Inactivation of the lysine binding sites of human plasminogen (hPg) reveals novel structural requirements for the tight hPg conformation, M-protein binding, and rapid activation
 Yetunde A. Ayinuola
Yetunde A. Ayinuola Francis J. Castellino
Francis J. Castellino- 1W. M. Keck Center for Transgene Research, Notre Dame, IN, United States
- 2Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN, United States
Accelerated activation of the human plasminogen zymogen (hPg) to two-chain active plasmin (hPm) is achieved following conformational changes induced by ligand-binding at the lysine-binding sites (LBSs) in four of the five hPg kringle domains. In this manner, pattern D skin-trophic strains of Group A streptococci (GAS), through the expression of surface plasminogen-binding M-protein (PAM), immobilize surface hPg, thereby enabling rapid hPg activation by GAS-secreted streptokinase (SK). Consequently, GAS enhances virulence by digesting extracellular and tight cellular junctional barriers using hPm activity. Many studies have demonstrated the singular importance of the kringle-2 domain of hPg (K2hPg) to PAM-binding using hPg fragments. Recently, we showed, using full-length hPg, that K2hPg is critical for PAM binding. However, these studies did not eliminate any modulatory effects of the non-K2hPg LBS on this interaction. Moreover, we sought to establish the significance of the intramolecular interaction between Asp219 of the LBS of K2hPg and its serine protease domain binding partner, Lys708, to conformational changes in hPg. In the current study, selective inactivation of the LBS of K1hPg, K4hPg, and K5hPg revealed that the LBS of these kringle domains are dispensable for hPg binding to PAM. However, the attendant conformational change upon inactivation of K4hPg LBS increased the affinity of hPg for PAM by an order of magnitude. This finding suggests that the native hPg conformation encloses PAM-binding exosites or sterically hinders access to K2hPg. While simultaneous inactivation of the LBS of K1hPg, K4hPg, and K5hPg inhibited hPg/SK association alongside hPg activation, the replacement of Lys708 generated a slight conformational change that optimally accelerated hPg activation. Thus, we accentuate disparate functions of hPg LBS and conclude, using intact proteins, that K2hPg plays a central role in regulating hPg activation.
Introduction
Activation of the single-chain human plasminogen (hPg) following the scission of its Arg561-Val562 bond to produce the two-chain proteolytically active plasmin (hPm) is an essential step in the process leading to the dissolution of the fibrin blood clot (Urano et al., 2018). In addition to this principal function, hPm exhibits a broad substrate specificity catalyzing the degradation/activation of extracellular matrix (ECM) proteins, such as collagen, fibronectin, matrix metalloproteinase, and collagenase (Wong et al., 1992; Andreasen et al., 2000; Wu et al., 2000; Miles et al., 2005). Moreover, many cell types, including blood, immune, and bacterial cells, are endowed with hPg receptors, enabling them to co-opt the proteolytic activity of hPm for their diverse functions (Bhattacharya et al., 2012; Plow et al., 2012). Consequently, hPm is thought to play a significant role in regulating ECM integrity, cell migration, and the outcome of bacterial infection.
hPm formation/activity is regulated by at least three mechanisms. One such regulatory mechanism is intrinsic to the hPg molecule, wherein this protein exists in a closed, slowly activatable conformation. hPg is a 791 amino acid residue, single-chain glycoprotein that exists in at least seven distinct domains. Residues Glu1-Lys77 serve the function of an activation peptide (AP) in the native hPg (Glu1-hPg). The AP domain is followed by five consecutive kringle domains (K1hPg–K5hPg), each of ∼80 residues, characterized by triple disulfide bonds and lysine binding sites (LBS). These LBS are used for ligand binding and are typically constituted by anionic, cationic, and hydrophobic centers that interact and coordinate with amino, carboxylate, and methylene groups of lysine (analogs and isosteres), respectively, in the order of K1hPg > K4hPg > K5hPg > K2hPg in decreasing binding affinity (McCance et al., 1994; McCance and Castellino, 1995). K3hPg LBS is inactive and has no measurable affinity for lysine due to the presence of Lys311 in place of Glu/Asp of the anionic center (Marti et al., 1994). The seventh and last domain, the serine protease (SP) domain, comprises residues Val562-Asn791, together with the AP domain, participate in intramolecular interactions that occupy the LBS of K2hPg, K4hPg, and K5hPg, retaining hPg in an activation-resistant closed conformation (Urano et al., 1988; Law et al., 2012). Disruption of these intramolecular interactions by limited proteolysis of Glu1-hPg through the action of preformed hPm that cleaves the AP domain to generate Lys78-Asn791 (Lys78-hPg) opens the hPg protein, making it more readily activatable (Markus et al., 1976; Urano et al., 1987a). Similarly, the binding of hPg to fibrin clot and cell surface receptors liberates the AP domain from the LBS of hPg, thereby relaxing its conformation (Miles and Parmer, 2013).
PAM, plasminogen-binding group A Streptococcus (GAS) M-protein, is a hPg receptor found on the surface of pattern D skin-trophic strains of S. pyogenes (Berge and Sjobring, 1993; Wistedt et al., 1995). GAS is a strict human pathogen responsible for various diseases, ranging from mild throat and skin infections to severe and life-threatening illnesses such as necrotizing fasciitis and toxic shock syndrome (Carapetis et al., 2005). We have demonstrated, with the aid of through-space lysine isosteres of PAM (Rios-Steiner et al., 2001; Yuan et al., 2019; Qiu et al., 2020), that the acquisition of hPg and its conversion to hPm by the endogenous GAS hPg activator, streptokinase (SK), assists GAS in the degradation of barriers to its dissemination (Vu et al., 2021). Importantly, we showed that PAM binding to K2hPg displaces the intramolecular interactions maintained by this domain, giving rise to a conformational switch that expedites streptokinase-mediated activation of hPg (Bhattacharya et al., 2014; Ayinuola et al., 2021). Likewise, the replacement of Asp219 of the anionic center of K2hPg LBS, (Ayinuola et al., 2021) while abolishing PAM-binding, produces a hPg with a relaxed conformation having a rapid and optimal activation potential (Ayinuola et al., 2021).
K2hPg is the weakest lysine-binding domain of hPg, and structural studies have shown that it does not interact with the AP domain that is classically known to maintain hPg in the closed conformation. Instead, K2hPg interacts with the SP domain, wherein Asp219 forms a salt bridge with Lys708 (Law et al., 2012). In the current study, we further investigated the significance of the interaction between Asp219 and Lys708, which is yet to be fully established, to the conformational and activation properties of hPg. Moreover, it is not yet known if the LBS of K1hPg, K4hPg, and K5hPg, in the intact hPg molecule, is important in enhancing or stabilizing the interaction between PAM and hPg. Herein, using site-directed mutagenesis of critical LBS residues, we investigated the influence of these residues on hPg conformation, activatability, and PAM-binding. These mutations also allowed us to highlight the residues of hPg involved in its interaction with SK and hPg activators, viz., urinary-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA).
Materials and methods
Construction, expression, and purification of recombinant proteins
Plasminogen
The cDNA encoding wild-type (WT) Glu1-hPg was inserted into multiple cloning sites of the Drosophila S2 parent expression plasmid, pMT-PURO, to generate hPg WT-pMT-PURO (Nilsen et al., 1999). A detailed description of the construction of the pMT-PURO plasmid has been provided elsewhere (Iwaki and Castellino, 2008). Five other variants of Glu1-hPg, viz., hPg [D139N], hPg [D413N], hPg [D518N], hPg [D139,413,518N], and hPg [K708A], were generated via primer-directed mutagenesis PCR using the primers listed in Table 1. As an example, to construct the plasmid encoding hPg [D139N], two PCR fragments were generated using hPg-pMT-PURO as a template. Primers K1f and K1LBSr generated fragment-1, while primers K1LBSf and K1r generated fragment-2. These fragments were subsequently joined in an overlapping PCR using primers K1f and K1r. All other variants, except for hPg [D139,413,518N]-pMT-Puro, where hPg [D413N]-pMT-PURO was used as the template, were similarly constructed using primers listed in Table 1. For each construct, the final PCR fragment was inserted into the pCR Blunt II TOPO vector (Invitrogen) and transformed into TOP10 Escherichia coli cells for propagation. After verifying the accuracy of the sequence, particularly the presence of the desired mutation by Sanger DNA sequencing, the hPg fragments in the PCR TOPO vectors were digested using appropriate restriction enzymes and ligated into WT-hPg-pMT-PURO, which was similarly digested. In the case of hPg [D139,413,518N], the digested fragment was ligated into hPg [D139N]-pMT-PURO. The construction of the plasmid coding for hPg [D219N] was previously reported (Ayinuola et al., 2021).
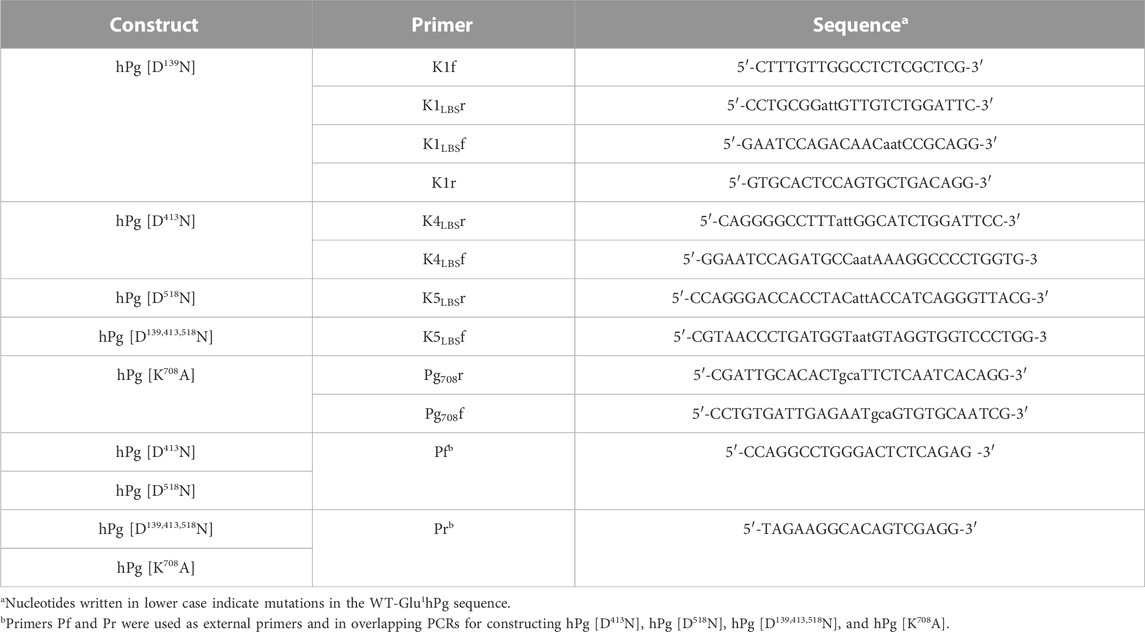
TABLE 1. Primers for construction of hPg variants.
Endotoxin-free preparations of the DNA constructs were used to transfect Drosophila S2 cells using TransIT-Insect Transfection Reagent (Mirus) with EX-CELL 420 serum-free medium (Sigma) supplemented with 10% fetal bovine serum (FBS). Cells that underwent successful transfection were selected using 10–15 μg/mL puromycin. Following selection and adaptation of the S2 cells to FBS-free medium, expression of the hPg variants was carried out in spinner flasks starting with ∼4.0 × 106 S2 cells in 200 mL of EX-CELL 420/0.1% pluronic acid/29250–39780 KIU aprotinin. Once the cell density reached ∼20 × 106, the culture volume was increased to 1 L, and the cells were allowed to grow to a density between 20–24 × 106 before the induction of hPg expression with 600 μM CuSO4. The culture was harvested 72–96 h post-induction. The supernatants, which contained the expressed proteins, were loaded onto individual lys-sepharose columns and purified as described by Deutsch and Mertz (1970). The purity of the hPg variants was ascertained by polyacrylamide gel electrophoresis, and molecular weights were estimated by the same method.
PAM and SK
The construction, expression, and purification of recombinant M-protein (PAM) and recombinant SK have been previously reported (Qiu et al., 2019; Zhang et al., 2012).
Analytical ultracentrifugation
Sedimentation coefficients (S020,w) of the hPg variants were determined by sedimentation velocity using the absorption optics (A280nm) of an analytical ultracentrifuge (Beckman Optima XL-I). The sample channels of three 2-channel centerpieces were loaded with 400 µL hPg variant at A280nm values of 0.175 (0.1 mg/mL), 0.35 (0.2 mg/mL), and 0.7 (0.4 mg/L), and the reference channels were filled with 420 μL sample buffer (50 mM sodium phosphate/100 mM NaCl, pH 7.4). The samples were subjected to high-speed centrifugation at 40,000 rpm, at 20°C, with a total of 250 scans recorded at A280nm every 3 min for ∼13 h. The data were analyzed by fitting into the continuous c(s) distribution model in Sedfit (Brown and Schuck, 2006). Protein and buffer parameters, which include partial specific volume, buffer density, and viscosity, were calculated from protein sequence and buffer composition using Sednterp (http://www.rasmb.org/sednterp). The effect of the lysine analog, ε-amino caproic acid (EACA), on S020,w of the hPg variants was similarly determined, except that the sample buffer contained, in addition to other components, 100 mM EACA.
Plasminogen activation
Activation of hPg to hPm was measured by coupling hPm formation to the hydrolysis of H-D-Val-l-Leu-l-Lys-p-nitroanilide (S2251; Chromogenix) in 10 mM Na-Hepes/150 mM NaCl, pH 7.4, at 25°C in a thermostated spectrophotometer. A typical assay mixture in the well of a 96-well microtiter plate contained 0.2 μM hPg, 0 or 0.25 μM PAM, and 0.25 mM S2251. SK (5 nM), human uPA (2.5 nM), or human tPA (5 nM) was finally added to accelerate the reaction. The hydrolysis of S2251 by hPm to generate p-nitroaniline was continuously monitored for 2 h at A405nm. The slope of the linear portion of the plot of A405nm vs. t2 (min2) was obtained using GraphPad Prism 9.0, and the initial velocity of hPm formation was calculated as described by Chibber et al. (1985).
Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were conducted in 10 mM Na-Hepes/150 mM NaCl, 3 mM EDTA/0.05% polysorbate-20, and pH 7.4 (HBS-EP+) running buffer at 25°C in a Biacore X100 (Cytiva). To determine the association (kon) and dissociation constants (koff) for the binding of the hPg variants to PAM, PAM was amine-coupled to the flow cell-2 (FC2) of a carboxymethyl dextran CM-5 chip to a response unit of 250 RU. Flow cell-1 (FC1) was treated as a blank, as described by Ayinuola et al. (2021). WT-hPg or its variants were diluted into HPS-EP+, at concentrations of 0.175–20 nM, and up to 800 nM of hPg [D219N] was injected into both FC1 and FC2 at a flow rate of 30 μL/s in a multicycle kinetic experiment with association and dissociation time set to 180 s and 300 s, respectively. The chip surface was regenerated between cycles by injecting 10 mM glycine-HCl, with pH 1.5. Signals from FC-1 were subtracted from those of FC-2, and the sensorgrams generated were analyzed by a 1:1 Langmuir binding model in BIAevaluation software version 3.0. The dissociation constant’s (KD) standard deviations were calculated from the experimental means of three independent experiments.
For the binding of hPg variants to streptokinase (SK), each hPg variant was immobilized by amine coupling on a CM-5 chip to a final response unit of ∼1200 RU, as described for PAM. Multicycle kinetic assays to determine the kon and koff values of these interactions were performed at SK concentrations ranging from 0.625 to 100 nM using HBS-EP+/50 μM NPGB (p-nitrophenyl-p’-guanidinobenzoate) as the analyte buffer. Association and dissociation times were 90 s and 120 s, respectively. Regeneration of the chip surface between cycles, and the data analysis, was performed as described for PAM.
Statistical analyses
Statistical analyses were performed by one-way analysis of variance (ANOVA) with Dunnett T3 multiple comparisons test, using GraphPad Prism 9.0. Probability values (p-values) considered significant were set at p < 0.05.
Results
Homogeneity and molecular weight of hPg variants
Four LBS variants, including hPg [D139N], hPg [D413N], hPg [D518N], and hPg [D139,413,518N], were used in this study to investigate the importance of lysine-binding kringle domains, in intact hPg, to the tight binding of hPg to PAM. In the first three variants, critical aspartic acid residues of K1hPg, K4hPg, and K5hPg were individually replaced by Asn, and all three were replaced in the last variant. We have previously shown that the replacement of Asp219 of K2hPg to provide hPg [D219N] eliminates the PAM/hPg interaction (Ayinuola et al., 2021). However, this mutation does not allow us to demonstrate whether the interaction between hPg and PAM is enhanced or stabilized by LBS of K1hPg, K4hPg, and/or K5hPg. Moreover, a fifth hPg variant, hPg [K708A], is of interest to examine the significance of the salt bridge formed between Asp219 of K2hPg and Lys708 of the SP domain in the conformation and activation of hPg. Thus, seven hPg variants, including WT-hPg and hPg [D219N] (Ayinuola et al., 2021), were used in this study.
As shown in Figure 1, all the hPg variants were purified to apparent homogeneity with yields between 25 and 55 mg/L of culture media, except for hPg [D139,413,518N], which gave a purified yield of ∼5 mg/L, primarily because of its poor affinity to sepharose-lysine. The low yield of this variant confirms that all the high-affinity LBS have been inactivated as expected. As shown in lanes 1 and 9 of Figure 1, Lys78-hPg exists at a molecular weight below that of WT-Glu1hPg due to the absence of the AP domain. Thus, it is evident that all of the LBS variants, including hPg [K708A], which migrates with relative mobility close to that of WT-hPg, exist in their Glu1-hPg form of ∼92 kDa (Ayinuola et al., 2021).
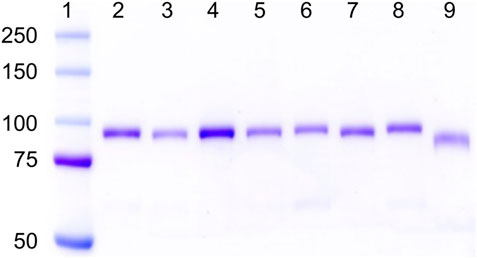
FIGURE 1. SDS-gel electrophoretograms of hPg variants on 8% Tris-glycine gel. Lane 1, pre-stained bands of protein ladder (Kaleidoscope, Bio-Rad); Lanes 2–9 show Coomassie-stained bands of hPg variants in the order of 2, WT-Glu1-hPg; 3, hPg [D139N]; 4, hPg [D219N]; 5, hPg [D413N]; 6, hPg [D518N]; 7, hPg [D139,413,518N]; 8, hPg [K708A]; and 9, Lys78-hPg.
Replacement of non-LBS Lys708 relaxes the conformation of Glu1-hPg
Since LBS regulates the conformation of hPg, all constructed variants were subjected to conformational analysis by the determination of sedimentation coefficient value, So20,w, a parameter dramatically reduced when hPg transitions from a tight closed (T) to a loose open (L) conformation (Brockway and Castellino, 1972). The results of the So20,w analyses are summarized in Table 2. As shown from the results obtained without EACA, WT-hPg and hPg [D139N] have identical So20,w values of ∼5.4 S, indicating that K1hPg may not play a major role in regulating the conformation of hPg. Other LBS variants sedimented more slowly with a reduction of ∼0.4–0.7 units in their So20,w values. The triple variant carrying a combined mutation of LBS residues in K1hPg, K4hPg, and K5hPg has the lowest So20,w value, implying that it is in a more relaxed conformation than any other variants. This is followed by hPg [D219N]. Of all the LBS variants, the replacement of Asp518 of the LBS of K5hPg resulted in the lowest degree of change in So20,w.
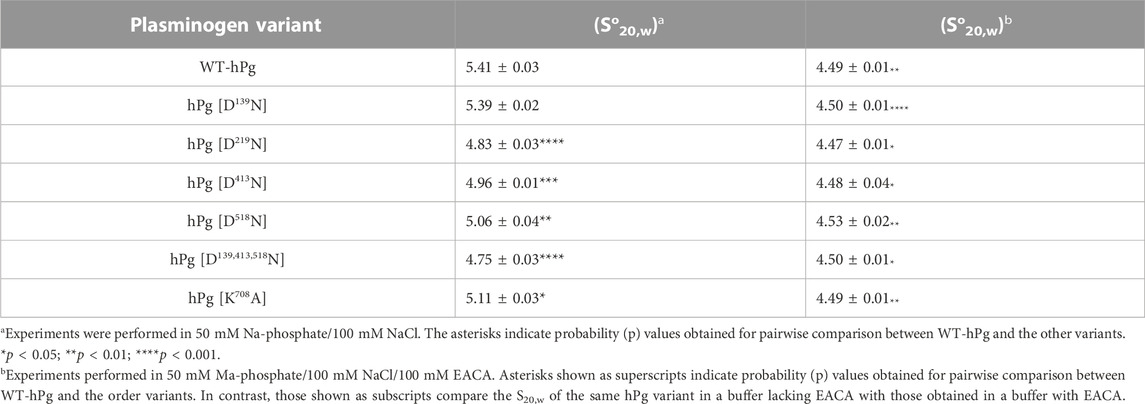
TABLE 2. So20,w values of the hPg variants.
Interestingly, the So20,w of hPg [K708A] is ∼0.3 unit lower than that of WT-hPg, demonstrating that this variant exists in a slightly more open conformation than Glu1-hPg. This suggests that the interaction between Lys708 and Asp219 modulates the hPg conformation. In the buffer containing EACA, all the hPg variants transitioned from either the closed conformation, as in hPg WT and hPg [D139N]; slightly relaxed as in hPgD518N and hPgK708A; or more relaxed as in hPgD219N, hPgD413N, and hPg D139,413,518N, to the fully relaxed and open conformation of ∼4.5 S, consistent with ∼1.0 S unit change characteristics of the transition of hPg from a closed to an open form (Brockway and Castellino, 1972).
Loss of Asp413 and Lys708 fuels rapid association of hPg and PAM
The association and dissociation curves for the binding of the hPg variants to immobilized PAM are given by the sensorgrams in Figure 2. Analyses of the sensorgrams showed that hPg variants hPg [D413N] and hPg [D139,413,518N] have ∼10X higher affinities for PAM compared to WT-hPg (Table 3). This is due to a fast on-rate upon the replacement of Asp413 of K4hPg. In addition the association rate of hPg [K708A] is 5X that of WT-hPg, leading to a ∼4-fold increase in its affinity for PAM. The replacement of LBS residues of K1hPg and K5hPg does not significantly alter the binding kinetics of hPg to PAM. Overall, the dissociation rates of the variants are about the same. These similar off-rates indicate that the altered LBS residues are not involved in stabilizing the hPg/PAM interaction.
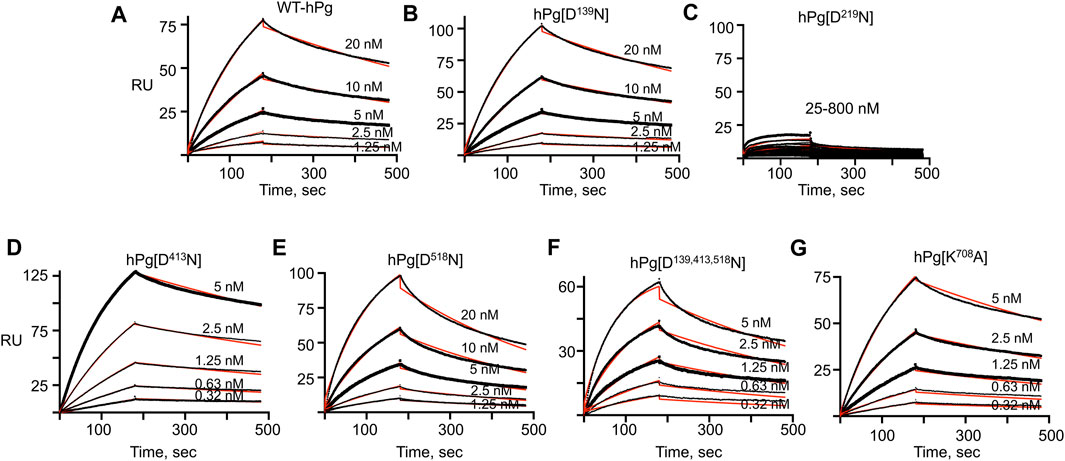
FIGURE 2. SPR sensorgrams of hPg variants’ interaction with PAM. PAM was amine-coupled to a CM-5 chip to a target level of 250 RU, and each hPg variant was injected on the chip in a multicycle kinetic experiment at the indicated concentrations. The black lines show the measured association (0–180 s) and dissociation curves (181–480 s) for the interaction of the hPg variants and PAM. The red lines show the best fit for the association and dissociation rate constants of the hPg variants to PAM using a 1:1 Langmuir model. (A) WT-hPg, (B) hPg[D139N], (C) hPg[D219N], (D) hPg[D413N], (E) hPg[D518N], (F) hPg[D139, 413, 518N] and (G) hPg[K708A].

TABLE 3. Binding constants of hPg variants/PAM as determined by SPR.
The LBS of hPg participates in functions other than maintenance of slowly activatable hPg conformation
Activation of hPg with SK
To expand the binding data obtained for the hPg/PAM interactions, we examined the stimulatory effect of PAM binding to hPg on the SK-mediated activation of the hPg variants. Consistent with the conformational differences observed between the variants, WT-hPg and hPg [D139N], both of which exist in the fully closed conformation, showed the slowest activation rates in an assay without PAM (Figure 3A). Upon the addition of PAM, the activation rates of these two variants were enhanced ∼4-fold. hPg [D518N] has a slightly relaxed conformation, which agrees with its somewhat faster activation rate than WT-hPg. As expected, based on the binding result of this variant with PAM, the addition of PAM generated more hPm per unit time. Three variants, hPg [D219N], hPg [D413N], and hPg [D139,413,518N], exist in the more relaxed conformation. Of these three variants, hPg [D219N] and hPg [D413N] showed the optimal activation rate possible with SK, and the addition of PAM did not increase their rates of activation. However, unexpectedly, the rate of hPm generation from hPg [D139,413,518N], although faster than that of WT-hPg, is inconsistent with its more relaxed conformation. Moreover, despite its tight binding to PAM, its activation rate is not responsive to the presence of PAM. Surprisingly, although hPg [K708A] shows a slightly relaxed conformation, it is optimally activatable, and PAM does not further stimulate its activation by SK. Overall, the activation profile of the hPg variants recapitulates the mechanism by which PAM enhances the rate of hPg activation in that PAM produces a conformational switch upon binding to hPg. hPg [D219N], hPg [D413N], and hPg [K708A] are already in the optimally activatable conformations required by SK, plausibly due to the activation loop that harbors the R561-V562 activation site being readily exposed in these conformations. Hence, the lack of binding of PAM to hPg [D219N] or its binding to hPg [D413N] and hPg [K708A] may not necessarily overcome additional conformational barriers to activating hPg.
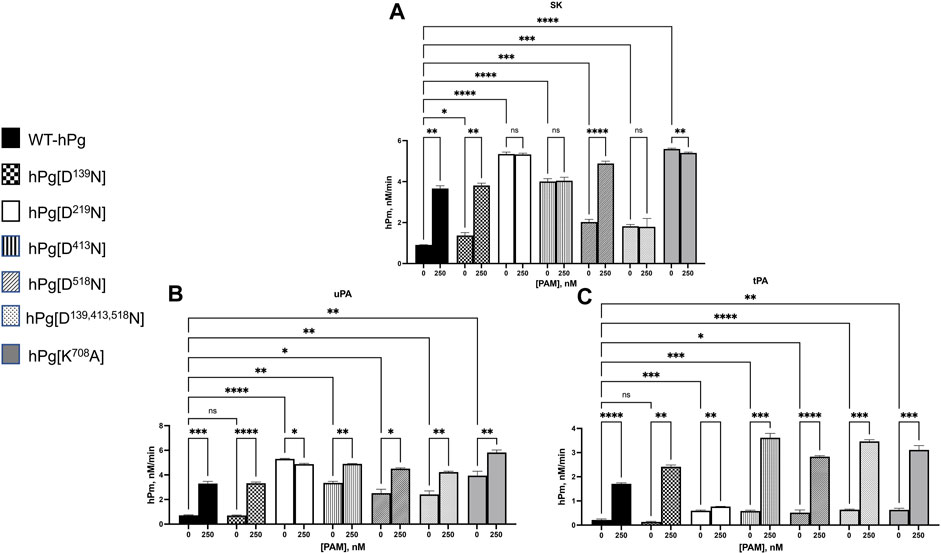
FIGURE 3. Activation of hPg variants by plasminogen activators at 25°C. Activation of hPg was coupled to the cleavage of chromogenic substrate S2251 (H-D-Val-L-Leu-L-Lys-p-nitroanilide) in 50 mM Na+-Hepes/100 mM NaCl, pH 7.4. Each assay mixture contained 200 nM hPg variant and 0.25 mM S2251, 0 or 250 nM PAM, and (A) 5 nM SK as the hPg activator; (B) 2.5 nM uPA as the hPg activator; and (C) 5 nM tPA as the hPg activator. The generation of p-nitroaniline from S2251 by the generated hPm was continuously measured at A405nm for 90 min. The rate function of hPg activation was calculated as the slope of the linear portions of A405nm vs. t2. Probability values obtained from pairwise comparisons between WT-hPg and the other variants at 0 nM and between each hPg variant at 0 and 250 nM PAM are shown. Here, * is p < 0.05, ** is p < 0.01, *** is p < 0.001, **** is p < 0.0001, and ns is not significant.
Activation by hPg activators, uPA and tPA
While it is clear from the AUC data, the SPR- and SK-mediated activation results show that individual mutations of LBS residues of K1hPg, K4hPg, and K5hPg do not negatively impact the hPg/PAM interaction. However, it is not clear why a combination of these mutations is less favorable to SK-mediated activation of hPg [D139,413,518N]. To better characterize this variant, we evaluated the activation rates of all the hPg variants using uPA and tPA as activators. Human uPA and tPA serve as valuable tools in this instance because they are serine proteases, and, unlike SK, they directly cleave the scissile R561-V562 bond in the activation loop of hPg. On the other hand, SK has no intrinsic proteolytic activity but functions similarly to a coenzyme forming a complex with hPg and generates protease activity. The SK-hPg complex, in turn, activates free hPg. Thus, mutations that diminish initial complex formation will hinder hPg activation.
The results for the uPA activation for WT-hPg, hPg [D139N], hPg [D219N], and hPg [D518N] (Figure 3B) followed the same trend observed using SK as the activator. However, the activation rates of hPg [D413N] and hPg [K708A], while still rapid, unlike the SK-mediated reactions, were further enhanced by the addition of PAM. Moreover, the rate of activation of hPg [D139,413,518N] is only slightly lower than those of hPg [D413N] and hPg [K708A] and is stimulated by PAM.
For tPA as an activator, Figure 3C shows that in assay mixtures devoid of PAM, tPA does not discriminate between a slight or a more relaxed hPg conformation. A small alteration that slightly opens the hPg molecule places hPg [D219N], hPg [D413N], hPg [D518N], hPg [D139,413,518N], and hPg [K708A] at the same activation potential with activation rates ∼3x faster than those of WT-hPg and hPg [D139N]. Upon the addition of PAM, the activation rates of all the hPg variants were further enhanced, except for hPg [D219N].
Largely, the activation of the hPg variants suggests that the replacement of the LBS residue of K5hPg, or the simultaneous replacement of LBS residues of K1hPg, K4hPg, and K5hPg, inhibits complex formation between SK and hPg. Moreover, the activation profiles highlight the difference in the mechanism of SK, uPA, and tPA activation.
To gain further insight into the reasons for the less favorable activation of hPg [D139,413,518N] compared to its other conformationally more relaxed counterparts, we determined the dissociation constants, KD, for the binding of the hPg variants to SK by SPR. The average KD value for the interaction is ∼0.3 nM for all the variants except for hPg [D139,413,518N], the KD of which 2.1 nM (Figure 4; Table 4), reflecting a ∼7X slower association rate with SK. Notably, none of the mutations created in the hPg variants altered the koff. Herein, it is evident that a key reason for the retarded activation of hPg D139,413,518N is the slow rate of forming the initial activator complex.
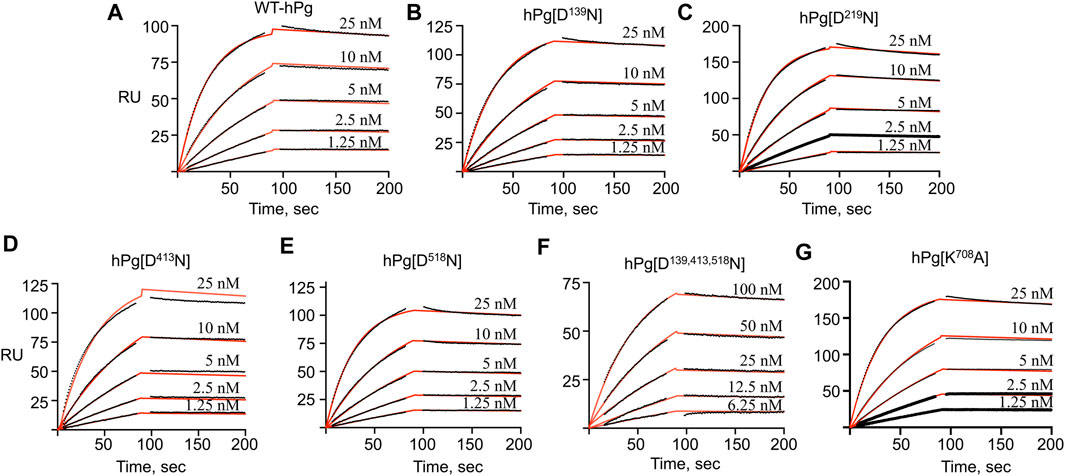
FIGURE 4. SPR sensorgrams for the binding of hPg variants to SK. hPg and hPg variants were amine-coupled to a CM-5 chip, and SK was injected on each chip at the indicated concentrations. The black lines showed the measured association (0–90 s) and dissociation (91–210 s) curves of the SK interaction with hPg. p-NPGB was added to prevent activation of the hPg bound to SK. Association and dissociation rate constants were obtained by fitting the data using a 1:1 Langmuir model, as shown by the red lines. (A) WT-hPg, (B) hPg[N139D], (C) hPg[D219N], (D) hPg[D413N], (E) hPg[D518N], (F) hPg[D139, 413, 518N] and (G) hPg[K708A].
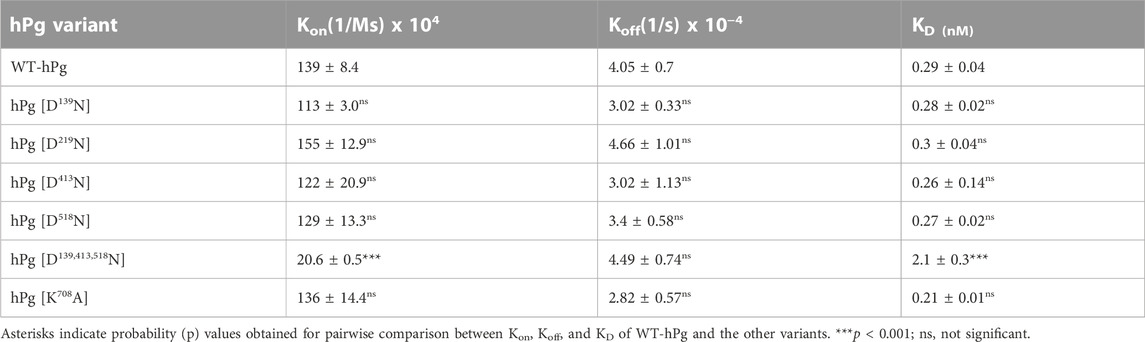
TABLE 4. Binding constants of hPg variants to SK as determined by SPR.
Discussion
The binding of hPg to cellular receptors plays a vital role in modulating hPg activation, chiefly due to conformational changes in hPg that accompany the binding events. Such conformational alterations are induced by competitive binding of the C-terminal lysine or lysine isosteres to the LBS of hPg, displacing the intramolecular interactions offered by the LBS that keep hPg in a tight activation-resistant conformation. Many pathogenic bacteria employ surface-associated hPg receptors to immobilize hPg on the cell surface. The multifunctional M-protein of group A S. pyogenes (GAS) functions in this capacity to indirectly or directly capture hPg on the GAS surface. Indirect hPg binding is achieved through initial high-affinity binding to fibrinogen, which in turn binds hPg (Herwald et al., 2004; Walker et al., 2005; Smeesters et al., 2010a; Smeesters et al., 2010b; Qiu et al., 2018). Direct hPg binding is mediated by the subclass of M-proteins called PAM, expressed only in pattern D skin-trophic GAS strains (Berge and Sjobring, 1993; Wistedt et al., 1995). PAM binds hPg with the aid of lysine isosteres, interacting with critical residues of K2hPg LBS (Rios-Steiner et al., 2001; Yuan et al., 2019). Binding experiments using individual kringle domains showed that K1hPg, K4hPg, and K5hPg do not participate in PAM/hPg interaction (Wistedt et al., 1998). In the current study, we explore the contributions of these kringle domains to hPg binding in the context of an intact hPg molecule. Our present study showed that hPg variants lacking critical aspartate residues of the anionic loci of K1hPg, K4hPg, and K5hPg bind tightly to PAM with very low nanomolar KD values, reiterating the non-involvement of these LBS in hPg binding by PAM. However, the observation that the replacements of Asp413, Lys708, and a combination of Asp413 and Asp518 produced conformational changes that allow rapid association of PAM and hPg suggests, on one hand, that the opening of the closed hPg conformation exposes residue exosites of K2hPg that facilitates the interaction. On the other hand, the data suggest that steric hindrance to K2hPg exists in the closed hPg conformation and that K2hPg is readily exposed for rapid PAM interaction in the opened hPg conformation.
In line with conformational perturbations that ensue following the interaction of hPg LBS with receptors, we demonstrated that the replacement of critical aspartate residues in homologous locations of K1hPg, K4hPg, and K5hPg, similar to our previous report on hPg [D219N] (K2hPg variant), deprived hPg of the critical anionic residue required to maintain its closed conformation in hPg [D413N] (K4hPg variant) and hPg [D518N] (K5hPg variant). However, hPg [D139N] (K1hPg variant) maintained a tight activation-resistant conformation regardless of the mutation introduced. These findings are consistent with the X-ray crystal structural study of hPg in which only K1hPg was shown to be positioned away from the hPg core, having no interaction with the AP or SP domain (Law et al., 2012). The implication is that although K1hPg has the highest lysine-binding affinity, it does not contribute to conformational changes that occur in hPg. Thus, receptors that strictly depend on K1hPg for interaction with hPg would not offer any stimulatory effect to hPg activation. This argument aligns with the reports that saturation of the high-affinity binding site of hPg by EACA or tranexamic acid does not induce a significant conformational change in hPg (Markus et al., 1978a; Markus et al., 1978b; Markus et al., 1979). An appreciable degree of conformational change was observed in hPg [D518N]. However, this is a slight shift in the conformation of hPg compared to the changes observed in hPg [D219N] and hPg [D413N]. Furthermore, the conformational change in hPg [D518N] only led to a 2X increase in hPg activation potential by SK, unlike a 4–5-fold increase observed upon the addition of PAM and in hPg [D219N] and hPg [D413N]. K5hPg has been shown to interact solely with the AP domain of hPg (Law et al., 2012). Moreover, a biophysical study indicated that benzamidine, a ligand that binds only at K5hPg LBS but not at K1hPg–K4hPg, does not induce the formation of the fully extended hPg conformation (Marshall et al., 1994). Consistent with these reports, our current findings confirm that K5hPg LBS is not involved in an intramolecular interaction that causes a global conformational change within hPg upon its removal. This signifies that the conformational change in hPg [D518N] does not influence the SP domain in a manner that significantly exposes the activation loop of hPg. However, the conformational impediments afforded by the interaction between the non-K5hPg LBS and SP domain to the activation of hPg [D518N] were overcome when PAM was added to its SK-catalyzed activation reaction mixture. This behavior of hPg [D518N] demonstrates why hPg [D219N], hPgD413N, and hP [gK708A] are readily activatable by SK, as explained below.
In the crystal structure of hPg, other than the interaction of Asp413 of K4hPg with Arg68 and Lys70 of the AP domain, an interface formed between K4hPg, the activation loop, and the SP domain positioned the inter-kringle loop of K3hPg and K4hPg in a manner that blocked the activation sequence (Law et al., 2012). Thus, the replacement of Asp413, as observed in hPg [D413N], will eliminate its interaction with the AP domain and plausibly alter the position of K3hPg/K4hPg inter-kringle loop, leading to rapid activation of hPg. In the case of K2hPg, two loops in the SP domain extensively interact with K2hPg. Moreover, Cl− coordinates multiple interactions between K2hPg and SP domain, and a salt bridge exists between Lys708 of the SP domain and Asp219/Glu221 of K2hPg LBS (Urano et al., 1987b; Law et al., 2012). These properties of K2hPg, highlighted by the crystal structure, agree with our findings that of all the replacements involving a single LBS residue, the substitution of Asp219 generated the largest conformational change that accelerated hPg activation. Thus, the elimination of the K2hPg/SP domain interaction appears to be the most critical event that optimally exposes the Arg561-Val562 activation site of hPg. This is especially evident in hPg [K708A], a variant exhibiting a minimal conformational change but optimally activatable by all hPg activators. First, the results of hPg [K708A] indicate that the extent of conformational change, even though necessary for rapid hPg activation, is not as significant as a change that exposes the activation loop. Second, it suggests that interactions between K2hPg and the SP domain are the most critical interactions that regulate hPg activation, and PAM-binding disrupts these interactions. Finally, the association rate for the binding of PAM to the hPg variants combined with the activation of hPg by SK suggests that the position of K2hPg is likely altered in hPg [D219N], hPg [D413N], and hPg [K708A], such as to expose the activation loop. Consequently, the binding of PAM to these variants will be redundant for the rate enhancement of hPg activation. A significant increase in the activation rates of these hPg variants observed upon the addition of PAM to their tPA-catalyzed reactions suggests that the presence of intact K2hPg LBS in hPg [D413N] and hPg [K708A] enabled hPg-PAM complex formation and, by extension, ternary complexes of hPg-PAM-tPA, which alters the catalysis by tPA.
Of all the hPg variants used in this study, hPg [D139,413,518N] exhibits the largest shift in So20,W value, implying that it exists in the most relaxed conformation. It would be expected that the activation rate of this variant will be similar to that of hPg [D413N]. However, the activation rate is indeed slower. A simple explanation for this is that the activation rate of hPg [D139,413,518N] reflects an interplay between an open conformation of hPg, wherein the activation loop is exposed, and the loss of essential residues of the LBS. Thus, even though the conformation is relaxed, for SK, the initial event of rapid complex formation is affected by the absence of important LBS residues. This is clearly seen in the SPR binding data of this variant with SK. In addition, we found that, unlike uPA- and tPA-catalyzed reactions, the lysine analog, EACA, would not stimulate the activation of hPg by SK. However, if a preformed complex of SK-hPg is used as the activator, EACA does stimulate the activation. This is consistent with earlier reports that EACA potently inhibits the binding of SK to hPg (Conejero-Lara et al., 1998; Lin et al., 2000), thereby strengthening the point that the slow activation rate of hPg [D139,413,518N] by SK is due to the inactivation of high-affinity LBS. The modulatory effect of the kringle domains of hPg on the interaction between hPg and SK was previously demonstrated by a binding saturation experiment, which revealed that both Glu1-hPg and mini-hPg (hPg devoid of K1hPg—K4hPg) possess equal binding affinity of ∼0.5 nM for SK, a value that is two orders of magnitude lower than that obtained with micro-hPg (hPg devoid of all five kringle domains). Therefore, it was suggested that K5hPg is the only kringle domain required for low nanomolar high-affinity interaction of hPg and SK (Lin et al., 2000). However, our current data showed that inactivation of either K1hPg, K4hPg, or K5hPg through the individual replacement of Asp139, Asp413, or Asp518 did not affect the interaction between hPg and SK, suggesting that in the absence of one high-affinity LBS, another can function to enable rapid interaction of SK and hPg. Based on a KD of 2.1 nM obtained for the interaction of SK and hPg [D139,413,518N], the activation mixture should contain ∼70% activator complex, albeit the observed rate is slower than expected for a 70% complex. Perhaps the replaced LBS residues are also involved in the interaction of the activator complex with the hPg substrate, consistent with enzyme kinetics studies which showed that lysine, EACA, and tranexamic acid are competitive inhibitors of hPg activation by uPA (Peltz et al., 1982; Wu et al., 2019). Hence, apart from its importance in the formation of SK/hPg activator complex, the LBSs seem to play an important role in the formation of enzyme-substrate complexes between plasminogen activators and hPg.
In summary, we show in the current study that K1hPg, K4hPg, and K5hPg do not directly influence the high-affinity interaction of full-length hPg with PAM, but conformational perturbations that relax the hPg molecule facilitate rapid binding with PAM. Inactivation of the LBS of K1hPg does not alter hPg conformation, and the loss of the K5hPg LBS does not induce a global conformational change in hPg. Substitution of critical K4hPg and K2hPg LBS residues places hPg at optimal activation potential, rapidly activatable by all hPg activators. However, the single replacement of Asp219 of K2hPg not only resulted in the largest conformational change of all the inactivated LBS, but the substitution of Lys708, a binding partner of Asp219, sufficiently rendered hPg optimally activatable. Thus, the order of increasing influence of LBS of the hPg kringle domains on its conformation and activatability is K1hPg < K5hPg < K4hPg < K2hPg. In conclusion, beyond a more relaxed extended hPg conformation, exposure of the activation loop, alongside the presence of appropriate LBS residues, is a prerequisite for rapid conversion of hPg to hPm by plasminogen activators.
Data availability statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Author contributions
YA performed experiments, generated the data, and wrote the initial draft of the manuscript; FC conceptualized the study and wrote the final draft manuscript.
Funding
These studies were supported by grant HL013423.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andreasen, P. A., Egelund, R., and Petersen, H. H. (2000). The plasminogen activation system in tumor growth, invasion, and metastasis. Cell. Mol. Life Sci. 57 (1), 25–40. doi:10.1007/s000180050497
Ayinuola, Y. A., Brito-Robinson, T., Ayinuola, O., Beck, J. E., Cruz-Topete, D., and Lee, S. W. (2021). Streptococcus co-opts a conformational lock in human plasminogen to facilitate streptokinase cleavage and bacterial virulence. J. Biol. Chem 296, 100099. doi:10.1074/jbc.RA120.016262
Berge, A., and Sjobring, U. (1993). PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem. 268 (34), 25417–25424. doi:10.1016/S0021-9258(19)74408-1
Bhattacharya, S., Ploplis, V. A., and Castellino, F. J. (2012). Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J. Biomed. Biotechnol. 2012, 482096. doi:10.1155/2012/482096
Bhattacharya, S., Liang, Z., Quek, A. J., Ploplis, V. A., Law, R., and Castellino, F., J. (2014). Dimerization is not a determining factor for functional high affinity human plasminogen binding by the group A streptococcal virulence factor PAM and is mediated by specific residues within the PAM a1a2 domain. J. Biol. Chem. 289 (31), 21684–21693. doi:10.1074/jbc.M114.570218
Brockway, W. J., and Castellino, F. J. (1972). Measurement of the binding of antifibrinolytic amino acids to various plasminogens. Arch. Biochem. Biophys. 151 (1), 194–199. doi:10.1016/0003-9861(72)90488-2
Brown, P. H., and Schuck, P. (2006). Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90 (12), 4651–4661. doi:10.1529/biophysj.106.081372
Carapetis, J. R., Steer, A. C., Mulholland, E. K., and Weber, M. (2005). The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5 (11), 685–694. doi:10.1016/S1473-3099(05)70267-X
Chibber, B. A. K., Morris, J. P., and Castellino, F. J. (1985). Effects of human fibrinogen and its cleavage products on activation of human plasminogen by streptokinase. Biochemistry 24 (14), 3429–3434. doi:10.1021/bi00335a006
Conejero-Lara, F., Parrado, J., Azuaga, A. I., Dobson, C. M., and Ponting, C. P. (1998). Analysis of the interactions between streptokinase domains and human plasminogen. Prot. Sci. 7 (10), 2190–2199. doi:10.1002/pro.5560071017
Deutsch, D. G., and Mertz, E. T. (1970). Plasminogen: Purification from human plasma by affinity chromatography. Science 170, 1095–1096. doi:10.1126/science.170.3962.1095
Herwald, H., Cramer, H., Morgelin, M., Russell, W., Sollenberg, U., Norrby-Teglund, A., et al. (2004). M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 116 (3), 367–379. doi:10.1016/s0092-8674(04)00057-1
Iwaki, T., and Castellino, F. J. (2008). A single plasmid transfection that offers a significant advantage associated with puromycin selection in Drosophila Schneider S2 cells expressing heterologous proteins. Cytotechnology 5 (1), 45–49. doi:10.1007/s10616-008-9129-0
Law, R. H. P., Caradoc-Davies, T., Cowieson, N., Horvath, A. J., Quek, A. J., Encarnacao, J. A., et al. (2012). The X-ray crystal structure of full-length human plasminogen. Cell. Rep. 1 (3), 185–190. doi:10.1016/j.celrep.2012.02.012
Lin, L. F., Houng, A. Y., and Reed, G. L. (2000). Epsilon amino caproic acid inhibits streptokinase-plasminogen activator complex formation and substrate binding through kringle-dependent mechanisms. Biochemistry 39 (16), 4740–4745. doi:10.1021/bi992028x
Markus, G., Evers, J. L., and Hobika, G. H. (1976). Activator activities of the transient forms of the human plasminogen-streptokinase complex during its proteolytic conversion to the stable activator complex. J. Biol. Chem. 251 (21), 6495–6504. doi:10.1016/S0021-9258(17)32975-7
Markus, G., DePasquale, J. L., and Wissler, F. C. (1978a). Quantitative determination of the binding of epsilon-aminocaproic acid to native plasminogen. J. Biol. Chem. 253, 727–732. doi:10.1016/S0021-9258(17)38163-2
Markus, G., Evers, J. L., and Hobika, G. (1978b). Comparison of some properties of native (glu) and modified (lys) human plasminogen. J. Biol. Chem. 253, 733–739. doi:10.1016/s0021-9258(17)38164-4
Markus, G., Priore, R. L., and Wissler, F. C. (1979). The binding of tranexamic acid to native (Glu) and modified (Lys) human plasminogen and its effect on conformation. J. Biol. Chem. 254 (4), 1211–1216. doi:10.1016/S0021-9258(17)34189-3
Marshall, J. M., Brown, A. J., and Ponting, C. P. (1994). Conformational studies of human plasminogen and plasminogen fragments: Evidence for a novel third conformation of plasminogen. Biochemistry 33 (12), 3599–3606. doi:10.1021/bi00178a017
Marti, D., Schaller, J., Ochensberger, B., and Rickli, E. E. (1994). Expression, purification and characterization of the recombinant kringle 2 and kringle 3 domains of human plasminogen and analysis of their binding affinity for omega-aminocarboxylic acids. Eur. J. Biochem. 219 (1-2), 455–462. doi:10.1111/j.1432-1033.1994.tb19959.x
McCance, S. G., and Castellino, F. J. (1995). Contributions of individual kringle domains toward maintenance of the chloride-induced tight conformation of human glutamic acid-1 plasminogen. Biochemistry 34 (29), 9581–9586. doi:10.1021/bi00029a035
McCance, S. G., Menhart, N., and Castellino, F. J. (1994). Amino acid residues of the kringle-4 and kringle-5 domains of human plasminogen that stabilize their interactions with omega-amino acid ligands. J. Biol. Chem. 269 (51), 32405–32410. doi:10.1016/S0021-9258(18)31649-1
Miles, L. A., and Parmer, R. J. (2013). Plasminogen receptors: The first quarter century. Semin. Thromb. Hemost. 39 (4), 329–337. doi:10.1055/s-0033-1334483
Miles, L. A., Hawley, S. B., Baik, N., Andronicos, N. M., Castellino, F. J., and Parmer, R. J. (2005). Plasminogen receptors: The sine qua non of cell surface plasminogen activation. Front. Biosci. 10, 1754–1762. doi:10.2741/1658
Nilsen, S. L., Miele, R., and Castellino, F. J. (1999). Expression of human plasminogen in Drosophila Schneider S2 cells. Prot. Express. Purif. 16 (1), 136–143. doi:10.1006/prep.1999.1045
Peltz, S. W., Hardt, T. A., and Mangel, W. F. (1982). Positive regulation of activation of plasminogen by urokinase: Differences in km for (glutamic acid)-plasminogen and lysine-plasminogen and effect of certain alpha, omega-amino acids. Biochemistry 21 (11), 2798–2804. doi:10.1021/bi00540a035
Plow, E. F., Doeuvre, L., and Das, R. (2012). So many plasminogen receptors: Why? J. Biomed. Biotechnol. 2012, 141806. doi:10.1155/2012/141806
Qiu, C., Yuan, Y., Zajicek, J., Liang, Z., Balsara, R. D., Brito-Robinson, T., et al. (2018). Contributions of different modules of the plasminogen-binding Streptococcus pyogenes M-protein that mediate its functional dimerization. J. Struct. Biol. 204 (2), 151–164. doi:10.1016/j.jsb.2018.07.017
Qiu, C., Yuan, Y., Liang, Z., Balsara, R. D., Lee, S. W., Ploplis, V. A., et al. (2019). Variations in the secondary structures of PAM proteins influence their binding affinities to human plasminogen. J. Struct. Biol. 206 (2), 193–203. doi:10.1016/j.jsb.2019.03.003
Qiu, C., Yuan, Y., Lee, S. W., Ploplis, V. A., and Castellino, F. J. (2020). A local alpha-helix drives structural evolution of streptococcal M-protein affinity for host human plasminogen. Biochem. J. 477 (9), 1613–1630. doi:10.1042/BCJ20200197
Rios-Steiner, J. L., Schenone, M., Mochalkin, I., Tulinsky, A., and Castellino, F. J. (2001). Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J. Mol. Biol. 308 (4), 705–719. doi:10.1006/jmbi.2001.4646
Smeesters, P. R., Drèze, P. A., Perez-Morga, D., Biarent, D., Van Melderen, L., and Vergison, A. (2010a). Group A Streptococcus virulence and host factors in two toddlers with rheumatic fever following toxic shock syndrome. Int. J. Infect. Dis. 14 (5), e403–e409. oi:. doi:10.1016/j.ijid.2009.06.025
Smeesters, P. R., McMillan, D. J., and Sriprakash, K. S. (2010b). The streptococcal M protein: A highly versatile molecule. Trends Microbiol. 18 (6), 275–282. doi:10.1016/j.tim.2010.02.007
Urano, T., Chibber, B. A. K., and Castellino, F. J. (1987a). The reciprocal effects of epsilon-aminohexanoic acid and chloride ion on the activation of human [Glu1]plasminogen by human urokinase. Proc. Natl. Acad. Sci. U. S. A. 84 (12), 4031–4034. doi:10.1073/pnas.84.12.4031
Urano, T., De Serrano, V. S., Chibber, B. A. K., and Castellino, F. J. (1987b). The control of the urokinase-catalyzed activation of human glutamic acid 1-plasminogen by positive and negative effectors. J. Biol. Chem. 262, 15959–15964. doi:10.1016/s0021-9258(18)47682-x
Urano, T., De Serrano, V. S., Gaffney, P. J., and Castellino, F. J. (1988). Effectors of the activation of human [Glu1]plasminogen by human tissue plasminogen activator. Biochemistry 27, 6522–6528. doi:10.1021/bi00417a049
Urano, T., Castellino, F. J., and Suzuki, Y. (2018). Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 16, 1487–1497. doi:10.1111/jth.14157
Vu, H. M., Hammers, D. E., Liang, Z., Nguyen, G. L., Benz, M. E., Moran, T. E., et al. (2021). Group A Streptococcus-induced activation of human plasminogen is required for keratinocyte wound retraction and rapid clot dissolution. Front. Cardiovasc. Med. 8 (8), 667554. doi:10.3389/fcvm.2021.667554
Walker, M. J., McArthur, J. D., McKay, F., and Ranson, M. (2005). Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 13 (7), 308–313. doi:10.1016/j.tim.2005.05.006
Wistedt, A. C., Ringdahl, U., Muller-Esterl, W., and Sjobring, U. (1995). Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol. Microbiol. 18 (3), 569–578. doi:10.1111/j.1365-2958.1995.mmi_18030569.x
Wistedt, A. C., Kotarsky, H., Marti, D., Ringdahl, U., Castellino, F. J., Schaller, J., et al. (1998). Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J. Biol. Chem. 273 (18), 24420–24424. doi:10.1074/jbc.273.38.24420
Wong, A. P., Cortez, S. L., and Baricos, W. H. (1992). Role of plasmin and gelatinase in extracellular matrix degradation by cultured rat mesangial cells. Am. J. Physiol. 236 (6), F1112–F1118. doi:10.1152/ajprenal.1992.263.6.F1112
Wu, Y. P., Siao, C. J., Lu, W. Q., Sung, T. C., Frohman, M. A., Milev, P., et al. (2000). The tissue plasminogen activator (TPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J. Cell. Biol. 148 (6), 1295–1304. doi:10.1083/jcb.148.6.1295
Wu, G., Mazzitelli, B. A., Quek, A. J., Veldman, M. J., Conroy, P. J., Caradoc-Davies, T. T., et al. (2019). Tranexamic acid is an active site inhibitor of urokinase plasminogen activator. Blood Adv. 3 (5), 729–733. doi:10.1182/bloodadvances.2018025429
Yuan, Y., Ayinuola, Y. A., Singh, D., Ayinuola, O., Mayfield, J. A., Quek, A., et al. (2019). Solution structural model of the complex of the binding regions of human plasminogen with its M-protein receptor from Streptococcus pyogenes. J. Struct. Biol. 208 (1), 18–29. doi:10.1016/j.jsb.2019.07.005
Zhang, Y., Liang, Z., Hsueh, H. T., Ploplis, V. A., and Castellino, F. J. (2012). Characterization of streptokinases from Group A Streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M-protein and cluster 2b streptokinase. J. Biol. Chem. 287 (50), 42093–42103. doi:10.1074/jbc.M112.417808
Keywords: plasminogen, plasminogen activation, streptococcal M-protein, streptokinase, protein mutagenesis, bacterial M-protein, Streptococcus pyogenes
Citation: Ayinuola YA and Castellino FJ (2023) Inactivation of the lysine binding sites of human plasminogen (hPg) reveals novel structural requirements for the tight hPg conformation, M-protein binding, and rapid activation. Front. Mol. Biosci. 10:1166155. doi: 10.3389/fmolb.2023.1166155
Received: 14 February 2023; Accepted: 16 March 2023;
Published: 04 April 2023.
Edited by:
Dmitriy Minond, Nova Southeastern University, United StatesReviewed by:
Ruby Hong Ping Law, Monash University, AustraliaIngrid Verhamme, Vanderbilt University Medical Center, United States
Copyright © 2023 Ayinuola and Castellino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francis J. Castellino, Fcastell@nd.edu