 Callum Beach1
Callum Beach1 David MacLean
David MacLean Monica M. Olcina
Monica M. Olcina- 1Department of Oncology, Medical Research Council Oxford Institute for Radiation Oncology, University of Oxford, Oxford, United Kingdom
- 2School of Cancer and Pharmaceutical Sciences, King’s College London, London, United Kingdom
The efficacy of radiotherapy, a mainstay of cancer treatment, is strongly influenced by both cellular and non-cellular features of the tumor microenvironment (TME). Tumor-associated macrophages (TAMs) are a heterogeneous population within the TME and their prevalence significantly correlates with patient prognosis in a range of cancers. Macrophages display intrinsic radio-resistance and radiotherapy can influence TAM recruitment and phenotype. However, whether radiotherapy alone can effectively “reprogram” TAMs to display anti-tumor phenotypes appears conflicting. Here, we discuss the effect of radiation on macrophage recruitment and plasticity in cancer, while emphasizing the role of specific TME components which may compromise the tumor response to radiation and influence macrophage function. In particular, this review will focus on soluble factors (cytokines, chemokines and components of the complement system) as well as physical changes to the TME. Since the macrophage response has the potential to influence radiotherapy outcomes this population may represent a drug target for improving treatment. An enhanced understanding of components of the TME impacting radiation-induced TAM recruitment and function may help consider the scope for future therapeutic avenues to target this plastic and pervasive population.
Introduction
Within neoplastic lesions, immune and mesenchymal cells interact with malignant tumor cells and influence many facets of tumor progression (1–3). Tumor-associated macrophages (TAMs) often make up a large proportion of the immune cell population within the TME. Macrophages are a highly plastic immune cell population, and their phenotypes are shaped by the microenvironments in which they reside (4, 5). In the context of cancer, macrophages are exploited by the tumor cells to adopt phenotypes which counterintuitively, help facilitate disease progression through providing a suitable microenvironment for the progression of multiple carcinomas (6). It is possible to consider the role of TAMs in tumor progression as occurring in phases (Figure 1) which include initial recruitment of TAM progenitors, subsequent polarization to an immunosuppressive phenotype and prevention of anti-tumor immune responses. TAMs can also facilitate angiogenesis to meet the metabolic demands of the cancer while assisting the passage of tumor cells into circulation and setting up the site for secondary tumor growth (7–9). Interestingly, the TAM population is phenotypically diverse to the extent that both pro- and anti-tumoral phenotypes of these cells can reside in the same tumor (10, 11). The prevalence of the TAM population correlates with poor patient prognosis in all cancers (except colorectal) (12–14) highlighting this population as a potential therapeutic target in cancer.
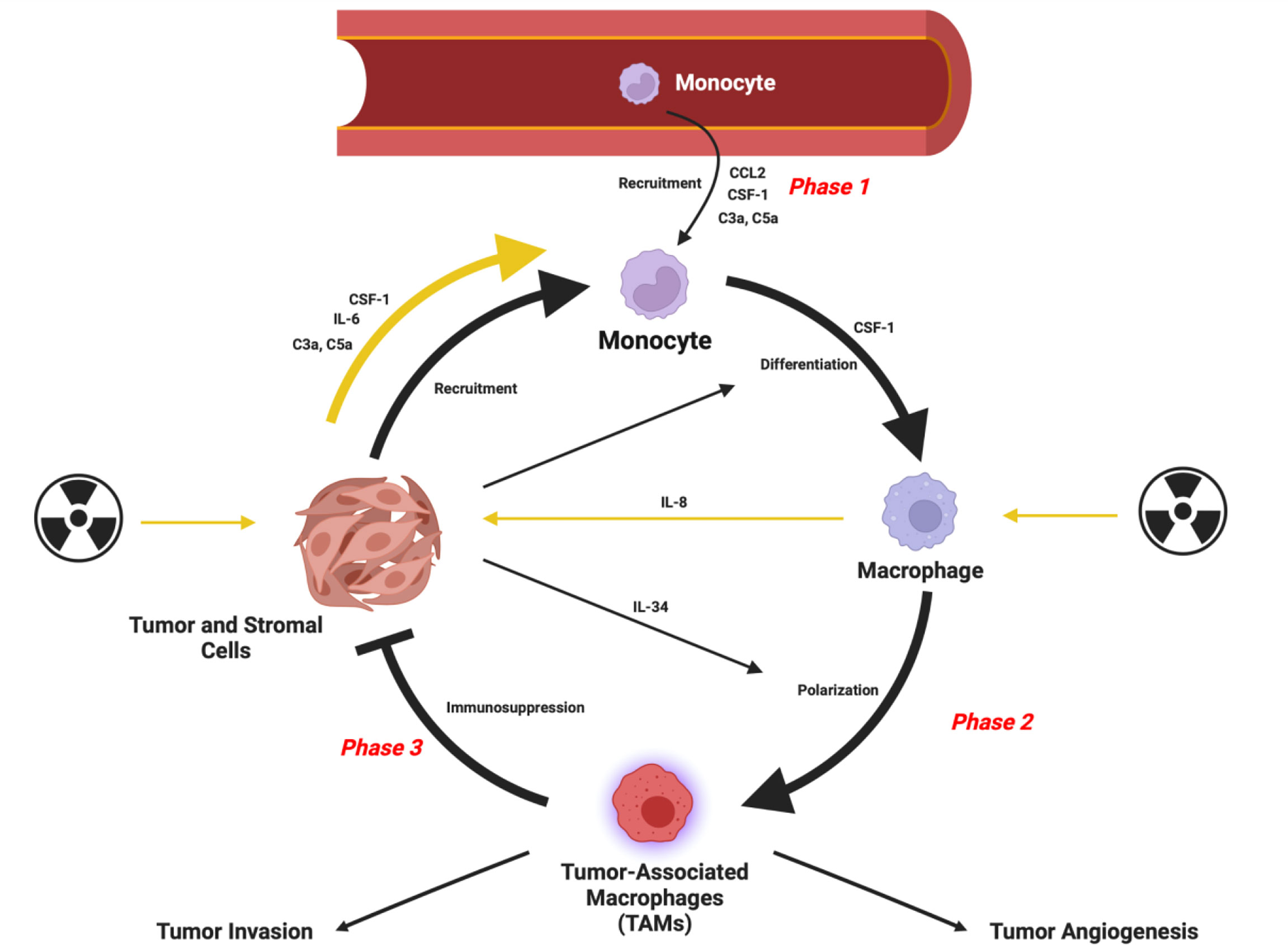
Figure 1 Schematic representation of the role of TAMs in tumor progression. Radiation can contribute to recruitment and polarization as indicated by the yellow arrows. Figure created in Biorender. Agreement number: RO24DGQPW4.
Radiotherapy is still a mainstay of cancer treatment for approximately 50% of all cancer patients. It is increasingly recognized that radiotherapy is a strong immune modulator, with the capacity to induce both pro- and anti-inflammatory processes (15, 16). As such, radiation can elicit macrophage recruitment into the tumor (17–20). TAM polarization away from tissue-protection and towards anti-tumoral/immunostimulatory functions could be a potential approach to boost the anti-cancer effects of radiotherapy and capitalize on the immune-stimulating effects of this treatment (16, 19). Here, the effect of radiation on TAM recruitment and polarization will be described. We will particularly focus on changes to soluble and physical components of the tumor microenvironment (TME) which may limit the positive effects of radiation on macrophage plasticity and highlight key examples that could be therapeutically targeted to improve radiation response.
Phase 1: Recruitment of TAMs
Recruitment overview
TAMs within the tumor are either present as tissue-resident macrophages or are formed after circulating monocytes are recruited and subsequently polarized into mature TAMs (21, 22). Resident macrophages are present during embryonic development and tend to exist in specific tissues such as Kupffer cells in the liver, and alveolar macrophages in the pulmonary alveolus of the lungs (23). These macrophages can provide a pro-tumorigenic niche and assist with initial tumor growth from a very early stage (24).
Soluble factors impacting TAM recruitment following radiotherapy
Soluble factors that mediate mobilization are critically associated with recruiting monocytes/macrophages to the TME (Figure 1). A well-documented signaling molecule involved in this process is chemokine (C-C motif) ligand 2 (CCL2, also known as monocyte chemoattractant protein 1; MCP1) (25–27). Radiotherapy is known to induce the expression of CCL2 within the TME (28, 29). Increased CCL2 expression can also be regulated by components of the humoral arm of innate immunity such as the complement system (30–32) and the long pentraxin PTX3 (33). Both of these innate immunity components appear to work in concert since PTX3 deficiency results in complement-dependent TAM recruitment in 3-Methylcholanthrene carcinogenesis models (33). Signaling of complement anaphylatoxins C3a and C5a through their respective receptors, C3aR and C5aR1, has been further demonstrated to result in TAM recruitment and polarization towards an immunosuppressive phenotype (30, 31). This includes reduced CD206 expression and upregulation of CD11c, major histocompatibility complex class II, CD80 and CD86 in TAMs from C3 and C3aR1-/- mice (32). Interestingly, expression of C3a, C5a and their receptors C3aR and C5aR1 is induced in melanoma murine tumors following irradiation (20 Gy) (34). Furthermore, complement inhibition at the level of C3 (with a CR2-Crry fusion protein) in combination with radiation has been demonstrated to enhance the numbers of macrophages with an M1-like phenotype (F4/80+, CD11c+, CD206-) in lymphoma tumor models (35).
In addition to chemokines and complement soluble factors, cytokines are also involved in the recruitment of monocytes/macrophages to the TME. Colony-stimulating factor 1 (CSF-1, also known as macrophage colony stimulating factor; M-CSF), which typically is associated with a differentiation/survival signal for monocyte/macrophages, also has chemotactic properties for the recruitment of these cells to a site of inflammation (36, 37). In several tumor types and murine models, radiation has been demonstrated to induce CSF-1 production which can facilitate macrophage recruitment (17, 18). Following irradiation of tumors the DNA damage-induced kinase ABL1 (c-Abl) is recruited into the nuclei of tumor cells to enhance CSF1 transcription (38). CSF-1 production is also induced in response to IL-8, which can be secreted by the macrophages themselves, contributing to a positive feedback axis further perpetuating macrophage recruitment. However, this axis is not necessarily macrophage-specific as cancer cells can also produce IL-8 themselves post-irradiation (39). IL-34 is a cytokine that shares its receptor with CSF-1, binding CSF1-R, and as such they have similar biological properties. Like CSF-1, IL-34 expression is induced after irradiation (40). This induction has also been demonstrated to promote monocyte recruitment to the TME and subsequent polarization to an immunosuppressive phenotype (41).
Furthermore, tumor cells produce IL-6 in response to radiation-induced damage which promotes monocytes/macrophage recruitment to the TME (42–44). In a double-edged role for IL-6, once monocyte recruitment occurs, the cytokine also blocks dendritic cell differentiation and promotes monocytes to differentiate towards a TAM-like cell with an immunosuppressive phenotype (6, 45).
Physical changes in the TME affecting TAM recruitment following radiotherapy
Hypoxia (low oxygen tension) is a common physical feature of the TME that arises due to insufficient oxygen supply to support rapidly growing tumors. Hypoxia is particularly relevant to radiotherapy since cells irradiated under reduced oxygen levels are more resistant to the lethal effects of radiation (46). Hypoxia-inducible factors (HIFs) are key to the transcriptional response to hypoxia. HIF heterodimers consist of an oxygen-sensitive subunit (HIF-1α, HIF-2α or HIF-3α), and a constitutively expressed HIF-β subunit. Under ambient oxygen concentrations, HIF-α subunits are continually degraded by ubiquitination and proteasomal degradation. However, under low oxygen tensions, HIF-α subunits are stabilized and trafficked to the nucleus where they modulate gene expression through binding hypoxia-responsive elements of specific genes associated with the hypoxic response (47–49). Both HIF-1α and HIF-2α can accumulate in macrophages exposed to hypoxic conditions in vitro (50, 51). In vivo, HIF-1α has been found to be essential for maintenance of appropriate cellular ATP pools necessary for myeloid cell motility and function (52). Furthermore, following tumor irradiation, nitric oxide (NO) generation in TAMs results in s-nitrosylation of HIF-1α at its oxygen-dependent degradation domain which prevents its destruction. Pharmacological inhibition of NO production is associated with reduced tumor growth following irradiation (53). Furthermore, studies using mice specifically lacking HIF-2α in myeloid cells have demonstrated reduced TAM infiltration in hepatocellular and colitis-associated colon carcinoma models through regulation of cytokine receptor CSF-1R and chemokine receptor CXCR4. Interestingly, this observed reduction in TAM infiltration was associated with reduced tumor cell proliferation (54). HIF-dependent induction of CCL2 also further supports monocyte/macrophage recruitment (55). A recent study has demonstrated that vascular endothelial growth factor-A (VEGF-A), another HIF-regulated gene, also plays a key role in both the recruitment of macrophages and the polarization toward an immunosuppressive phenotype as shown by the increase of the marker CD163 (56).
Extracellular matrix
The extracellular matrix (ECM), which constitutes the protein scaffold around the tumor and stromal cells, has a role in providing a platform for innate immune cell infiltration, with many of its components and post-degradation fragments sharing the ability to recruit monocytes. Much focus has been directed to proteolytic fragments of the ECM which have been demonstrated to represent endogenous ligands for binding and activating toll-like receptors (TLRs). The release of glycosaminoglycan hyaluronan (HA) after irradiation of the tumor has been documented (57). HA can also play a role in facilitating macrophage infiltration into the tumor stroma through an interaction with the HA receptor CD44 expressed by macrophages (58). Monocytes/TAMs recruited by the CD44:HA axis have an immunosuppressive phenotype. This is facilitated by the upregulation of IL-10 expression while concurrently downregulating NF-κB signaling (59).
In addition to HA, latent TGF-β (an inactive form of the cytokine) is also released by the ECM post-irradiation. Once activated, TGF-β has a potent influence on TAM recruitment. This can occur directly through enhanced integrin expression and type IV collagenase secretion (60) and indirectly through the upregulation of CXCR4 on monocytes, with perivascular fibroblast expression of CXCL12 attracting the monocytes to the tumor bed (61).
Additionally, damaging the ECM leads to macrophage recruitment due to the attraction of immunosuppressive TAMs through the scavenger receptor CD206 (mannose receptor). This allows the phagocytosis and degradation of collagen fragments to form a strong chemoattractant for macrophages (62, 63). This leads to a feedback loop where initial radiation-induced damage to the ECM leads to recruitment of TAMs that themselves facilitate a continuous wound-healing state within the tumor site, further increasing monocyte/TAM recruitment. In a similar fashion, elastin fragments generated by the activity of macrophage-derived MMPs (9 and -12) have been demonstrated to act as chemotactic factors for monocytes, creating a positive feedback loop which increases the prevalence of TAMs in the TME (64).
Phase 2: Macrophage polarization
Polarization overview
Previously, monocyte polarization into mature macrophages was thought to be binary, with TAMs either acting as inflammatory or immunosuppressive agents within the stroma (65). However, it is becoming increasingly clear that, once polarized, the TAMs phenotypically fall on a spectrum (4). Data, mostly gathered from in vitro studies, has indicated that polarization on this spectrum may depend on the presence of specific factors such as IL-4, IL-10, IL-13, IFNγ, and lipopolysaccharide (LPS) (66, 67) (Figure 2). Once these factors bind to their respective receptor, monocytes undergo polarization and maturation into more specialised TAM phenotypes through downstream signal transduction pathways altering transcription within these cells (68). Recently, it has been identified that TAM polarization can be refined to a three-way polarization program in a spontaneous murine model of breast cancer (11). This three-way program is broadly split into an alternatively-activated-like, angiogenic/immunosuppressive, and inflammatory phenotypic specialization of these cells (11).
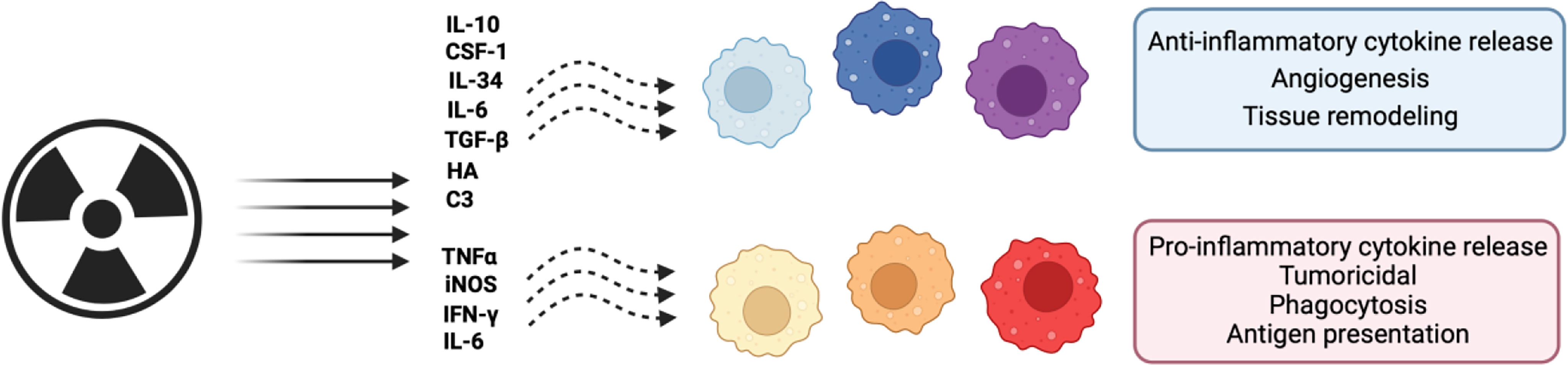
Figure 2 Schematic representation of the effects of radiation on macrophage polarization. Macrophages can adopt both pro- and anti-tumoral phenotypes across a spectrum of possible polarization states. Shown are the effects of radiation on these phenotypes and effector molecules. Figure created in Biorender. Agreement number: LH24DGQ49Q.
Pathways involved in radiation-induced polarization
Following irradiation, macrophage polarization towards either pro- or anti-inflammatory sides of the spectrum may be dependent on irradiation dose and which transcription factors are formed to drive downstream gene expression (69, 70). NF-κB is a key modulator of macrophage polarization and NF-κB p65-p50 heterodimers can initiate transcription of pro-inflammatory genes such as TNFα, IL1β, IL6, IL12, IFNγ and CXCL10 (70). Increased p65/RelA expression following 2 Gy irradiation of the RAW264.7 macrophage cell line or CD11b+ peritoneal macrophages, is associated with increased levels of inducible nitric oxide synthase (iNOS, which is an M1-associated marker) (71). Low dose (2 Gy) whole body irradiation has also been demonstrated to induce iNOS, and concurrently reduce M2-associated markers such as Ym-1 and Fizz-1 in peritoneal macrophages. iNOS expressing TAMs in turn appear important for effector T-cell recruitment into the tumor through vascular normalization (69). Irradiation of human monocyte-derived macrophages with 2, 6 or 10 Gy, results in increased RelB expression which is accompanied by reduced expression of anti-inflammatory genes (such as CD163, and IL-10) (72). Conversely, loss of NF-κB p50 expression has been associated with a pro-inflammatory macrophage phenotype including enhanced TNFα and reduced IL10 expression in bone marrow-derived macrophages incubated with both LPS and irradiated 4T1 cancer cells (10 Gy) (17) (Figure 2).
Enhanced radiation-induced NF-κB signaling can occur following activation of the apical DNA damage kinase, ATM. ATM-dependent NF-κB activation occurs following ubiquitination of NEMO (NF-κB essential modulator) which releases the cytoplasmic p50-p65 heterodimer allowing its translocation to the nucleus to act as a transcriptional activator (73).
ATM activation can also occur downstream of reactive oxygen species (ROS) production. NADPH oxidase 2 (NOX2)-dependent ROS production was reported to be important in ATM-dependent polarization of macrophages towards a pro-inflammatory phenotype through regulation of IRF5 at the mRNA and post-translational level. Therapeutically targeting other DNA damage response components, such as poly (ADP-ribose) polymerase (PARP) also appeared to activate macrophages towards a pro-inflammatory phenotype following increased ATM and IRF5 activation (74). Importantly enhanced expression of iNOS+CD68+ and NOX2+CD68+ TAMs was observed in resected specimens of rectal cancer patients with good responses to neoadjuvant radiotherapy (74). A recent study also suggested that targeting the angiogenic factor, fibroblast growth factor 2 (FGF2), in combination with radiotherapy can increase the iNOS+/CD206+ TAM ratio and improve tumor responses following fractionated radiotherapy (75). These data suggest that FGF2 could be considered as a therapeutic target to be exploited in combination with radiotherapy.
Examples of potential barriers to effective polarization by radiation
As previously mentioned, radiotherapy induces the expression of CCL2 within the TME (28, 29). CCL2 acts to shift the recruited monocytes towards a more immunosuppressive phenotypic type directly by downregulating polarization-related gene expression and indirectly via T helper 2 cells (Th2) releasing anti-inflammatory cytokines such as IL-4, IL-6 and IL-10 (76). In a preclinical pancreatic ductal adenocarcinoma model, the inhibition of CCL2 in isolation had little impact on tumor growth unless used in combination with radiotherapy (77). It was found that irradiation of the tumor caused a significant increase in CCL2 production and radiation-dependent recruitment of monocytes/macrophages (77). Inhibiting this CCL2/CCR2 recruitment axis led to a decrease in tumor growth and vascularity (77). Additionally, the inhibition of CCL2 led to a decrease in TAM presence and a decrease in metastasis (78). This decrease in metastasis was caused by CCL2 inhibition reducing the production of CCL3 by immunosuppressive TAMs thereby reducing the ability of these macrophages to assist with tumor intravasation (78).
There has also been a lot of interest in therapeutically targeting CSF-1 signaling to modulate macrophage polarization following irradiation in a variety of cancers. In glioblastoma tumor models, CSF-1R inhibition delays recurrence following irradiation by reducing radiation-induced monocyte recruitment and differentiation to immunosuppressive TAMs (40). Interestingly, TAM survival in the context of CSF-1R inhibition appears to be facilitated by granulocyte-macrophage CSF (GM-CSF) and IFNγ (79). Altered TAM polarization and a reduction in macrophage migration was also seen in a preclinical prostate cancer model (38). Furthermore, in preclinical colorectal and pancreatic models, macrophage depletion using CSF-1 blocking antibodies, enhances the effectiveness of combined radiotherapy and immune checkpoint inhibitor (anti-PD-L1) treatment suggesting that macrophages act to hinder productive anti-tumor immune functions of radiotherapy (19).
Complement activation and signaling of complement anaphylatoxins through their respective receptors can also impact macrophage polarization. This is relevant in the context of radiotherapy since irradiation has been found to increase the local tumor expression of several complement factors in murine models (following 5 and 20 Gy irradiation) and in patient samples (treated with 1.5-2 Gy) (34). Of note, in the TME, the presence of stromal CD34high fibroblasts expressing high levels of central complement component C3 (which when cleaved will result in C3a production) may also support the recruitment of macrophages with immunosuppressive phenotypes and results in attenuation of T-cell mediated responses (80). Interestingly, C3aR activation in TAMs can occur following intracellular production of C3a by tumor cells; and activation of PI3Kγ signaling downstream of C3aR activation contributes to suppression of anti-tumor responses (81). The effects of irradiation on intracellular C3a or C5a levels across tumor cells, however, is still unclear. Previously published work suggested that the presence of C5a and C3a might be essential for effective tumor radiation responses (34). However, the well-documented impact of C3a and C5a on macrophage recruitment and polarization towards immunosuppression may indicate that targeting the C3a-C3aR or C5a-C5aR signaling axes might prove to be beneficial in certain contexts. In combination with anti-PD-1 blocking antibodies, blocking C5a/C5aR1 signaling has indeed proven effective at improving primary and metastatic disease in lung tumor models (82). Similarly, in the B16-F10 melanoma model, blocking the PD-1/PDL-1 axis alongside C3a-C3aR or C5a-C5aR resulted in improved tumor control (83). The effects of radiotherapy in combination with immune checkpoint and C5a/C5aR1 inhibition, however, has yet to be determined.
The use of TGF-β inhibition in combination with PD-1/PD-L1 inhibition has also found success in a multitude of clinical trials, with phase two trials commencing in non-small cell lung (NCT03631706), triple negative breast (NCT03579472), colorectal (NCT03724851), and pancreatic (NCT02734160) cancers. A summary of additional recent clinical trials combining radiotherapy and macrophage targeting is shown in Table 1. Interestingly, combining TGF-β and PD-1 inhibition with radiotherapy in a preclinical colorectal cancer model demonstrated improved survival plus reduced tumor growth (84). Additionally, this study demonstrated a reduction in TAM recruitment to both primary tumors as well as non-irradiated bilateral lesions (84).
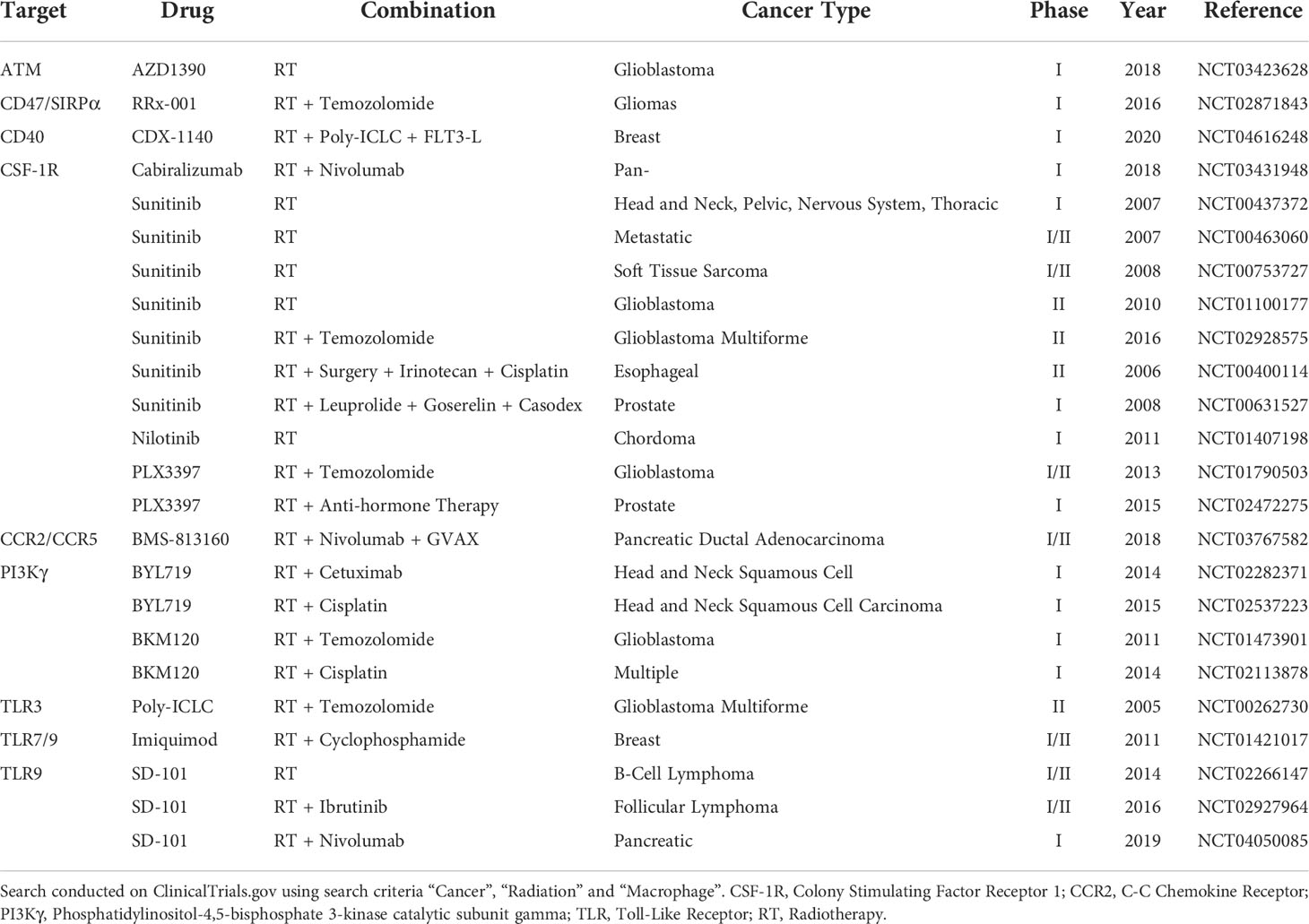
Table 1 Table summarizing latest clinical trials combining radiotherapy and approaches which may impact macrophage recruitment or function.
Conclusion
Effectively modulating the immunostimulatory effects of radiation has the enticing potential to improve local and distant tumor control (85). Given the relatively high numbers of macrophages in the TME (relative to other cell types) and the enhanced macrophage recruitment observed following irradiation, it is likely that combination therapies will have to consider how to polarize this immune population to the pro-inflammatory, tumoricidal side of the spectrum (86). Indeed, investigation into targeting TAMs is currently at the forefront of cancer immunotherapies and, a greater understanding of mechanisms of recruitment and pro-tumor activity of these macrophages may provide new therapeutic opportunities to improve the efficacy of existing treatments (39). Targeting the soluble factor-receptor axes interactions that may pose a barrier to the most effective polarization could be considered. For example, CSF1-CSF1R, C5a-C5aR1, FGF2 or TGFβ/TGFβR blockade in combination with immune checkpoint inhibitors such as PD1/PDL-1 could be promising strategies (19, 84). Further research into the effect of different radiation doses and fractionation regimes on macrophage recruitment and plasticity will help optimize the timing and nature of the most effective combination therapies. A consideration of the effect of an altered macrophage response to normal tissue toxicity following radiotherapy will also be important since maximal therapeutic benefit relies on effective tumor control with minimal normal tissue toxicity.
Author contributions
Conceptualization: CB and MO. Writing original draft: CB, DMac, DMaj, and MO. Writing review and editing: CB, DMac, DMaj, JA, and MO. Resources: JA and MO. Supervision: JA and MO. Funding acquisition: JA and MO. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Medical Research Council (MC_UU_00001/10). C.B is supported by an International Accelerator Award, ACRCelerate funded by Cancer Research UK (A26825 and A28223). J.N.A. is the recipient of a Cancer Research Institute/Wade F.B. Thompson CLIP grant (CRI3645) and Cancer Research UK grant DCRPGF\100009.
Acknowledgments
We apologies to all the authors that we could not cite due to space constraints. This work was supported by the Medical Research Council (MC_UU_00001/10). C.B is supported by an International Accelerator Award, ACRCelerate funded by Cancer Research UK (A26825 and A28223). J.N.A. is the recipient of a Cancer Research Institute/Wade F.B. Thompson CLIP grant (CRI3645) and Cancer Research UK grant DCRPGF\100009.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaminska K, Szczylik C, Bielecka Z, Bartnik E, Porta C, Lian F, et al. The role of the cell–cell interactions in cancer progression. J Cell Mol Med (2015) 19(2):283–96. doi: 10.1111/jcmm.12408
2. Hiam-Galvez K, Allen B, Spitzer M. Systemic immunity in cancer. Nat Rev Cancer (2021) 21:345–59. doi: 10.1038/s41568-021-00347-z
3. Liang W, Chen X, Zhang S, Fang J, Chen M, Xu Y, et al. Mesenchymal stem cells as a double-edged sword in tumor growth: focusing on MSC-derived cytokines. CMBL (2021) 26(3):1–25. doi: 10.1186/s11658-020-00246-5
4. Murray P, Allen J, Biswas S, Fisher E, Gilroy D, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
5. Liu J, Gao M, Yang Z, Zhao Y, Guo K, Sun B, et al. Macrophages and metabolic reprograming in the tumor microenvironment. Front Oncol (2022) 12(795159). doi: 10.3389/fonc.2022.795159
6. Muliaditan T, Caron J, Opzoomer J, Kosti P, Georgouli M, Gordon P, et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat Commun (2018) 9(2951):1–15. doi: 10.1038/s41467-018-05346-7
7. Stockmann C, Doedens A, Weidermann A, Zhang N, Takeda N, Greenberg J, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature (2008) 456(7223):814–8. doi: 10.1038/nature07445
8. Bosiljcic M, Cederberg R, Hamilton M, LePard N, Harbourne B, Collier J, et al. Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs. Breast Cancer Res (2019) 21(103):1–16. doi: 10.1186/s13058-019-1189-x
9. Erler J, Bennewith K, Cox T, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell (2009) 15(1):35–44. doi: 10.1016/j.ccr.2008.11.012
10. Wang Y, Lin Y-X, Qiao S-L, An H-W, Ma Y, Qiao Z-Y, et al. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials (2017) 112:153–63. doi: 10.1016/j.biomaterials.2016.09.034
11. Opzoomer J, Anstee J, Dean I, Hill E, Bouybayoune I, Caron J, et al. Macrophages orchestrate the expansion of a proangiogenic perivascular niche during cancer progression. Sci Adv (2021) 9(2951):1–15. doi: 10.1101/2020.10.30.361907
12. Zhang Q-W, et al. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS One (2012) 7(12):1–14. doi: 10.1371/journal.pone.0050946
13. Wang C, Lin Y, Zhu H, Zhou Y, Mao F, Huang X, et al. The prognostic and clinical value of tumor-associated macrophages in patients with breast cancer: A systematic review and meta-analysis. Front Oncol (2022) 12(905846). doi: 10.3389/fonc.2022.905846
14. Larionova I, Tuguzbaeva G, Ponomaryova A, Stakheyeva M, Cherdyntseva N, Pavlov V, et al. Tumor-associated macrophages in human breast, colorectal, lung, ovarian and prostate cancers. Front Oncol (2020) 10(566511). doi: 10.3389/fonc.2020.566511
15. Appleton E, Hassan J, Wah Hak C-C, Sivamanoharan N, Wilkins A, Samson A, et al. Kickstarting immunity in cold tumours: Localised tumour therapy combinations with immune checkpoint blockade. Front Immunol (2021) 12(754436). doi: 10.3389/fimmu.2021.754436
16. Moon E, Petersson K, Olcina M. The importance of hypoxia in radiotherapy for the immune response, metastatic potential and FLASH-RT. Int J Radiat. Biol (2022) 98(3):439–51. doi: 10.1080/09553002.2021.1988178
17. Crittenden M, Cottam B, Savage T, Nguyen C, Newell P, Gough M, et al. Expression of NF-κB p50 in tumor stroma limits the control of tumors by radiation therapy. PLoS One (2012) 7(6):1–10. doi: 10.1371/journal.pone.0039295
18. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum R-R, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. (2014) 124(2):687–95. doi: 10.1172/JCI67313
19. Jones K, Tiersma J, Yuzhalin A, Gordon-Weeks A, Buzzelli J, Im J, et al. Radiation combined with macrophage depletion promotes adaptive immunity and potentiates checkpoint blockade. EMBO Mol Med (2018) 10(e9342):1–16. doi: 10.15252/emmm.201809342
20. Agn G-O, et al. Inhibition of mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA (2010) 107(18):8363–8. doi: 10.1073/pnas.0911378107
21. Franklin R, Liao W, Sarkar A, Kim M, Bivona M, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344(6186):921–5. doi: 10.1126/science.1252510
22. Zhu Y, Herndon J, Sojka D, Kim K-W, Knolhoff B, Zuo C C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity (2017) 47(2):323–338. doi: 10.1016/j.immuni.2017.07.014
23. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol (2017) 15(53):1–18. doi: 10.1186/s12915-017-0392-4
24. Casanova-Acebes M, Dalla E, Leader A, LeBerichel J, Nikolic J, Morales B, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature (2021) 595(7868):578–84. doi: 10.1038/s41586-021-03651-8
25. Mizutani K, Sud S, McGregor N, Martinovski G, Rice B, Craig M, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia (2009) 11(11):1235–42. doi: 10.1593/neo.09988
26. Stiener J, Murphy E. Importance of chemokine (CC-motif) ligand 2 in breast cancer. Int J Biol Markers (2012) 27(3):179–85. doi: 10.5301/JBM.2012.9345
27. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Kaiser L, et al. CCL2 recruits inflammatory monocytes to facilitate breast tumor metastasis. Nature (2012) 475(7355):222–5. doi: 10.1038/nature10138
28. Bravata V, Minafra L, Forte G I, Cammarata F, Russo G, Di Maggio FM, et al. Cytokine profile of breast cell lines after different radiation doses. Int J Radiat. Biol (2017) 93(11):1217–26. doi: 10.1080/09553002.2017.1362504
29. Wang P, Guo F, Han L, Wang X, Li J, Guo Y, et al. X-ray-induced changes in the expression of inflammation-related genes in human peripheral blood. Int J Mol Sci (2014) 15(11):19516–34. doi: 10.3390/ijms151119516
30. Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J Biol Chem (2015) 290(17):10667–76. doi: 10.1074/jbc.M114.612622
31. Czermak B, Sarma V, Bless N, Schmal H, Friedl H, Ward P, et al. In vitro and in vivo dependency of chemokine generation on C5a and TNF-alpha. J Immunol (1999) 162(4):2321–5.
32. Magrini E, Di Marco S, Mapelli S, Perucchini C, Pasqualini F, Donato A, et al. Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression. Nat Cancer. (2021) 2(2):218–32. doi: 10.1038/s43018-021-00173-0
33. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell (2015) 160(4):700–14. doi: 10.1016/j.cell.2015.01.004
34. Surace L, Lysenko V, Fontana A, Cecconi V, Janssen H, Bicvic A, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. (2015) 42(4):767–77. doi: 10.1016/j.immuni.2015.03.009
35. Elvington M, Scheiber M, Yang X, Lyons K, Jacqmin D, Wadsworth C, et al. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell Rep (2014) 8(3):818–30. doi: 10.1016/j.celrep.2014.06.051
36. Sauter K, Waddell L, Lisowski Z, Young R, Lefevre L, Davis G, et al. Macrophage colony-stimulating factor (CSF1) controls monocyte production and maturation and the steady-state size of the liver in pigs. Am J Physiol Gastrointest Liver Physiol (2016) 311(3):533–47. doi: 10.1152/ajpgi.00116.2016
37. Pyonteck S, Gadea B, Wang H-W, Gocheva V, Hunter K, Tang L-H, et al. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene (2017) 39(11):1459–1467. doi: 10.1038/onc.2011.337
38. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Microenviron. (2013) 73(9):2782–94. doi: 10.1158/0008-5472.CAN-12-3981
39. Ge Z DS. The crosstalk between tumor-associated macrophages (TAMs) and tumor cells and the corresponding targeted therapy. Front Oncol (2020) 10(590941). doi: 10.3389/fonc.2020.590941
40. Stafford J, Hirai T, Deng L, Chernikova S, Urata K, West B, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncology (2016) 18(6):797–806. doi: 10.1093/neuonc/nov272
41. Segaliny A, Mohamadi A, Dizier B, Lokajczyk A, Brion R, Lanel R, et al. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitmen. Int J Cancer (2017) 137(1):73–85. doi: 10.1002/ijc.29376
42. Zhou T, Zhou YL, Qian MJ, Fang YZ, Ye S, Xin WX, et al. Interleukin-6 induced by YAP in hepatocellular carcinoma cells recruits tumor-associated macrophages. J Pharmacol Sci (2018) 138(2):89–95. doi: 10.1016/j.jphs.2018.07.013
43. Zhang J, Nakatsugawa S, Niwa O, Ju GZ, Liu SZ. Ionizing radiation-induced IL-1 alpha, IL-6 and GM-CSF production by human lung cancer cells. Chin Med J (Engl). (1994) 107(9):653–7.
44. Yamanaka R, Tanaka R, Yoshida S. Effects of irradiation on cytokine production in glioma cell lines. Neurol Med Chir (1993) 33(11):744–8. doi: 10.2176/nmc.33.744
45. Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. (2007) 110(13):4319–30. doi: 10.1182/blood-2007-02-072587
46. Hammond E, Asselin M-C, Forster D, O'Connor J-P-B, Senra J-M, Williams K-J, et al. The meaning, measurement and modification of hypoxia in the laboratory and the clinic. Clin Oncol (R. Coll Radiol.) (2014) 26(5):277–88. doi: 10.1016/j.clon.2014.02.002
47. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (2001) 292(5516):464–8. doi: 10.1126/science.1059817
48. Jaakola P, Mole D, Tian Y, Wilson M, Gielbert J, Gaskell S, et al. Targeting of HIF-alpha to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292(5516):468–72. doi: 10.1126/science.1059796
49. Wang G, Semenza G. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem (1995) 270(3):1230–7. doi: 10.1074/jbc.270.3.1230
50. Griffiths L, Binley K, Iqball S, Kan O, Maxwell P, Ratcliffe P, et al. The macrophage – a novel system to deliver gene therapy to pathological hypoxia. Gene Ther (2000) Volume 7:255–62. doi: 10.1038/sj.gt.3301058
51. Burke B, Tang N, Corke K, Tazzyman D, Ameri K, Wells M, et al. Expression of HIF-1α by human macrophages: implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J Pathol (2002) 196(2):204–12. doi: 10.1002/path.1029
52. Cramer T, Yamanishi Y, Clausen B, Forster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57. doi: 10.1016/S0092-8674(03)00154-5
53. Li F, Sonveaux P, Rabbani Z, Liu S, Yan B, Huang Q, et al. Regulation of HIF-1α stability through s-nitrosylation. Mol Cell (2007) 26(1):63–74. doi: 10.1016/j.molcel.2007.02.024
54. Imtiyaz H, Williams E, Hickey M, Patel S, Durham A, Yuan L-Y, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. JCI (2010) 120(8):2699–714. doi: 10.1172/JCI39506
55. Lin N, Simon M. Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. J Clin Invest. (2016) 126(10):3661–71. doi: 10.1172/JCI84426
56. Wheeler K, Jena M, Pradham B, Nayak N, Das S, Hsu C-D, et al. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS One (2018) 13(1):1–18. doi: 10.1371/journal.pone.0191040
57. Li Y, Rahmanian M, Widstrom C, Lepperdinger G, Frost G, Heldin P, et al. Irradiation-induced expression of hyaluronan (HA) synthase 2 and hyaluronidase 2 genes in rat lung tissue accompanies active turnover of HA and induction of types I and III collagen gene expression. Am J Respir Cell Mol Biol (2000) 23(3):411–8. doi: 10.1165/ajrcmb.23.3.4102
58. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res (2010) 70(18):7073–83. doi: 10.1158/0008-5472.CAN-09-4687
59. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget (2016) 7(32):52294–306. doi: 10.18632/oncotarget.10561
60. Wahl SAJ, Weeks B, Wong H, Klotman P. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc Natl Acad Sci USA (1993) 90(10):4577–81. doi: 10.1073/pnas.90.10.4577
61. Arwert E, Harney A, Entenberg D, Wang Y, Sahai E, Pollard J, et al. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep (2018) 23(5):1239–48. doi: 10.1016/j.celrep.2018.04.007
62. Madsen D, Leonard D, Masedunskas A, Moyer A, Jessen H, Jurgensen H, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor–mediated pathway. JCB (2013) 202(6):951–66. doi: 10.1083/jcb.201301081
63. O'Brien J, Lyons T, Monks J, Lucia M, Wilson R, Hines L, et al. Alternatively activated macrophages and collagen remodeling characterize the postpartum involuting mammary gland across species. Am J Pathol (2010) 176(3):1241–55. doi: 10.2353/ajpath.2010.090735
64. Houghton A, Grisolano J, Baumann M, Kobayashi D, Hautamaki R, Nehring L, et al. Macrophage elastase (Matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res (2006) 66(12):6149–55. doi: 10.1158/0008-5472.CAN-04-0297
65. Mills C, Kincaid K, Alt J, Heilman M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
66. Mosser D, Edwards J. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
67. Xue J, Schmidt S, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
68. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol (2011) Volume 11:750–61. doi: 10.1038/nri3088
69. Klug F, Prakash H, Huber P, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS⁺/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24(5):589–602. doi: 10.1016/j.ccr.2013.09.014
70. Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front Immunol (2017) 8(826). doi: 10.3389/fimmu.2017.00828
71. Prakash H, Kulg F, Nadella V, Mazumdar V, Schmitz-Winnethal H, Umansky L, et al. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: lesson from insulinoma. J Carcinog (2016) 37(3):301–13. doi: 10.1093/carcin/bgw007
72. Pinto A, Pinto M, Cardoso A, Monteiro C, Pinto M, Maia A, et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci Rep (2016) 6:1–15. doi: 10.1038/srep18765
73. Huang T, Wuerzberger-Davis S, Wu Z-H, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell (2003) 115(5):565–76. doi: 10.1016/S0092-8674(03)00895-X
74. Wu Q, Allouch A, Paoletti A, Leteur C, Mirjolet C, Martins I, et al. NOX2-dependent ATM kinase activation dictates pro-inflammatory macrophage phenotype and improves effectiveness to radiation therapy. Cell Death Diff. (2017) 24(9):1632–44. doi: 10.1038/cdd.2017.91
75. Im J, Buzzelli JN, Jones K, Franchini F, Gordon-Weeks A, Marklec B, et al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun (2020) 11(1):4064. doi: 10.1038/s41467-020-17914-x
76. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and m-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol (2014) 192(8):3858–67. doi: 10.4049/jimmunol.1302821
77. Kalbasi A, Komar C, Tooker G, Liu M, Lee J, Gladney W, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res (2017) 23(1):137–48. doi: 10.1158/1078-0432.CCR-16-0870
78. Kitamura T, Qian B-Z, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. JEM (2015) 212(7):1043–59. doi: 10.1084/jem.20141836
79. Pyontek S, Akkari L, Schuhmacher A, Bowman R, Sevenich L, Quail D, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) Volume 19:1264–72. doi: 10.1038/nm.3337
80. Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, et al. Single-cell RNA sequencing reveals a dynamic stromal niche that supports tumor growth. Cell Rep (2020) 31(7):1–25. doi: 10.1016/j.celrep.2020.107628
81. Haoran Z, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res (2019) 7(2):193–207. doi: 10.1158/2326-6066.CIR-18-0272
82. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares M, Agoretta J, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov (2017) 7(7):694–703. doi: 10.1158/2159-8290.CD-16-1184
83. Wang Y, Sun S-N, Liu Q, Yu Y-Y, Guo Wang JK. Autocrine complement inhibits IL10-dependent T-cell-Mediated antitumor immunity to promote tumor progression. Cancer Discovery (2016) 6(9):1022–35. doi: 10.1158/2159-8290.CD-15-1412
84. Rodriguez-Ruiz M, Rodriguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, et al. TGFβ blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Transl Cancer Res (2019) 18(3):621–31. doi: 10.1158/1535-7163.MCT-18-0558
85. Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones K, Garcia-Martinez E, Rudqvist N-P, et al. Barriers to radiation-induced In situ tumor vaccination. Front Immunol (2017) 8(229). doi: 10.3389/fimmu.2017.00229
Keywords: radiotherapy, tumor microenvironment, hypoxia, extracellular matrix, macrophage polarization, macrophage recruitment, tumor associated macrophages (TAM), complement system
Citation: Beach C, MacLean D, Majorova D, Arnold JN and Olcina MM (2022) The effects of radiation therapy on the macrophage response in cancer. Front. Oncol. 12:1020606. doi: 10.3389/fonc.2022.1020606
Received: 16 August 2022; Accepted: 12 September 2022;
Published: 29 September 2022.
Edited by:
Marco Tigano, Thomas Jefferson University, United StatesReviewed by:
Clément Anfray, University of Milan, ItalyCopyright © 2022 Beach, MacLean, Majorova, Arnold and Olcina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica M. Olcina, monica.olcina@oncology.ox.ac.uk