 Amirali B. Bukhari
Amirali B. Bukhari Gordon K. Chan
Gordon K. Chan Armin M. Gamper
Armin M. Gamper- Department of Oncology, Cross Cancer Institute, University of Alberta, Edmonton, AB, Canada
Cancer cells typically heavily rely on the G2/M checkpoint to survive endogenous and exogenous DNA damage, such as genotoxic stress due to genome instability or radiation and chemotherapy. The key regulator of the G2/M checkpoint, the cyclin-dependent kinase 1 (CDK1), is tightly controlled, including by its phosphorylation state. This posttranslational modification, which is determined by the opposing activities of the phosphatase cdc25 and the kinase Wee1, allows for a more rapid response to cellular stress than via the synthesis or degradation of modulatory interacting proteins, such as p21 or cyclin B. Reducing Wee1 activity results in ectopic activation of CDK1 activity and drives premature entry into mitosis with unrepaired or under-replicated DNA and causing mitotic catastrophe. Here, we review efforts to use small molecule inhibitors of Wee1 for therapeutic purposes, including strategies to combine Wee1 inhibition with genotoxic agents, such as radiation therapy or drugs inducing replication stress, or inhibitors of pathways that show synthetic lethality with Wee1. Furthermore, it become increasingly clear that Wee1 inhibition can also modulate therapeutic immune responses. We will discuss the mechanisms underlying combination treatments identifying both cell intrinsic and systemic anti-tumor activities.
Wee1, the Cell Cycle, and the DNA Damage Response
The cellular genome is exposed to insults by several endogenous (reactive oxygen species, DNA replication errors) as well as exogenous (chemical mutagens, ionizing radiation, ultraviolet light) DNA damaging factors. Ionizing radiation from cosmic radiations or medical treatments (X-ray scans or radiation therapy) can generate base lesions as well as single and double-strand DNA breaks. Additionally, cancer chemotherapeutics can intentionally induce a variety of DNA lesions, including inter- and intra-strand cross-links arising from drugs like cisplatin or Mitomycin C. To ensure safe passage of the genomic material to the next generation, all organisms have evolved mechanisms – collectively termed the DNA damage response (DDR) – to detect DNA damage and to activate a signaling cascade to promote repair, including via cell cycle checkpoint activation (1), or in the case of extensive DNA damage to trigger mechanisms to either permanently exit the cell cycle (senescence) or undergo programmed cell death (apoptosis), presumably preventing cells from accumulating mutations and resulting in the development of cancer.
The DNA damage response and the cell cycle are intimately linked through cell cycle checkpoints, “control mechanisms enforcing dependency in the cell cycle” (2). Of the four cell cycle checkpoints, only the spindle checkpoint in mitosis is not clearly linked to pathways activated by DNA damage. As most cells in a human are in G1 (G0) phase, the G1/S checkpoint will prevent most normal cells to enter the cell cycle after DNA damage. The pathways initiated by the apical kinases Ataxia Telangiectasia-mutated (ATM) (3, 4) and Ataxia telangiectasia and Rad3 related (ATR) (5) relay the damage signal to downstream effectors, including the tumor suppressor p53, a central node in the DNA damage response (6). These two kinases also play an important role in the S phase checkpoint (7–9), which ensuring accurate replication, and for the G2/M checkpoint (7, 10). The latter checkpoint prevents cells with damaged or under-replicated DNA to enter mitosis, an event which poses a high risk of chromosome aberrations (11). As all checkpoints are governed by cyclin-dependent kinases (CDKs), all DNA damage pathways ultimately converge on the regulation of the CDK activity. A dysregulated cell cycle is able to lead to DNA damage and genomic instability is a hallmark of cancer (12).
The Kinase Wee1, a Gatekeeper at Several Cell Cycle Checkpoints
Wee1 is a tyrosine kinase originally discovered in Schizosaccharomyces pombe (13). Human Wee1 was subsequently discovered as a crucial regulator of the G2/M checkpoint (14). The primary structure of Wee1 is composed of an amino-terminal regulatory domain, a kinase domain, and a short C-terminal domain. The N-terminal domain coordinates signals to shuttle Wee1 into and out of the nucleus (15, 16). Wee1 contains four cyclin binding motifs, RxL1, RxL2, RxL3, and RxL4, to facilitate interaction with CDK (15) (Figure 1A).
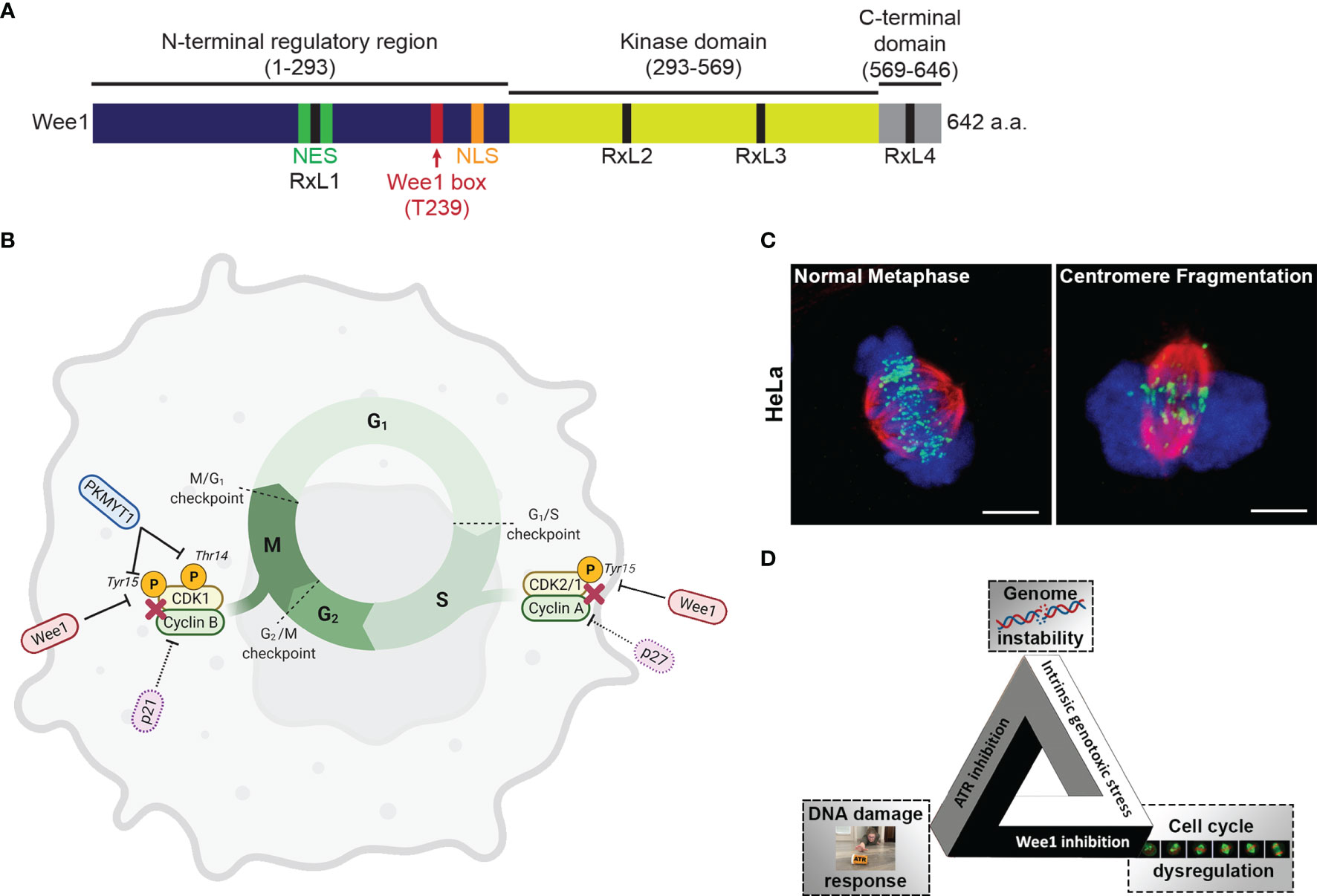
Figure 1 (A) The 642 amino acid long protein kinase Wee1 contains a N-terminal regulatory domain (dark blue), a kinase domain (yellow green), and a short C-terminal domain (gray). The diagram also shows a nuclear localization sequence (NLS; orange), a nuclear export sequence (NES; green), a highly conserved regulatory Wee1 box (red), and four cyclin binding motifs (RxL1, RxL2, RxL3, and RxL4; black). (B) Regulation of the cell cycle via phosphorylation of Cyclin-Dependent Kinases (CDKs) by Wee1 and the related protein kinase MYT1. (C) Images of HeLa cells in metaphase undergoing unperturbed mitosis or centromere fragmentation due to mitotic catastrophe as a result of premature entry into mitosis. (chromosomes, blue; tubulin, red; centromeres, green) (D) The fateful triangle underlying the conditional synthetic lethality observed in cancer cells leading to selective killing by combined ATR and Wee1 inhibition.
The Wee kinase family comprises three serine/threonine kinases: Wee1, PKMYT1, and Wee2. In mammalian cells, Wee1 and PKMYT1 (protein kinase membrane-associated tyrosine/threonine 1; also known as Myt1) have a vital role in regulating the G2/M transition (17) (Figure 1B). Wee2 (or Wee1B) is only expressed in germ cells, where it prevents premature restart of oocyte meiosis prior to ovulation and permits metaphase II exit at fertilization (18). PKMYT1 functions as an essential component of an organelle-based cell cycle checkpoint to prevent CDK1-induced premature fragmentation of Golgi and the endoplasmic reticulum during the G2 phase (19). PKMYT1 negatively regulates CDK1 activity by phosphorylation on both threonine 14 and tyrosine 15 (20, 21) - unlike Wee1, which only phosphorylates CDK1 on tyrosine 15 rendering CDK1 inactive (22, 23). In the absence of DNA damage, CDK1 is dephosphorylated by the Cell division cycle 25 (Cdc25c) phosphatase resulting in CDK1/cyclin B activation and initiation of mitotic events (24). In the unperturbed cell cycle Polo-like kinase 1 (PLK1) phosphorylates Wee1 at the G2/M transition, which targets Wee1 for degradation via the ubiquitin proteasome system (25). PLK1 also phosphorylates and activates the phosphatase cdc25 resulting in CDK1 activation (25, 26). Furthermore, Wee1 has a role in regulating replication dynamics during S phase. During S phase, initiation of replication results in the firing of many replication origins triggered by the action of DBF4-Dependent cdc7 kinase (DDK) and CDK2, the main S phase CDK (26, 27). Wee1 and cdc25 control CDK2 activity by regulating the phosphorylation status at tyrosine 15 (28). Importantly, Wee1 downregulation triggers a DNA damage response resulting in DNA replication stalling and reduced replication fork speed and causes cells to accumulate in S phase (29). It was proposed that in unperturbed cells, Wee1 protects replication forks and prevents generation of DNA damage by inhibiting the Mus81 endonuclease (29). Several studies have indicated that Wee1 levels are regulated by non-coding RNAs, which could impact cellular sensitivity to genotoxic agents (30).
Cancers often have a deregulated G1 checkpoint. As a result, they are heavily reliant on the G2/M checkpoint for survival and mitosis. Consequently, Wee1 is often highly expressed in many cancers including breast (31, 32) and lung (31) cancers, glioma (33), melanoma (34), leukemia (35, 36), osteosarcoma (37), and squamous cell carcinoma (38). As most cancer therapies aim to induce lethal amounts of DNA damage in cancer cells, Wee1 overexpression promotes cancer cell survival (and resistance) by reinforcing DNA damage checkpoints and preventing mitotic catastrophe (33). The key role of Wee1 in regulating the G2/M checkpoint in response to DNA damage has made it an attractive target for cancer therapy. Despite its appeal, to date only one selective and highly potent small molecule Wee1 inhibitor, AZD1775 (also known as Adavosertib or MK-1775) (39), has been widely reported and is being evaluated against various advanced cancers in phase I/II clinical trials either as a monotherapy (40–42) or in combination with other chemotherapies (40, 43, 44). Yet several new inhibitors are being developed or are already making it into the clinic (see below).
Wee1 Inhibition and Mitotic Catastrophe
Mitotic catastrophe is a major mode of tumor cell death following genotoxic treatments including irradiation (45). Mitotic catastrophe is loosely defined as cell death that occurs during or following an aberrant mitosis (46, 47). While its molecular mechanism is unclear, increasing evidence points to the involvement of caspases (46, 48). Wee1 knockout and the loss of Cdk1 T14 and Y15 phosphorylation causes ectopic Cdk1 activity, uncontrolled mitotic entry and cell death (14, 49–51). Similarly, Wee1 inhibition with AZD1775 or siRNA-mediated knockdown of Wee1 results in premature mitotic entry, prolonged mitotic arrest and mitotic catastrophe (33, 52–56). Furthermore, ectopic activation of Cdk1 and activation of the Mus81 endonuclease complex in S phase results in stalled DNA replication forks and DNA damage (29, 57). The ectopic Cdk1 activity induces replication stress and fork collapse through the depletion of dNTPs and aberrant replication origin firing (58–60), as Cdk1 phosphorylation of the ribonucleotide reductase subunit RRM2 induces its ubiquitin mediated degradation during DNA synthesis resulting in a 70% drop in dNTPs (60). Since Cdk1 activity induces chromosome condensation, ectopic Cdk1 activity also promotes premature chromosome condensation (61–63), generates torsional strain to the DNA backbone and results in DNA breakage (64, 65). Centromeres are late replicating due to a lower prevalence of replication origins and are prone to breakage during premature condensation or cleavage by the Mus81 endonuclease complex (64, 66). In a process known as checkpoint adaptation, cells with damaged DNA eventually escape the S and G2 checkpoint and enter mitosis prematurely (67). Checkpoint adaptation in both lower eukaryotes and mammalian cells has been consistently linked to the Plk1 [reviewed in (68)], the kinase phosphorylating Wee1 and promotes its ubiquitin mediated degradation through the SCFβTrCP pathway (69, 70). Underlining the importance of this coordinated timing of kinases, Wee1 inhibition and subsequent premature entry into mitosis in the presence of under-replicated chromosomes can result in centromere fragmentation, a morphological marker of mitotic catastrophe (53, 71) (Figure 1C). Conversely, Wee1 overexpression can promote cell survival by reinforcing the DNA damage checkpoints and preventing mitotic catastrophe (33).
Wee1 inhibition by AZD1775 has been shown to induce in vitro and in vivo synergistic tumor cell killing with several DNA damaging therapies including IR (55) and chemotherapeutics like cisplatin, paclitaxel doxorubicin, 5-fluorouracil, and gemcitabine (53, 72–74). Given the role of p53 in regulating the G1 cell cycle checkpoint, treatment with AZD1775 has been reported to selectively target cancers harboring p53 mutations or loss of gene function (39, 75). Having said that, a few studies have also shown that AZD1775 sensitizes cancer cells to DNA damaging therapies independent of p53 status (76–78). Additionally, DNA damaging agents that specifically interfere with DNA synthesis and arrest cells in S-phase show high synergy with AZD1775 (59, 72). Overall, these preclinical studies support that AZD1775 has antitumor effects in a wide range of tumors both as a monotherapy and in combination with other chemotherapeutics.
Wee1 Inhibitors in the Clinic
There are 60 clinical trials listed on clinicaltrials.gov (accessed January 2022) for AZD1775 where it is being evaluated against a wide range of cancer types including breast cancer, cervical cancer, leukemia, lung cancer, ovarian cancer, pancreatic cancer, pediatric and adult brain tumors (For a list of completed and active clinical trials with Wee1 inhibitors, see Table 1). Findings of phase I clinical trials show that AZD1775 is relatively well tolerated with acceptable toxicity profiles both as a single agent and in combination with other therapies (42). As a monotherapy, the maximum tolerated dose was determined as 225 mg, which was administered orally to ovarian cancer patients in five doses (2 twice per day, 1 once a day) per week over 3 weeks (42). The dose limiting toxicities included hematologic events, nausea, vomiting, and fatigue (40, 42). Interestingly, two of nine patients harboring BRCA1 mutation recorded partial response, but unexpectedly none of the patients with documented p53 mutation exhibited a response (42). Early indications from a phase II trial evaluating AZD1775 plus carboplatin in p53 mutant ovarian cancer refractory or therapy-resistant patients show encouraging antitumor activity with one (5%) complete response and eight (38%) partial responses (43). Moreover, the overall response rate (43%) far exceeded the results that could be expected with second-line single agent treatments (11% to 21%) (43). A recent clinical trial evaluated the efficacy of AZD1775 as a monotherapy given once-daily as 5 days on and 2 days off in a 21 day cycle to patients (n = 42) with advanced solid tumors (79). The recommended phase II dose was determined as 300 mg, with most common toxicities including gastrointestinal and hematologic adverse effects. The dose-limiting toxicities included grade 4 hematologic toxicity and grade 3 fatigue (79). Six patients (14%; four ovarian and two endometrial cancers) confirmed partial response as the best response. Interestingly, one patient who progressed rapidly was found to have a Wee1 tumor mutation and potential compensatory PKMYT1 overexpression (79) (see below).
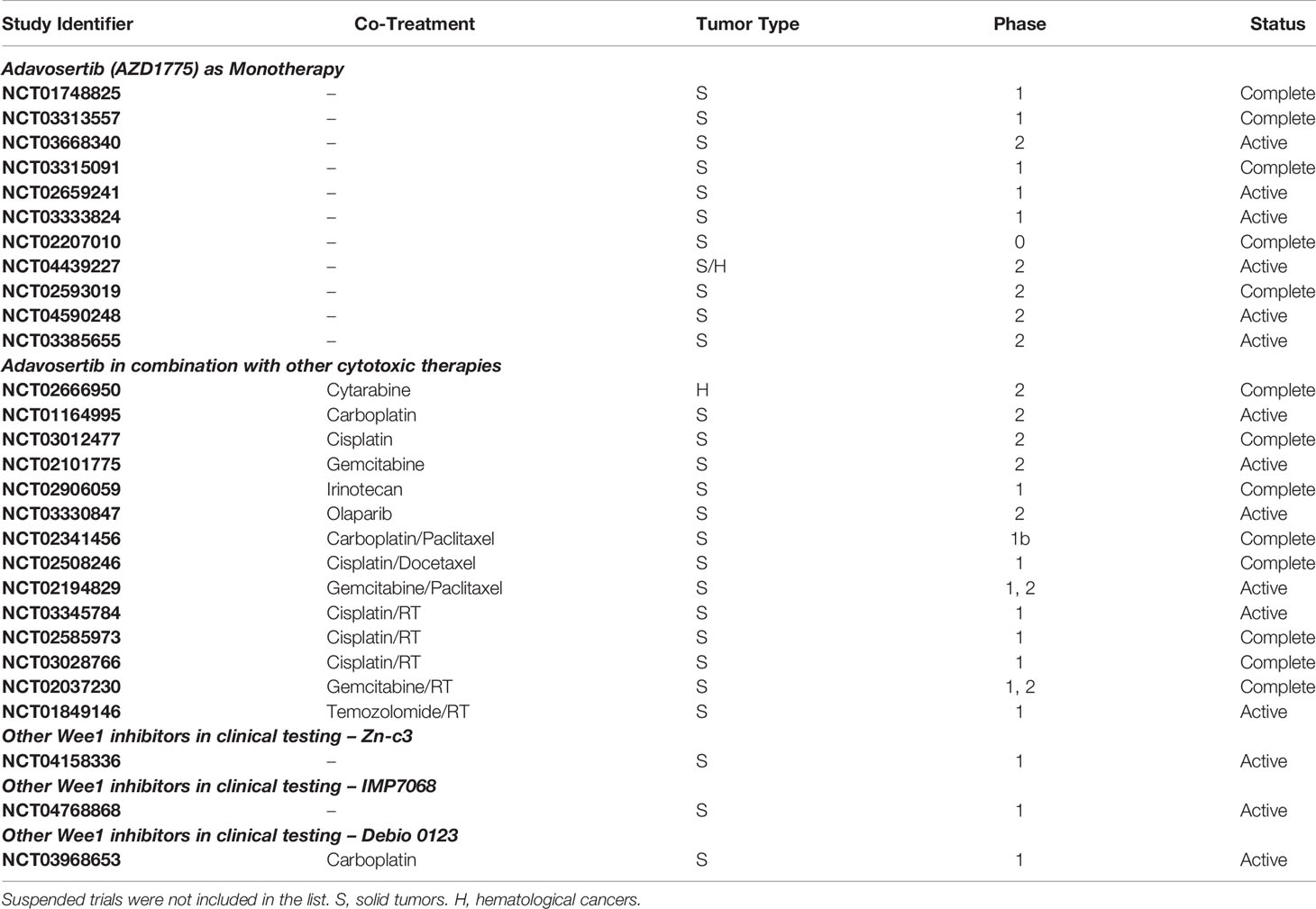
Table 1 Wee1 inhibitors in clinical trials.
While AZD1775 is the most promising Wee1 inhibitor undergoing phase II clinical testing to date, its toxicity profile limits its use to intermittent dosing, potentially impacting clinical efficacy. Recently, Zentalis Pharmaceuticals reported the development of ZN-c3, a selective small molecule orally active bioavailable inhibitor of Wee1. Compared to AZD1775, which at higher concentrations also inhibits PLK1, a negative regulator of Wee1, ZN-c3 showed much higher selectivity for Wee1 over other kinases (80). Moreover, ZN-c3 demonstrated similar efficacy to AZD1775 in vivo. The expected superior safety profiles of ZN-c3 has enabled its quick transition to phase I/II clinical testing either as a monotherapy or in combination with other chemotherapies (NCT04158336). Similarly, a Wee1 inhibitor developed by Debiopharm, Debio 0123, is being tested in a phase I study (NCT03968653).
Early results for ZN-c3 of the phase I dose-escalation (25 mg to 450 mg) trial (NCT04158336) reported 300 mg as the recommended phase II dose (81). Of the 16 patients with post-baseline tumor assessments, 5 patients showed indications of stable disease, and 2 patients showed partial response. Interestingly, both the partial response patients had stage IV metastatic disease (colorectal cancer and ovarian cancer, respectively) and had undergone several lines of therapy (81). Importantly, ZN-c3 was reported to have higher selectivity and better safety profiles compared to AZD1775, making it particularly well suited for combination therapies (82). Out of the 39 patients involved in the trial, 30 experienced mild to moderate symptoms like nausea, diarrhea, vomiting, and fatigue (81). In addition to this, ZN-c3 is also undergoing clinical testing in combination with other chemotherapeutics like carboplatin, doxorubicin, paclitaxel, and gemcitabine in patients with platinum-resistant ovarian cancer (NCT04516447).
Other Wee1 inhibitors in clinical trials are Debio 0123 by the French company Debiopharm and, the most recent addition, IMP7068, developed by Impact Therapeutiucs in China. Schrödinger and Nuvation Bio have preclinical compounds that might soon advance soon in the pipeline as well (SDGR2 and NUV-569, respectively).
Radiosensitization by Wee1 Inhibition
The G2/M checkpoint constitutes an important safeguard for preventing cells with damaged or under-replicated DNA to enter mitosis, particularly in cancer cells which often have an abrogated G1 checkpoint due to aberrations in p53 signaling, caused e.g. by mutations in the p53 gene, viral proteins or MDM2 overexpression. It is therefore not surprising that the first described Wee1 inhibitor, (non-selective) PD0166285, was tested in combination with ionizing radiation and was found to radiosensitize, i.e. to kill more efficiently, cancer cells in a p53-dependent manner (83). Studies with AZD1775 (39), which unlike PD0166285 does not inhibit the related kinase PKMYT1 as well, confirmed that inhibition of Wee1 leads to radiosensitization of a variety of cancer cells and increased radiation-induced tumor delay in mouse models (55). Since then several studies have shown that combining ionizing radiation with inhibition of Wee1 by AZD1775 increased cell death or clonogenic death of cells derived from a variety of cancers, including of the lung, breast, prostate, esophagus, cervix, liver, brain, and pancreas (55, 84–93). Yet it is not always clear in the mentioned studies whether the cooperativity was synergistic or just additive. (Only in the former case it would be appropriate to call the effect radiosensitizing.) Importantly, several preclinical studies in mice also showed increased tumor delay when radiation was combined with Wee1 inhibition (55, 85–87, 90–93) and several clinical trials are currently examining the efficacy of AZD1775 with radiation therapy (sometimes in combination with chemotherapy). Phase I trials produced promising results (94, 95), although the combination with cisplatin prompted the need for toxicity–related dosing adjustments (95). The initial proposal that p53 status was an essential biomarker for the radiosensitization by Wee1 inhibition (55) was put into doubt by subsequent studies (89). A likely explanation is that p53 status-independent defects of the G1 checkpoint could make cancer cells reliant on the G2/M checkpoint. Indeed, cyclin E overexpression renders cancer cells sensitive to Wee1 inhibition (96). Furthermore, several other mechanisms could lead to increased replication stress in cancer cells which would synergize with radiation and Wee1 inhibitor-mediated replication stress to endanger the survival of cancer cells entering mitosis. In this regard, the exact cellular mechanisms underlying the radiosensitization by Wee1 inhibition are still to be determined. For example, is the main reason for the cooperativity due to Wee1 inhibition lowering the G2/M transition threshold or increasing replication stress on top of ionizing radiation-induced DNA damage? Wee1 inhibition also suppresses homologous recombination (97), an important repair pathway particularly for radiation-induced double strand breaks. In the G2 phase homologous recombination could repair even complex DNA damage and DNA structures resulting from stalled or collapsed replication forks. The contribution of inhibition of these Wee1 roles, as well as known and yet to be identified crosstalk with other cellular pathways [e.g. autophagy (93)], to radiosensitization is likely specific to the cell type or even to the subpopulation (given tumor heterogenicity). Of importance for the clinic – and unfortunately much less characterized - is the heterogeneity in the radiosensitization of normal cells (between cell type and between persons) by inhibitors of cell cycle regulators. Of particular concern for normal tissue complications are potential deleterious effects of combining Wee1 inhibitors and radiation on stem cells, which often rely on an intricate crosstalk between external signaling factors and the cell cycle machinery to regulate their differentiation potential (98). As tissue homeostasis is usually dependent on tissue specific stem cells, the impact of Wee1 inhibition in the clinic must also be seen in the context of the heterogeneity in the radiation response within the stem cell compartment and plasticity (reviewed in (99)). Radiosensitizers are only useful for cancer therapy if they improve the therapeutic index in the current highly conformal treatment plans in radiation oncology.
Synthetic Lethality With Wee1 Inhibition
Synthetic lethality refers to an interaction between two genes when the perturbation of either gene alone is viable but the simultaneous perturbation of both genes (gene functions) leads to cell death. A well-known example is deficiency of homologous recombination proteins BRCA1 or BRCA2 causing cancer cell sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors (100, 101). This distinctive synthetic lethality led to a strong interest in therapeutic approaches targeting cancer cells with other deficiencies in DNA damage response (DDR) pathways by inhibition of the alternative DDR pathway. Yet as this approach only targets cells with a specific defect in the DDR, unless loss of gene function leads to the cell-of-origin or occurs early in carcinogenesis, in which case the gene defect would be in all or most tumor cells, it is bound to only affect a subset of populations within a heterogeneous tumor. For example, the loss of Ataxia Telangiectasia-mutated (ATM) frequently observed in a variety of cancers (102) is likely due to a driver mutation occurring at an early stage during lung carcinogenesis (103). It therefore is expected that most cancer cells in those tumors will be killed by targeted drugs showing high efficacy in the background of defective ATM (104). Yet except for the ATM and p53 pathways (alterations in ATM, CHEK2, p53, MDM2), the majority of driver mutations in DNA damage response and repair genes were found to be subclonal in non-small cell lung cancer (103). It is to be expected that targeting an evolutionary late occurring gene defect, even if found in the subpopulation that constitutes the bulk of the cancer cells, would lead to the selection for the subpopulation with the functional gene and treatment resistance to follow. Furthermore, even in a homogenous population with a common DDR defect resistance can arise by reactivation of the defective pathway, as was observed in some PARP inhibitor resistant breast cancers (105). Conditional synthetic lethality refers to synthetic lethality observed only under certain circumstances, such as genetic background or metabolic state of cells or cellular environment (106). An approach to build synthetic lethality around cancer-intrinsic characteristics has the potential to decrease the probability for the tumors to acquire resistance. In that regard, one of the most common features of cancer cells is oncogene-induced DNA damage (107, 108), often leading to levels of replication and mitotic stress not observed in normal proliferating cells. This tumor-specific property makes it an ideal selective condition to base a synthetic lethality on to achieve a favorable therapeutic index.
An example of a successful attempt to establish a fateful triangle for cancer cells in a conditional synthetic lethality approach is the combination of inhibitors of Wee1 and of the kinase Ataxia telangiectasia and Rad3 related (ATR). This multi-pronged attack takes advantage of three features of cancer cells to selectively target them: genomic instability, dysregulated cell cycle and the reliance on particular DDR pathways for survival (Figure 1D). In a 2008 review discussing genomic instability, a designated hallmark of cancer (12), Halazonetis, Gourgoulis, and Bartek pointed out that, based on their data and the literature on observations in many cancer cell lines and precancerous and cancerous lesions from patients, “the presence of DNA damage was a feature that could distinguish precancerous lesions and cancers from normal tissues, irrespective of their proliferation rate” (108). DNA damage (genotoxic stress) is therefore a fundamental characteristic of cancer cells, unlike some other hallmarks of cancer which, due to tumor heterogeneity, may not manifest in every tumor or every tumor cell.
ATR is an apical kinase in the DDR and is activated by replication protein A (RPA)-coated single-stranded DNA, structures that can arise from stalled replication forks or resected DNA double-strand breaks (1). Not surprisingly, ATR plays a crucial role in the response to replication stress – likely the reason for it being an essential gene (109, 110). As a result, cancer cells rely on functional ATR signaling, particularly as other DNA damage response pathways are lost (such as the p53 and/or ATM pathway). This is exemplified by the importance of ATR signaling for the survival of cancer cells to ionizing radiation (5). Unsurprisingly, ATR activity is often upregulated in cancer cells (111, 112), including in cancer stem cells (113). ATR regulates Chk1 activity by phosphorylation of Chk1 kinase at serines 317 and 345 (1). Chk1 in turn targets Cdc25, the phosphatase removing inhibitory phospho-groups from cyclin-dependent kinases (CDKs), for degradation by phosphorylation-dependent ubiquitination. Because CDKs, particularly CDK1 and CDK2, regulate entry into mitosis and replication origin firing, Chk1 activation thereby prevents cell cycle progression (114). Thus, ATR/Chk1 signaling initiated at structures containing single-stranded DNA controls the S and G2 phase cell cycle checkpoints in mammalian cells (114). Importantly, the phosphorylation state of CDKs 1 and 2 (and thus their inhibition) is regulated by the balance between the kinase activity of Wee1 (and Myt1) and the phosphatase activity of Cdc25. The observed synergistic effects of Wee1 and ATR inhibition (71, 115) on cancer cell killing are surely in grant part due to the lowering of the threshold for CDK activation by combining inhibiting the constitutive phosphorylation and preventing checkpoint activation by the ATR/CHK1/Cdc25 axis, as combined AZD1775 and AZD6738 treatment leads to mitotic catastrophe in cancer cells (71). Yet both Wee1 and ATR regulate other cellular aspects that will play a role, including their activities during replication: For example, the above mentioned role of Wee1 during S phase, including replication fork protection, as well as reportedly in timing the entry into S phase (116) are perturbed by AZD1775 and lead to substantial replication stress. ATR on the other hand, besides the many functions during unperturbed replication (117), also regulates DNA damage repair by promoting extensive DNA end-resection needed for homologous recombination (5, 118, 119). By utilizing the reversibility of Wee1 and ATR inhibition, we characterized the contributions of inactivation of each kinase and during different phases of the cell cycle, thus studying how abrogation of ATR and Wee1 activity cooperatively leads to cell death caused by mitotic defects (71). The findings are compatible with a model, where synergistic killing by ATR and Wee1 inhibitors is based on an increase in the DNA damage level while simultaneously lowering the DNA damage response capacity leading to mitotic catastrophe. This is achieved by Wee1 inhibition-induced DNA damage during replication, abrogation of ATR-mediated S phase checkpoint activation, inhibition of ATR-dependent homologous recombination, and amplified by increased entry into mitosis with defective genomes due to combined inhibition of ATR and Wee1. As high replication stress in cancer cells - due to the high level of baseline DNA damage per se, but also to the resulting exhaustion of factors needed for both repair and replication, such as RPA (120) – contrasts from the stress in normal cells, even in highly proliferative tissues, and cancer cells often have an increased reliance on the G2/M checkpoint, a therapeutic window is achieved in an example of a conditional synthetic lethality.
Several studies have also investigated whether defects in specific pathways sensitize to Wee1 inhibition. A recent study in basal-like breast cancer cells suggests that loss of PTEN may be one of the strongest markers of Wee1 inhibitor sensitivity (121). This might not come as a surprise, given the role PTEN plays in replication progression and several studies showing that PTEN loss increases replication stress (122–124). A recent study also showed that HPV16 positivity sensitizes head and neck squamous cell carcinomas to Wee1 inhibition by a mechanism involving a circuit linking CDK1 and FOXM1 (125), a master transcriptional regulator of mitotic genes (126). Fitting with the scheme of vulnerability to Wee1 inhibition based on an already dysregulated cell cycle, an unbiased screen identified several S phase genes as determinants for AZD1775 sensitivity (127). Related, another screen identified defects in the Fanconi anemia pathway and in homologous recombination, mechanisms needed for effective DNA replication particularly in the background of increased DNA damage, as sensitizing to Wee1 inhibition (128). A strategy of releasing tumor cells from a cell cycle block into a phase where the cells are sensitive to Wee1 inhibition was used in a preclinical study with sarcoma. The combined sequential treatment with the CDK4/6 inhibitor Palbociclib and AZD1775 showed at least additive effects on tumor growth (129). In a model for the clinical scenario of breast cancer cells resistant to endocrine therapy and CDK4/6 inhibitors, derived long-term estrogen deprived endocrine resistant cell lines were found to be more resistant to CDK4/6 inhibitors, but more sensitive to AZD1775 or Wee1 knockdown than their parental cell lines (130). An interesting observed synergistic interaction was found between AZD1775 and A1155463 in cancer cells from a genetically engineered animal model for triple negative breast cancers (131). A1155463 is an inhibitor of the anti-apoptotic BCL-XL protein (132). The drug combination also showed efficacy in vivo, but unfortunately the authors did not report the effect of the individual drugs in their mouse model (131). AZD1775 was also found to synergize with the PARP inhibitor olaparib in a xenograft model for triple negative breast cancer (133).
Resistance Mechanisms to Wee1 Inhibition
Obvious candidate resistance mechanisms to Wee1 inhibitors include reversal of expression profiles of genes that are the base for Wee1 inhibitor vulnerability. For example, while cyclin E overexpression sensitizes cancer cells to AZD1775, reducing cyclin E levels has the opposite effect (96). A mechanism of acquired AZD1775 resistance observed both in vitro and in vivo is via the upregulation of PKMYT1 (53). As mentioned, Wee1 and the related kinase PKMYT1 exhibit functionally redundant roles in the inhibition of CDK1/cyclin B, the mitosis promoting complex (134–136). Yet compared to Wee1, PKMYT1 is much less studied in the context of cancer biology. This might be due to reports that inhibition or knockdown of Wee1 alone is sufficient to abrogate the S- and G2/M DNA damage checkpoints and that the loss of PKMYT1 neither affects the timing of mitosis nor abrogates DNA damage checkpoints in the presence of Wee1 (56, 137–139). On the other hand, combined knockdown of Wee1 and PKMYT1 causes more HeLa cells to enter mitosis with damaged DNA compared to Wee1 knockdown alone (56), PKMYT1 knockdown enhances AZD1775 induced cell killing in cell lines derived from brain metastases (140), and PKMYT1 is essential for cell survival in a subset of glioblastoma cells that have downregulated Wee1 expression (141). The protective mechanism by PKMYT1 upregulation leading to AZD1775 resistance was found to be due to compensatory inhibition of ectopic CDK1 activity by PKMYT1, allowing cells to escape mitotic catastrophe, the mode of cell death induced by Wee1 inhibition (53).
It was proposed that cancer stem cells, which often show increased chemo- and radiation resistance compared to bulk cancer cells and due to their cellular plasticity and tumor initiating capability can lead to tumor relapse (142), could be targeted by Wee1 inhibition (143). Only a few studies have examined the efficacy of Wee1 inhibition – alone or in combination - in the eradication of cancer stem cells. Early findings that Wee1 inhibition by the unspecific inhibitor PD0166285 radiosensitizes glioma stem cells (CD133 enriched glioma neurospheres) (33) were contradicted by a study using AZD1775 (and glioma cell lines enriched for neuronal stem cells) (92). In contrast, another study found that glioma stem cells (unlike neuronal progenitor cells) were sensitive to Wee1 inhibition alone (141). Our studies in breast cancer showed that breast cancer stem cells were less sensitive to AZD1775 compared to bulk cancer cells, which could be due to reduced drug uptake or decreased reliance on Wee1 signaling. Interestingly, combined Wee1 and ATR inhibition was as toxic to cancer stem cells as to bulk breast cancer cells, potentially explaining the antimetastatic effect of the combination treatment (71). To our knowledge, this was the first report of a higher drug synergy observed in cancer stem cells compared to bulk cancer cells, compensating for the reduced sensitivity of cancer stem cells to the individual drugs. A recent study found that trastuzumab resistant breast cancer cell lines were enriched in cancer stem cells, but on average showed greater sensitive to AZD1775. AZD1775 treatment disrupted stem like properties in the tested trastuzumab resistant breast cancer cell lines (144). These studies indicate that insights into the role of Wee1 in cancer stem cell maintenance and the associated correlation with drug resistance could have a significant impact in the clinic.
As the ongoing clinical trials will provide data and samples from patients treated with AZD1775 (Adavosertib), not only will predictive biomarkers be identified, but it will also become clearer which are the preferred pathways for resistance acquisition to single agent therapies. This in turn will provide important clues for improved treatment plans with combination therapies.
Wee1 Inhibition - Beyond Cell-Intrinsic Cytotoxicity
The interaction between tumor cells and immune cells plays a determining role not only during carcinogenesis, where the survival of transformed cells is based on immune evasion, but also in cancer therapy, where the immune system is a key factor in achieving local and systemic tumor control. Several pathways involved in both DNA damage repair/signalling and immunity indicate that the immune system and the DNA damage response (DDR) have coevolved, resulting in processes with overlapping enzymatic networks. Examples range from prokaryotic defense systems, such as the antiviral CRISPR machinery, to the complex mammalian immune stimulation and maturation processes, such as class-switch recombination. A classic case in point is the discovery in 1995 that the lack of DNA-PK caused both extreme radiosensitivity and severe combined immunodeficiency (145, 146). Since then several cellular links between proteins in the DNA damage response and immune signaling have been uncovered. Of particular interest is the stimulator of interferon genes (STING) pathway, that can be activated by cyclic GMP-AMP synthase (cGAS) binding to DNA fragments and the subsequent production of the allosteric modulator of STING, the small messenger molecule cGAMP (147). This pathway was discovered as an important defense mechanism against DNA viruses, but was later found to get activated by DNA damage in the nucleus or mitochondria as well (148, 149). Besides cGAS, which binds to the DNA backbone, STING activation by DNA fragments might also involve the recognition of DNA ends by DNAPK (150) and/or the MRN complex (151). Kinases downstream of STING, TANK binding kinase 1 and IκB kinase, induce the transcription of genes involved in the innate immune response, such as interferons, interleukins and TNF, via the transcription factors IRF3 and NFκB (152). Several studies have shown that exogenous and endogenous genotoxic stress can induce the expression of interferon-stimulated genes, including stress due the loss of genes involved in the DNA damage response [reviewed in (153)]. Furthermore, activation of the apical DDR kinases ATM and ATR can also lead to upregulation of PD-L1 (via the STAT1 and STAT3 pathway) (154) and natural killer group 2D (NKG2D) ligands (155, 156).
Besides these DNA damage-induced changes in surviving cells, therapy-induced cell death itself can have a big immunomodulatory effect. The Nomenclature Committee on Cell Death defines immunogenic cell death (ICD) as “a functionally peculiar form of regulated cell death that is sufficient to activate an adaptive immune response specific for endogenous (cellular) or exogenous (viral) antigens expressed by dying cells” (45). Besides the release of antigenic determinants, such as neoepitopes, dying tumor cells also can lead to a local release of damage-associated molecular patterns and cytokines resulting in local effects on immune cell trafficking and activation. The observation that inhibition of Wee1 increases replication stress as well as the likelihood of untimely entry into mitosis, raising the possibility of DNA structures activating the STING pathway as well as mitotic catastrophe, make Wee1 inhibition a good candidate drug to increase the antitumor immune response. This makes Wee1 inhibition especially attractive to be combined with radiation therapy, as the latter is well known to be particularly inducive to ICD and Wee1 inhibitors are, as discussed previously, also radiosensitizing (39, 89, 157). Indeed preclinical studies have shown immune stimulating effects of Wee1 inhibition in combination with irradiation (158, 159). The exact mechanisms underlying the increased anti-tumor immunity, including the extent interferon signaling is involved, are still unclear. Of note, a recent study showed that inhibition of Wee1 alone failed to induce a type I interferon response, despite increasing DNA double strand breaks, cytosolic DNA, and micronuclei – all cellular phenotypes previously correlated with STING pathway activation (160).
Conclusion
In conclusion, Wee1 inhibitors show great potential to make an impact in the clinic for the therapy of several cancer types. While some concerns have arisen from phase I/II clinical trials regarding potential side effects, it remains to be seen whether newer Wee1 inhibitors with supposedly higher kinase selectivity show an improved safety profile. Yet the most promising path are combination therapies allowing lower dosing of the Wee1 inhibitor than in monotherapy. Furthermore, optimization of the treatment plans, such as intermittent dosing of the Wee1 inhibitor, might improve the drug tolerance.
Regarding the kinase itself, still many questions remain to be elucidated on the biological role of Wee1, which revealed itself to be a multifaceted player during several phases of the cell cycle. Of special interest are the redundant and divergent roles of Wee1 and the related kinase PKMYT1, in normal tissues and in various cancer types.
Author Contributions
AB, GC, and AG wrote and reviewed the manuscript together. All authors contributed to the article and approved the submitted version.
Funding
ABB is supported by Alberta Cancer Foundation’s Dr. Cyril M. Kay Graduate Scholarship. GKC and AMG are funded by the Canadian Institute of Health Research and the Natural Sciences and Engineering Research Council of Canada, as well as the Cancer Research Society and Women and Children’s Health Research Institute (AMG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge the many researchers who contributed to our growing knowledge of the biology of Wee1 and the DDR in general and whose work we could not cite in this non-exhaustive overview of the field. We thank members of the Chan and Gamper labs for images used in the figures, and Kenaan Ramji for help in proofreading the manuscript. We also thank Stéphanie La France for her assistance with an image used in the figure. Some illustrations were generated using BioRender.
References
1. Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play With Knives. Mol Cell (2010) 40(2):179–204. doi: 10.1016/j.molcel.2010.09.019
2. Hartwell LH, Weinert TA. Checkpoints: Controls That Ensure the Order of Cell Cycle Events. Science (1989) 246(4930):629–34. doi: 10.1126/science.2683079
3. Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, et al. A Mammalian Cell Cycle Checkpoint Pathway Utilizing P53 and GADD45 Is Defective in Ataxia-Telangiectasia. Cell (1992) 71(4):587–97. doi: 10.1016/0092-8674(92)90593-2
4. Bakkenist CJ, Kastan MB. DNA Damage Activates ATM Through Intermolecular Autophosphorylation and Dimer Dissociation. Nature (2003) 421(6922):499–506. doi: 10.1038/nature01368
5. Gamper AM, Rofougaran R, Watkins SC, Greenberger JS, Beumer JH, Bakkenist CJ. ATR Kinase Activation in G1 Phase Facilitates the Repair of Ionizing Radiation-Induced DNA Damage. Nucleic Acids Res (2013) 41(22):10334–44. doi: 10.1093/nar/gkt833
6. Vogelstein B, Lane D, Levine AJ. Surfing the P53 Network. Nature (2000) 408(6810):307–10. doi: 10.1038/35042675
7. Painter RB, Young BR. Radiosensitivity in Ataxia-Telangiectasia: A New Explanation. Proc Natl Acad Sci USA (1980) 77(12):7315–7. doi: 10.1073/pnas.77.12.7315
8. Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a Replication-Dependent Chromatin-Binding Protein Required for the DNA Replication Checkpoint. Curr Biol (2000) 10(24):1565–73. doi: 10.1016/S0960-9822(00)00855-1
9. Gamper AM, Choi S, Matsumoto Y, Banerjee D, Tomkinson AE, Bakkenist CJ. ATM Protein Physically and Functionally Interacts With Proliferating Cell Nuclear Antigen to Regulate DNA Synthesis. J Biol Chem (2012) 287(15):12445–54. doi: 10.1074/jbc.M112.352310
10. Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, et al. Overexpression of a Kinase-Inactive ATR Protein Causes Sensitivity to DNA-Damaging Agents and Defects in Cell Cycle Checkpoints. EMBO J (1998) 17(1):159–69. doi: 10.1093/emboj/17.1.159
11. Wilhelm T, Said M, Naim V. DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes (Basel) (2020) 11(6):646–74. doi: 10.3390/genes11060642
12. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
13. Thuriaux P, Nurse P, Carter B. Mutants Altered in the Control Co-Ordinating Cell Division With Cell Growth in the Fission Yeast Schizosaccharomyces Pombe. Mol Gen Genet (1978) 161(2):215–20. doi: 10.1007/BF00274190
14. Heald R, McLoughlin M, McKeon F. Human Wee1 Maintains Mitotic Timing by Protecting the Nucleus From Cytoplasmically Activated Cdc2 Kinase. Cell (1993) 74(3):463–74. doi: 10.1016/0092-8674(93)80048-J
15. Li C, Andrake M, Dunbrack R, Enders GH. A Bifunctional Regulatory Element in Human Somatic Wee1 Mediates Cyclin A/Cdk2 Binding and Crm1-Dependent Nuclear Export. Mol Cell Biol (2010) 30(1):116–30. doi: 10.1128/MCB.01876-08
16. Squire CJ, Dickson JM, Ivanovic I, Baker EN. Structure and Inhibition of the Human Cell Cycle Checkpoint Kinase, Wee1A Kinase: An Atypical Tyrosine Kinase With a Key Role in CDK1 Regulation. Structure (2005) 13(4):541–50. doi: 10.1016/j.str.2004.12.017
17. Schmidt M, Rohe A, Platzer C, Najjar A, Erdmann F, Sippl W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules (2017) 22(12). doi: 10.3390/molecules22122045
18. Oh JS, Susor A, Conti M. Protein Tyrosine Kinase Wee1B Is Essential for Metaphase II Exit in Mouse Oocytes. Science (2011) 332(6028):462–5. doi: 10.1126/science.1199211
19. Villeneuve J, Scarpa M, Ortega-Bellido M, Malhotra V. MEK1 Inactivates Myt1 to Regulate Golgi Membrane Fragmentation and Mitotic Entry in Mammalian Cells. EMBO J (2013) 32(1):72–85. doi: 10.1038/emboj.2012.329
20. Liu F, Stanton JJ, Wu Z, Piwnica-Worms H. The Human Myt1 Kinase Preferentially Phosphorylates Cdc2 on Threonine 14 and Localizes to the Endoplasmic Reticulum and Golgi Complex. Mol Cell Biol (1997) 17(2):571–83. doi: 10.1128/MCB.17.2.571
21. Booher RN, Holman PS, Fattaey A. Human Myt1 Is a Cell Cycle-Regulated Kinase That Inhibits Cdc2 But Not Cdk2 Activity. J Biol Chem (1997) 272(35):22300–6. doi: 10.1074/jbc.272.35.22300
22. McGowan CH, Russell P. Cell Cycle Regulation of Human WEE1. EMBO J (1995) 14(10):2166–75. doi: 10.1002/j.1460-2075.1995.tb07210.x
23. Do K, Doroshow JH, Kummar S. Wee1 Kinase as a Target for Cancer Therapy. Cell Cycle (2013) 12(19):3159–64. doi: 10.4161/cc.26062
24. Donzelli M, Draetta GF. Regulating Mammalian Checkpoints Through Cdc25 Inactivation. EMBO Rep (2003) 4(7):671–7. doi: 10.1038/sj.embor.embor887
25. Lindqvist A, Rodriguez-Bravo V, Medema RH. The Decision to Enter Mitosis: Feedback and Redundancy in the Mitotic Entry Network. J Cell Biol (2009) 185(2):193–202. doi: 10.1083/jcb.200812045
26. Labib K. How do Cdc7 and Cyclin-Dependent Kinases Trigger the Initiation of Chromosome Replication in Eukaryotic Cells? Genes Dev (2010) 24(12):1208–19. doi: 10.1101/gad.1933010
27. Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell (2011) 146(1):80–91. doi: 10.1016/j.cell.2011.06.012
28. Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, et al. Regulators of Cyclin-Dependent Kinases Are Crucial for Maintaining Genome Integrity in S Phase. J Cell Biol (2010) 188(5):629–38. doi: 10.1083/jcb.200905059
29. Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, et al. Wee1 Controls Genomic Stability During Replication by Regulating the Mus81-Eme1 Endonuclease. J Cell Biol (2011) 194(4):567–79. doi: 10.1083/jcb.201101047
30. Aranza-Martinez A, Sanchez-Perez J, Brito-Elias L, Lopez-Camarillo C, Cantu de Leon D, Perez-Plasencia C, et al. Non-Coding RNAs Associated With Radioresistance in Triple-Negative Breast Cancer. Front Oncol (2021) 11:752270. doi: 10.3389/fonc.2021.752270
31. Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, et al. Integrated Functional, Gene Expression and Genomic Analysis for the Identification of Cancer Targets. PloS One (2009) 4(4):e5120. doi: 10.1371/journal.pone.0005120
32. Murrow LM, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A. Identification of WEE1 as a Potential Molecular Target in Cancer Cells by RNAi Screening of the Human Tyrosine Kinome. Breast Cancer Res Treat (2010) 122(2):347–57. doi: 10.1007/s10549-009-0571-2
33. Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In Silico Analysis of Kinase Expression Identifies WEE1 as a Gatekeeper Against Mitotic Catastrophe in Glioblastoma. Cancer Cell (2010) 18(3):244–57. doi: 10.1016/j.ccr.2010.08.011
34. Magnussen GI, Holm R, Emilsen E, Rosnes AK, Slipicevic A, Florenes VA. High Expression of Wee1 is Associated With Poor Disease-Free Survival in Malignant Melanoma: Potential for Targeted Therapy. PloS One (2012) 7(6):e38254. doi: 10.1371/journal.pone.0038254
35. Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, et al. RNAi Screening of the Kinome With Cytarabine in Leukemias. Blood (2012) 119(12):2863–72. doi: 10.1182/blood-2011-07-367557
36. Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, et al. Integrated Genomic Analyses Identify WEE1 as a Critical Mediator of Cell Fate and a Novel Therapeutic Target in Acute Myeloid Leukemia. Leukemia (2012) 26(6):1266–76. doi: 10.1038/leu.2011.392
37. PosthumaDeBoer J, Wurdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, et al. WEE1 Inhibition Sensitizes Osteosarcoma to Radiotherapy. BMC Cancer (2011) 11:156. doi: 10.1186/1471-2407-11-156
38. Magnussen GI, Hellesylt E, Nesland JM, Trope CG, Florenes VA, Holm R. High Expression of Wee1 Is Associated With Malignancy in Vulvar Squamous Cell Carcinoma Patients. BMC Cancer (2013) 13:288. doi: 10.1186/1471-2407-13-288
39. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-Molecule Inhibition of Wee1 Kinase by MK-1775 Selectively Sensitizes P53-Deficient Tumor Cells to DNA-Damaging Agents. Mol Cancer Ther (2009) 8(11):2992–3000. doi: 10.1158/1535-7163.MCT-09-0463
40. Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol (2016) 34(36):4371–80. doi: 10.1200/JCO.2016.67.5991
41. Sanai N, Li J, Boerner J, Stark K, Wu J, Kim S, et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin Cancer Res (2018) 24(16):3820–8. doi: 10.1158/1078-0432.CCR-17-3348
42. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol (2015) 33(30):3409–15. doi: 10.1200/JCO.2014.60.4009
43. Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J Clin Oncol (2016) 34(36):4354–61. doi: 10.1200/JCO.2016.67.5942
44. Mendez E, Rodriguez CP, Kao MC, Raju S, Diab A, Harbison RA, et al. A Phase I Clinical Trial of AZD1775 in Combination With Neoadjuvant Weekly Docetaxel and Cisplatin Before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res (2018) 24(12):2740–8. doi: 10.1158/1078-0432.CCR-17-3796
45. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
46. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death Through a Tragedy: Mitotic Catastrophe. Cell Death Differ (2008) 15(7):1153–62. doi: 10.1038/cdd.2008.47
47. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic Catastrophe: A Mechanism for Avoiding Genomic Instability. Nat Rev Mol Cell Biol (2011) 12(6):385–92. doi: 10.1038/nrm3115
48. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19(1):107–20. doi: 10.1038/cdd.2011.96
49. Duda H, Arter M, Gloggnitzer J, Teloni F, Wild P, Blanco MG, et al. A Mechanism for Controlled Breakage of Under-Replicated Chromosomes During Mitosis. Dev Cell (2016) 39(6):740–55. doi: 10.1016/j.devcel.2016.11.017
50. Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, et al. Premature Activation of Cdk1 Leads to Mitotic Events in S Phase and Embryonic Lethality. Oncogene (2019) 38(7):998–1018. doi: 10.1038/s41388-018-0464-0
51. Tominaga Y, Li C, Wang RH, Deng CX. Murine Wee1 Plays a Critical Role in Cell Cycle Regulation and Pre-Implantation Stages of Embryonic Development. Int J Biol Sci (2006) 2(4):161–70. doi: 10.7150/ijbs.2.161
52. De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 Kinase Targeting Combined With DNA-Damaging Cancer Therapy Catalyzes Mitotic Catastrophe. Clin Cancer Res (2011) 17(13):4200–7. doi: 10.1158/1078-0432.CCR-10-2537
53. Lewis CW, Jin Z, Macdonald D, Wei W, Qian XJ, Choi WS, et al. Prolonged Mitotic Arrest Induced by Wee1 Inhibition Sensitizes Breast Cancer Cells to Paclitaxel. Oncotarget (2017) 8(43):73705–22. doi: 10.18632/oncotarget.17848
54. Mak JP, Man WY, Chow JP, Ma HT, Poon RY. Pharmacological Inactivation of CHK1 and WEE1 Induces Mitotic Catastrophe in Nasopharyngeal Carcinoma Cells. Oncotarget (2015) 6(25):21074–84. doi: 10.18632/oncotarget.4020
55. Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, et al. MK-1775, a Novel Wee1 Kinase Inhibitor, Radiosensitizes P53-Defective Human Tumor Cells. Clin Cancer Res (2011) 17(17):5638–48. doi: 10.1158/1078-0432.CCR-11-0650
56. Chow JP, Poon RY. The CDK1 Inhibitory Kinase MYT1 in DNA Damage Checkpoint Recovery. Oncogene (2013) 32(40):4778–88. doi: 10.1038/onc.2012.504
57. Alexander JL, Orr-Weaver TL. Replication Fork Instability and the Consequences of Fork Collisions From Rereplication. Genes Dev (2016) 30(20):2241–52. doi: 10.1101/gad.288142.116
58. Beck H, Nahse-Kumpf V, Larsen MS, O'Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome Through Control of Replication Initiation and Nucleotide Consumption. Mol Cell Biol (2012) 32(20):4226–36. doi: 10.1128/MCB.00412-12
59. Hauge S, Naucke C, Hasvold G, Joel M, Rodland GE, Juzenas P, et al. Combined Inhibition of Wee1 and Chk1 Gives Synergistic DNA Damage in S-Phase Due to Distinct Regulation of CDK Activity and CDC45 Loading. Oncotarget (2017) 8(7):10966–79. doi: 10.18632/oncotarget.14089
60. Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell (2015) 28(5):557–68. doi: 10.1016/j.ccell.2015.09.015
61. Langan TA, Gautier J, Lohka M, Hollingsworth R, Moreno S, Nurse P, et al. Mammalian Growth-Associated H1 Histone Kinase: A Homolog of Cdc2+/CDC28 Protein Kinases Controlling Mitotic Entry in Yeast and Frog Cells. Mol Cell Biol (1989) 9(9):3860–8. doi: 10.1128/mcb.9.9.3860-3868.1989
62. Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA, et al. In Vitro Disassembly of the Nuclear Lamina and M Phase-Specific Phosphorylation of Lamins by Cdc2 Kinase. Cell (1990) 61(4):591–602. doi: 10.1016/0092-8674(90)90471-P
63. Seibert M, Kruger M, Watson NA, Sen O, Daum JR, Slotman JA, et al. CDK1-Mediated Phosphorylation at H2B Serine 6 Is Required for Mitotic Chromosome Segregation. J Cell Biol (2019) 218(4):1164–81. doi: 10.1083/jcb.201806057
64. Beeharry N, Rattner JB, Caviston JP, Yen T. Centromere Fragmentation Is a Common Mitotic Defect of S and G2 Checkpoint Override. Cell Cycle (2013) 12(10):1588–97. doi: 10.4161/cc.24740
65. El Achkar E, Gerbault-Seureau M, Muleris M, Dutrillaux B, Debatisse M. Premature Condensation Induces Breaks at the Interface of Early and Late Replicating Chromosome Bands Bearing Common Fragile Sites. Proc Natl Acad Sci USA (2005) 102(50):18069–74. doi: 10.1073/pnas.0506497102
66. Madan K, Allen JW, Gerald PS, Latt SA. Fluorescence Analysis of Late DNA Replication in Mouse Metaphase Chromosomes Using BUdR and 33258 Hoechst. Exp Cell Res (1976) 99(2):438–44. doi: 10.1016/0014-4827(76)90604-2
67. Kubara PM, Kerneis-Golsteyn S, Studeny A, Lanser BB, Meijer L, Golsteyn RM. Human Cells Enter Mitosis With Damaged DNA After Treatment With Pharmacological Concentrations of Genotoxic Agents. Biochem J (2012) 446(3):373–81. doi: 10.1042/BJ20120385
68. Verma N, Franchitto M, Zonfrilli A, Cialfi S, Palermo R, Talora C. DNA Damage Stress: Cui Prodest? Int J Mol Sci (2019) 20(5):1073. doi: 10.3390/ijms20051073
69. van Vugt MA, Bras A, Medema RH. Polo-Like Kinase-1 Controls Recovery From a G2 DNA Damage-Induced Arrest in Mammalian Cells. Mol Cell (2004) 15(5):799–811. doi: 10.1016/j.molcel.2004.07.015
70. Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, et al. M-Phase Kinases Induce Phospho-Dependent Ubiquitination of Somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA (2004) 101(13):4419–24. doi: 10.1073/pnas.0307700101
71. Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J Clin Invest (2019) 129(3):1329–44. doi: 10.1172/JCI122622
72. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced Mitotic Entry of S-Phase Cells as a Therapeutic Strategy Induced by Inhibition of WEE1. Cancer Discov (2012) 2(6):524–39. doi: 10.1158/2159-8290.CD-11-0320
73. Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a Small Molecule Wee1 Inhibitor, Enhances Anti-Tumor Efficacy of Various DNA-Damaging Agents, Including 5-Fluorouracil. Cancer Biol Ther (2010) 9(7):514–22. doi: 10.4161/cbt.9.7.11115
74. Zheng H, Shao F, Martin S, Xu X, Deng CX. WEE1 Inhibition Targets Cell Cycle Checkpoints for Triple Negative Breast Cancers to Overcome Cisplatin Resistance. Sci Rep (2017) 7:43517. doi: 10.1038/srep43517
75. Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a Potent Wee1 Inhibitor, Synergizes With Gemcitabine to Achieve Tumor Regressions, Selectively in P53-Deficient Pancreatic Cancer Xenografts. Clin Cancer Res (2011) 17(9):2799–806. doi: 10.1158/1078-0432.CCR-10-2580
76. Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, et al. Inhibition of Wee1 Sensitizes Cancer Cells to Antimetabolite Chemotherapeutics In Vitro and In Vivo, Independent of P53 Functionality. Mol Cancer Ther (2013) 12(12):2675–84. doi: 10.1158/1535-7163.MCT-13-0424
77. Harris PS, Venkataraman S, Alimova I, Birks DK, Balakrishnan I, Cristiano B, et al. Integrated Genomic Analysis Identifies the Mitotic Checkpoint Kinase WEE1 as a Novel Therapeutic Target in Medulloblastoma. Mol Cancer (2014) 13:72. doi: 10.1186/1476-4598-13-72
78. Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M. MK1775, a Selective Wee1 Inhibitor, Shows Single-Agent Antitumor Activity Against Sarcoma Cells. Mol Cancer Ther (2012) 11(1):174–82. doi: 10.1158/1535-7163.MCT-11-0529
79. Takebe N, Naqash AR, O'Sullivan Coyne G, Kummar S, Do K, Bruns A, et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-Daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients With Advanced Solid Tumors. Clin Cancer Res (2021) 27(14):3834–44. doi: 10.1158/1078-0432.CCR-21-0329
80. Huang PQ, Boren BC, Hegde SG, Liu H, Unni AK, Abraham S, et al. Discovery of ZN-C3, a Highly Potent and Selective Wee1 Inhibitor Undergoing Evaluation in Clinical Trials for the Treatment of Cancer. J Med Chem (2021) 64(17):13004–24. doi: 10.1021/acs.jmedchem.1c01121
81. Tolcher A, Mamdani H, Chalasani P, Meric-Bernstam F, Gazdoiu M, Makris L, et al. Abstract CT016: Clinical Activity of Single-Agent ZN-C3, an Oral WEE1 Inhibitor, in a Phase 1 Dose-Escalation Trial in Patients With Advanced Solid Tumors. Cancer Res (2021) 81(13 Supplement):CT016. doi: 10.1158/1538-7445.AM2021-CT016
82. Li J, Boren B, Huang PQ, Bunker KD, Doñate F, Samatar AA. Abstract 1965: Discovery of ZN-C3, a Potent Wee-1 Inhibitor With a Differentiated Pharmacologic and Kinase Selectivity Profile. Cancer Res (2021) 81(13 Supplement):1965. doi: 10.1158/1538-7445.AM2021-1965
83. Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radiosensitization of P53 Mutant Cells by PD0166285, a Novel G(2) Checkpoint Abrogator. Cancer Res (2001) 61(22):8211–7.
84. Gill SJ, Wijnhoven PWG, Fok JHL, Lloyd RL, Cairns J, Armenia J, et al. Radiopotentiation Profiling of Multiple Inhibitors of the DNA Damage Response for Early Clinical Development. Mol Cancer Ther (2021) 20(9):1614–26. doi: 10.1158/1535-7163.MCT-20-0502
85. Yang L, Shen C, Pettit CJ, Li T, Hu AJ, Miller ED, et al. Wee1 Kinase Inhibitor AZD1775 Effectively Sensitizes Esophageal Cancer to Radiotherapy. Clin Cancer Res (2020) 26(14):3740–50. doi: 10.1158/1078-0432.CCR-19-3373
86. Lee YY, Cho YJ, Shin SW, Choi C, Ryu JY, Jeon HK, et al. Anti-Tumor Effects of Wee1 Kinase Inhibitor With Radiotherapy in Human Cervical Cancer. Sci Rep (2019) 9(1):15394. doi: 10.1038/s41598-019-51959-3
87. Parsels LA, Karnak D, Parsels JD, Zhang Q, Velez-Padilla J, Reichert ZR, et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization With Combined WEE1 and PARP Inhibitors. Mol Cancer Res (2018) 16(2):222–32. doi: 10.1158/1541-7786.MCR-17-0455
88. Richer AL, Cala JM, O'Brien K, Carson VM, Inge LJ, Whitsett TG. WEE1 Kinase Inhibitor AZD1775 Has Preclinical Efficacy in LKB1-Deficient Non-Small Cell Lung Cancer. Cancer Res (2017) 77(17):4663–72. doi: 10.1158/0008-5472.CAN-16-3565
89. Cuneo KC, Morgan MA, Davis MA, Parcels LA, Parcels J, Karnak D, et al. Wee1 Kinase Inhibitor AZD1775 Radiosensitizes Hepatocellular Carcinoma Regardless of TP53 Mutational Status Through Induction of Replication Stress. Int J Radiat Oncol Biol Phys (2016) 95(2):782–90. doi: 10.1016/j.ijrobp.2016.01.028
90. Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, Phillips J, et al. Targeting Wee1 for the Treatment of Pediatric High-Grade Gliomas. Neuro Oncol (2014) 16(3):352–60. doi: 10.1093/neuonc/not220
91. Caretti V, Hiddingh L, Lagerweij T, Schellen P, Koken PW, Hulleman E, et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol Cancer Ther (2013) 12(2):141–50. doi: 10.1158/1535-7163.MCT-12-0735
92. Sarcar B, Kahali S, Prabhu AH, Shumway SD, Xu Y, Demuth T, et al. Targeting Radiation-Induced G(2) Checkpoint Activation With the Wee-1 Inhibitor MK-1775 in Glioblastoma Cell Lines. Mol Cancer Ther (2011) 10(12):2405–14. doi: 10.1158/1535-7163.MCT-11-0469
93. Suzuki M, Anko M, Ohara M, Matsumoto KI, Hasegawa S, et al. Radiation-Induced Autophagy in Human Pancreatic Cancer Cells Is Critically Dependent on G2 Checkpoint Activation: A Mechanism of Radioresistance in Pancreatic Cancer. Int J Radiat Oncol Biol Phys (2021) 111(1):260–71. doi: 10.1016/j.ijrobp.2021.04.001
94. Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J Clin Oncol (2019) 37(29):2643–50. doi: 10.1200/JCO.19.00730
95. Chera BS, Sheth SH, Patel SA, Goldin D, Douglas KE, Green RL, et al. Phase 1 Trial of Adavosertib (AZD1775) in Combination With Concurrent Radiation and Cisplatin for Intermediate-Risk and High-Risk Head and Neck Squamous Cell Carcinoma. Cancer (2021) 37(29):2643–50. doi: 10.1002/cncr.33789
96. Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin Cancer Res (2018) 24(24):6594–610. doi: 10.1158/1078-0432.CCR-18-1446
97. Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, et al. Forced Activation of Cdk1 via Wee1 Inhibition Impairs Homologous Recombination. Oncogene (2013) 32(24):3001–8. doi: 10.1038/onc.2012.296
98. Liu L, Michowski W, Kolodziejczyk A, Sicinski P. The Cell Cycle in Stem Cell Proliferation, Pluripotency and Differentiation. Nat Cell Biol (2019) 21(9):1060–7. doi: 10.1038/s41556-019-0384-4
99. McBride WH, Schaue D. Radiation-Induced Tissue Damage and Response. J?Pathol (2020) 250(5):647–55. doi: 10.1002/path.5389
100. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific Killing of BRCA2-Deficient Tumours With Inhibitors of Poly(ADP-Ribose) Polymerase. Nature (2005) 434(7035):913–7. doi: 10.1038/nature03443
101. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature (2005) 434(7035):917–21. doi: 10.1038/nature03445
102. Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, et al. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers (Basel) (2020) 12(3):917–21. doi: 10.3390/cancers12030687
103. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med (2017) 376(22):2109–21. doi: 10.1056/NEJMoa1616288
104. Jette NR, Radhamani S, Arthur G, Ye R, Goutam S, Bolyos A, et al. Combined Poly-ADP Ribose Polymerase and Ataxia-Telangiectasia Mutated/Rad3-Related Inhibition Targets Ataxia-Telangiectasia Mutated-Deficient Lung Cancer Cells. Br J Cancer (2019) 121(7):600–10. doi: 10.1038/s41416-019-0565-8
105. Fugger K, Hewitt G, West SC, Boulton SJ. Tackling PARP Inhibitor Resistance. Trends Cancer (2021) 7(12):1102–18. doi: 10.1016/j.trecan.2021.08.007
106. O'Neil NJ, Bailey ML, Hieter P. Synthetic Lethality and Cancer. Nat Rev Genet (2017) 18(10):613–23. doi: 10.1038/nrg.2017.47
107. Lecona E, Fernandez-Capetillo O. Replication Stress and Cancer: It Takes Two to Tango. Exp Cell Res (2014) 329(1):26–34. doi: 10.1016/j.yexcr.2014.09.019
108. Halazonetis TD, Gorgoulis VG, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science (2008) 319(5868):1352–5. doi: 10.1126/science.1140735
109. Brown EJ, Baltimore D. ATR Disruption Leads to Chromosomal Fragmentation and Early Embryonic Lethality. Genes Dev (2000) 14(4):397–402. doi: 10.1101/gad.14.4.397
110. de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, et al. Targeted Disruption of the Cell-Cycle Checkpoint Gene ATR Leads to Early Embryonic Lethality in Mice. Curr Biol (2000) 10(8):479–82. doi: 10.1016/S0960-9822(00)00447-4
111. Parikh RA, Appleman LJ, Bauman JE, Sankunny M, Lewis DW, Vlad A, et al. Upregulation of the ATR-CHEK1 Pathway in Oral Squamous Cell Carcinomas. Genes Chromosomes Cancer (2014) 53(1):25–37. doi: 10.1002/gcc.22115
112. Abdel-Fatah TM, Middleton FK, Arora A, Agarwal D, Chen T, Moseley PM, et al. Untangling the ATR-CHEK1 Network for Prognostication, Prediction and Therapeutic Target Validation in Breast Cancer. Mol Oncol (2015) 9(3):569–85. doi: 10.1016/j.molonc.2014.10.013
113. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444(7120):756–60. doi: 10.1038/nature05236
114. Sorensen CS, Syljuasen RG. Safeguarding Genome Integrity: The Checkpoint Kinases ATR, CHK1 and WEE1 Restrain CDK Activity During Normal DNA Replication. Nucleic Acids Res (2012) 40(2):477–86. doi: 10.1093/nar/gkr697
115. Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, et al. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia (2018) 20(5):478–88. doi: 10.1016/j.neo.2018.03.003
116. Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, Bakkenist CJ. WEE1 Kinase Inhibitor AZD1775 Induces CDK1 Kinase-Dependent Origin Firing in Unperturbed G1- and S-Phase Cells. Proc Natl Acad Sci USA (2019) 116(48):23891–3. doi: 10.1073/pnas.1915108116
117. Simoneau A, Zou L. An Extending ATR-CHK1 Circuitry: The Replication Stress Response and Beyond. Curr Opin Genet Dev (2021) 71:92–8. doi: 10.1016/j.gde.2021.07.003
118. Kibe T, Zimmermann M, de Lange T. TPP1 Blocks an ATR-Mediated Resection Mechanism at Telomeres. Mol Cell (2016) 61(2):236–46. doi: 10.1016/j.molcel.2015.12.016
119. Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell (2017) 65(2):336–46. doi: 10.1016/j.molcel.2016.12.007
120. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell (2013) 155(5):1088–103. doi: 10.1016/j.cell.2013.10.043
121. Brunner A, Suryo Rahmanto A, Johansson H, Franco M, Viiliainen J, Gazi M, et al. PTEN and DNA-PK Determine Sensitivity and Recovery in Response to WEE1 Inhibition in Human Breast Cancer. Elife (2020) 9:1088–103. doi: 10.7554/eLife.57894
122. He J, Kang X, Yin Y, Chao KS, Shen WH. PTEN Regulates DNA Replication Progression and Stalled Fork Recovery. Nat Commun (2015) 6:7620. doi: 10.1038/ncomms8620
123. Wang G, Li Y, Wang P, Liang H, Cui M, Zhu M, et al. PTEN Regulates RPA1 and Protects DNA Replication Forks. Cell Res (2015) 25(11):1189–204. doi: 10.1038/cr.2015.115
124. Feng J, Liang J, Li J, Li Y, Liang H, Zhao X, et al. PTEN Controls the DNA Replication Process Through MCM2 in Response to Replicative Stress. Cell Rep (2015) 13(7):1295–303. doi: 10.1016/j.celrep.2015.10.016
125. Diab A, Gem H, Swanger J, Kim HY, Smith K, Zou G, et al. FOXM1 Drives HPV+ HNSCC Sensitivity to WEE1 Inhibition. Proc Natl Acad Sci USA (2020) 117(45):28287–96. doi: 10.1073/pnas.2013921117
126. Alvarez-Fernandez M, Medema RH. Novel Functions of FoxM1: From Molecular Mechanisms to Cancer Therapy. Front Oncol (2013) 3:30. doi: 10.3389/fonc.2013.00030
127. Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, et al. A Haploid Genetic Screen Identifies the G1/S Regulatory Machinery as a Determinant of Wee1 Inhibitor Sensitivity. Proc Natl Acad Sci USA (2015) 112(49):15160–5. doi: 10.1073/pnas.1505283112
128. Aarts M, Bajrami I, Herrera-Abreu MT, Elliott R, Brough R, Ashworth A, et al. Functional Genetic Screen Identifies Increased Sensitivity to WEE1 Inhibition in Cells With Defects in Fanconi Anemia and HR Pathways. Mol Cancer Ther (2015) 14(4):865–76. doi: 10.1158/1535-7163.MCT-14-0845
129. Francis AM, Alexander A, Liu Y, Vijayaraghavan S, Low KH, Yang D, et al. CDK4/6 Inhibitors Sensitize Rb-Positive Sarcoma Cells to Wee1 Kinase Inhibition Through Reversible Cell-Cycle Arrest. Mol Cancer Ther (2017) 16(9):1751–64. doi: 10.1158/1535-7163.MCT-17-0040
130. Fallah Y, Demas DM, Jin L, He W, Shajahan-Haq AN. Targeting WEE1 Inhibits Growth of Breast Cancer Cells That Are Resistant to Endocrine Therapy and CDK4/6 Inhibitors. Front Oncol (2021) 11:681530. doi: 10.3389/fonc.2021.681530
131. Lamballe F, Ahmad F, Vinik Y, Castellanet O, Daian F, Muller AK, et al. Modeling Heterogeneity of Triple-Negative Breast Cancer Uncovers a Novel Combinatorial Treatment Overcoming Primary Drug Resistance. Adv Sci (Weinh) (2021) 8(3):2003049. doi: 10.1002/advs.202003049
132. Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a Potent and Selective BCL-XL Inhibitor With in Vivo Activity. ACS Med Chem Lett (2014) 5(10):1088–93. doi: 10.1021/ml5001867
133. Ha DH, Min A, Kim S, Jang H, Kim SH, Kim HJ, et al. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci Rep (2020) 10(1):9930. doi: 10.1038/s41598-020-66018-5
134. Ayeni JO, Varadarajan R, Mukherjee O, Stuart DT, Sprenger F, Srayko M, et al. Dual Phosphorylation of Cdk1 Coordinates Cell Proliferation With Key Developmental Processes in Drosophila. Genetics (2014) 196(1):197–210. doi: 10.1534/genetics.113.156281
135. Okumura E, Fukuhara T, Yoshida H, Hanada Si S, Kozutsumi R, Mori M, et al. Akt Inhibits Myt1 in the Signalling Pathway That Leads to Meiotic G2/M-Phase Transition. Nat Cell Biol (2002) 4(2):111–6. doi: 10.1038/ncb741
136. Palmer A, Gavin AC, Nebreda AR. A Link Between MAP Kinase and P34(Cdc2)/Cyclin B During Oocyte Maturation: P90(Rsk) Phosphorylates and Inactivates the P34(Cdc2) Inhibitory Kinase Myt1. EMBO J (1998) 17(17):5037–47. doi: 10.1093/emboj/17.17.5037
137. Coulonval K, Kooken H, Roger PP. Coupling of T161 and T14 Phosphorylations Protects Cyclin B-CDK1 From Premature Activation. Mol Biol Cell (2011) 22(21):3971–85. doi: 10.1091/mbc.e11-02-0136
138. Nakajima H, Yonemura S, Murata M, Nakamura N, Piwnica-Worms H, Nishida E. Myt1 Protein Kinase Is Essential for Golgi and ER Assembly During Mitotic Exit. J Cell Biol (2008) 181(1):89–103. doi: 10.1083/jcb.200708176
139. Wells NJ, Watanabe N, Tokusumi T, Jiang W, Verdecia MA, Hunter T. The C-Terminal Domain of the Cdc2 Inhibitory Kinase Myt1 Interacts With Cdc2 Complexes and Is Required for Inhibition of G(2)/M Progression. J Cell Sci (1999) 112( Pt 19):3361–71. doi: 10.1242/jcs.112.19.3361
140. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical Evaluation of the WEE1 Inhibitor MK-1775 as Single-Agent Anticancer Therapy. Mol Cancer Ther (2013) 12(8):1442–52. doi: 10.1158/1535-7163.MCT-13-0025
141. Toledo CM, Ding Y, Hoellerbauer P, Davis RJ, Basom R, Girard EJ, et al. Genome-Wide CRISPR-Cas9 Screens Reveal Loss of Redundancy Between PKMYT1 and WEE1 in Glioblastoma Stem-Like Cells. Cell Rep (2015) 13(11):2425–39. doi: 10.1016/j.celrep.2015.11.021
142. Pattabiraman DR, Weinberg RA. Tackling the Cancer Stem Cells - What Challenges do They Pose? Nat Rev Drug Discov (2014) 13(7):497–512. doi: 10.1038/nrd4253
143. Ronco C, Martin AR, Demange L, Benhida R. ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. Medchemcomm (2017) 8(2):295–319. doi: 10.1039/C6MD00439C
144. Sand A, Piacsek M, Donohoe DL, Duffin AT, Riddell GT, Sun C, et al. WEE1 Inhibitor, AZD1775, Overcomes Trastuzumab Resistance by Targeting Cancer Stem-Like Properties in HER2-Positive Breast Cancer. Cancer Lett (2020) 472:119–31. doi: 10.1016/j.canlet.2019.12.023
145. Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, et al. DNA-Dependent Kinase (P350) as a Candidate Gene for the Murine SCID Defect. Science (1995) 267(5201):1178–83. doi: 10.1126/science.7855601
146. Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS 3rd, Barron GM, et al. Absence of P350 Subunit of DNA-Activated Protein Kinase From a Radiosensitive Human Cell Line. Science (1995) 267(5201):1183–5. doi: 10.1126/science.7855602
147. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21(9):548–69. doi: 10.1038/s41577-021-00524-z
148. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548(7668):466–70. doi: 10.1038/nature23470
149. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548(7668):461–5. doi: 10.1038/nature23449
150. Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA Sensor for IRF-3-Dependent Innate Immunity. Elife (2012) 1:e00047. doi: 10.7554/eLife.00047
151. Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA Damage Sensor MRE11 Recognizes Cytosolic Double-Stranded DNA and Induces Type I Interferon by Regulating STING Trafficking. Proc Natl Acad Sci USA (2013) 110(8):2969–74. doi: 10.1073/pnas.1222694110
152. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat Rev Genet (2019) 20(11):657–74. doi: 10.1038/s41576-019-0151-1
153. Reislander T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell (2020) 80(1):21–8. doi: 10.1016/j.molcel.2020.07.026
154. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat Commun (2017) 8(1):1751. doi: 10.1038/s41467-017-01883-9
155. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-Dependent Up-Regulation of DNAM-1 and NKG2D Ligands on Multiple Myeloma Cells by Therapeutic Agents Results in Enhanced NK-Cell Susceptibility and Is Associated With a Senescent Phenotype. Blood (2009) 113(15):3503–11. doi: 10.1182/blood-2008-08-173914
156. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA Damage Pathway Regulates Innate Immune System Ligands of the NKG2D Receptor. Nature (2005) 436(7054):1186–90. doi: 10.1038/nature03884
157. Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, et al. Combined Inhibition of Wee1 and PARP1/2 for Radiosensitization in Pancreatic Cancer. Clin Cancer Res (2014) 20(19):5085–96. doi: 10.1158/1078-0432.CCR-14-1038
158. Patel P, Sun L, Robbins Y, Clavijo PE, Friedman J, Silvin C, et al. Enhancing Direct Cytotoxicity and Response to Immune Checkpoint Blockade Following Ionizing Radiation With Wee1 Kinase Inhibition. Oncoimmunology (2019) 8(11):e1638207. doi: 10.1080/2162402X.2019.1638207
159. Wang B, Sun L, Yuan Z, Tao Z. Wee1 Kinase Inhibitor AZD1775 Potentiates CD8+ T Cell-Dependent Antitumour Activity via Dendritic Cell Activation Following a Single High Dose of Irradiation. Med Oncol (2020) 37(8):66. doi: 10.1007/s12032-020-01390-w
Keywords: kinase, DNA damage response (DDR), cell cycle, cancer therapy, Wee1, synthetic lethality
Citation: Bukhari AB, Chan GK and Gamper AM (2022) Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 12:828684. doi: 10.3389/fonc.2022.828684
Received: 03 December 2021; Accepted: 21 January 2022;
Published: 17 February 2022.
Edited by:
Peter McHugh, University of Oxford, United KingdomReviewed by:
Vijay Menon, Yale University, United StatesJulio Morales, University of Oklahoma, United States
Copyright © 2022 Bukhari, Chan and Gamper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armin M. Gamper, gamper@ualberta.ca