SHROOM4 Variants Are Associated With X-Linked Epilepsy With Features of Generalized Seizures or Generalized Discharges
 Wen-Jun Bian
Wen-Jun Bian Zong-Jun Li
Zong-Jun Li Jie Wang
Jie Wang Sheng Luo
Sheng Luo  Bing-Mei Li
Bing-Mei Li Liang-Di Gao
Liang-Di Gao Na He
Na He Yong-Hong Yi
Yong-Hong Yi- Institute of Neuroscience and Department of Neurology, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
Objective: SHROOM4 gene encodes an actin-binding proteins, which plays an important role in cytoskeletal architecture, synaptogenesis, and maintaining gamma-aminobutyric acid receptors-mediated inhibition. SHROOM4 mutations were reported in patients with the Stocco dos Santos type of X-linked syndromic intellectual developmental disorder (SDSX; OMIM# 300434). In this study, we investigated the association between SHROOM4 and epilepsy.
Methods: Trios-based whole-exome sequencing was performed in a cohort of 320 cases with idiopathic generalized epilepsy or idiopathic partial epilepsy. Protein modeling was used to assess the damaging effects of variations.
Results: Six hemizygous missense SHROOM4 variants, including c.13C > A/p. Pro5Thr, c.3236C > T/p.Glu1079Ala, c.3581C > T/p.Ser1194Leu, c.4288C > T/p.Arg1430Cys, c.4303G > A/p.Val1435Met, c.4331C > T/p.Pro1444Leu, were identified in six cases with idiopathic epilepsy without intellectual disability. All patients presented with features of generalized seizures or generalized discharges. These hemizygous variants had no or extremely low allele frequencies in controls and showed statistically higher frequency in the case cohort than controls. All variants were predicted to alter hydrogen bond with surrounding amino acids or decreased protein stability. The SHROOM4 variants reported in patients with SDSX were mostly destructive or duplicative variants; in contrast, the SHROOM4 variants were all missense variants, suggesting a potential genotype-phenotype correlation. The two missense variants associated with SDSX were located in the middle of SHROOM4 protein, whereas variants associated with idiopathic epilepsy were located around the N-terminal PDZ domain and the C-terminal ASD2 domain.
Significance: SHROOM4 was potentially a candidate pathogenic gene of idiopathic epilepsy without intellectual disability. The genotype-phenotype correlation and sub-regional effect helps understanding the mechanism underlying phenotypic variation.
Introduction
SHROOM4 gene (OMIM* 300579) (also known as KIAA1202 gene) resides on Xp11.22 and encodes a member of the actin-binding proteins of shroom family (Hagens et al., 2006), which contain a PDZ and an ASD2 domain. The SHROOM4 protein is broadly distributed, including the brain and predominantly during embryonic and adult period. SHROOM4 and members of this protein family have been shown to localize at the cytoskeleton, and play a role in neurulation, cellular architecture, actin remodeling, ion channel function, and synaptogenesis (Hagens et al., 2006; Zapata et al., 2017). SHROOM4 mutations have been demonstrated to be associated with Stocco dos Santos type of X-linked syndromic intellectual developmental disorder (SDSX; OMIM# 300166) (Froyen et al., 2007; Honda et al., 2010; Armanet et al., 2015; Farwell et al., 2015). Knockdown of SHROOM4 in rat severely impairs gamma-aminobutyric acid (GABA) receptors activity causing increased anxiety-like behavior and susceptibility to seizures (Zapata et al., 2017). However, the relationship between SHROOM4 gene mutations and epilepsy is not determined.
In this study, we performed trio-based whole-exome sequencing (WES) in a cohort of patients with idiopathic epilepsy. Six novel hemizygous missense variants of SHROOM4 were identified in six unrelated cases with epilepsy with generalized seizures or generalized discharges on electroencephalography (EEG). To understand the mechanism of phenotypic variation, we analyzed the genotype-phenotype correlation and the sub-regional effect of SHROOM4 variants.
Materials and Methods
Patients
A total of 320 cases (trios) with epilepsy without acquired causes (idiopathic epilepsies) were recruited in this study from the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University in China between January 2013 and December 2020. The complete pedigree and clinical data of the probands were collected, including age, gender, age of seizure onset, type, course, and frequency of seizure, family history, therapy, prognosis, general and neurological examination, long-term video EEG, and brain magnetic resonance imaging (MRI). Epilepsy syndromes and epileptic seizures were diagnosed according to the criteria of the Commission on Classification and Terminology of the ILAE (1981, 1989, 2001, 2010, and 2017). Idiopathic generalized epilepsy were diagnosed based on a range of seizure types including absence, spasms, myoclonus, clonic, atonic, tonic, and tonic-clonic seizures, supported by typically generalized discharges on EEG. Idiopathic partial epilepsy had partial seizures and features of bilateral discharge or tendency of generalized discharge. EEG examinations showed focal abnormalities with features of idiopathic epilepsies, including shifting, bilateral, or multiple focal discharges with normal backgrounds. Patients with acquired causes were excluded. All subjects were followed up for at least one year.
This project was approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University and was conducted according to the guidelines of the International Committee of Medical Journal Editors regarding patients’ consent for research or participation. Written informed consent was obtained from the patients and their legal guardians.
Whole-Exome Sequencing and Genetic Analysis
Peripheral blood samples were obtained from the probands, parents, and other available family members to determine the origin of the identified genetic variants. Genomic DNA was extracted as previously reported (Wang et al., 2018; Shi et al., 2019). Trio-based Whole-Exome Sequencing (WES) was performed with the Illumina HiSeq 2500/4000 platform by BGI-Shenzhen (Shenzhen, China). Deep sequencing data were aligned to the reference GRCh37 build (hg19) and variants were called according to the standard procedures as previously reported (Shi et al., 2019). A case-by-case analytical approach was used to identify candidate causative variants in each trio. Generally, the rare variants with a minor allele frequency < 0.005 were first prioritized in the 1000 Genomes Projects, Exome Aggregation Consortium, and Genome Aggregation Database (gnomAD) (Genomes Project et al., 2015; Karczewski et al., 2020). Next, potentially pathogenic mutations were retained, including frameshift, nonsense, canonical splice site, initiation codon, and missense mutations predicted as being damaging by in silico tools1. Finally, potential disease-causing variants were screened under following five models: (1) epilepsy-associated gene model; (2) de novo dominant inheritance model; (3) autosomal recessive inheritance model, including compound heterozygous and homozygous variants; (4) X-linked inheritance model; (5) co-segregation model. To identify novel epilepsy-associated genes, the known epilepsy-associated genes (Wang et al., 2017) were not analyzed in the present study. Genes with repetitively identified de novo variants, bi-allelic variants, hemizygous variants, or variants with segregations, were selected for further studies to define the gene-disease association. SHROOM4 appeared as one of the candidate genes with recurrent hemizygous variants in this cohort. All variants in SHROOM4 were annotated based on transcript NM_020717.5. Positive findings and the variant origination were validated by Sanger sequencing.
Mutation Analysis
Protein modeling was conducted via Iterative Threading ASSEmbly Refinement (I-TASSER) software to predict the effect of candidate variants on molecular structure (Zhang, 2008; Yang and Zhang, 2015). Three-dimensional protein structure and hydrogen bonds alteration were visualized and analyzed by using PyMOL 1.7.
I-Mutant server was used to predict protein stability alteration caused by single nucleotide mutations-related amino acid change (Capriotti et al., 2005). Protein stability was measured with free energy change value (DDG, kcal/mol).
To evaluate the genotype–phenotype correlation, SHROOM4 variants were systematically reviewed through PubMed database and human gene mutation database up to December 2021.
Statistical Analysis
IBM SPSS Statistics 19 was used for statistical analysis. Fisher’s exact test was applied to access the allele frequencies of SHROOM4 variants in the cohort of this study and the control populations, and the proportions of missense variants between epilepsy and intellectual disability. A p value of < 0.05 was considered to be statistically significant.
Results
Identification of SHROOM4 Variants
Six hemizygous missense variants in SHROOM4, including c.13C > A/p.Pro5Thr, c.3236C > T/p.Glu1079Ala, c.3581C > T/p.Ser1194Leu, c.4288C > T/p.Arg1430Cys, c.4303G > A/p.Val1435Met, and c.4331C > T/p.Pro1444Leu, were identified in six unrelated cases with idiopathic epilepsy (Figure 1 and Table 1). All of the hemizygous missense variants were inherited from their asymptomatic mothers, consisted with a classical X-linked recessive (XLR) inheritance pattern (Figures 1A,B).
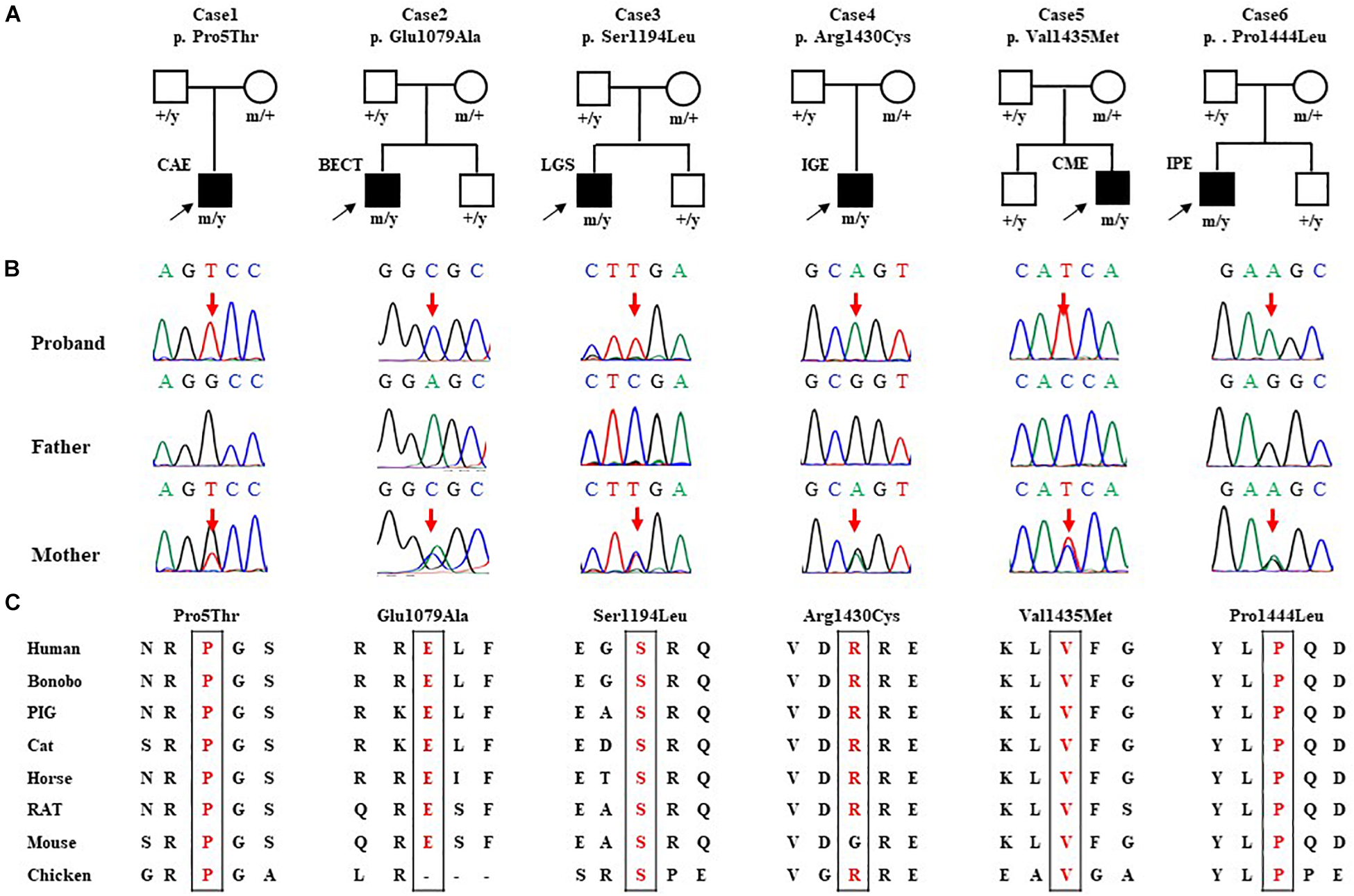
Figure 1. Genetic data of cases with SHROOM4 variants. (A) Pedigrees of the six cases with SHROOM4 mutations and their corresponding phenotypes. (B) DNA sequence chromatogram of the SHROOM4 mutations. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the six missense mutations shows that the residues Pro5, Glu1079, Ser1194, Arg1430, Val1435, and Pro1444 are highly conserved across species.
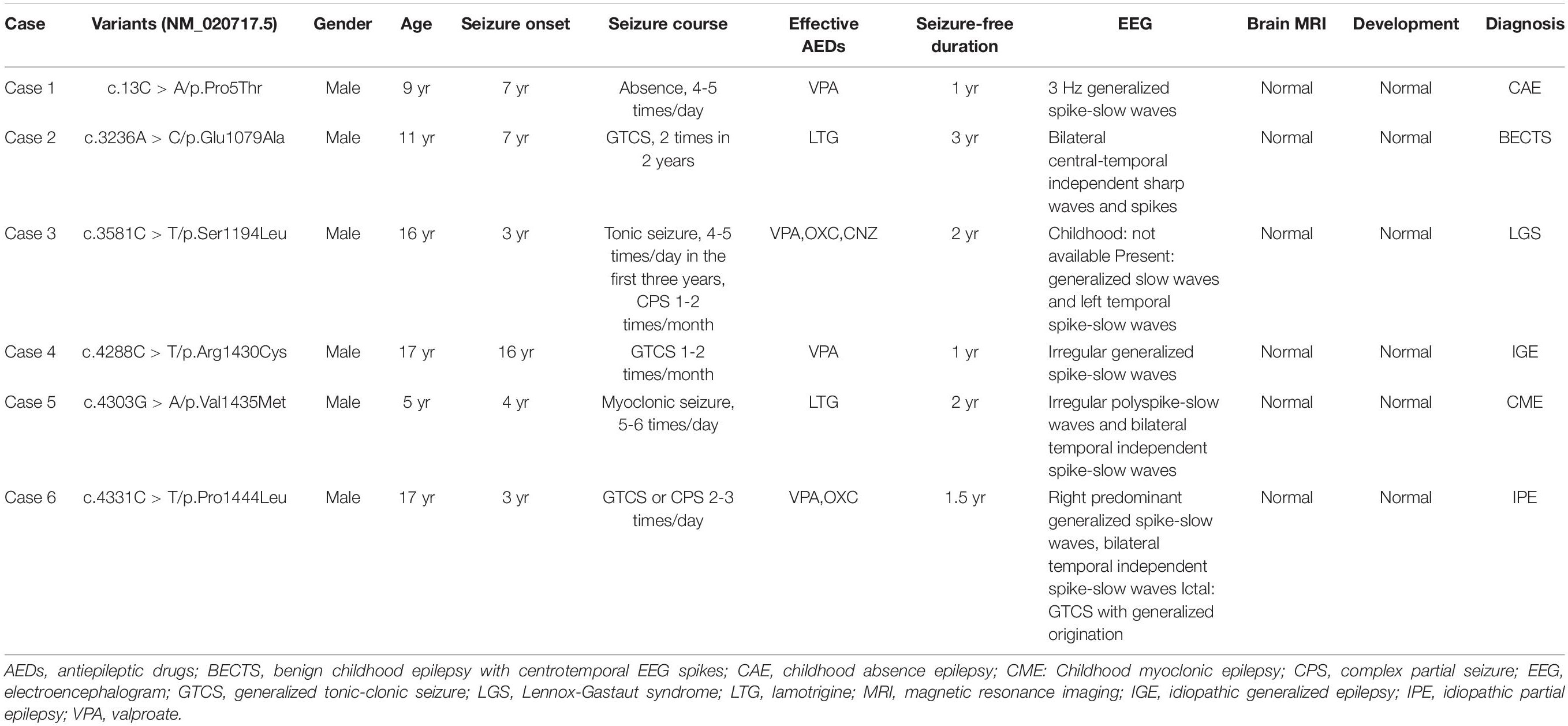
Table 1. Clinical features of the individuals with SHROOM4 variants.
No hemizygote of these variants were found in the controls of gnomAD-all populations, except the variant Ser1194Leu with an extremely low frequency (1/87,776) (Table 2). All of the hemizygous variants were not found in the controls of gnomAD-East Asian populations.
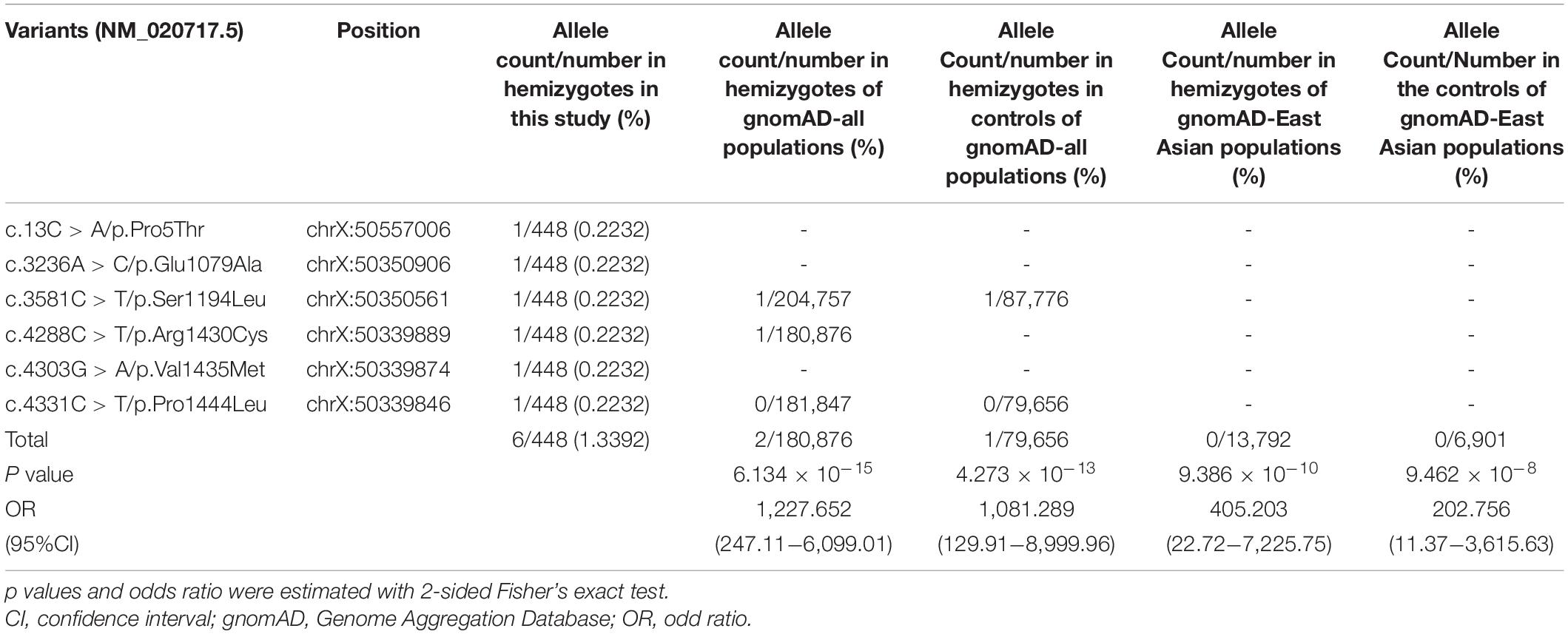
Table 2. Analysis of the aggregate frequency of SHROOM4 variants identified in this study.
The aggregate frequency of the hemizygous variants in this cohort was significantly higher than that in controls (Table 2), including the gnomAD-all population (6/448 vs. 2/180,876; p = 6.134 × 10–15), the controls of gnomAD-all population (vs. 1/79,656; p = 4.273 × 10–13), the gnomAD-East Asian population (vs. 0/13,792, p = 9.386 × 10–10), and the controls of the gnomAD-East Asian population (vs. 0/6,901, p = 9.462 × 10–8).
All SHROOM4 variants identified in this study were predicted to be damaging by one of the silico tools (Supplementary Table 1). Protein sequence alignment indicated that five of six variants are located at residues that are highly conserved across species (Figure 1C). The Arg1430 was less conserved but was predicted to be conserved by GERP (score = 4.15) and phyloP (score = 2.907) (Supplementary Table 2). None of the six affected patients had pathogenic or likely pathogenic variants in genes known to be associated with epileptic phenotypes (Wang et al., 2017).
Clinical Features
The clinical features of the six cases with SHROOM4 variants were summarized in Table 1. The onset age of seizures ranged from 3 years to 16 years old, with a median age of onset of 5.5 years. The patient of case 1 was diagnosed as childhood absence epilepsy (CAE) characterized by absence seizure and 3 Hz generalized spike-slow waves on EEGs (Figure 2A). The patients of case 3, case 4, and case 5 were diagnosed as generalized epilepsy, including Lennox-Gastaut syndrome (LGS), idiopathic generalized epilepsy (IGE), and childhood myoclonic epilepsy (CME); and the three cases had both generalized and focal discharge features on EEGs (Figures 2C−E). The patients of case 2 and case 6 were diagnosed as partial epilepsy, i.e., benign childhood epilepsy with centrotemporal EEG spikes (BECTS) and idiopathic partial epilepsy (IPE), but their EEGs had generalized discharges (asymmetric) (Figures 2B,F). In a word, these patients mainly present with generalized epilepsy, or idiopathic partial epilepsy with generalized seizures or generalized discharge on EEGs. All patients showed normal development. Their brain MRI were normal. These patients all presented good responses to antiepileptic drug and achieved seizure-free.
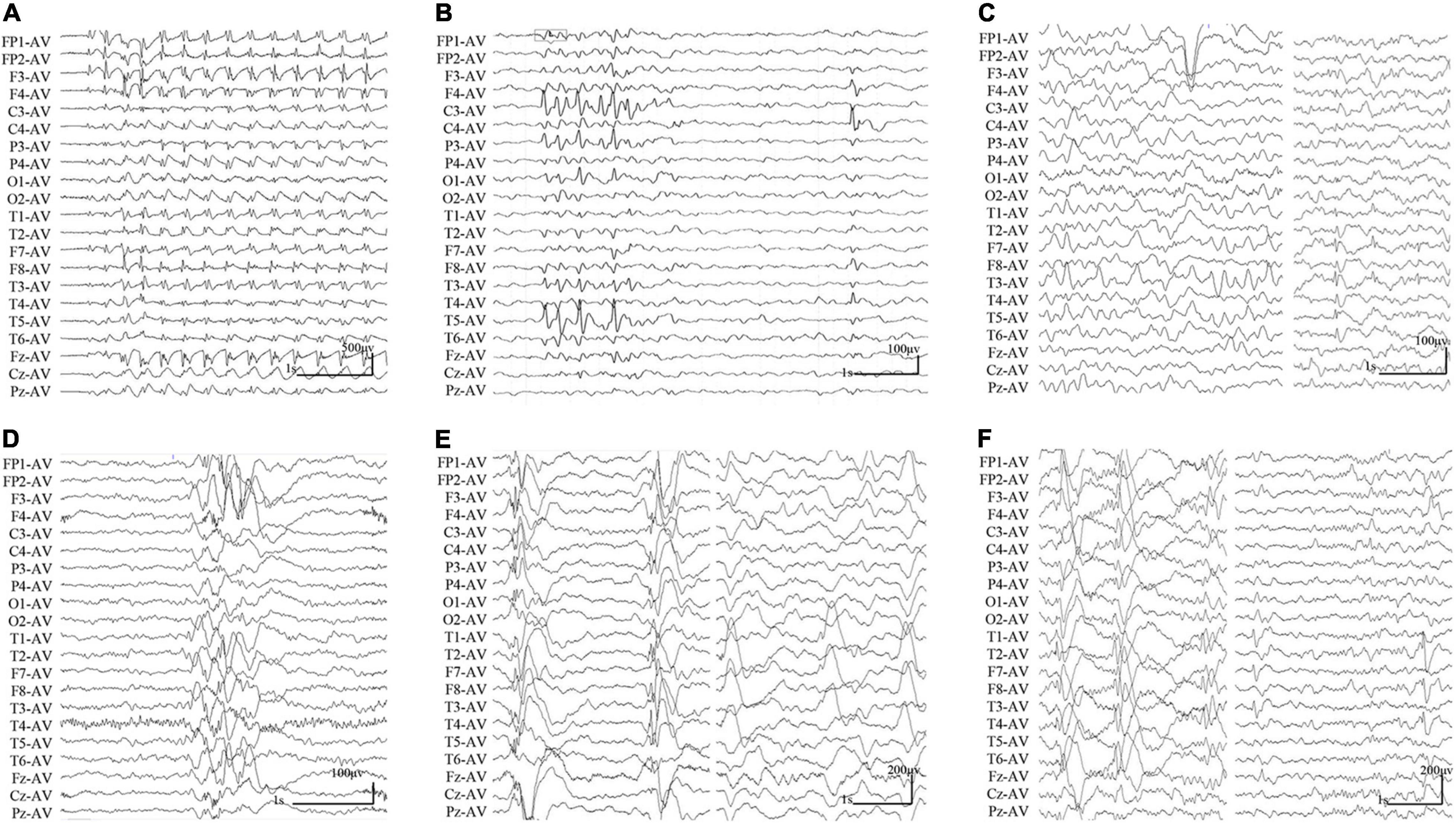
Figure 2. Changes of interictal EEGs and MRI in the cases with SHROOM4 variants. (A) Interictal EEG of case 1 showed 3 Hz generalized spike-slow waves (obtained at the age of 7 years). (B) Interictal EEG of case 2 showed bilateral central-temporal independent sharp waves and spikes (obtained at the age of 7 years). (C) Interictal EEG of case 3 showed generalized slow waves and left temporal spike-slow waves (obtained at the age of 14 years). (D) Interictal EEG of case 4 showed irregular generalized spike-slow waves (obtained at the age of 16 years). (E) Interictal EEG of case 5 showed irregular polyspike-slow waves and bilateral temporal independent spike-slow waves (obtained at the age of 4 years). (F) Interictal EEG of case 6 showed right predominant generalized spike-slow waves, bilateral temporal independent spike-slow waves (obtained at the age of 3 years).
Molecular and Molecular Sub-Regional Effects of the SHROOM4 Variants
The SHROOM4 protein contains two evolutionarily conserved domains, i.e., an N-terminal PDZ domain, and a C-terminal ASD2 domain (Hagens et al., 2006; Yoder and Hildebrand, 2007). In the present study, variant Pro5Thr was located in the N-terminal PDZ domain, while Glu1079Ala, Ser1194Leu, Arg1430Cys, Val1435Met, and Pro1444Leu were located in or near to the C-terminal ASD2 domain (Figure 3A).
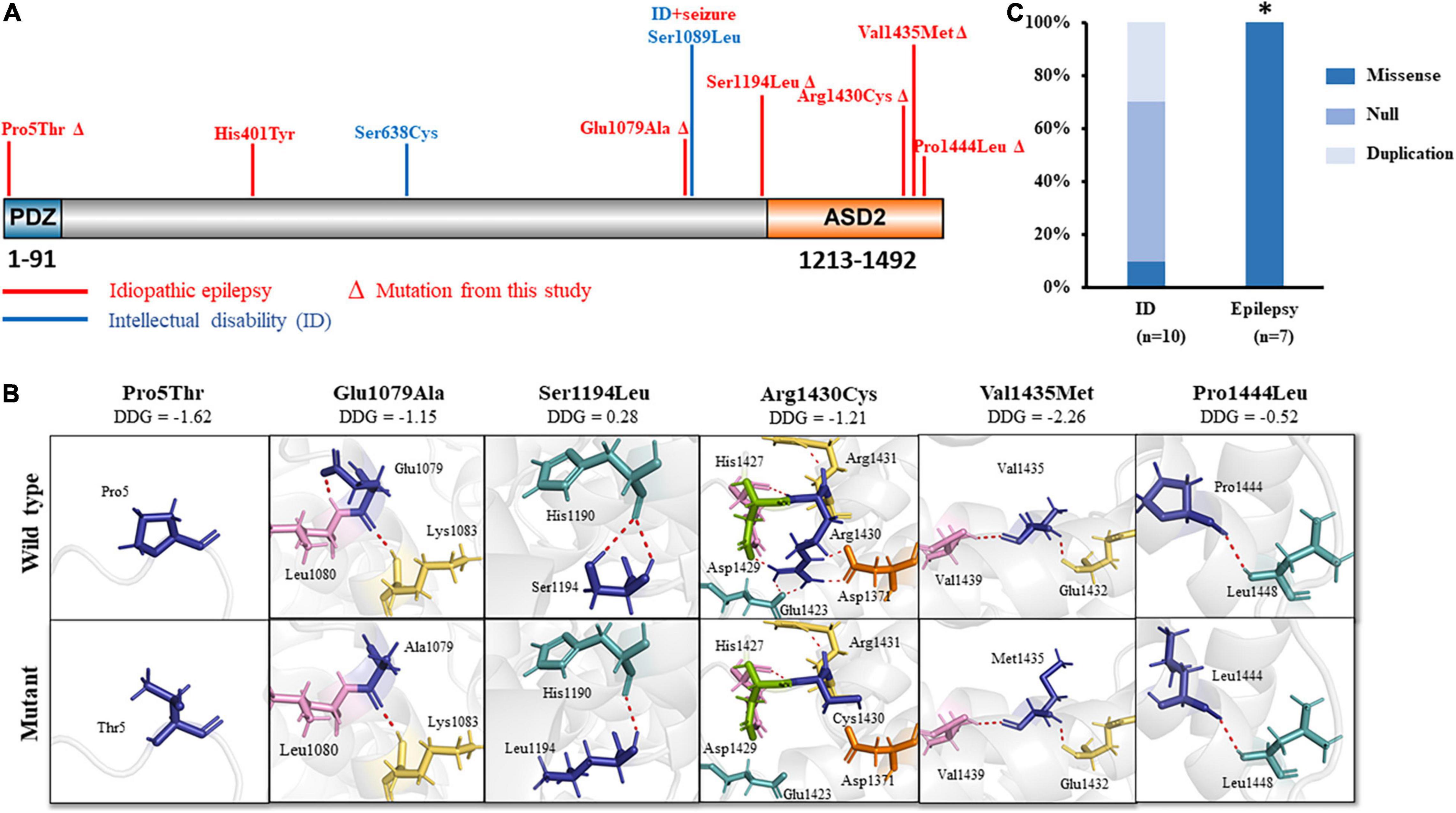
Figure 3. Schematic illustration of SHROOM4 variants. (A) Linear schematic of missense SHROOM4 mutations and their locations on SHROOM4 protein. Mutations associated with epilepsy were shown in red color. Mutations associated with ID were shown in blue. ΔThe mutation was found in this study (B) Changes of hydrogen bonds and free energy change value (DDG) of the mutations. (C) The proportion of missense mutations in epilepsy and ID. *The proportion of missense mutations in epilepsy is significantly higher than that in ID.
The molecular effect of the missense variants was analyzed by using I-TASSER for protein modeling and PyMOL 1.7 for visualization (Figure 3B). Three of the variants, including Glu1079Ala, Ser1194Leu, and Arg1430Cys, changed hydrogen bonds with the surrounding residues. Originally, Glu1079 formed two hydrogen bonds with residues Leu1080 and Lys1083, respectively. When glutamine was replaced by alanine, the hydrogen bonds with Leu1080 was destroyed. Ser1194 formed two hydrogen bonds with His1190. When serine was replaced by leucine, one of the hydrogen bond was destroyed. Arg1430 formed seven hydrogen bonds with residues Asp1371, Glu1423, His1427, Asp1429, and Arg1431, respectively. When arginine was replaced by cystine, five hydrogen bonds with Asp1371, Glu1423, and Asp1429 were destroyed, and only the hydrogen bonds with His1427 and Arg1431 were kept.
The other three variants, including Pro5Thr, Pro1444Leu, and Val1435Met, have no changed in hydrogen bonds, but they were predicted to decrease the protein stability significantly (ΔΔG values < −0.5 kcal/mol) (Figure 3B).
Genotype-Phenotype Correlation
Previously, 12 SHROOM4 variants have been reported, including 3 missense (Hagens et al., 2006; Farwell et al., 2015; Routier et al., 2019), 2 duplications (Froyen et al., 2007; Isrie et al., 2012), and 7 destructive variants (1 nonsense, 3 gross deletion variants (Honda et al., 2010; Armanet et al., 2015; Danyel et al., 2019; Heide et al., 2020), and 3 complex rearrangements (Hagens et al., 2006; Dong et al., 2021)) (Supplementary Table 2). Eleven of the variants associated with intellectual disability (ID), of which 9 were destructive mutations or duplications (Figure 3C). Among the three missense variants, one missense variant was associated with ID (Farwell et al., 2015) and one missense variant was associated with ID and seizures (Hagens et al., 2006). Additional one missense variant (His401Tyr) was reported to be associated with myoclonic-atonic epilepsy (MAE) (Routier et al., 2019). These variants are all hemizygous. In this study, all hemizygous variants were missense variants, which were associated with idiopathic epilepsy without ID (Figure 3A). The proportion of missense variants in epilepsy (7/7) was significantly higher than that in ID (1/10) (p < 0.001) (Figure 3C), suggesting a genotype-phenotype correlation.
Discussion
SHROOM4 gene encodes an actin-binding proteins, which plays an important role in cytoskeletal architecture, synaptogenesis, and maintaining GABAB receptor-mediated inhibition. Hemizygous missense variants were identified in six cases with idiopathic epilepsy without intellectual disability (Figure 1A). All patients presented with features of generalized seizures or generalized discharges. These hemizygous variants had no or extremely low allele frequencies in controls and showed statistically higher frequency in the case cohort than controls (Table 2). All variants were predicted to alter hydrogen bond with surrounding amino acids or decreased protein stability. This study suggests that SHROOM4 is potentially a candidate causative gene of X-link epilepsy with features of generalized seizures or generalized discharges.
The SHROOM4 gene is widely expressed in the brain2. It plays a critical role in regulating dendritic spine morphology and controls the cell surface expression and intracellular trafficking of GABAB receptor (Zapata et al., 2017), which inhibit neuronal activity through G protein-coupled second-messenger systems (Euro, 2014; Yoo et al., 2017). In rat, Shrm4 were found to influence hippocampal excitability by modulating tonic inhibition in dentate gyrus granule cells. Knockdown of Shrm4 cause increased susceptibility to seizures (Zapata et al., 2017). Previously, a study identified a hemizygous missense variant in a family with ID and seizures (Hagens et al., 2006). Recently, a missense variant with unknown origin was also found in a male with MAE (Routier et al., 2019). However, the relationship between SHROOM4 mutations and epilepsy remains uncertain. In this study, we identified six novel hemizygous missense variants in six unrelated cases with idiopathic epilepsy but without ID, suggesting that SHROOM4 is potentially a candidate gene of epilepsy.
Previous studies have shown that mutations of the GABA receptors, such as, GABRA1, GABRA5, GABRA6, GABRB3, GABRD, GABRG2, were associated with idiopathic generalized epilepsy (Cossette et al., 2002; Hirose, 2006; Moller et al., 2017; Lee et al., 2018; May et al., 2018). Mutations of the GABAB receptor cause generalized epilepsy by impairing inhibitory network neurodevelopment (Samarut et al., 2018). Recent study found that knockdown of Shrm4 severely impairs GABAB receptor-mediated inhibition and thus potentially associated with generalized epilepsy. In the present study, patients with SHROOM4 variants presented mainly generalized epilepsy, such as CAE, MAE, LGS, IGE, and those with partial seizures also had bilateral or generalized discharge or generalized seizures (Table 1 and Figure 1), potentially suggesting an association between SHROOM4 variants and generalized epilepsy. The patients with SHROOM4 mutations showed good responses to proper antiepileptic treatment and got seizure free, in spite of frequent daily seizures in several cases. These findings suggested that the establishment of SHROOM4-epilepsy association would be potentially significant in management of the patients with SHROOM4 mutations.
Previously, 11 variants were reported in patients with ID (Ng et al., 2004), 9 of which were destructive mutations or duplications (Figure 3C and Supplementary Table 2). Only two of those variants were missense variants. In contrast, epilepsy-associated variants were all missense variants (Figure 3C), suggesting a genotype-phenotype correlation.
SHROOM4 gene encodes a member of the Shroom family, which contains an N-terminal PDZ domain and a C-terminally positioned motif termed ASD2 (Hagens et al., 2006; Yoder and Hildebrand, 2007). The PDZ domain interacts with C terminus of GABAB receptors, while the ASD2 domain is capable of inducing myosin II-dependant changes in cell shape. The central portion of the protein appears to be an actin targeting sequence and mediated Shrm4 localization (Yoder and Hildebrand, 2007). The current data demonstrated that two missense variants located in or closed to N-terminal PDZ domain were associated with generalized epilepsy; two missense variants located in the middle of protein were associated with ID, one of which was located near to C-terminal ASD2 domain and associated with epilepsy (Hagens et al., 2006; Farwell et al., 2015); missense variants located in or near to C-terminal ASD2 domain were associated with epilepsy with both focal and generalized seizures or discharges. These evidences suggested a possible molecular sub-regional effect of SHROOM4 variants, as that in several genes reported previously (Wang et al., 2018; Liu et al., 2020; Tang et al., 2020). However, further studies are required to determine the function details of each region and their association with different phenotypes.
This study has several limitations. Knockdown, i.e., LOF of Shrm4, in rat resulted in severely impaired synaptogenesis and reduced GABAB receptor-mediated inhibition, causing susceptibility to seizures. The probability of being LOF intolerant (pLI) score was high for SHROOM4 (pLI = 0.997), suggesting that SHROOM4 is intolerant to LOF variants. However, the specific mechanism of epileptogenesis of the SHROOM4 variants remains unknown; and further experimental studies are required to determine the functional consequence of the variants. Additionally, although these missense variants were associated with generalized epilepsy, the patients presented different epilepsy syndromes. The mechanism of phenotype variation warrants further studies.
In conclusion, we identified six novel SHROOM4 hemizygous missense variants in epilepsy patients with features of generalized seizures or generalized discharges. Further analysis revealed that a potential genotype-phenotype correlation and sub-regional molecular implication of SHROOM4 variants. This study potentially extends the spectrum of diseases phenotype associated with SHROOM4 variants and helps to understand the mechanisms of phenotypic variation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of The Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
Y-HY designed the study, administered the project, and revised the manuscript. W-JB completed collection of the clinical data, analyzed the data, and draft of the manuscript. Z-JL analyzed the data and drafted of the manuscript. SL, NH, L-DG, B-ML, and JW contributed to data analysis and interpretation. B-ML and L-DG performed data analysis and provided technical assistance. All authors have read and approved the final manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (Grant No. 81870903 to Y-HY), the National Natural Science Foundation of China (Grant No. 81971216 to NH), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515011048 to NH), Science and Technology Project of Guangzhou (Grant No. 201904010292 to NH), and UCB Pharma Ltd. Joint Science Research Foundation of China Association Against Epilepsy (Grant Nos. 2020006B to NH and CU-2022-027 to W-JB). The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Our sincere thanks to Professor Wei-Ping Liao for editing this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2022.862480/full#supplementary-material
Footnotes
References
Armanet, N., Metay, C., Brisset, S., Deschenes, G., Pineau, D., Petit, F. M., et al. (2015). Double Xp11.22 deletion including SHROOM4 and CLCN5 associated with severe psychomotor retardation and Dent disease. Mol. Cytogenet. 8:8. doi: 10.1186/s13039-015-0107-x
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310. doi: 10.1093/nar/gki375
Cossette, P., Liu, L., Brisebois, K., Dong, H., Lortie, A., Vanasse, M., et al. (2002). Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 31, 184–189. doi: 10.1038/ng885
Danyel, M., Suk, E. K., Raile, V., Gellermann, J., Knaus, A., and Horn, D. (2019). Familial Xp11.22 microdeletion including SHROOM4 and CLCN5 is associated with intellectual disability, short stature, microcephaly and Dent disease: a case report. BMC Med. Genom. 12:6. doi: 10.1186/s12920-018-0471-6
Dong, Z., Chau, M. H. K., Zhang, Y., Dai, P., Zhu, X., Leung, T. Y., et al. (2021). Deciphering the complexity of simple chromosomal insertions by genome sequencing. Hum. Genet. 140, 361–380. doi: 10.1007/s00439-020-02210-x
Euro, E.-R. E. S. C. (2014). De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies.. Am. J. Hum. Genet. 95, 360–370. doi: 10.1016/j.ajhg.2014.08.013
Farwell, K. D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E. C., Tippin Davis, B., et al. (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586. doi: 10.1038/gim.2014.154
Froyen, G., Van Esch, H., Bauters, M., Hollanders, K., Frints, S. G., Vermeesch, J. R., et al. (2007). Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum. Mutat. 28, 1034–1042. doi: 10.1002/humu.20564
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Hagens, O., Dubos, A., Abidi, F., Barbi, G., Van Zutven, L., Hoeltzenbein, M., et al. (2006). Disruptions of the novel KIAA1202 gene are associated with X-linked mental retardation. Hum. Genet. 118, 578–590. doi: 10.1007/s00439-005-0072-2
Heide, S., Spentchian, M., Valence, S., Buratti, J., Mach, C., Lejeune, E., et al. (2020). Prenatal exome sequencing in 65 fetuses with abnormality of the corpus callosum: contribution to further diagnostic delineation. Genet. Med. 22, 1887–1891. doi: 10.1038/s41436-020-0872-8
Hirose, S. (2006). A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 70, (Suppl. 1), S206–S217. doi: 10.1016/j.eplepsyres.2005.12.007
Honda, S., Hayashi, S., Imoto, I., Toyama, J., Okazawa, H., Nakagawa, E., et al. (2010). Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis. J. Hum. Genet. 55, 590–599. doi: 10.1038/jhg.2010.74
Isrie, M., Froyen, G., Devriendt, K., de Ravel, T., Fryns, J. P., Vermeesch, J. R., et al. (2012). Sporadic male patients with intellectual disability: contribution of X-chromosome copy number variants. Eur. J. Med. Genet. 55, 577–585. doi: 10.1016/j.ejmg.2012.05.005
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Lee, C. G., Lee, J., and Lee, M. (2018). Multi-gene panel testing in Korean patients with common genetic generalized epilepsy syndromes. PLoS One 13:e0199321. doi: 10.1371/journal.pone.0199321
Liu, L., Chen, Z. R., Xu, H. Q., Liu, D. T., Mao, Y., Liu, H. K., et al. (2020). DEPDC5 Variants Associated Malformations of Cortical Development and Focal Epilepsy With Febrile Seizure Plus/Febrile Seizures: the Role of Molecular Sub-Regional Effect. Front. Neurosci. 14:821. doi: 10.3389/fnins.2020.00821
May, P., Girard, S., Harrer, M., Bobbili, D. R., Schubert, J., Wolking, S., et al. (2018). Rare coding variants in genes encoding GABAA receptors in genetic generalised epilepsies: an exome-based case-control study. Lancet Neurol. 17, 699–708. doi: 10.1016/S1474-4422(18)30215-1
Moller, R. S., Wuttke, T. V., Helbig, I., Marini, C., Johannesen, K. M., Brilstra, E. H., et al. (2017). Mutations in GABRB3: from febrile seizures to epileptic encephalopathies. Neurology 88, 483–492. doi: 10.1212/WNL.0000000000003565
Ng, D., Thakker, N., Corcoran, C. M., Donnai, D., Perveen, R., Schneider, A., et al. (2004). Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat. Genet. 36, 411–416. doi: 10.1038/ng1321
Routier, L., Verny, F., Barcia, G., Chemaly, N., Desguerre, I., Colleaux, L., et al. (2019). Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: is there a major genetic factor? Clin. Genet. 96, 254–260. doi: 10.1111/cge.13581
Samarut, E., Swaminathan, A., Riche, R., Liao, M., Hassan-Abdi, R., Renault, S., et al. (2018). gamma-Aminobutyric acid receptor alpha 1 subunit loss of function causes genetic generalized epilepsy by impairing inhibitory network neurodevelopment. Epilepsia 59, 2061–2074. doi: 10.1111/epi.14576
Shi, Y. W., Zhang, Q., Cai, K., Poliquin, S., Shen, W., Winters, N., et al. (2019). Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 142, 3028–3044. doi: 10.1093/brain/awz250
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2020). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief Bioinform. 21, 1776–1786. doi: 10.1093/bib/bbz115
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, J. Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X. R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Yang, J., and Zhang, Y. (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181. doi: 10.1093/nar/gkv342
Yoder, M., and Hildebrand, J. D. (2007). Shroom4 (Kiaa1202) is an actin-associated protein implicated in cytoskeletal organization. Cell Motil. Cytoskel. 64, 49–63. doi: 10.1002/cm.20167
Yoo, Y., Jung, J., Lee, Y. N., Lee, Y., Cho, H., Na, E., et al. (2017). GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann. Neurol. 82, 466–478. doi: 10.1002/ana.25032
Zapata, J., Moretto, E., Hannan, S., Murru, L., Longatti, A., Mazza, D., et al. (2017). Epilepsy and intellectual disability linked protein Shrm4 interaction with GABABRs shapes inhibitory neurotransmission. Nat. Commun. 8:14536. doi: 10.1038/ncomms14536
Keywords: epilepsy, SHROOM4 gene, whole-exome sequencing, intellectual disability, genotype-phenotype correlation, sub-regional effect
Citation: Bian W-J, Li Z-J, Wang J, Luo S, Li B-M, Gao L-D, He N and Yi Y-H (2022) SHROOM4 Variants Are Associated With X-Linked Epilepsy With Features of Generalized Seizures or Generalized Discharges. Front. Mol. Neurosci. 15:862480. doi: 10.3389/fnmol.2022.862480
Received: 26 January 2022; Accepted: 25 April 2022;
Published: 17 May 2022.
Edited by:
Qian Chen, Massachusetts Institute of Technology, United StatesReviewed by:
Xiaofeng Yang, Bioland Laboratory, ChinaNigel CK Tan, National Neuroscience Institute (NNI), Singapore
Copyright © 2022 Bian, Li, Wang, Luo, Li, Gao, He and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Hong Yi, yyh168@sina.com
†ORCID: Wen-Jun Bian, orcid.org/0000-0002-7175-1210; Yong-Hong Yi, orcid.org/0000-0002-6075-2015
‡These authors have contributed equally to this work