 Nathan T. Bain
Nathan T. Bain Yang Wang
Yang Wang Surein Arulananda
Surein Arulananda- 1Department of Medical Oncology, Monash Health, Clayton, VIC, Australia
- 2School of Clinical Sciences, Faculty of Medicine, Monash University, Clayton, VIC, Australia
- 3Centre for Cancer Research, Hudson Institute of Medical Research, Clayton, VIC, Australia
- 4Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
Targeted therapy with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) is an effective treatment for EGFR-mutant non-small-cell lung cancer (NSCLC), however most patients invariably relapse after a period of minimal residual disease (MRD). This mini-review explores the mechanistic pathways leading to tumour dormancy, cellular senescence and epigenetic changes involving YAP/TEAD activation. We describe the various approaches of utilising TKIs in combination with agents to intensify initial depth of response, enhance apoptosis and target senescence-like dormancy. This mini-review will also highlight the potential novel therapies under development targeting MRD to improve outcomes for patients with EGFR-mutant NSCLC.
Introduction
Lung cancer, the second most commonly diagnosed cancer in 2020, remains the leading cause of global cancer death with an estimated 1.8 million deaths worldwide (1). Early-stage non-small-cell lung cancer (NSCLC) is managed by curative intent treatments such as surgical resection or ablative radiotherapy (2). Depending on histological and nodal staging, this is followed in most cases by a course of adjuvant systemic chemotherapy and/or immunotherapy to reduce the risk of cancer recurrence (3). Following complete surgical resection, the additional survival benefit conferred by platinum-based adjuvant chemotherapy increases with pathological stage (4, 5). Nevertheless, recurrence rates remain high with 30% to 55% of patients with NSCLC developing recurrence (6). This is largely attributed to a propensity for NSCLC to persist within the body following curative and adjuvant treatments in the form of minimal residual disease (MRD): micrometastatic deposits or circulating tumour cells (7, 8). Detecting MRD has become standard in some haematological malignancies, and liquid biopsy for circulating tumour DNA (ctDNA) is an active area of interest in several solid tumour types (9). However, the biology driving MRD in solid cancers seems to be highly variable between solid tumour types, with some clinically important differences recently associated with the presence of oncogenic driver mutations.
Oncogenic driver mutations have emerged as key therapeutic targets. EGFR is a receptor tyrosine kinase involved in extracellular growth factor signalling which is associated with growth, proliferation and survival (10). EGFR mutations are common and are seen in 10-15% of North American/European NSCLC, but up to 30-50% of East Asian patients.(ref) They are characteristically seen in pulmonary adenocarcinoma with bronchoalveolar features, particularly in patients without a history of smoking. EGFR mutations such as exon 19 deletion and L858R substitution lead to dysregulated EGFR activation and anti-apoptotic signalling can be achieved by mutation within the four exons (18-21) encoding the part of the tyrosine kinase domain clustered around the ATP-binding pocket of the enzyme.
Like other oncogenic mutations, EGFR mutations display “clonal” behaviour in vivo: they arise within individual cells which propagate and diverge into different lineages which undergo a quasi-Darwinian process where tumour cell populations descended from a progenitor or ‘clone” acquire successive somatic mutations that confer a selective advantage (11). This inevitably results in solid tumours which display highly heterologous cell populations. Exposure to systemic anti-cancer treatments has the effect of a selective pressure selecting for cancer cell lineages which contain or develop treatment resistance. As treatment resistance is only present in a small proportion of treatment-naïve cancer cells, initial clinical response can often be quite promising. Unfortunately, these responses lack durability, either over time or in the absence of ongoing systemic treatment. An archetypal example of this pattern can be seen in the response of EGFR-mutant NSCLC to tyrosine kinase inhibitors.
In advanced EGFR-mutant NSCLC, gefitinib (12), and erlotinib (13) were the first TKIs to demonstrate improved progression free survival (PFS) in comparison with platinum doublet chemotherapy. Osimertinib, a third generation TKI with improved binding activity, demonstrated high response rates as well as superior PFS compared to gefitinib and erlotinib in the advanced disease setting, and is the current standard-of-care therapy (14). Interestingly, while long term responders were observed in both the osimertinib and gefitinib/erlotinib groups, the survival rates for the FLAURA study at 3 years was 54% in the osimertinib group (15). Unfortunately, despite good initial response to treatment, TKIs do not in most patients create a durable response. This pattern of failure suggests that the effect of TKIs on EGFR-mutant NSCLC is more likely to be suppression rather than eradication. This distinction between cancer suppression and cancer eradication, calls into question the true value of TKIs in the adjuvant setting where the elimination (not suppression) of MRD is the therapeutic goal.
In the setting of completely resected EGFR-mutant NSCLC, gefitinib demonstrated superior PFS compared to standard platinum doublet adjuvant chemotherapy (16). However, subsequent follow-up showed that this initial response did not translate into an improvement in overall survival (17). The landmark ADAURA study (18) comparing a three-year course of adjuvant osimertinib versus placebo in completely resected stage IB to IIIA EGFR-mutant NSCLC demonstrated an early advantage in disease free survival of 89% vs 52% at 24 months with an overall hazard ratio (HR) 0.2 (99% CI, 0.14 to 0.30). Despite this significant difference, questions have been raised about clinical significance of disease-free survival as an endpoint, as well as the variable use of adjuvant chemotherapy between treatment groups (19). Overall survival data is eagerly awaited. It may well be that the effect of TKI treatment in the adjuvant setting turns out to be one of MRD-suppression rather than MRD-eradication.
It is important to bear in mind that the persistence of MRD to TKI treatment is distinct from bona fide treatment resistance which is classically characterised as the development or presence of specific mutations such as T790M EGFR mutations. These persister cells on the other hand do not by necessity harbour such mutations and are able to maintain viability throughout TKI treatment through other, less well understood mechanisms. It has been observed that EGFR-mutant tumour cells can enter a drug-tolerant state reminiscent of cellular senescence that enables ongoing survival predominantly through resistance to or inhibition of apoptosis (20).
Apoptosis and senescence in EGFR-mutant cancer cells
Apoptosis is an inducible, stepwise process of programmed cell death which can be classified into two broad pathways. The extrinsic pathway which is initiated by a class of cell membrane proteins known as death receptors, and the intrinsic pathway which is initiated through elaborate intracellular processes which invariably converge at the mitochondrial outer membrane. This leads to permeabilization and the subsequent release of cytochrome c into the cytoplasm. The release of cytochrome c from the mitochondria is stimulated by pro-apoptotic members of the BCL-2 family (i.e., BAX, BAK, BIM, BMF, BID and BAD) and inhibited by pro-survival members of the same family, such as BCL-2, BCL- XL and MCL1. Importantly, the efficacy of TKIs in EGFR-mutant NSCLC is reliant upon their ability to induce apoptosis by modulating the expression of members of the BCL-2 family (21). The pro-apoptotic protein BIM is phosphorylated by several cell survival pathways including ERK1/2 and MAPK1. The knockdown of BIM by small interfering RNA was observed to attenuate apoptosis induced by EGFR TKIs in cancer cell lines in vitro (22). Patients with EGFR-mutant NSCLC harbouring a BIM deletion polymorphism exhibit greater resistance to TKI treatment (23). An important pre-clinical study showed that a primary means by which cancer cells may evade apoptosis both in vitro and in vivo is by entering into a state of dormancy or senescence (24).
Cellular senescence is broadly defined as a viable, non-proliferative state akin to cellular dormancy. Replicative senescence is attributed to the progressive loss of protective telomeric DNA with mitotic cellular division and is mediated predominantly through the actions of p53 (25). Inducible senescence is a more varied phenomenon and can result from exposure to noxious and/or oncogenic stimuli such as ionising radiation or oxidative stress. One unique form of inducible senescence is oncogene-induced cellular senescence (OIS), which was initially demonstrated in vitro by the transfection of oncogenic HRAS V12 into murine fibroblasts which produced a strong anti-proliferative effect associated with activation of p16INK4A and p19ARF (26). The cell signalling pathways mediating OIS are complex but both pRB and p53 are involved in maintaining proliferative arrest (27). Rather than being maladaptive, senescence in this context is thought to be anti-oncogenic. Upregulation of oncogenes such as RAS and RAF have been observed to induce senescence in several in vitro models (28). However, senescence may also be playing an anti-therapeutic role in the context of anti-cancer therapies. TKIs such as gefitinib have also been observed to induce cellular senescence in malignant cells both in vitro and ex vivo (29). The escape/evasion of EGFR inhibition poses a hard problem to current TKI treatments targeting EGFR-mutant NSCLC.
Therapeutic strategies to enhance Osimertinib response
Targeting senescence-like cell dormancy
A novel area of significant interest is targeting cancer cell dormancy itself (24). An EGFR-mutant lung cancer cell line exposed to EGFR/MEK inhibition in vitro through a combination of osimertinib and trametinib (via a DMSO-containing solution of 100 nM and 30 nM of the former and latter agents respectively) induced a widespread apoptotic response. A small proportion of cells were able to persist throughout this die-off, independent of ERK signalling, by entering a stable dormant state and were still detectable after fifteen weeks of treatment. After drug washout, these dormant cells were observed to proliferate and recolonize the wells within a matter of days. This reversible dormant state shared several characteristics in common with cellular senescence including senescence-associated beta-galactosidase staining, flattened morphology typical of senescent cells, and H3K9Me3-positive nuclear foci.
The establishment and maintenance of dormancy was found to be closely associated with YAP/TEAD/Hippo activity as shown in Figure 1. Dormant EGFR-mutant NSCLC cells expressed significant enrichment of YAP/TEAD gene expression signature, which was identified using RNA sequencing. Increased YAP activity and decreased ERK1/2 activity was observed in a patient-derived xenograft in vivo model from tumour tissue sampled from a patient with EGFR-mutant NSCLC who had undergone partial response following treatment with osimertinib and selumetinib (24). This study went on to demonstrate that co-treatment of EGFR-mutant NSCLC cell lines with osimertinib plus XAV939, a tankyrase inhibitor that indirectly inhibits YAP signalling, reduced both the abundance of dormant cells as well as the regrowth of cells following washout. In three separate YAP1 knockout EGFR-mutant NSCLC cell lines, cell dormancy following osimertinib exposure was completely abolished. Co-targeting cell dormancy in combination with EGFR/MEK inhibition as a means of bypassing apoptosis evasion is a promising direction for future therapeutics. This could be accomplished in several ways. For instance, YAP inhibition may be accomplished by direct inhibitors such as MFF, or indirect inhibitors such as the tankyrase inhibitor XAV939. As well, the most effective timing of YAP inhibition will need to be established. YAP inhibitors used upfront in conjunction with EGFR/MEK inhibitors may exhibit different efficacy or toxicity compared to treating with EGFR/MEK inhibitors first and adding YAP inhibitors later when dormancy has been established.
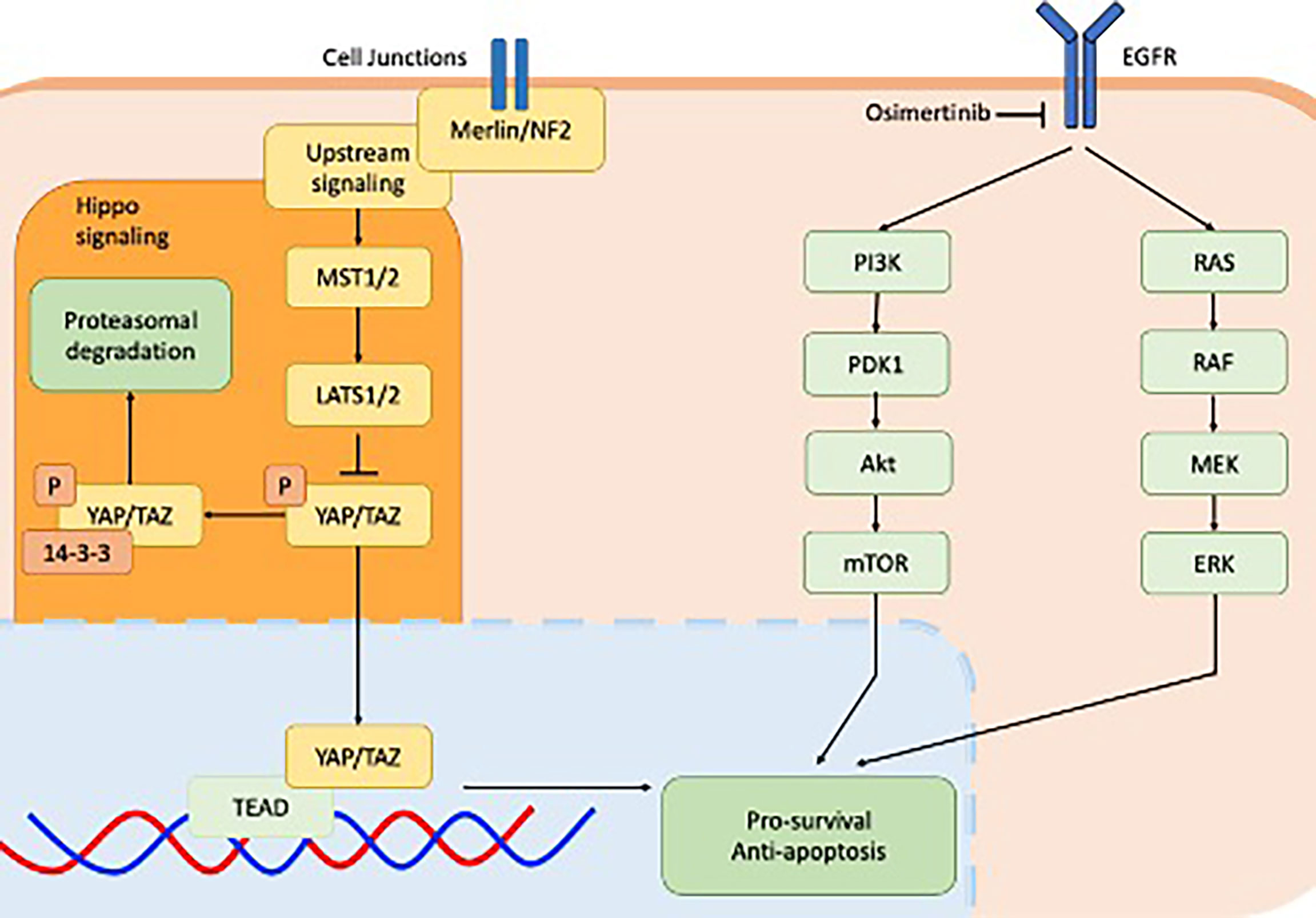
Figure 1 Cell signalling pathways implicated in EGFR-mutant non-small-cell lung cancer including downstream signalling of EGFR receptor via the PI3K-AKT pathway (green) and MAPK pathway (green) and the YAP/TEAD hippo signalling pathway downstream of Merlin/NF2. Both pathways converge on the apoptosis pathway.
Targeting apoptosis
If TKI escape/evasion is occurring through the attenuation of the upstream, pro-apoptotic proteins such as BMF and BIM, then direct co-stimulation of apoptotic effectors may potentiate TKI treatments. Venetoclax is a selective and potent inhibitor of BCL-2 that binds to the BH3 domain of BCL-2 and disrupts its ability to interact with the pro-apoptotic protein BIM, thereby inducing apoptosis (30). As a monotherapy, venetoclax has been found to inhibit the growth of high BCL-2-expressing small cell lung cancer in vitro (30). Similar pre-clinical work has shown BH3-mimetics targeting BCL-XL and BCL-2 are also effective agents against pleural mesothelioma (31). Perhaps most interesting, the combination of pro-apoptotic BH3-mimetics with additional anti-cancer agents has been observed to produce synergistic anti-tumour effects in several studies. Venetoclax combined with osimertinib significantly enhances apoptosis in EGFR-mutant NSCLC cells with acquired osimertinib resistance (32). As well, a strong anti-tumour effect was induced through the combination of ABT-737, a BH3 mimetic which displaces the anti-apoptotic protein BCL-XL, and cisplatin, was observed in a murine NSCLC model (33). Similarly, navitoclax, a BCL-2 inhibitor, was found to work synergistically with 5-fluorouracil in oesophageal cancer cells in vitro (34). Potentiating apoptosis may be a reliable way of amplifying or even restoring the efficacy of several anti-cancer regiments.
Trials
Intensifying TKI regimens
Various approaches are currently underway to address MRD by combining EGFR TKIs with other therapeutic agents. One such strategy is combining EGFR TKIs with platinum-pemetrexed chemotherapy. The FLAURA2 study is currently underway and attempts to answer this question of whether osimertinib used in combination with chemotherapy would deepen MRD in the first line setting and delay the development of resistance (NCT04035486). The TAKUMI trial randomised sixty-two patients who had developed T790M resistance mutation after first line EGFR-TKI therapy to either osimertinib with combination carboplatin-pemetrexed versus osimertinib alone and found that there was no significance difference in median PFS (35). In regards to safety, a meta-analysis of combination chemotherapy with first-generation TKIs found overall increased toxicities most notably myelosuppression and gastrointestinal side effects (36). As well, no specific pattern of toxicity leading to dose modification or discontinuation was observed, although one patient discontinued study treatments due to pneumonitis (37).
Co-targeting EGFR and MEK inhibition is a promising area of research interest given pre-clinical trials have shown efficacy compared to single agent alone. Osimertinib combined with a MEK or ERK inhibitor enhanced apoptosis and prevented the emergence of osimertinib resistance in vitro by enhancing osimertinib-induced apoptosis, the prevention of ERK1/2 reactivation, and inhibited the emergence of resistance in osimertinib-sensitive models known to acquire resistance via both T790M-dependent and T790M-independent mechanisms (38, 39). The clinical application of this has yielded mixed results. There have been case studies showing combination osimertinib and MEK inhibitor trametinib have led to an increased response after progression with osimertinib (40, 41), whilst in another study the sequential addition of a MEK inhibitor trametinib in a patient population already pre-treated with prior TKI therapy did not demonstrate efficacy highlighting the importance of patient selection (42). The findings of a phase II study evaluating the combination of osimertinib with MEK 1/2 inhibitor selumetinib will undoubtably be informative as to the in vivo efficacy of this combination (NCT03392246).
The strategy of TKI-immunotherapy combinations has been largely limited by the lack of data on the clear benefit and the high rates of toxicities namely interstitial lung disease. Combination osimertinib and durvalumab was associated with a high incidence (38%) of interstitial lung disease (43), and in the TATTON study; the same combination was shown to cause significant toxicities with grade 3 or higher adverse events reported in almost half of patients and interstitial lung disease reported in 22% leading to early discontinuation (44).
Future perspectives
The current landscape of trials in the EGFR-mutant NSCLC space primarily involves osimertinib in combination with agents active against downstream pathways and acquired resistance mechanisms as shown in Table 1. While fourth generation EGFR-TKIs are in development to target the most commonly seen emergent mutations C797S and C797X after third line EGFR-TKI treatment, the authors speculate that these approaches are reactionary and invariably other resistance mechanisms will develop (NCT04862780), (NCT05153408), (NCT05394831). Aurora kinase A activation, another acquired resistance mechanism is shown to play a role in drug resistance to EGFR-TKI treatment and is also the subject of early phase trials involving the addition of Alisertib (NCT04085315) (NCT04479306) (NCT05017025). Other early phase trials are evaluating inhibition of the downstream signalling pathway PI3K/mTOR with sapanisertib (NCT04479306) and MET inhibition with bispecific antibody Amivantamab (NCT02609776) (NCT04965090).
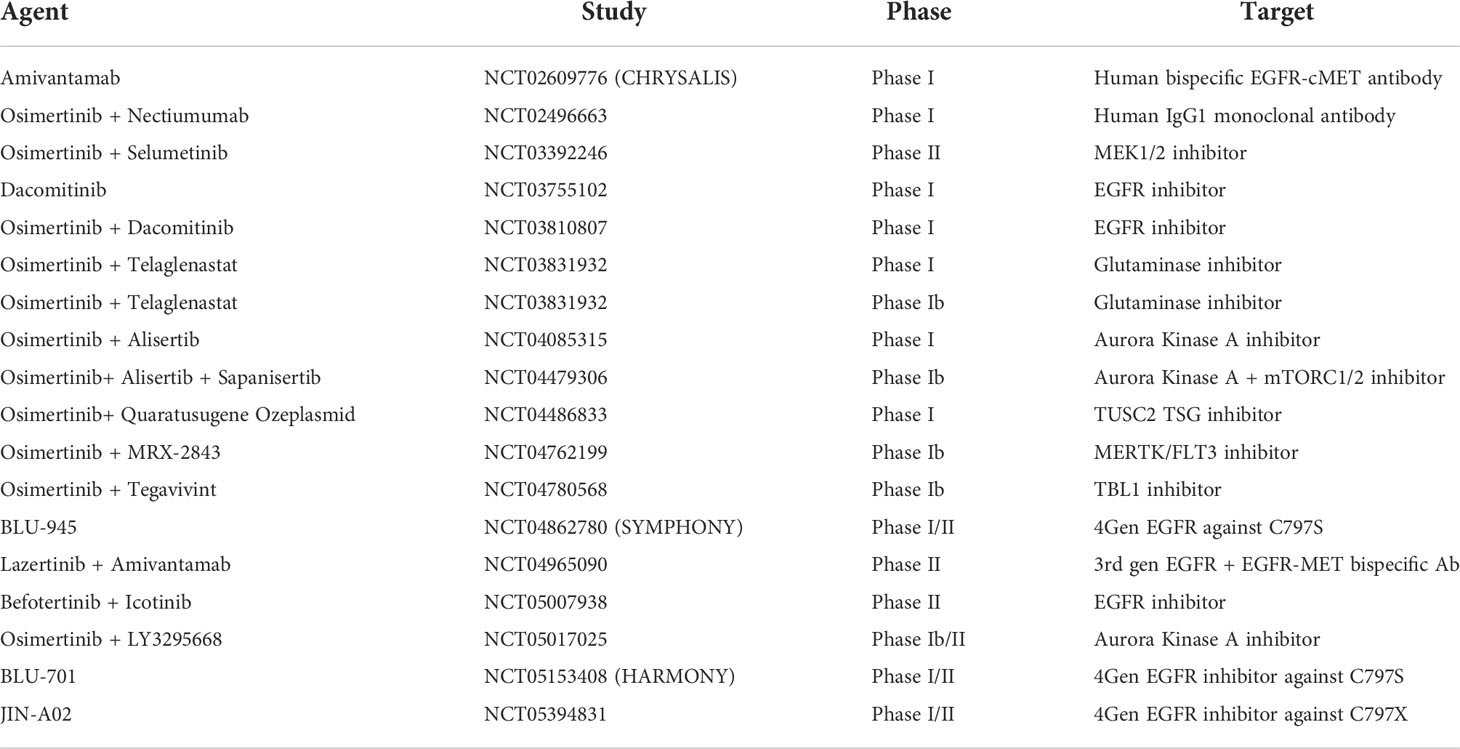
Table 1 Currently recruiting EGFR mutation non-small-cell lung cancer trials.
Relatively few trials are underway that directly address MRD and the evasion of apoptosis. This may be owing to the novelty of the proposed mechanisms involved, as well as the paucity of literature demonstrating a clear role for apoptosis-targeting agents in lung cancer. To date, the phase II study of combination osimertinib with MEK1/2 inhibitor selumetinib (NCT03392246) is the only trial currently underway that is evaluating whether this combination may prevent the emergence of acquired resistance.
Conclusions
EGFR-TKI therapy remains the mainstay of first-line treatment for EGFR-mutant NSCLC however this rarely leads to cure as acquired resistance invariably develops. The understanding of the MRD state that develops after initial exposure to EGFR-TKIs such as YAP/TEAD provide an insight into how tumour cells escape from initial apoptosis.
The pre-clinical success of osimertinib and XAV939 demonstrates a promising alternative of targeting multiple pathways proactively in the first line setting to deepen MRD that may circumvent the development of drug resistance and prove to be beneficial than targeting the acquired resistance pathways that develop post osimertinib exposure. Further trials are required to develop a more effective treatment strategy and evaluate the efficacy and safety of upfront combination or sequential targeted therapies. The ongoing discovery of more targetable resistance mechanisms are likely to continue to reshape the future treatment of EGFR-mutant NSCLC. However, a strategy which involves optimal EGFR inhibition and eliminating these MRD cells which have senescent properties, is potentially the key to unlocking the ‘holy grail’ of cure in this disease.
Author contributions
Conceptualisation, SA and NB. Supervision, SA. Writing, review, and editing, NB, YW, and SA.
Acknowledgments
SA would like to acknowledge the Royal Australasian College of Physicians for the Robert Maple-Brown Research Establishment Fellowship as post-doctoral fellowship support.
Conflict of interest
SA Speaker fees: Merck-Sharpe & Dohme, Astra Zeneca, Roche, and Bristol-Myers Squibb. Travel support: Astra Zeneca, Roche, Merck-Sharpe & Dohme. Advisory boards: Boehringer Ingelheim, Roche. Non-financial aid: Astra Zeneca, Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Ball D, Mai GT, Vinod S, Babington S, Ruben J, Kron T, et al. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): A phase 3, open-label, randomised controlled trial. Lancet Oncol (2019) 20:494–503. doi: 10.1016/S1470-2045(18)30896-9
3. Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet (2021) 398:1344–57. doi: 10.1016/S0140-6736(21)02098-5
4. Douillard J-Y, Tribodet H, Aubert D, Shepherd FA, Rosell R, Ding K, et al. Adjuvant cisplatin and vinorelbine for completely resected non-small cell lung cancer: Subgroup analysis of the lung adjuvant cisplatin evaluation. J Thorac Oncol (2010) 5:220–8. doi: 10.1097/JTO.0b013e3181c814e7
5. Pignon J-P, Tribodet H, Scagliotti GV, Douillard J-Y, Shepherd FA, Stephens RJ, et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE collaborative group. J Clin Oncol (2008) 26:3552–9. doi: 10.1200/JCO.2007.13.9030
6. Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res (2014) 3:242–9. doi: 10.3978/j.issn.2218-6751.2013.12.05
7. Chudacek J, Bohanes T, Klein J, Benedikova A, Srovnal J, Szkorupa M, et al. Detection of minimal residual disease in lung cancer. Biomed Pap (2014) 158:189–93. doi: 10.5507/bp.2013.019
8. Frisone D, Friedlaender A, Addeo A. The role and impact of minimal residual disease in NSCLC. Curr Oncol Rep (2021) 23:136. doi: 10.1007/s11912-021-01131-w
9. Tie J, Cohen JD, Lahouel K, Lo SN, Wang Y, Kosmider S, et al. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N Engl J Med (2022) 386:2261–72. doi: 10.1056/NEJMoa2200075
10. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7:169–81. doi: 10.1038/nrc2088
11. Kohsaka S, Petronczki M, Solca F, Maemondo M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: Implications for therapeutic sequencing. Future Oncol (2019) 15:637–52. doi: 10.2217/fon-2018-0736
12. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–Small-Cell lung cancer to gefitinib. N Engl J Med (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
13. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13:239–46. doi: 10.1016/S1470-2045(11)70393-X
14. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–Small-Cell lung cancer. N Engl J Med (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
15. Ramalingam SS, Gray JE, Ohe Y, Cho BC, Vansteenkiste J, Zhou C, et al. Osimertinib vs comparator EGFR-TKI as first-line treatment for EGFRm advanced NSCLC (FLAURA): Final overall survival analysis. Ann Oncol (2019) 30:v914–5. doi: 10.1093/annonc/mdz394.076
16. Zhong W-Z, Wang Q, Mao W-M, Xu S-T, Wu L, Shen Y, et al. Gefitinib versus vinorelbine plus cisplatin as adjuvant treatment for stage II–IIIA (N1–N2) EGFR-mutant NSCLC (ADJUVANT/CTONG1104): A randomised, open-label, phase 3 study. Lancet Oncol (2018) 19:139–48. doi: 10.1016/S1470-2045(17)30729-5
17. Zhong W-Z, Wang Q, Mao W-M, Xu S-T, Wu L, Wei Y-C, et al. Gefitinib versus vinorelbine plus cisplatin as adjuvant treatment for stage II-IIIA (N1-N2) EGFR-mutant NSCLC: Final overall survival analysis of CTONG1104 phase III trial. J Clin Oncol (2021) 39:713–22. doi: 10.1200/JCO.20.01820
18. Wu Y-L, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR -mutated non–Small-Cell lung cancer. N Engl J Med (2020) 383:1711–23. doi: 10.1056/NEJMoa2027071
19. Addeo A, Banna GL, Friedlaender A. ADAURA: Mature enough for publication, not for prime time. Oncologist (2021) 26:266–8. doi: 10.1002/onco.13637
20. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med (2016) 22:262–9. doi: 10.1038/nm.4040
21. Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, et al. BIM expression in treatment-naïve cancers predicts responsiveness to kinase inhibitors. Cancer Discovery (2011) 1:352–65. doi: 10.1158/2159-8290.CD-11-0106
22. Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PloS Med (2007) 4:e315. doi: 10.1371/journal.pmed.0040315
23. Ng KP, Hillmer AM, Chuah CTH, Juan WC, Ko TK, Teo ASM, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med (2012) 18:521–8. doi: 10.1038/nm.2713
24. Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell (2020) 37:104–122.e12. doi: 10.1016/j.ccell.2019.12.006
25. Fakhri S, Zachariah Moradi S, DeLiberto LK, Bishayee A. Cellular senescence signaling in cancer: A novel therapeutic target to combat human malignancies. Biochem Pharmacol (2022) 199:114989. doi: 10.1016/j.bcp.2022.114989
26. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
27. Francica P, Aebersold DM, Medová M. Senescence as biologic endpoint following pharmacological targeting of receptor tyrosine kinases in cancer. Biochem Pharmacol (2017) 126:1–12. doi: 10.1016/j.bcp.2016.08.022
28. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol (2008) 20:150–5. doi: 10.1016/j.ceb.2008.01.007
29. Hotta K, Tabata M, Kiura K, Kozuki T, Hisamoto A, Katayama H, et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol Rep (2007) 17:313–17. doi: 10.3892/or.17.2.313
30. Lochmann TL, Floros KV, Naseri M, Powell KM, Cook W, March RJ, et al. Venetoclax is effective in small-cell lung cancers with high BCL-2 expression. Clin Cancer Res (2018) 24:360–9. doi: 10.1158/1078-0432.CCR-17-1606
31. Arulananda S, O’Brien M, Evangelista M, Jenkins LJ, Poh AR, Walkiewicz M, et al. A novel BH3-mimetic, AZD0466, targeting BCL-XL and BCL-2 is effective in pre-clinical models of malignant pleural mesothelioma. Cell Death Discovery (2021) 7:1–9. doi: 10.1038/s41420-021-00505-0
32. Liu Z, Gao W. Synergistic effects of bcl-2 inhibitors with AZD9291 on overcoming the acquired resistance of AZD9291 in H1975 cells. Arch Toxicol (2020) 94:3125–36. doi: 10.1007/s00204-020-02816-0
33. Kim EY, Jung JY, Kim A, Chang YS, Kim SK. ABT-737 synergizes with cisplatin bypassing aberration of apoptotic pathway in non-small cell lung cancer. Neoplasia (2017) 19:354–63. doi: 10.1016/j.neo.2017.02.008
34. Chen Q, Song S, Wei S, Liu B, Honjo S, Scott A, et al. ABT-263 induces apoptosis and synergizes with chemotherapy by targeting stemness pathways in esophageal cancer. Oncotarget (2015) 6:25883–96. doi: 10.18632/oncotarget.4540
35. Tanaka K, Asahina H, Kishimoto J, Miyata Y, Uchida T, Watanabe K, et al. Osimertinib versus osimertinib plus chemotherapy for non–small cell lung cancer with EGFR (T790M)-associated resistance to initial EGFR inhibitor treatment: An open-label, randomised phase 2 clinical trial. Eur J Cancer (2021) 149:14–22. doi: 10.1016/j.ejca.2021.02.019
36. Wu Q, Luo W, Li W, Wang T, Huang L, Xu F. First-generation EGFR-TKI plus chemotherapy versus EGFR-TKI alone as first-line treatment in advanced NSCLC with EGFR activating mutation: A systematic review and meta-analysis of randomized controlled trials. Front Oncol (2021) 11:598265. doi: 10.3389/fonc.2021.598265
37. Planchard D, Feng P-H, Karaseva N, Kim S-W, Kim TM, Lee CK, et al. Osimertinib plus platinum–pemetrexed in newly diagnosed epidermal growth factor receptor mutation-positive advanced/metastatic non-small-cell lung cancer: safety run-in results from the FLAURA2 study. ESMO Open (2021) 6:100271. doi: 10.1016/j.esmoop.2021.100271
38. Gu J, Yao W, Shi P, Zhang G, Owonikoko TK, Ramalingam SS, et al. MEK or ERK inhibition effectively abrogates emergence of acquired osimertinib resistance in the treatment of EGFR-mutant lung cancers. Cancer (2020) 126:3788–99. doi: 10.1002/cncr.32996
39. Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR -mutant lung cancer. Cancer Discovery (2015) 5:960–71. doi: 10.1158/2159-8290.CD-15-0063
40. Meng P, Koopman B, Kok K, ter Elst A, Schuuring E, van Kempen LC, et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer (2020) 146:358–61. doi: 10.1016/j.lungcan.2020.05.036
41. Ribeiro MFSA, Knebel FH, Bettoni F, Saddi R, Sacardo KP, Canedo FSNA, et al. Impressive response to dabrafenib, trametinib, and osimertinib in a metastatic EGFR-mutant/BRAF V600E lung adenocarcinoma patient. NPJ Precis Oncol (2021) 5:5. doi: 10.1038/s41698-021-00149-4
42. Luo J, Makhnin A, Tobi Y, Ahn L, Hayes SA, Iqbal A, et al. Erlotinib and trametinib in patients with EGFR-mutant lung adenocarcinoma and acquired resistance to a prior tyrosine kinase inhibitor. JCO Precis Oncol (2021), 5:55–64. doi: 10.1200/PO.20.00315
43. Ahn MJ, Yang J, Yu H, Saka H, Ramalingam S, Goto K. 136O: osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: Results from the TATTON phase ib trial. J Thorac Oncol (2016) 11:S115. doi: 10.1016/S1556-0864(16)30246-5
Keywords: Non-small-cell lung cancer, minimal residual disease, EGFR, apoptosis, senescence
Citation: Bain NT, Wang Y and Arulananda S (2022) Minimal residual disease in EGFR-mutant non-small-cell lung cancer. Front. Oncol. 12:1002714. doi: 10.3389/fonc.2022.1002714
Received: 25 July 2022; Accepted: 31 August 2022;
Published: 21 September 2022.
Edited by:
Ashleigh Poh, Olivia Newton-John Cancer Research Institute, AustraliaReviewed by:
Suad Abdirahman, Peter MacCallum Cancer Centre, AustraliaKa Yee Fung, The University of Melbourne, Australia
Copyright © 2022 Bain, Wang and Arulananda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Surein Arulananda, surein.arulananda@monash.edu