 Mark Makarov
Mark Makarov Liliia Kushnireva
Liliia Kushnireva Michele Papa
Michele Papa Eduard Korkotian
Eduard Korkotian- 1Department of Neurobiology, Weizmann Institute of Science, Rehovot, Israel
- 2Department of Mental and Physical Health and Preventive Medicine, University of Campania "Luigi Vanvitelli", Caserta, Italy
This review uncovers the intricate relationship between presenilins, calcium, and mitochondria in the context of Alzheimer’s disease (AD), with a particular focus on the involvement of presenilin mutations in mitochondrial dysfunction. So far, it is unclear whether the impairment of mitochondrial function arises primarily from damage inflicted by β-amyloid upon mitochondria or from the disruption of calcium homeostasis due to presenilins dysfunctions. The roles of presenilins in mitophagy, autophagy, mitochondrial dynamics, and many other functions, non-γ-secretase related, also require close attention in future research. Resolution of contradictions in understanding of presenilins cellular functions are needed for new effective therapeutic strategies for AD.
Introduction
Presenilins (PS) are a family of highly conserved transmembrane proteins located in endoplasmic reticulum (ER), endosomes, and lysosomes, at ER-mitochondria contact sites, nuclear envelope, Golgi apparatus, but primarily in compartments near the cell surface (Area-Gomez et al., 2009; Escamilla-Ayala et al., 2020; Hernandez-Sapiens et al., 2022). The two homologs of PS, PS-1, and PS-2 are expressed ubiquitously in the brain and peripheral tissue, but have both overlapping and unique functions. PS is the catalytic core of the γ-secretase complex, which performs intramembrane proteolytic removal of many transmembrane proteins, including Notch-1, CD44, Syndecan, Delta, Jagged, and ErbB4, involved in various pathological processes (Escamilla-Ayala et al., 2020; Hernandez-Sapiens et al., 2022). However, presenilins are primarily known for their role in sequentially cleaving the amyloid precursor protein (APP) to generate β-amyloid (Aβ) peptides of various lengths. There are over 300 mutations in the presenilins genes, many of which result in excessive production of the most hydrophobic/aggregate form of Aβ42, leading to neurodegeneration and dementia in familial Alzheimer’s disease (FAD). Other mutations, as PS1 L286V or M146V and PS2 N141I, may increase the likelihood of apoptosis independently of amyloid via mitochondrial deficiency or calcium imbalance (Czech et al., 2000; Filadi and Pizzo, 2019). In addition to the amyloid hypothesis, various other theories have been proposed to explain the pathogenesis of Alzheimer’s disease (AD), including those involving tau protein accumulation, cholinergic dysfunction, viral infections, neuroinflammation, and the mitochondrial cascade hypothesis (Liu et al., 2019). Ca2+ signaling dysregulation emerges as a common underlying factor linking most of these theories (Filadi and Pizzo, 2019).
The mitochondrial cascade hypothesis
Of particular interest, the mitochondrial cascade hypothesis has prompted investigations of the impact of presenilin mutations on γ-secretase activity, not only in relation to amyloidogenic processing, but also with regard to non-amyloid substrates and γ-secretase-independent functions of presenilins. These diverse functions encompass the modulation of calcium homeostasis, mitochondrial function, autophagy, and mitophagy. The hypothesis suggests a relative primacy of mitochondrial dysfunction over amyloid accumulation in the pathogenesis of AD and is currently subject to debate (Makarov and Korkotian, 2023). While the deleterious effects of amyloid accumulation are well-established, the question arises as to whether a single amyloid accumulation event is sufficient to elicit subsequent pathological consequences, and whether the amyloid hypothesis is excessively emphasized. Disruption of mitochondrial function due to impaired mitophagy may represent a more pivotal mechanism. It has been documented that compromised mitochondrial function promotes increased production of Aβ in AD models (Leuner et al., 2012), while induction of mitophagy has shown potential for alleviating disease manifestations (Lautrup et al., 2019; Lee et al., 2021). Presenilins participate in mitophagy through various pathways, including interaction with PINK1/Parkin proteins for mitophagy regulation (Quinn et al., 2020; Saito et al., 2022), and control of gene expression associated with PARK2-dependent autophagy through transcription factor interactions (Checler et al., 2017). Presenilins also exert influence on the regulation of autophagy. For instance, mutated PS2 has been found to impede autophagy at the stage of autophagosome-lysosome fusion, attributed to disruptions in Ca2+ homeostasis (Fedeli et al., 2019). The elucidation of the precise role of presenilins in mitophagy and autophagy represents a dynamic area of investigation, with potential implications for advancing comprehension of AD pathogenesis as well as the broader cellular functions of presenilins.
Effect of presenilins on mitochondrial dynamic
The functional roles of presenilins extend to the regulation of mitochondrial dynamics, encompassing processes, such as movement, fusion, fission, and distribution within neurons (Han et al., 2021). Precise and coordinated movement of mitochondria in both anterograde and retrograde directions is crucial for fulfilling the metabolic demands of cell regions, particularly those located distally from the soma, such as axon terminals and distal dendrites. Significantly, specific PS1 mutations, including M146V, I143T, and D9, have been found to impair kinesin-based anterograde axonal transport by inducing excessive activation of glycogen synthase kinase 3β (GSK3β), which plays a key role in the transmission of regulatory and proliferative signals in cells. Increased phosphorylation of kinesin light chain (KLC) by GSK3β promotes the dissociation of kinesin-I from membrane-bound organelles (MBOs), leading to diminished motility of mitochondria and synaptic vesicles. In fact, cultured hippocampal neurons expressing the PS1 M146V mutation exhibited reduced density of both mitochondria and synaptic vesicles (Wang et al., 2020). It has been established that the anterograde transport of mitochondria is the most vulnerable to Aβ oligomers (Correia et al., 2016). However, the precise consequences of presenilin mutations on the intricate anterograde transport process mediated by kinesin motors in dendrites have yet to be fully elucidated. Additionally, PS1 mutations also disrupt retrograde transport—a critical mechanism for sustaining synaptic plasticity and facilitating the removal of damaged mitochondria. The impairment of dynein-mediated transport, responsible for the transportation of cargo proteins to the soma for degradation, leads to the formation of obstructions, thereby compromising neuronal signaling and the transport of molecules and organelles (Kimura et al., 2012).
The process of mitochondrial fusion plays a crucial role in maintaining plasticity mechanisms, as well as in response to stress conditions. Mitochondrial “fusion” is a physical connection of the outer and inner mitochondrial membranes of two initially separate mitochondria promotes enhanced oxidative phosphorylation processes and dilution of damaged mitochondrial DNA (Gao and Hu, 2021). Conversely, the “fission” mechanism is in which mitochondria divide into two separate organelles serves to generate new mitochondria and aid in the removal of impaired ones. In AD, a notable increase in fragmented mitochondria is observed, which is associated with mutations in presenilins. Post-mortem analyses of AD patient tissues frequently exhibit elevated levels of dynamin-related protein 1 (Drp1), a key protein involved in mitochondrial fission, along with decreased expression of fusion proteins. While Drp1-mediated fission is essential for maintaining mitochondrial function and facilitating their sequestration within autophagosomes, excessive fission may disrupt their interaction with the endoplasmic reticulum (ER) at mitochondria-associated membranes (MAMs; Shields et al., 2021). Drp1-caused overfission leads to abnormally small mitochondria with impaired bioenergetics: the ability to buffer calcium and produce ATP, which contributes to synapse damage and further loss of neurons (Wang et al., 2020).
The role of presenilins in calcium homeostasis
Presenilins also exhibit localization in MAM contact sites, where they play a direct role in mediating communication and calcium transfer between ER and mitochondria. Mechanisms underlying the impact of presenilin mutations on MAM contacts, ER-mitochondrial crosstalk, and mitochondrial reactive oxygen species (ROS) production remain poorly understood (Hedskog et al., 2013). Studies using primary cortical neurons from PS1 knockout mice have demonstrated elevated calcium release from the ER mediated by inositol 1,4,5-trisphosphate (IP3) receptors. PS1 M146L and PS2 N141I mutations prolong the opening time of IP3 channels, leading to increased Ca2+ leakage permeability (Cheung et al., 2008). Number and function of ryanodine receptors (RyR) are also increased in mouse models containing mutations PS1 M146V and PS2 N141I (Supnet and Bezprozvanny, 2011). At the same time, there is evidence suggesting that transmembrane domains 7 and 9 of PS1 have been implicated in pore-forming of an ionic conduction as Ca2+ − leak channel (Zhang et al., 2013). However, the PS1-M146V, PS2-N141I, and some others mutations disrupt or abolish this leak, resulting in ER Ca2+ overload (Nelson et al., 2007). The optimal width of the MAM is crucial for the proper transport of Ca2+ between the ER and mitochondria (Zhang et al., 2021). A reduction in the distance between the ER and mitochondria within MAM regions has been observed in the context of Alzheimer’s disease, potentially serving as a compensatory mechanism resulting from diminished Ca2+ leakage (Perez-Leanos et al., 2021). Furthermore, presenilins are also capable of physically interacting with the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), which actively transports Ca2+ from the cytosol to the ER, thereby helping to maintain low resting levels of cytosolic Ca2+. PS2-mediated calcium sequestration by SERCA pumps is vital for refilling ER calcium stores (Green et al., 2008; Fedeli et al., 2019).
Presenilin mutations dysregulate mitochondria with calcium
Paradoxically, while evidence indicates that loss of leakage function of presenilins may lead to Ca2+ ER overload, both overexpression of normal presenilins and some FAD-related mutations in them have been associated with increased Ca2+ release from the ER, depletion of Ca2+ stores, and an increase in cytosolic Ca2+ (Nelson et al., 2007; Galla et al., 2020). Other FAD mutations, on the contrary, reduce IP3-mediated increase in cytosolic Ca2+ (Galla et al., 2020). Dysregulation of calcium signaling observed in both scenarios has deleterious consequences for mitochondrial energy functions (Korkotian et al., 2019). Decreased Ca2+ ER-leakage caused by presenilin mutants leads to ER overload and hence exaggerated Ca2+ release upon cell stimulation (Zampese et al., 2011). By increasing Ca2+ release through the ER membrane, PS1 mutations can enhance Ca2+ signaling at synaptic terminals, leading to dysfunction and degeneration in aging and amyloid accumulation in AD (Mattson, 2010).
Presenilins play a vital role in mediating interactions between the mitochondria-associated endoplasmic reticulum (ER) membranes, with a particular emphasis in PS2. PS2 functions as a positive regulator of the calcium connection between ER and mitochondria (Filadi et al., 2016; Korkotian et al., 2019). The compromised signaling of local cytosolic Ca2+ resulting from the aforementioned overload of calcium stores within the ER and spine apparatus, can diminish the capacity to attract, immobilize, and retain mitochondria beneath synapses that exhibit high ATP demand. This, in turn, leads to a diminished ability to form stable synapses. Conversely, excessive levels of calcium transferred from the ER to mitochondria-associated ER membranes can give rise to mitochondrial Ca2+ overload, leading to the decoupling of the oxidative phosphorylation chain and subsequent reduction in ATP production (Kushnireva and Korkotian, 2022). Strikingly, some studies report that mitochondrial Ca2+ uptake in FAD-linked PS2 mutants is reduced as a result of decreased ER Ca2+, causing a blunted increase in cytosolic Ca2+ after stimulation (Rossini et al., 2021). These effects also depend on the ability of PS2 to partially block SERCA activity and induce hyperactivation of IP3 receptors (Rossini et al., 2021).
An increased number of MAM sites has been observed in cellular and animal models of AD as a compensatory mechanism against decreased ER-Ca2+ for improving Ca2+ transfer from ER to mitochondria (Green et al., 2008), as well as result of accumulation of C99 and ceramides (Pera et al., 2017). In addition, the presence of FAD-linked PS2 mutations leads to increased juxtaposition of ER-mitochondria (Zampese et al., 2011). However, the coexisting exaggerated release of Ca2+ via RyR- and IP3 receptors flowing into mitochondria stimulates oxidative phosphorylation, which leads to increased production of ROS, mitochondrial degradation, and neurodegeneration (Toglia et al., 2016; Ryan et al., 2020). This same excess release that increases cytosolic calcium levels also interferes with the induction of long-term potentiation and mediates some of the cytotoxic effects of β-amyloid (Yu et al., 2021). Thus, the impairment of calcium homeostasis induced by presenilins affects the development of neurodegeneration in many controversial and unclear ways. The most vulnerable in this case are mitochondrial functions, including those associated with their maintenance of synaptic plasticity. Calcium dysregulation and mitochondrial injury can be claimed to be causative factors of neurodegeneration in AD, being common mechanisms implicated in the onset of both genetic and sporadic forms of this age-related pathology (Figure 1).
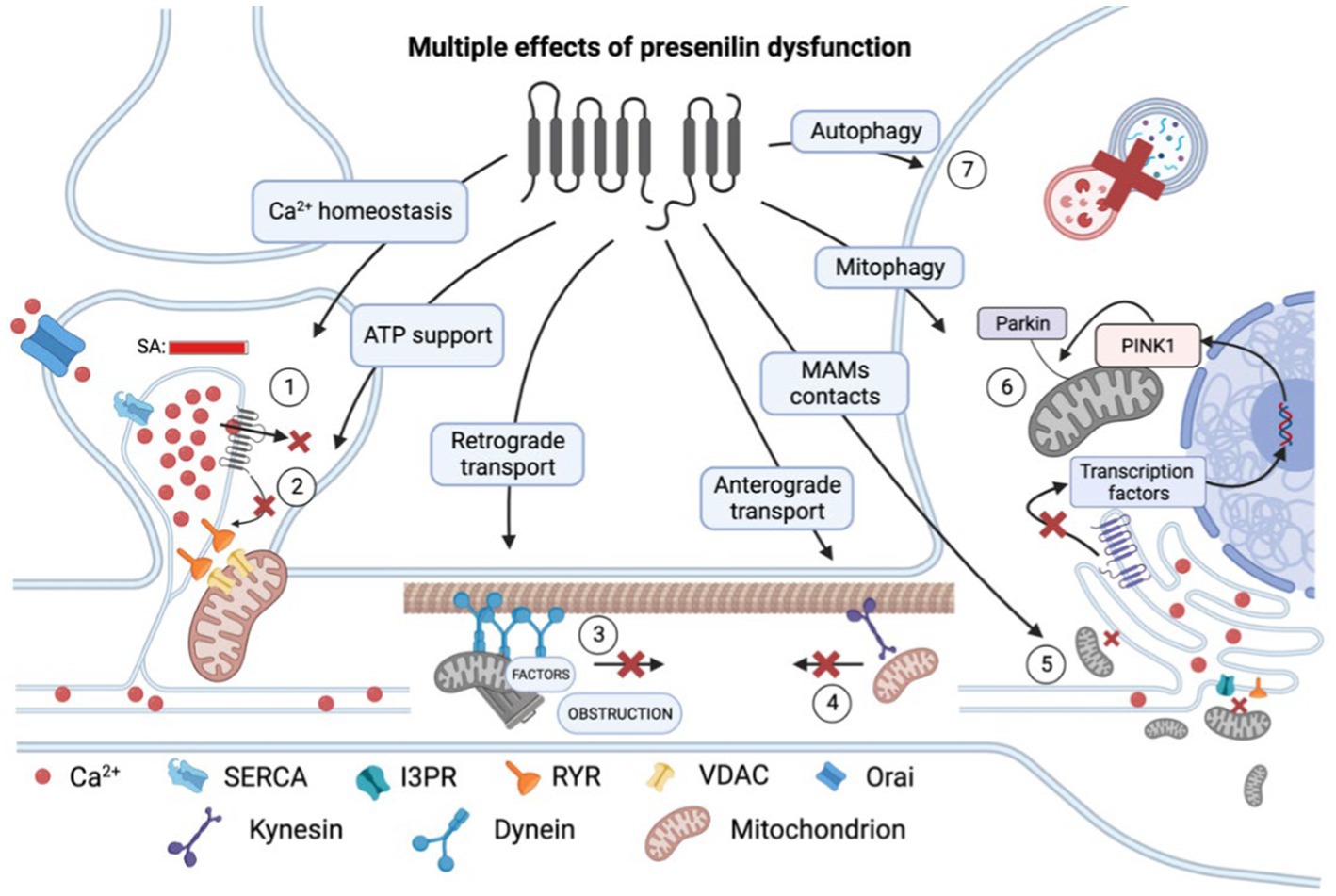
Figure 1. Multiple effects of presenilin (PS) dysfunction. Presenilin mutations result in an overload of calcium (Ca2+) within the spine apparatus (SA) due to decreased leakage via (1) pore of PS itself and (2) reduced influence of PS on RYR channels. This excessive calcium accumulation leads to impaired calcium conduction into mitochondria and decreased ATP production and therefore energy support of postsynaptic processes. (3) Presenilin mutations disrupt retrograde transport and cause dendrite obstruction. (4) Presenilin mutations lead to a reduction in connections between kinesin and membrane-bound organelles, such as mitochondria. (5) Mutations in presenilin result in extensive mitochondrial fission and impaired mitochondria-ER contact at mitochondria-associated membranes (MAMs). (6) Presenilin influences mitophagy by participating in the activation of transcription factors of PINK1 genes, which are essential for recruiting ubiquitin ligase PARKIN to the mitochondrial membrane and establishing a connection with the phagophore membrane. (7) Dysregulation of calcium homeostasis hampers autophagy by impairing lysosomal fusion. RYR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; and VDAC, voltage-dependent anion channels. In the figure, a red cross denotes the inhibition of a process.
Presenilin in astrocytes
The association between presenilins and Alzheimer’s disease extends beyond their impact on neurons, as emerging evidence suggests their involvement in astrocytes and the perisynaptic region of tripartite synapses. Postulated that astrocytes, when damaged, release inflammatory factors that contribute to the progression of neurodegeneration, with gliosis being an early symptom (Carter et al., 2012; Virtuoso et al., 2023). Astrocytes may also play a role in synaptic loss, as studies have shown an association between PS1&PS2 knockout and astrogliosis resulting in neuroinflammation (Peng et al., 2022). The effect of presenilin in astrocytic mitochondria remains inconclusive, as Contino et al. (2021) found no influence on the mitochondria of cultured neurons and astrocytes. However, presenilin deficiency has been shown to induce hyperfunction of the mitochondria-associated endoplasmic reticulum membranes (MAMs). In this regard, MAM-mediated synthesis of phospholipids and cholesterol significantly increased in astrocytes treated with apolipoprotein E, which is implicated in Alzheimer’s disease (Tambini et al., 2016).
Discussion
The primary cause of mitochondrial dysfunction in the pathogenesis of FAD remains elusive. Specifically, it is unclear whether the impairment of mitochondrial function arises primarily from damage inflicted by β-amyloid upon mitochondria or from the disruption of calcium homeostasis due to the dysfunctional presenilins. The independent role of presenilins in calcium homeostasis and their effect on the quality of mitochondria also requires deep study. In the article, we have mentioned the accumulated contradictions regarding the mechanism of the influence of mutant presenilins on ATP synthesis through disruption of the ER, SA, and MAM in particular. The reason for these contradictions can be both the use of calcium indicators with different sensitivities in experiments, and the variability of the multiple established effects at rest and in cellular activation. Connections of certain presenilin mutations with the impaired local regulation of intracellular calcium in SA (which is functionally different from the ER), will help to resolve the existing contradictions and identify the root causes of mitochondrial dysfunction in neurodegeneration. Accurate determination of the role played by presenilin in the mitochondrial cascade would provide valuable guidance to researchers in their quest for therapeutic targets and the development of treatment and prevention strategies.
While the role of mutant presenilins is unquestionable, it remains uncertain whether their presence alone is sufficient and which factors contribute to the implementation of FAD. Disturbances in calcium homeostasis and associated mitochondrial dysfunction are anticipated to have significant implications in aging and degeneration, making it an area of paramount importance for research. Additionally, further elucidation is necessary regarding the multiple functions of presenilins in various brain structures and cell types that may contribute to particular mitochondrial dynamics and bioenergetics. Given the apparent failure of drug therapies targeting at amyloid peptide clearance, it is imperative to focus on alternative hypotheses regarding the etiology of AD to identify effective therapeutic interventions.
Author contributions
EK: conceptualization and writing-review and editing. MM and LK: literature research and analysis and writing. MM: visualization. EK and MP: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by #NEXTGENERATIONEU (NGEU) and funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006)—A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022).
Acknowledgments
The authors thank Menahem Segal for constructive comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Area-Gomez, E., de Groof, A. J., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Carter, B. S., Meng, F., and Thompson, R. C. (2012). Glucocorticoid treatment of astrocytes results in temporally dynamic transcriptome regulation and astrocyte-enriched mRNA changes in vitro. Physiol. Genomics 44, 1188–1200. doi: 10.1152/physiolgenomics.00097.2012
Checler, F., Goiran, T., and Alves da Costa, C. (2017). Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: implication for neurodegenerative diseases. Autophagy 13, 2004–2005. doi: 10.1080/15548627.2017.1363950
Cheung, K. H., Shineman, D., Müller, M., Cardenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58, 871–883. doi: 10.1016/j.neuron.2008.04.015
Contino, S., Suelves, N., Vrancx, C., Vadukul, D. M., Payen, V. L., Stanga, S., et al. (2021). Presenilin-deficient neurons and astrocytes display normal mitochondrial phenotypes. Front. Neurosci. 14:586108. doi: 10.3389/fnins.2020.586108
Correia, S. C., Perry, G., and Moreira, P. I. (2016). Mitochondrial traffic jams in Alzheimer's disease—pinpointing the roadblocks. Biochim. Biophys. Acta 1862, 1909–1917. doi: 10.1016/j.bbadis.2016.07.010
Czech, C., Tremp, G., and Pradier, L. (2000). Presenilins and Alzheimer's disease: biological functions and pathogenic mechanisms. Prog. Neurobiol. 60, 363–384. doi: 10.1016/s0301-0082(99)00033-7
Escamilla-Ayala, A. A., Sannerud, R., Mondin, M., Poersch, K., Vermeire, W., Paparelli, L., et al. (2020). Super-resolution microscopy reveals majorly mono- and dimeric presenilin1/γ-secretase at the cell surface. elife 9:e56679. doi: 10.7554/eLife.56679
Fedeli, C., Filadi, R., Rossi, A., Mammucari, C., and Pizzo, P. (2019). PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 15, 2044–2062. doi: 10.1080/15548627.2019.1596489
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2016). Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of Mitofusin 2. Cell Rep. 15, 2226–2238. doi: 10.1016/j.celrep.2016.05.013
Filadi, R., and Pizzo, P. (2019). Defective autophagy and Alzheimer's disease: is calcium the key? Neural Regen. Res. 14, 2081–2082. doi: 10.4103/1673-5374.262584
Galla, L., Redolfi, N., Pozzan, T., Pizzo, P., and Greotti, E. (2020). Intracellular calcium dysregulation by the Alzheimer's disease-linked protein Presenilin 2. Int. J. Mol. Sci. 21:770. doi: 10.3390/ijms21030770
Gao, S., and Hu, J. (2021). Mitochondrial fusion: the machineries in and out. Trends Cell Biol. 31, 62–74. doi: 10.1016/j.tcb.2020.09.008
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 181, 1107–1116. doi: 10.1083/jcb.200706171
Han, J., Park, H., Maharana, C., Gwon, A. R., Park, J., Baek, S. H., et al. (2021). Alzheimer's disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 11, 8855–8873. doi: 10.7150/thno.59776
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc. Natl. Acad. Sci. U. S. A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hernandez-Sapiens, M. A., Reza-Zaldívar, E. E., Márquez-Aguirre, A. L., Gómez-Pinedo, U., Matias-Guiu, J., Cevallos, R. R., et al. (2022). Presenilin mutations and their impact on neuronal differentiation in Alzheimer's disease. Neural Regen. Res. 17, 31–37. doi: 10.4103/1673-5374.313016
Kimura, N., Okabayashi, S., and Ono, F. (2012). Dynein dysfunction disrupts intracellular vesicle trafficking bidirectionally and perturbs synaptic vesicle docking via endocytic disturbances a potential mechanism underlying age-dependent impairment of cognitive function. Am. J. Pathol. 180, 550–561. doi: 10.1016/j.ajpath.2011.10.037
Korkotian, E., Meshcheriakova, A., and Segal, M. (2019). Presenilin 1 regulates [Ca2+]i and mitochondria/ER interaction in cultured rat hippocampal neurons. Oxidative Med. Cell. Longev. 2019, 7284914–7284967. doi: 10.1155/2019/7284967
Kushnireva, L., and Korkotian, E. (2022). Mitochondria-Endoplasmic Reticulum Interaction in Central Neurons In Updates on Endoplasmic Reticulum. London, UK: IntechOpen.
Lautrup, S., Lou, G., Aman, Y., Nilsen, H., Tao, J., and Fang, E. F. (2019). Microglial mitophagy mitigates neuroinflammation in Alzheimer's disease. Neurochem. Int. 129:104469. doi: 10.1016/j.neuint.2019.104469
Lee, H. J., Jung, Y. H., Choi, G. E., Kim, J. S., Chae, C. W., Lim, J. R., et al. (2021). Urolithin a suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 28, 184–202. doi: 10.1038/s41418-020-0593-1
Leuner, K., Schütt, T., Kurz, C., Eckert, S. H., Schiller, C., Occhipinti, A., et al. (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 16, 1421–1433. doi: 10.1089/ars.2011.4173
Liu, P. P., Xie, Y., Meng, X. Y., and Kang, J. S. (2019). History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct. Target. Ther. 4:29. doi: 10.1038/s41392-019-0063-8
Makarov, M., and Korkotian, E. (2023). Differential role of active compounds in Mitophagy and related neurodegenerative diseases. Toxins 15:202. doi: 10.3390/toxins15030202
Mattson, M. P. (2010). ER calcium and Alzheimer's disease: in a state of flux. Sci. Signal. 3:pe10. doi: 10.1126/scisignal.3114pe10
Nelson, O., Tu, H., Lei, T., Bentahir, M., De Strooper, B., and Bezprozvanny, I. (2007). Familial Alzheimer disease–linked mutations specifically disrupt ca 2+ leak function of presenilin 1. J. Clin. Invest. 117, 1230–1239. doi: 10.1172/JCI30447
Peng, W., Xie, Y., Liao, C., Bai, Y., Wang, H., and Li, C. (2022). Spatiotemporal patterns of gliosis and neuroinflammation in presenilin 1/2 conditional double knockout mice. Front. Aging Neurosci. 14:966153. doi: 10.3389/fnagi.2022.966153
Pera, M., Larrea, D., Guardia-Laguarta, C., Montesinos, J., Velasco, K. R., Agrawal, R. R., et al. (2017). Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 36, 3356–3371. doi: 10.15252/embj.201796797
Perez-Leanos, C. A., Romero-Campos, H. E., Dupont, G., and Gonzalez-Velez, V. (2021). Reduction of ER-mitochondria distance: a key feature in Alzheimer's and Parkinson's disease, and during Cancer treatment. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2021, 4412–4415. doi: 10.1109/EMBC46164.2021.9631090
Quinn, P. M. J., Moreira, P. I., Ambrósio, A. F., and Alves, C. H. (2020). PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 8:189. doi: 10.1186/s40478-020-01062-w
Rossini, M., García-Casas, P., Filadi, R., and Pizzo, P. (2021). Loosening ER-mitochondria coupling by the expression of the Presenilin 2 loop domain. Cells 10:1968. doi: 10.3390/cells10081968
Ryan, K. C., Ashkavand, Z., and Norman, K. R. (2020). The role of mitochondrial calcium homeostasis in Alzheimer's and related diseases. Int. J. Mol. Sci. 21:9153. doi: 10.3390/ijms21239153
Saito, T., Kodani, E., and Gottlieb, R. A. (2022). Presenilin-2 regulates Mitophagy via promoting intracellular translocation of Parkin to mitochondria. Circulation 146:A12308. doi: 10.1161/circ.146.suppl_1.12308
Shields, L. Y., Li, H., Nguyen, K., Kim, H., Doric, Z., Garcia, J. H., et al. (2021). Mitochondrial fission is a critical modulator of mutant APP-induced neural toxicity. J. Biol. Chem. 296:100469. doi: 10.1016/j.jbc.2021.100469
Supnet, C., and Bezprozvanny, I. (2011). Presenilins function in ER calcium leak and Alzheimer's disease pathogenesis. Cell Calcium 50, 303–309. doi: 10.1016/j.ceca.2011.05.013
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Toglia, P., Cheung, K. H., Mak, D. O., and Ullah, G. (2016). Impaired mitochondrial function due to familial Alzheimer's disease-causing presenilins mutants via ca(2+) disruptions. Cell Calcium 59, 240–250. doi: 10.1016/j.ceca.2016.02.013
Virtuoso, A., De Luca, C., Korai, S. A., Papa, M., and Cirillo, G. (2023). Neuroinflammation and glial activation in the central nervous system: a metabolic perspective. Neural Regen. Res. 18, 1025–1026. doi: 10.4103/1673-5374.355754
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30. doi: 10.1186/s13024-020-00376-6
Yu, W., Jin, H., and Huang, Y. (2021). Mitochondria-associated membranes (MAMs): a potential therapeutic target for treating Alzheimer's disease. Clin. Sci. (Lond.) 135, 109–126. doi: 10.1042/CS20200844
Zampese, E., Fasolato, C., Kipanyula, M. J., Bortolozzi, M., Pozzan, T., and Pizzo, P. (2011). Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. U. S. A. 108, 2777–2782. doi: 10.1073/pnas.1100735108
Zampese, E., Fasolato, C., Pozzan, T., and Pizzo, P. (2011). Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun. Integr. Biol. 4, 357–360. doi: 10.4161/cib.4.3.15160
Zhang, S., Zhang, M., Cai, F., and Song, W. (2013). Biological function of Presenilin and its role in AD pathogenesis. Translat. Neurodegen. 2, 1–13. doi: 10.1186/2047-9158-2-15
Keywords: presenilins, Alzheimer’s disease, mitochondria associated membranes, spine apparatus, neurodegeneration
Citation: Makarov M, Kushnireva L, Papa M and Korkotian E (2023) Presenilins and mitochondria—an intriguing link: mini-review. Front. Neurosci. 17:1249815. doi: 10.3389/fnins.2023.1249815
Edited by:
Heather Jane Mortiboys, The University of Sheffield, United KingdomReviewed by:
Punya Sachdeva, Amity University, IndiaCláudia Pereira, University of Coimbra, Portugal
Copyright © 2023 Makarov, Kushnireva, Papa and Korkotian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduard Korkotian, eduardkorkotian@gmail.com
†These authors have contributed equally to this work