.18632/oncotarget.10659
95. Chen J, Jin Y, Li S, Qiao C, Peng X, Li Y, et al. Patient-Derived Xenografts Are a Reliable Preclinical Model for the Personalized Treatment of Epithelial Ovarian Cancer. Front Oncol (2021) 11:744256:. doi: 10.3389/fonc.2021.744256
96. Shin HY, Lee EJ, Yang W, Kim HS, Chung D, Cho H, et al. Identification of Prognostic Markers of Gynecologic Cancers Utilizing Patient-Derived Xenograft Mouse Models. Cancers (2022) 14(3):829. doi: 10.3390/cancers14030829
97. Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. Current Status and Perspectives of Patient-Derived Xenograft Models in Cancer Research. J Hematol Oncol (2017) 10(1):106. doi: 10.1186/s13045-017-0470-7
98. Byrne AT, Alférez DG, Amant F, Annibali D, Arribas J, Biankin AV, et al. Interrogating Open Issues in Cancer Precision Medicine With Patient-Derived Xenografts. Nat Rev Cancer (2017) 17(4):254–68. doi: 10.1038/nrc.2016.140
99. Pavía-Jiménez A, Tcheuyap VT, Brugarolas J. Establishing a Human Renal Cell Carcinoma Tumorgraft Platform for Preclinical Drug Testing. Nat Protoc (2014) 9(8):1848–59. doi: 10.1038/nprot.2014.108
100. Tracey AT, Murray KS, Coleman JA, Kim K. Patient-Derived Xenograft Models in Urological Malignancies: Urothelial Cell Carcinoma and Renal Cell Carcinoma. Cancers (2020) 12(2):439. doi: 10.3390/cancers12020439
101. Rangarajan A, Weinberg RA. Comparative Biology of Mouse Versus Human Cells: Modelling Human Cancer in Mice. Nat Rev Cancer (2003) 3(12):952–9. doi: 10.1038/nrc1235
102. Sobczuk P, Brodziak A, Khan MI, Chhabra S, Fiedorowicz M, Wełniak-Kamińska M, et al. Choosing The Right Animal Model for Renal Cancer Research. Trans Oncol (2020) 13(3):100745. doi: 10.1016/j.tranon.2020.100745
103. Day CP, Merlino G, Van Dyke T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell (2015) 163(1):39–53. doi: 10.1016/j.cell.2015.08.068
104. Shao X, Somlo S, Igarashi P. Epithelial-Specific Cre/lox Recombination in the Developing Kidney and Genitourinary Tract. J Am Soc Nephrol JASN (2002) 13(7):1837–46. doi: 10.1097/01.asn.0000016444.90348.50
105. Hou W, Ji Z. Generation of Autochthonous Mouse Models of Clear Cell Renal Cell Carcinoma: Mouse Models of Renal Cell Carcinoma. Exp Mol Med (2018) 50(4):1–10. doi: 10.1038/s12276-018-0059-4
106. Espana-Agusti J, Warren A, Chew SK, Adams DJ, Matakidou A. Loss of PBRM1 Rescues VHL Dependent Replication Stress to Promote Renal Carcinogenesis. Nat Commun (2017) 8(1):2026. doi: 10.1038/s41467-017-02245-1
107. Gu Y-F, Cohn S, Christie A, McKenzie T, Wolff N, Do QN, et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discovery (2017) 7(8):900–17. doi: 10.1158/2159-8290.cd-17-0292
108. Wang S-S, Gu Y-F, Wolff N, Stefanius K, Christie A, Dey A, et al. Bap1 is Essential for Kidney Function and Cooperates With Vhl in Renal Tumorigenesis. Proc Natl Acad Sci (2014) 111(46):16538–43. doi: 10.1073/pnas.1414789111
109. Bailey ST, Smith AM, Kardos J, Wobker SE, Wilson HL, Krishnan B, et al. MYC Activation Cooperates With Vhl and Ink4a/Arf Loss to Induce Clear Cell Renal Cell Carcinoma. Nat Commun (2017) 8(1):15770. doi: 10.1038/ncomms15770
110. Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular Tumors in Livers With Targeted Inactivation of the Von Hippel–Lindau Tumor Suppressor. Proc Natl Acad Sci (2001) 98(4):1583–8. doi: 10.1073/pnas.98.4.1583
111. Rankin EB, Tomaszewski JE, Haase VH. Renal Cyst Development in Mice With Conditional Inactivation of the Von Hippel-Lindau Tumor Suppressor. Cancer Res (2006) 66(5):2576–83. doi: 10.1158/0008-5472.can-05-3241
112. Pritchett TL, Bader HL, Henderson J, Hsu T. Conditional Inactivation of the Mouse Von Hippel-Lindau Tumor Suppressor Gene Results in Wide-Spread Hyperplastic, Inflammatory and Fibrotic Lesions in the Kidney. Oncogene (2015) 34(20):2631–9. doi: 10.1038/onc.2014.197
113. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated Molecular Analysis of Clear-Cell Renal Cell Carcinoma. Nat Genet (2013) 45(8):860–7. doi: 10.1038/ng.2699
114. Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE, Anagnostopoulos A, et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res (2019) 47(Database issue):D801–D6. doi: 10.1093/nar/gky1056
Keywords: preclinical models, cell culture, organoid, xenograft, microfluidics, renal cell carcinoma
Citation: Shapiro DD, Virumbrales-Muñoz M, Beebe DJ and Abel EJ (2022) Models of Renal Cell Carcinoma Used to Investigate Molecular Mechanisms and Develop New Therapeutics. Front. Oncol. 12:871252. doi: 10.3389/fonc.2022.871252
Received: 08 February 2022; Accepted: 10 March 2022;
Published: 07 April 2022.
Edited by:
Thierry Massfelder, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Kevin Dzobo, University of Cape Town, South AfricaFrans Schutgens, University Medical Center Utrecht, Netherlands
Mariangela Santorsola, University of Bologna, Italy
Sounak Sahu, National Cancer Institute at Frederick (NIH), United States
Vui King Vincent-Chong, University at Buffalo, United States
Copyright © 2022 Shapiro, Virumbrales-Muñoz, Beebe and Abel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: E. Jason Abel, abel@urology.wisc.edu