A Novel NAD Signaling Mechanism in Axon Degeneration and its Relationship to Innate Immunity
 Eleanor L. Hopkins
Eleanor L. Hopkins Weixi Gu
Weixi Gu Bostjan Kobe2
Bostjan Kobe2  Michael P. Coleman
Michael P. Coleman- 1John van Geest Centre for Brain Repair, Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 2School of Chemistry and Molecular Biosciences, Institute for Molecular Bioscience and Australian Infectious Diseases Research Centre, The University of Queensland, Brisbane, QLD, Australia
Axon degeneration represents a pathological feature of many neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease where axons die before the neuronal soma, and axonopathies, such as Charcot-Marie-Tooth disease and hereditary spastic paraplegia. Over the last two decades, it has slowly emerged that a central signaling pathway forms the basis of this process in many circumstances. This is an axonal NAD-related signaling mechanism mainly regulated by the two key proteins with opposing roles: the NAD-synthesizing enzyme NMNAT2, and SARM1, a protein with NADase and related activities. The crosstalk between the axon survival factor NMNAT2 and pro-degenerative factor SARM1 has been extensively characterized and plays an essential role in maintaining the axon integrity. This pathway can be activated in necroptosis and in genetic, toxic or metabolic disorders, physical injury and neuroinflammation, all leading to axon pathology. SARM1 is also known to be involved in regulating innate immunity, potentially linking axon degeneration to the response to pathogens and intercellular signaling. Understanding this NAD-related signaling mechanism enhances our understanding of the process of axon degeneration and enables a path to the development of drugs for a wide range of neurodegenerative diseases.
Introduction
Neurons are the longest cells in the body; this structure lends itself to their function of transmitting signals around the body to react to changing stimuli, but their length leaves them vulnerable to injury and other stresses. As they cannot rely solely on diffusion of cell survival factors over distances up to 1 m, they have to actively transport a multitude of molecules along their axons in an energy-intensive process (De Vos et al., 2008). Changes in metabolism can disrupt axon transport and lead to degeneration, as can physical injury such as axon transection or crush (Coleman, 2005). Chemicals such as vincristine and paclitaxel are also known to induce degeneration by inhibiting axonal transport, limiting their use as cancer chemotherapeutics (Lapointe et al., 2013; Turkiew et al., 2017; Geisler et al., 2019a). Recently, it has been shown that neuroinflammation and necroptosis can also induce axon degeneration (Ko et al., 2020).
The first stage of many neurodegenerative diseases is axon degeneration, precisely because of their vulnerability to damage (Stokin et al., 2005; Cheng et al., 2010; Loring and Thompson, 2020). Diseases involving axon loss, such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) (van Rheenen et al., 2016) and peripheral neuropathies, contribute greatly to global disease burden, and are examples of such diseases where axon degeneration is involved in disease progression (Stokin et al., 2005; Geisler et al., 2016; Coleman and Höke, 2020).
We review here the well-understood and widely-occurring mechanism, programmed axon degeneration (or Wallerian degeneration), in which NAD (nicotinamide adenine dinucleotide) and related biology are known to play a central role. A large number of disease models in animals and cell culture, and a growing list of human disorders involve axon loss through this mechanism (Conforti et al., 2014; Coleman and Höke, 2020; Loreto et al., 2020b).
NAD is a molecule found in all the kingdoms of life. It exists in either oxidized (NAD+, electron acceptor) or reduced form (NADH, electron donor). Under physiological conditions, the cytoplasmic NAD+-to-NADH ratio is from 1 to 700, while in mitochondria, this ratio is maintained at 7–8 (Ying, 2008). In cells, NAD is dynamically regulated through continuous biosynthesis, consumption and recycling. In mammals, NAD is synthesized from three main pathways. The de novo, Preiss-Handler and salvage pathways use tryptophan, nicotinic acid (NA) and nicotinamide (Nam), respectively to generate NAD. NAD-consuming enzymes include sirtuins, PARPs (poly(ADP–ribose) polymerases), CD38 (cluster of differention 38) (Navarro et al., 2021) and SARM1 (sterile alpha and Toll/interleukin-1 receptor motif-containing 1) (Essuman et al., 2017). NAD is recognized for its ability to carry electrons in redox reactions (Warburg and Christian, 1936; Nikiforov et al., 2015), and plays an important role in various physiological processes including energy metabolism, DNA repair and transcriptional regulation (Ziegler and Oei, 2001; Luo and Kraus, 2012; Cantó et al., 2015), and in pathological processes associated with neurodegeneration, cancer and inflammation (Demarest et al., 2019; Lautrup et al., 2019). Emerging evidence reveals that NAD depletion is found during mitochondrial dysfunction, impaired DNA repair and inflammation, while an NAD increase enhances metabolic fitness (Lautrup et al., 2019).
NAD links two key players in the programmed axon degeneration pathway: the proteins NMNAT2 (nicotinamide mononucleotide adenylyltransferase 2) catalyzing the synthesis of NAD from NMN (nicotinamide mononucleotide) and ATP (adenosine triphosphate), and SARM1 whose multiple activities include hydrolysis of NAD into Nam and ADPR (adenosine diphosphate ribose), cyclization of NAD into cADPR (cyclic ADPR) and base exchange of the nicotinamide group for free bases (Figure 1) (Conforti et al., 2014; Zhao et al., 2019; Coleman and Höke, 2020). The activity of NMNAT2 is essential for axonal survival, while SARM1 activation can kill most cells but is blocked in healthy axons (Gilley and Coleman, 2010; Osterloh et al., 2012; Gerdts et al., 2013; Gilley et al., 2015). NAD also limits SARM1’s activity through blocking its activation (Gilley and Coleman, 2010; Essuman et al., 2017; Jiang et al., 2020; Sporny et al., 2020; Figley et al., 2021). Recently, NMN has been shown to be an activator of SARM1, indicating that NMNAT2 has a dual action in preventing SARM1 activation by converting an activator, NMN, into a molecule that opposes activation, NAD (Di Stefano et al., 2015; Zhao et al., 2019; Bratkowski et al., 2020; Figley et al., 2021).
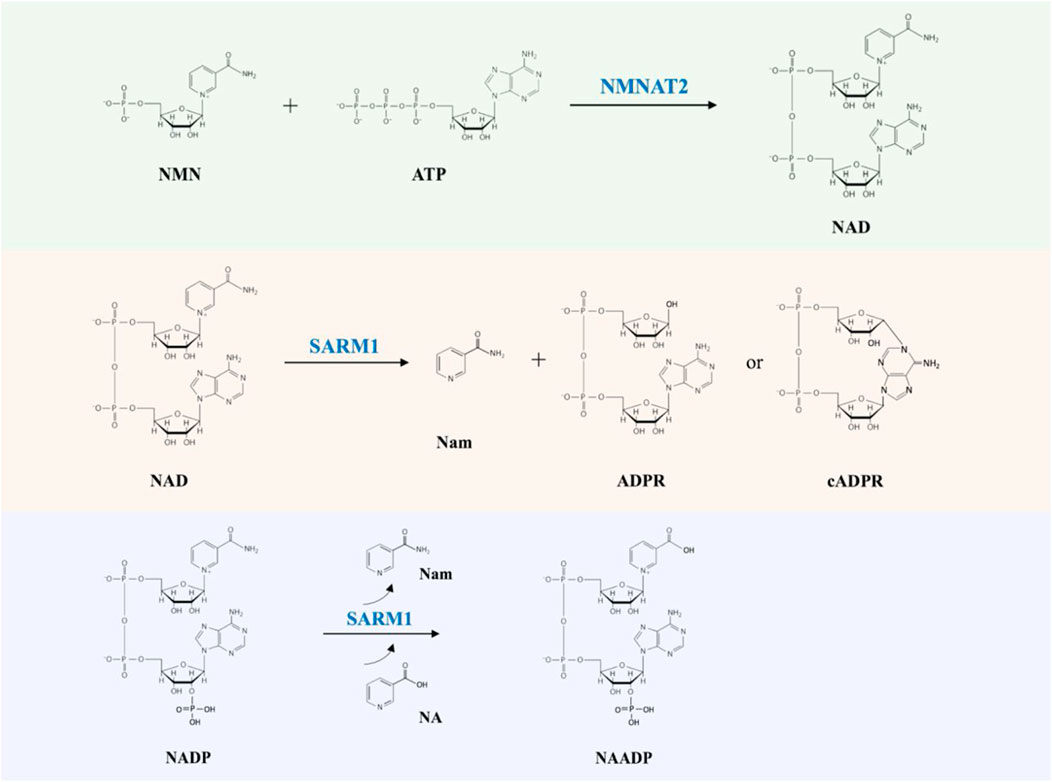
FIGURE 1. The NMNAT2 and SARM1 catalyzed reactions. NMNAT2 catalyzes the synthesis of NAD from NMN and ATP. SARM1 cleaves NAD into Nam and either ADPR or cADPR; SARM1 catalyzes the exchange of Nam of NADP with NA (nicotinic acid) to produce NAADP (nicotinic acid adenine dinucleotide phosphate).
Intriguingly, SARM1 is not only involved in axon degeneration, but also in innate immunity (Carty and Bowie, 2019). Its structure contains a TIR (Toll/interleukin-1 receptor) domain, which is commonly seen in Toll-like receptors (TLRs) in the innate immune system (O’Neill et al., 2013; Ve et al., 2015). As removing SARM1 delays axon degeneration (Osterloh et al., 2012), it is seen as a good candidate for therapies (Loring and Thompson, 2020). It is currently unclear, though, whether targeting SARM1 could lead to innate immune consequences (Uccellini et al., 2020).
In this review, the mechanisms of axon degeneration are thoroughly examined, while detailing the importance of NAD and related metabolites as signaling molecules. We also discuss whether the role of SARM1 in innate immunity indicates important roles in the response to pathogens and axonal damage.
Axon Degeneration Research from 1850 to Today
Axons have been known to degenerate upon injury since Waller’s observations of lesioned nerves of frogs in 1850 (Waller, 1851); however, it was long thought to be a passive process. This process of axon fragmentation is now known as Wallerian degeneration (Figure 2), and differs from neuronal death by apoptosis (Geden and Deshmukh, 2016). While Waller’s work demonstrated the gross degeneration of the distal stump of nerves after transection, it was later work by Ramón y Cajal, in 1928 that detailed the histological changes taking place for the first time (Ramón y Cajal, 1991 English translation).
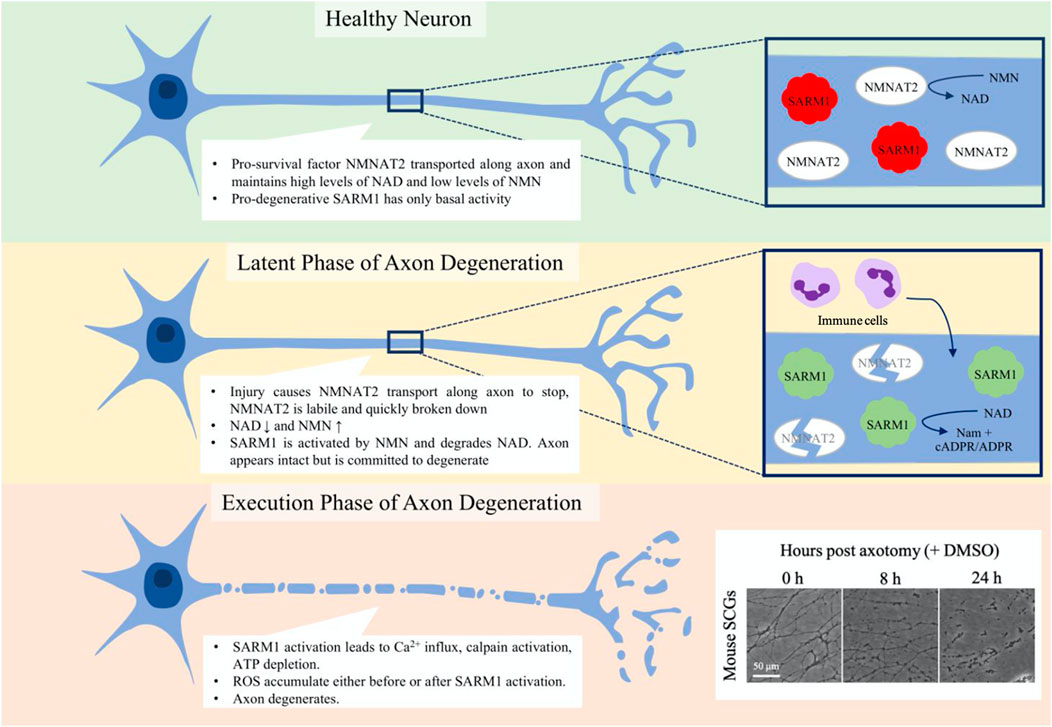
FIGURE 2. Phases of programmed axon degeneration. Programmed axon degeneration is split into two main phases, the latent phase, where the axons are morphologically normal but are committed to degeneration, and the execution phase, where the axon undergoes gross swelling and granulation as it breaks down. In healthy neurons, NMNAT2 pro-survival factor converts NMN to NAD, maintaining NAD and NMN levels that keep SARM1 pro-degenerative factor inactive. After injury, in the latent phase NMNAT2 starts to be broken down and cannot be transported along the axon. NAD levels thus lower and NMN levels rise, leading to the activation of SARM1. Immune cells are seen to enter the nerve at this stage. After up to 36 h, the axons progress to the execution phase, where Ca2+ levels increase, calpains are activated and the axon breaks down. Mouse superior cervical ganglia (SCGs) are often used as an assay for axon degeneration. The box on the bottom corner shows axon degeneration progressing over 24 h post-axotomy. At 8 h axons are committed to degenerate and some have started visibly breaking down. By 24 h, all axons are degenerating and much of what is seen is axon debris.
Axon degeneration can be divided into two phases: the latent phase and the execution phase (Figure 2). The latent phase lasts up to 36 h wherein the axons appear normal and for much of this time retain the ability to conduct an action potential, although the axon becomes committed to degenerate (Tsao et al., 1999; Beirowski et al., 2004; Beirowski et al., 2005; Conforti et al., 2014). This is also the stage in which the immune system is first seen to be involved; tissue-resident macrophages are activated and bone-marrow derived macrophages, neutrophils and phagocytes infiltrate into the nerve (Perkins and Tracey, 2000; Sta et al., 2014; Zigmond and Echevarria, 2019; Boissonnas et al., 2020; Ydens et al., 2020; Chen et al., 2021). There is also an injury-associated increase in the levels of cytokines (Yi et al., 2017). The execution stage becomes apparent once the axon morphology starts to change visibly but molecular execution steps may precede this. The axons fragment and the compound action potential is lost as they become non-functional (Beirowski et al., 2004; Beirowski et al., 2005; Conforti et al., 2014; Sta et al., 2014). Neutrophils may contribute in the early stages of the execution of degeneration by phagocytosing axonal debris (Lindborg et al., 2017), whilst macrophages clear myelin and axonal debris and later are involved in the axon regeneration process (Boissonnas et al., 2020).
Molecules that actively regulate axonal survival and degeneration have long been hypothesized (Lubińska, 1977; Lubińska, 1982). In 1982, Lubińska first proposed that an ‘axonal trophic factor’ is responsible for axon survival and it was likely transported along axons to inhibit Wallerian degeneration, but was unable to identify it (Lubińska, 1982). This unnamed ‘axonal trophic factor’ fits the properties of the protein NMNAT2, and NMNAT2 remains the best candidate for the proposed molecule in Lubińska’s studies (Gilley and Coleman, 2010). Recently, the loss-of-function genetic screens in invertebrates (Drosophila) and vertebrates (mouse) identified SARM1, which was shown to be a central executioner in Wallerian degeneration; its deficiency protects severed axons for weeks (Osterloh et al., 2012; Gerdts et al., 2013) and permanently rescues the lethal axon growth deficit of Nmnat2 null mice (Gilley et al., 2017).
Wallerian Degeneration Slow Mice
The discovery of WldS (Wallerian degeneration slow) mice, a strain in which injured axons survive 10-fold longer than in wild-type mice (Lunn et al., 1989), challenged the hypothesis that Wallerian degeneration is a passive process and revolutionized the study of axon degeneration (Coleman and Höke, 2020). The WldS mouse strain arose from a spontaneous intrachromosomal triplication event (Coleman et al., 1998) that results in the formation of a unique chimeric protein, WLDS. The WLDS protein was shown to be a fusion of a 70-amino acid N-terminal fragment of ubiquitin ligase UBE4B joined by a unique short linker (Wld18) to NMNAT1 (nicotinamide mononucleotide adenylyltransferase 1) (Conforti et al., 2000; Mack et al., 2001). While NMNAT1 is normally localized to the nucleus of neurons, when fused to UBE4B in WLDS, it is partially relocalized to the axons (Babetto et al., 2010). WLDS confers a concentration-dependent axon protection (Mack et al., 2001) and was later found to act by maintaining axonal NMNAT activity if NMNAT2, an axonal protein essential for axon survival, is degraded upon axon injury or is absent for other reasons (Araki et al., 2004; Babetto et al., 2010; Gilley and Coleman, 2010; Gilley et al., 2013; Loreto et al., 2020b). The serendipitous discovery of WldS mice uncovered one of non-redox roles for NAD and provided the initial evidence that NMNATs and NAD signaling have a role in axon regulation.
Regulation of Axon Degeneration by Nicotinamide Adenine Dinucleotide-Mediated Signaling
In damaged axons and nerves, the level of NAD is known to decrease and the presence of WLDS prevents this decrease suggesting it has an axonal site of action (Wang et al., 2005; Di Stefano et al., 2015), even though it was difficult to detect in axons (Mack et al., 2001). Additional evidence for this came from findings that shifting WLDS or NMNAT1 from nuclei, where these proteins are most abundant, into the cytoplasm (Beirowski et al., 2009; Sasaki et al., 2009a) and subsequently more specifically into axons (Babetto et al., 2010) provided stronger axon protection. Finally, chemical-genetic methods were used to manipulate the stability of axonal WLDS after separating axons from the soma to definitively prove that it has to be present, and stable, in axons to protect them (Cohen et al., 2012; Wang et al., 2015) (Figure 3).
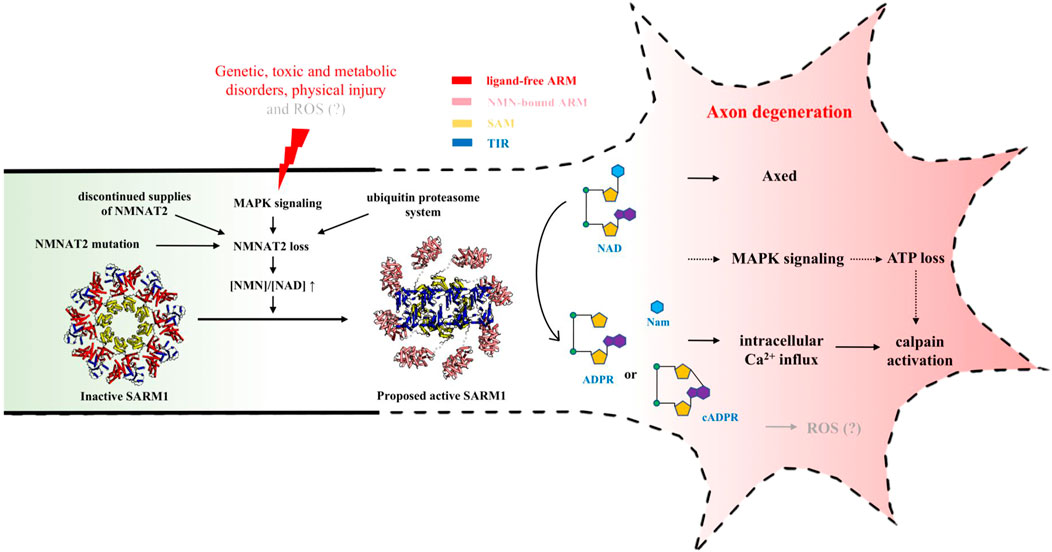
FIGURE 3. NAD-mediated signaling regulating injury-induced axon degeneration. Upon injury, NMNAT2 loss results in an increased ratio of NMN-to-NAD, which is a signal to activate SARM1 from the inactive state (PDB: 7LD0) to active state (PDB (ARM): 7LCZ; PDB (TIR): 6O0R). Active SARM1 cleaves NAD into Nam and either ADPR or cADPR, initiating the pathways downstream, including intracellular Ca2+ influx, ATP loss, Axed activation and eventually axon loss.
Nicotinamide Mononucleotide Adenylyltransferase 2
In axons, NAD synthesis relies predominantly on NMNAT2, a labile NMNAT isoform (Gilley and Coleman, 2010). At least in primary neuronal cultures, NMNAT2 appears to be largely synthesized in the cell bodies and transported constantly to axons, where it is required for maintenance of axonal health; axotomy causes its axonal levels to decline rapidly (Gilley and Coleman, 2010). In primary neurons, the majority of NMNAT2 is associated with membranes (Summers et al., 2018), which requires NMNAT2 to be modified by palmitoylation (Milde et al., 2013). Palmitoylated NMNAT2 is relatively unstable, compared to cytosolic non-palmitoylated NMNAT2 (Milde et al., 2013). Depletion of NMNAT2 is sufficient to trigger axon degeneration, which can be counteracted by overexpressing NMNAT2, WldS or other NMNAT isoforms before axons are damaged (Sasaki et al., 2009b; Yahata et al., 2009; Gilley and Coleman, 2010; Yan et al., 2010; Coleman and Höke, 2020), demonstrating its neuroprotective role. Interestingly, protection of axons by NMNAT seems not to depend on an elevated basal level of NAD in axons, but on its enzymatic activity of NAD synthesis after axonal injury (Di Stefano et al., 2015).
CD38 and PARP1 are two important enzymes that consume NAD (Malavasi et al., 2008). In mice, deficiency in CD38 alone, or deficiency in both CD38 and PARP1, leads to an increase of NAD in brain lysates and cultured DRG (dorsal root ganglion) neurons, but axons of these mutated DRG neurons undergo a similar rate of injury-induced degeneration to that seen in the wild-type neurons (Sasaki et al., 2009b).
NMNAT2 is shown to be lost rapidly and spontaneously in damaged neurons as a result of natural turnover before axon degeneration (∼2 h) (Gilley and Coleman, 2010). Axon injury can cause a failure in NMNAT2 transport and so the constant supply of wild-type NMNAT2 into axons is disrupted. The remaining NMNAT2 is regulated by two independent pathways, the MAPK (mitogen-activated protein kinase) signaling pathway and the ubiquitin proteasome system (Milde et al., 2013; Walker et al., 2017; Summers et al., 2018). MAPK signaling promotes the degradation of the palmitoylated NMNAT2 via the MAP3K (MAPK kinase kinase) DLK (dual leucine zipper kinase) and LZK (leucine zipper kinase) (Summers et al., 2018). The non-palmitoylated NMNAT2 is degraded by an E3 ubiquitin ligase complex (Summers et al., 2018). In Drosophila, the Highwire (Hiw) E3 ubiquitin ligase has been shown to promote the rapid loss of dNmnat upon injury (Xiong et al., 2012), while its orthologue MYCBP2 (MYC binding protein 2), also known as PHR1 (PAM-Highwire-Rpm-1) E3 ubiquitin ligase promotes NMNAT2 loss in mice (Babetto et al., 2013). A recent serine/threonine-linked ubiquitination mechanism of MYCBP2 suggests ways in which it could be inhibited as a potential therapy in axonal and other disorders (Mabbitt et al., 2020).
NMNAT2 loss is the initial event and an important node in the axon degeneration pathway; thus, understanding the mechanism of NMNAT2 loss and what its loss causes will further our understanding of the axon degeneration process.
Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1
Around 2012, loss-of-function genetic screens for axon protection after injury were performed in Drosophila olfactory receptor neuron axons (Osterloh et al., 2012) and mouse DRG neurons (Gerdts et al., 2013), both with follow-up in vivo studies in mice; both screens identified the protein SARM1, or its Drosophila orthologue dSarm, to be involved in the axon degeneration process. Loss of SARM1 provides axonal protection upon injury for weeks in vivo, while expression of wild-type SARM1 in Sarm1−/− neurons restores the phenotype of rapid injury-induced axon degeneration (Osterloh et al., 2012; Gerdts et al., 2013). Moreover, TIR-1, the SARM1 orthologue in C. elegans (Caenorhabditis elegans), has recently been shown to inhibit regeneration of damaged motor axons via the NSK-1/ASK1 MAPK pathway, that is independent on its role in axon degeneration (Julian and Byrne, 2020). This dual role of SARM1 in both axon degeneration and regeneration indicates that inhibiting SARM1 can be a way to block axon loss making it a therapeutic candidate for drug development to slow or stop aberrant Wallerian degeneration in several neurodegenerative diseases.
SARM1 has been initially described as one of the adaptor proteins in TLR signaling (O’Neill et al., 2003; Carty et al., 2006). It is a highly conserved protein consisting of an N-terminal ARM (armadillo repeat motif) domain, two central tandem SAM (sterile-alpha motif) domains and a C-terminal TIR domain, responsible for auto-inhibition, oligomerization and NAD hydrolysis, respectively (Mink et al., 2001; O’Neill et al., 2003; Kim et al., 2007; Gerdts et al., 2013; Essuman et al., 2017; Horsefield et al., 2019). In solution, SARM1 exists as ring-shaped octamers, with oligomerization mediated by the SAM domains (Horsefield et al., 2019; Sporny et al., 2019; Bratkowski et al., 2020; Jiang et al., 2020; Sporny et al., 2020; Figley et al., 2021; Shen et al., 2021). In neurons where SARM1 shows the greatest abundance, SARM1 is mostly present in the axons (Osterloh et al., 2012). In healthy axons, SARM1 is held in an inactive state by the auto-inhibitory ARM domains, by means of a direct interaction with and separation of the TIR domains away from each other (Bratkowski et al., 2020; Jiang et al., 2020; Sporny et al., 2020; Figley et al., 2021; Shen et al., 2021). Upon injury and consequent NMNAT2 loss, this auto-inhibition is relieved, permitting the C-terminal TIR domains to self-associate to hydrolyze NAD into Nam, and either ADPR or cADPR, along with other activities, and axons degenerate (Essuman et al., 2017; Horsefield et al., 2019). Although the mechanism of SARM1 activation is still not fully understood, the discovery that SARM1 is a self-regulated NADase (Essuman et al., 2017), together with the discovery of NMNAT2 loss upon injury (Gilley and Coleman, 2010), is particularly important, because it not only solves the puzzle why the activation of SARM1 leads to NAD destruction, but also further provides the evidence for the key role of axonal NAD- and NADP-related metabolism in this death pathway of axon degeneration.
Nicotinamide Adenine Dinucleotide Depletion vs Nicotinamide Mononucleotide Accumulation Hypotheses
NMNAT2 loss is known to be an event upstream of SARM1 activation (Gilley et al., 2015; Summers et al., 2020). The direct results of NMNAT2 loss are a decrease of the product NAD and an accumulation of the substrate NMN (Di Stefano et al., 2015; Gilley et al., 2015). Therefore, two hypotheses have been proposed to explain the mechanism of the NMNAT2 loss-induced SARM1 activation, the “NAD depletion hypothesis” and the “NMN accumulation hypothesis.”
The “NAD depletion hypothesis” proposes a feed-forward model, where the depletion of NMNAT2’s product NAD is a trigger for SARM1 activation (Gerdts et al., 2016). The studies by Sasaki et al. (2016) found that cytNMNAT1, an engineered NMNAT1 mutant that only targets the cytoplasm and axons, protects axons by blocking the SARM1-mediated NAD depletion, rather than through NAD synthesis. Because NMNAT2 is predominantly expressed in axons, it was inferred that it is likely that NMNAT2 also blocks SARM1-mediated NAD depletion by a similar mechanism in axons. This is consistent with the mouse knockout experiment, in which the embryonically lethal phenotype of Nmnat2 null mice was rescued by a knockout of SARM1 in the double Nmnat2 null and Sarm1−/− mice (Gilley et al., 2013; Gilley et al., 2015). The recently published cryo-electron microscopy (EM) structures of the NAD-bound SARM1, combined with functional data, provide evidence to support the ‘NAD depletion hypothesis’ (Jiang et al., 2020; Sporny et al., 2020). However, this hypothesis could not explain the effects of modulating NMN described below.
Conforti, Coleman and colleagues proposed an alternative model, in which NMN accumulation upon injury-induced NMNAT2 loss may be a trigger for SARM1 activation in damaged axons (Di Stefano et al., 2015; Di Stefano et al., 2017). To provide support for this hypothesis, they used two independent methods to keep NMN at low levels, with and without altering NAD levels, and studied the effect on axon degeneration. The first method involved the inhibition of NAMPT (nicotinamide phosphoribosyl transferase), an NMN synthesizing enzyme, using the inhibitor FK866 (Sasaki et al., 2009b; Di Stefano et al., 2015); the second method involved exogenous expression of bacterial NMN deamidase, an NMN utilizing enzyme, without altering NAD levels (Di Stefano et al., 2015; Di Stefano et al., 2017). It was found that both methods of blocking the increase of NMN resulted in axon protection, similar to that seen in the WldS phenotype, more modestly with FK866 but as strong as WldS with NMN deamidase. Further research to study the mechanism demonstrated that NMN indeed accumulated in the distal axons during Wallerian degeneration (Di Stefano et al., 2015; Sasaki et al., 2016) and its accumulation induced the influx of extracellular Ca2+, which depended on the appearance of SARM1 and was sufficient to initiate Wallerian degeneration (Loreto et al., 2015). Consistently, fluorescence-based NADase assays in vitro illustrated that NMN was indeed an activator of SARM1 (Zhao et al., 2019; Bratkowski et al., 2020; Figley et al., 2021). Moreover, NMN analogues including CZ48, a synthetic cell-permeable molecule (Zhao et al., 2019), and vacor mononucleotide (VMN), a metabolite of neurotoxin vacor (Loreto et al., 2020a), could also activate SARM1 NADase activity, the latter also inducing axon degeneration. However, this hypothesis could also not explain the experimental data that raising the level of both NMN and NAD together protects axons from degeneration (Wang et al., 2015; Sasaki et al., 2016).
Because neither hypothesis could explain all the observations, it was hypothesized that both of NMN and NAD, or NMN-to-NAD ratio, regulate SARM1 activity (Di Stefano et al., 2017). In most cells, the NAD levels are known to be much higher than NMN levels (Formentini et al., 2009; Mori et al., 2014). In the brain, NMN-to-NAD ratio is estimated to be around 0.02 ([NAD]/[NMN] ≈ 49), based on the analysis of the extracts from C57BL/6 mice brain immediately frozen post mortem (Mori et al., 2014). Although it is still not clear how much this ratio changes in axons after injury, when NMNAT2 is lost, NMN accumulation and NAD depletion lead to an increased ratio of NMN-to-NAD, which could be a signal for SARM1 activation. Recently, both NMN and NAD were demonstrated to interact directly with the ARM domain of SARM1 (Jiang et al., 2020; Figley et al., 2021). Using cell-, NMR (nuclear magnetic resonance)- and structure-based assays, Figley et al. further demonstrated that SARM1 acts as a metabolic sensor activated by an increasing NMN-to-NAD ratio (Figley et al., 2021). In the proposed model, NAD competes with NMN for the binding site in the regulatory ARM domain, prevents the NMN-induced compaction of this domain and hence promotes the auto-inhibition of the protein (Figure 3) (Figley et al., 2021). Further work is still required to fully understand the mechanism of the relief of auto-inhibition and the activation of NAD-cleavage activity of SARM1.
Pathways Downstream of Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1
In 2015, Loreto et al. showed that an increase of intra-axonal Ca2+ occurred downstream of SARM1, prior to Wallerian degeneration (Loreto et al., 2015). They argued that this Ca2+ increase belongs to the second Ca2+ influx after injury, and could be a causative event marking the execution phase of Wallerian degeneration. Active SARM1 cleaves NAD into Nam and either ADPR or cADPR (Essuman et al., 2017; Horsefield et al., 2019), resulting in local NAD destruction and eventually axon degeneration (Gerdts et al., 2015). Both ADPR and cADPR are powerful agents that mobilize Ca2+ by targeting the TRPM2 (transient receptor potential melastatin 2) and RyR (ryanodine receptor), respectively (Perraud et al., 2001; Guse, 2004). Consistently, preventing extracellular Ca2+ influx using ryanodine receptor antagonist ryanodine confers axonal protection from NMN-induced degeneration (Loreto et al., 2015). One of the possible pathways downstream of SARM1 activation is the activation of neuronal calpains, the Ca2+-activated nonlysosomal cysteine proteases that play a role in neurodegenerative events of traumatic brain injury (TBI) (Saatman et al., 2010). It has been shown that calpain activity is increased within minutes in the animal model of TBI (Büki et al., 1999). Consistently, the endogenous calpain inhibitor calpastatin, which inhibits Wallerian degeneration in vivo and in vitro, is depleted upon physical injury (Yang et al., 2013). Future work is needed to investigate if the change of Ca2+ through the SARM1 pathway is necessary and sufficient to activate calpains. SARM1 activation also results in a local energy deficit, inducing pathological axon degeneration. Using traumatic injury as a model, Yang et al. showed that SARM1 is required for the activation of a MAPK cascade involving MEKK4, MLK2, DLK, MKK4, MKK7, JNK1, JNK2, and JNK3, to trigger axon degeneration (Yang et al., 2015). This SARM1-dependent MAPK pathway results in ATP depletion prior to calpain activation and axon destruction, demonstrating that this MAPK cascade is also one of the pathways downstream of SARM1. However, the mechanism of how SARM1 activates the MAPK pathway and how this pathway causes ATP depletion remains obscure. Another possible mediator downstream of SARM1 activation is Axundead (Axed), which was identified in a genetic screen in glutamatergic sensory neurons from the Drosophila wing (Neukomm et al., 2017). Axed loss-of-function leads to a complete block of SARM1-induced axon degeneration, suggesting that SARM1 executes its pro-degenerative activity through an Axed-dependent pathway (Neukomm et al., 2017). However, much is still required to be learned regarding to the roles of the Axed orthologues in other species. Finally, there is accumulation of ROS (reactive oxygen species) that occurs either before or after SARM1 activation whose origin and function in axon degeneration requires further study (Press and Milbrandt, 2008; Summers et al., 2014).
Over the past two decades, the NAD-related signaling mechanism associated with the axon degeneration pathway has been slowly being elucidated (Figure 3). It is now known that it starts with the loss of the axon survival factor NMNAT2 through different possible pathways, which results in an increased ratio of NMN-to-NAD due to NMN accumulation and NAD depletion in axons; this leads to the activation of SARM1 NADase activity, local NAD destruction, intracellular Ca2+ influx, ATP loss, Axed activation and eventually axon loss, although the potential causative roles of some of the later steps require further clarification. Despite several questions remaining, studying this novel NAD metabolism in axons will enhance our understanding of the degenerative mechanism of axons, and also facilitate the development of drugs for a wide range of neurodegenerative diseases.
Nicotinamide Adenine Dinucleotide Signaling and the Immune System
Roles of Toll/Interleukin-1 Receptor Domain-Mediated NADase Activity in the Plant Immune System
SARM1 TIR domain-mediated NAD hydrolysis requires a catalytic glutamate residue at position 642 (E642) in the human protein. An analogous glutamate is also present in plant TIR domain-containing immune receptors, such as Linum usitatissimum L6 (E135), Muscadinia rotundifolia RUN1 (E100), and Arabidopsis thaliana RBA1 (E86) and RPP1 (E164), suggesting there may be NADase activity by the TIR domains involved in plant immune responses; this suggestion has been experimentally confirmed (Horsefield et al., 2019; Wan et al., 2019). In plants, NLRs (nucleotide-binding (NB), leucine-rich repeat (LRR) receptors) detect pathogen effectors and induce immune responses, often characterized by localized cell death (hypersensitive response) to restrict the pathogens to the infection sites (Dangl et al., 2013). Plant NLRs typically consist of an N-terminal TIR domain or coiled-coil (CC) domain, a central NB and oligomerization domain (NOD) and a C-terminal LRR domain, responsible for signaling, oligomerization and recognition of pathogen effectors, respectively (Maekawa et al., 2011; Burdett et al., 2019). Once TIR-NLRs are activated, the central NOD undergoes a conformational change to an oligomerization-prone state, bringing the TIR domains together to facilitate NAD hydrolysis. The recently determined cryo-EM structures of two TIR-NLRs in their active states, Arabidopsis thaliana RPP1 and Nicotiana benthamiana ROQ1, revealed the protein forms a tetramer, suggesting a minimum functional unit of four TIR domains for NADase activity (Ma et al., 2020; Martin et al., 2020). In both proteins, the TIR domains are arranged through two interfaces, the symmetrical “AE interface,” consisting of the αA helix and the neighboring αE helix, and the asymmetrical “BE interface,” also referred to as the “BB-loop interface” (the BB-loop connects the βB and αB secondary structure elements) (Ma et al., 2020; Martin et al., 2020). Analogous interfaces are also observed in the crystal structure of human SARM1 TIR domain, and mutagenesis showed that the residues mediating these interfaces are important for NADase activity (Horsefield et al., 2019). These similar interactions between the TIR domains illustrate a common mechanism of the TIR domain-mediated NAD cleavage. Interestingly, NAD hydrolysis by plant TIR domains produces a non-canonical variant of cADPR (v-cADPR) (Wan et al., 2019). Future work is required to understand the specificity of the products and the role of this v-cADPR.
Roles of Toll/Interleukin-1 Receptor Domain-Mediated NADase Activity in the Prokaryotic Immune System
TIR domain-mediated NADase activity is also found in the bacterial and archaeal TIR-domain containing proteins (Tcps), such as Staphylococcus aureus TirS, Echerichia coli TcpC and Theionarchaea archaeon TcpA (Essuman et al., 2018). Like the TIR domains in SARM1 and plant proteins, NAD hydrolysis activity of prokaryotic TIRs also relies on a catalytic glutamate residue (e.g. E216 in TirS, E244 in TcpC and E267 in TcpA). Interestingly, in the available crystal structures of bacterial TIR domains, these glutamates are not found in a location structurally similar to the location in SARM1 and plant TIR domains. Bacterial and archaeal TIR domains from different species also show differences in terms of reaction kinetics and types of the products (Essuman et al., 2018).
In bacteria and archaea, diverse antiphage defense systems have evolved to protect themselves from attack by viruses. Genes encoding these systems are prone to cluster in genomic defense islands (Makarova et al., 2013). A recent study indicated that some bacterial Tcps are found in the defense islands important for anti-phage defense (Doron et al., 2018). The authors named this prokaryotic Tcp defense system as the Thoeris defense system. The Thoeris system is broadly distributed in prokaryotic genomes, encodes an NAD-binding protein ThsA and a TIR domain-containing protein ThsB (Doron et al., 2018). Using Bacillus subtilis expressing the Thoeris system (MSX-D12), ThsB has been found to produce a v-cADPR that activates ThsA upon detection of phage infection (Ofir et al., 2021). Activated ThsA further hydrolyzes NAD, causing cell suicide, probably by depleting cellular NAD (Ofir et al., 2021).
In addition, plant TIR domains, SARM1 TIR domain and Tcps also cleave NADP (nicotinamide adenine dinucleotide phosphate) (Horsefield et al., 2019; Wan et al., 2019; Zhao et al., 2019; Essuman et al., 2018), but only SARM1 TIR domain has been shown to have a base-exchange activity (Figure 1) (Zhao et al., 2019). Further studies are required to explain the mechanism of the similar and different functions between different TIR domains.
Roles of Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1 in Innate Immunity
The role of NAD metabolism in the immune response has been reviewed in a number of papers (Singhal and Cheng, 2019; Navarro et al., 2021; Navas and Carnero, 2021). It is important to highlight that CD38, predominantly expressed in immune cells, acts as the main enzyme digesting cellular NAD under basal conditions (Camacho-Pereira et al., 2016; Navarro et al., 2021). CD38 has been shown to be involved in NAD cleavage at the inflammation site to maintain T-cell survival (Adriouch et al., 2007). Its expression is increased during infections by a variety of pathogens (Adekambi et al., 2015) and aging (Chini et al., 2020; Covarrubias et al., 2020). CD38 expression is induced by senescence-associated inflammation during aging, which contributes to age-related NAD decline in macrophages (Chini et al., 2020; Covarrubias et al., 2020), as knockout of CD38 prevents this decline and leads to enhanced metabolic health in mice (Camacho-Pereira et al., 2016). Although axon degeneration-related SARM1 NADase activity has been extensively characterized in neurons (rather than immune cells), it still remains unclear if this enzymatic activity plays a role also in the immune system. However, SARM1 has initially been found to be involved in the innate immunity pathways through the ability of its TIR domain to interact with other proteins, rather than its NADase activity (O’Neill et al., 2003; Panneerselvam and Ding, 2015; Carty and Bowie, 2019). TIR domains involved in innate immunity pathways form higher-order assemblies and carry out their signaling function through a mechanism termed signaling by co-operative assembly formation (SCAF) (Nimma et al., 2017; Vajjhala et al., 2017; Nanson et al., 2019).
Negative Regulation by Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1 in Toll-Like Receptor Signaling
TLRs are an important set of germline-encoded pattern-recognition receptors in the innate immune system; they recognize pathogen-associated molecular patterns, including microbial lipids, lipoproteins and nucleic acids (Kawai and Akira, 2010; Thompson et al., 2011). Once activated, TLRs dimerize to create an intracellular TIR-domain signaling scaffold, which then recruits TIR domain-containing adaptor proteins to transfer signals downstream (O’Neill et al., 2013). Six adaptor proteins have been identified in TLR signaling pathways; they are MyD88 (myeloid differentiation primary response gene 88), MAL (MyD88 adaptor-like protein), TRIF (TIR domain-containing adaptor protein-including interferon β), TRAM (TRIF-related adaptor molecule), SARM1 and BCAP (B-cell adaptor for PI3K). Unlike the four key cytosolic adaptor proteins (MyD88, MAL, TRIF and TRAM), SARM1 is not required for TLR signaling, but regulates this signaling through its interaction with other adaptor proteins.
In humans, SARM1 negatively regulates TRIF-dependent TLR3 and TLR4 signaling, hence inactivating the NF-κB (nuclear factor-κB) response (Carty et al., 2006). Using co-immunoprecipitation, yeast two-hybrid assays and GST pull-down assays, it has been shown that SARM1 inhibition of TRIF was achieved through a direct TIR-TIR domain interaction (Carty et al., 2006). Moreover, the BB loop in the SARM1 TIR domain may participate in the interaction, as the BB-loop mutant G601A disrupted SARM1 interaction with TRIF, and its inhibition of the lipopolysaccharide-regulated expression of inflammatory cytokines (Carlsson et al., 2016). The ARM domain plays an inhibitory role, because SARM1 truncation to remove the ARM domain causes a more potent inhibition in TRIF-dependent signaling (Carty et al., 2006). Consistently, in mice, SARM1 expression has been shown to increase during the infection of macrophages with Burkholderia pseudomallei, leading to the inhibition of TRIF and hence decreasing IFNβ (β interferon) production (Pudla et al., 2011). In pigs, the SARM1 orthologue negatively regulates NF-κB in a TRIF-dependent pathway during the infection with porcine reproductive and respiratory syndrome virus (Zhou et al., 2013). In horseshoe crabs, Carcinoscorpius rotundicauda, the SARM1 orthologue is shown to downregulate TRIF-dependent NF-κB activation during the infection with Pseudomonas aeruginosa (Belinda et al., 2008). These results demonstrate the functional conservation of SARM1 as a negative regulator in TRIF-dependent TLR signaling.
Besides inhibiting the NF-κB response, SARM1 also inhibits TRIF- and MyD88-regulated activation of AP1 (activator protein 1) through TIR-TIR domain interactions (Carlsson et al., 2016), or/and direct involvement in inhibiting the phosphorylation of MAPKs via an unknown mechanism (Peng et al., 2010). The ARM domain also self-regulates SARM1 activity in this pathway, as full-length SARM1 is less efficient in this inhibition than the SAM-TIR domains-only-containing fragment. Although it is still not clear if the NADase activity of SARM1 plays any roles in these processes, the ARM domain removal permits self-association of the TIR domains and leads to a more efficient inhibition, suggesting that the SARM1 TIR assembly may facilitate the interaction with the TIR domains of other adaptor proteins, hence likely interfering with the downstream signaling.
Up-Regulatory Role of Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1 in Cytokine and Chemokine Production
SARM1 has also been shown to positively regulate the production of cytokines and chemokines. In the liver of mice suffering from non-alcoholic fatty liver disease (NAFLD) induced by a high fat diet (HFD), SARM1 shows a significant increase in both mRNA and protein levels. SARM1 knockout in these mice leads to a reduction in inflammatory response (a decrease in IL-1β (interleukin-1β), IL-6, TNF-α (tumor necrosis factor α) and MCP-1 (monocyte chemotactic protein-1)) through inactivating TLR4, 7 and 9 signaling, and the NF-κB pathway, supporting a positively regulatory role of SARM1 in cytokine and chemokine production in TLR signaling pathways (Pan and An, 2018). Interestingly, TNF-α also seems to function upstream of SARM1. The Yang lab has recently shown that TNF-α mediates axonal loss via SARM1 in mice liver (Liu et al., 2021), consistent with the finding that TNF-α induces SARM1-dependent axon degeneration in sensory neurons (Ko et al., 2020).
In mice, the neuronal immune response to traumatic axonal injuries was investigated using the in vivo model of murine sensory neurons, showing that SARM1 is required for the expression of chemokines Ccl2, Ccl7, and Ccl12, and the cytokine Csf1, through a SARM1-JNKs-cJun pathways, and demonstrating an important role of SARM1 in the inflammatory gene expression in the central nervous system (CNS) (Wang et al., 2018). A number of studies were also performed to investigate the physiological role of SARM1 in the CNS against microbial infection, showing that infecting the CNS of Sarm1−/− mice with vesicular stomatitis virus (VSV, commonly used as a model for neurotropic viral infection), but not Listeria monocytogenes, Mycobacterium tuberculosis and influenza virus, restricts viral infection by decreasing cytokine production, suggesting an up-regulatory role of SARM1 in cytokine production (Hou et al., 2013). Interestingly, it has also been shown that Sarm1−/− mice were more susceptible to West Nile virus (WNV) infection in a brain-specific manner. Despite a similar reduction of TNF-α observed in Sarm1−/− mice, the increased cell death occurred during WNV infection but not VSV infection (Szretter et al., 2009). Although further studies are required to understand this difference, the result indicates that SARM1 contributes to the immune responses during microbial infection in the CNS, linking viral pathogenesis-induced neuronal injury and innate immunity. Because SARM1 NADase activity is required for cell death, it is likely that this enzyme activity also plays a role in this process.
Similar mouse studies were also performed to study the role of SARM1 in the peripheral immune system, by examining the cytokine or chemokine responses to TLR ligands or viral infection in the macrophages of Sarm1−/− mice (Gürtler et al., 2014; Uccellini et al., 2020). Using bone marrow-derived macrophages from wild-type and Sarm1−/− mice, the Bowie lab demonstrated that SARM1 promotes gene induction of Ccl5 through recruiting the RNA polymerase II to the Ccl5 promoter by an unknown mechanism, following TLR4 and TLR7 stimulation (Gürtler et al., 2014). However, the recent data from the García-Sastre lab showed that the deficiency in the production of chemokines, such as Ccl5, Ccl3 and Ccl4, was due to the background effects of the knockout strain 129. These chemokine loci are so close to the Sarm1 gene, that they remain linked to the null mutation in subsequent crosses to C57BL/6. Uccellini et al. (2020) also corrected their previous finding of the important role of SARM1 in VSV infection in the CNS (Hou et al., 2013). These reports indicate a need to re-evaluate the role of SARM1 in the immune responses during microbial infection using methods or resources independent of the 129-derived null strain (Uccellini et al., 2020).
Roles of Sterile Alpha and Toll/Interleukin-1 Receptor Motif-Containing 1 Beyond Toll-Like Receptor Signaling
In some species, SARM1 has been reported to have innate immunity functions beyond TLR signaling. Inflammasome activation upon infection and injury result in either cytokine release (without cell death) or pyroptosis (Broz and Dixit, 2016). In mice, SARM1 has been shown to negatively regulate the NLRP3 (NLR family pyrin domain containing 3) inflammasome–dependent caspase-1 activation, hence reducing the release of IL-1β (Carty et al., 2019). On the other hand, SARM1 has also been shown to mediate mitochondrial depolarization (MDP), which is required for optimal pyroptosis and a key to distinguishing from other NLRP3 activators causing cytokine release (Carty et al., 2019). In C. elegans, inactivation by RNA interference of TIR-1, the human SARM1 orthologue, decreased worm survival when subjected to fungal and bacterial infections (Carole et al., 2004). This is because TIR-1 is required for the expression of antimicrobial peptide genes (Carole et al., 2004). TIR-1 has also been shown to play a significant role in specifying the asymmetric odorant receptor expression through NSY-1 (ASK1) MAP3K signaling. In this context, TIR-1 functions downstream of a voltage-gated calcium channel and calcium-calmodulin-dependent protein kinase II (CaMKII), UNC-43. It interacts directly with UNC-43 at a high level of Ca2+ and localizes NSY-1 to post-synaptic regions of AWC (amphid wing “C”) axons, silencing str-2 gene (a putative chemoreceptor gene) expression and hence leading to a AWCOFF phenotype (default state) (Chuang and Bargmann, 2005).
Links to Diseases and Future Therapies
There are few drugs currently on the market that treat neurodegenerative diseases effectively; most either manage the symptoms or slow disease progression, and there are no therapies yet that can reverse the damage done (Durães et al., 2018). This is where research into Wallerian degeneration, in particular SARM1, could prove vital. Wallerian degeneration is a well characterized, druggable pathway that is involved in a number of neurodegenerative diseases (Coleman and Höke, 2020). Wallerian degeneration has been shown to be a cause of axonal death in a number of disease models, including peripheral neuropathies such as those induced by chemotherapy or diabetes (Lapointe et al., 2013; Cashman and Höke, 2015; Geisler et al., 2016; Feldman et al., 2017; Turkiew et al., 2017), traumatic brain injury (Henninger et al., 2016; Ziogas and Koliatsos, 2018), some motor neuron disorders (Ferri et al., 2003; Veriepe et al., 2015; White et al., 2019), Parkinson’s disease (Sajadi et al., 2004; Hasbani and O’Malley, 2006), as well as eye diseases such as glaucoma (Howell et al., 2007; Beirowski et al., 2008; Wang et al., 2013; Zhu et al., 2013; Williams et al., 2017a; Williams et al., 2017b; Fernandes et al., 2018). Loss of SARM1 has been shown to protect mice from peripheral neuropathies (Geisler et al., 2016; Turkiew et al., 2017) and TBI-induced axon loss (Henninger et al., 2016), although there is still a debate on the contribution of SARM1 to ALS (Fogh et al., 2014; Veriepe et al., 2015; Peters et al., 2018).
By targeting SARM1, a drug could be developed that not only slows the progression of one disease, but potentially many that involve axon degeneration. Acute injury, such as chemotherapy-induced peripheral neuropathy (CIPN), is a particularly good initial target for a SARM1 inhibitor, as it is a major reason for limiting the doses of chemotherapies, and the neuropathy onsets only after chemotherapy starts (Cashman and Höke, 2015; Simon and Watkins, 2018; Coleman and Höke, 2020; Bosanac et al., 2021). Ideally, an inhibitor would be given throughout the course of chemotherapy to slow the progression of CIPN or even prophylactically to prevent the neuronal injury from starting.
Another question currently being addressed is whether people have mutations in genes encoding any of the proteins involved in Wallerian degeneration, and if so, do they cause or act as risk factors for neurological diseases? The first evidence of humans with mutations in NMNAT2 was found in 2019, with biallelic NMNAT2 loss-of-function mutations found in sisters with polyneuropathy (Huppke et al., 2019). Furthermore, stillborn fetuses with biallelic NMNAT2 null mutations were also reported (Lukacs et al., 2019). Following earlier human studies implicating this pathway in ALS by GWAS association to the SARM1 chromosomal locus and loss of potential SARM1 regulator STMN2 in human induced pluripotent stem cell (hiPSC)-derived motor neurons (Fogh et al., 2014; van Rheenen et al., 2016; Klim et al., 2019; Melamed et al., 2019), recent work has revealed that human SARM1 variant alleles that hyperactivate SARM1 NADase function and enhance neuronal vulnerability are enriched in patients with sporadic ALS, hereditary spastic paraplegia and other motor nerve disorders (Bloom et al., 2021; Gilley et al., 2021). Whether further mutations to NMNAT2, SARM1 or other proteins involved in the Wallerian degeneration pathway represent risk factors for other neurological diseases in living humans, is yet to be seen. It has been hypothesized that such mutations, affecting the enzymatic activity of NMNAT2 or SARM1, could be considered risk factors for neurodegenerative diseases (Coleman and Höke, 2020).
There are several potential ways to target Wallerian degeneration, but at present these remain in preclinical development. Gene therapy where SARM1 dominant-negative mutants are transfected into neurons using AAV (adeno-associated virus), robustly protected axons from degeneration both in cut DRGs in vitro and cut sciatic nerve in vivo in mice (Geisler et al., 2019b). Antisense oligonucleotides that target SARM1 and lower SARM1 levels in axons by more than 50% were also shown to slow degeneration in vitro (Gould et al., 2021). Furthermore, the first small molecule inhibitors of SARM1 have been discovered and are shown to slow axon degeneration in DRGs to the same level as Sarm1−/− (Hughes et al., 2021; Li et al., 2021). Interestingly, the Hughes et al. paper indicated that the inhibitor could be added up to 3 h after injury with the same level of protection, and could even protect axons fated to degenerate through rotenone exposure and promote recovery from the latent stage of degeneration back towards healthier axons (Hughes et al., 2021). Small-molecule inhibitors of SARM1 have recently been tested in hiPSC-derived motor neurons, protecting the axons from degeneration at 16h post-axotomy and in vivo, where there was a partial protection of axonal function in mouse models of CIPN (Bosanac et al., 2021; Hughes et al., 2021). These different approaches to tackling axon degeneration and their successes so far in preclinical models are encouraging for the prospects of clinical testing in the near future.
Conclusions and Future Questions
Axons are eliminated through programmed axon degeneration, a subcellular self-destruction program that can be activated in necroptosis, genetic, toxic metabolic disorders, physical injury, in addition to neuroinflammation. The central executioner of this process is SARM1, an enzyme with NADase, NADPase and base-exchange activities, whose activation is regulated by an upstream NAD synthesizing enzyme, NMNAT2. Emerging structural and functional data for SARM1 indicates allosteric regulation through its N-terminal ARM domain sensing the NMNAT2-mediated change of NMN-to-NAD ratio. Both the regulatory and catalytic sites in SARM1 in the ARM and TIR domains, respectively, represent paths to therapeutic intervention.
SARM1 also plays a role in innate immunity, including regulating TLR signaling, cytokine and chemokine production and gene expression for antimicrobial peptides, and inducing cell death. It is important to be cautious when changing the activity of SARM1, as it may have off-target effects on the immune system of patients. Gilley et al. have demonstrated that NMNAT2gtE/gtE; Sarm1−/− mice live as long as the wild-type ones, so the act of knocking out the proteins themselves does not shorten lifespan (Gilley et al., 2015). However, the mice are kept in specific pathogen-free conditions, so the effects of the changes in the immune response in Sarm1−/− mice are currently uncertain. Reassuringly though, the work by Uccellini et al. has shown that many of the supposed immune system effects of SARM1 could be due to passenger mutations in Sarm1−/− mouse lines, rather than through SARM1 activity (Uccellini et al., 2020).
Despite the questions surrounding SARM1’s potential involvement in immunity, it remains a promising target for drug development. The possibility of slowing or even halting axon degeneration in a number of neurodegenerative diseases, such as AD, PD, TBI, ALS and peripheral neuropathies, could be incredibly impactful, as these diseases contribute greatly to the global disease burden. Both activators and inhibitors of SARM1 are already known, as is its three-dimensional structure. Basic research continues to investigate how SARM1 is activated and will be crucial in understanding how to inhibit its NADase activity. Excitingly, initial small molecule screens have shown that inhibition of SARM1 is possible and that it does delay axon degeneration in vitro. It is likely that further investigation into these initial small molecule inhibitors will move into in vivo work and potential new drugs to treat neurodegenerative diseases will be developed. If the rate of discovery in this promising field continues on its current trajectory, it is likely that great strides towards the improvement of human health will be made in the near future–an exciting prospect for all.
Author Contributions
EH and WG wrote the manuscript, with input from BK and MC. All authors read and approved the manuscript.
Funding
This work was funded by Australian National Health and Medical Research Council (NHMRC 1160570) and Australian Research Council (ARC; Laureate Fellowship FL180100109) to BK. WG and BK received funding from Disarm Therapeutics, a wholly-owned subsidiary of Eli Lilly & Co., Cambridge, MA, United States. EH was funded by Biotechnology and Biological Sciences Research Council (BBSRC), part of UK Research and Innovation, and Astra Zeneca plc, Cambridge, United Kingdom. MC is funded by the John and Lucille van Geest Foundation.
Conflict of Interest
BK is shareholder of and consultant to Disarm Therapeutics, a wholly-owned subsidiary of Eli Lilly & Co., Cambridge, MA, United States. MC and EH are recipients of grant funding from AstraZeneca Ltd for academic research projects and MC is a consultant for Nura Bio. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the MC and BK labs for helpful discussions. EH would like to express thanks in particular to Elisa Merlini for providing SCG degeneration images.
References
Adekambi, T., Ibegbu, C. C., Cagle, S., Kalokhe, A. S., Wang, Y. F., Hu, Y., et al. (2015). Biomarkers on Patient T Cells Diagnose Active Tuberculosis and Monitor Treatment Response. J. Clin. Invest. 125, 1827–1838. doi:10.1172/JCI77990
Adriouch, S., Hubert, S., Pechberty, S., Koch-Nolte, F., Haag, F., and Seman, M. (2007). NAD+ Released During Inflammation Participates in T Cell Homeostasis by Inducing ART2-Mediated Death of Naive T Cells In Vivo. J. Immunol. 179, 186–194. doi:10.4049/jimmunol.179.1.186
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 305, 1010–1013. doi:10.1126/science.1098014
Babetto, E., Beirowski, B., Janeckova, L., Brown, R., Gilley, J., Thomson, D., et al. (2010). Targeting NMNAT1 to Axons and Synapses Transforms its Neuroprotective Potency In Vivo. J. Neurosci. 30, 13291–13304. doi:10.1523/JNEUROSCI.1189-10.2010
Babetto, E., Beirowski, B., Russler, E. V., Milbrandt, J., and DiAntonio, A. (2013). The Phr1 Ubiquitin Ligase Promotes Injury-Induced Axon Self-Destruction. Cell Rep. 3, 1422–1429. doi:10.1016/j.celrep.2013.04.013
Beirowski, B., Adalbert, R., Wagner, D., Grumme, D. S., Addicks, K., Ribchester, R. R., et al. (2005). The Progressive Nature of Wallerian Degeneration in Wild-Type and Slow Wallerian Degeneration (WldS) Nerves. BMC. Neurosci. 6, 6. doi:10.1186/1471-2202-6-6
Beirowski, B., Babetto, E., Coleman, M. P., and Martin, K. R. (2008). TheWldSgene Delays Axonal but not Somatic Degeneration in a Rat Glaucoma Model. Eur. J. Neurosci. 28, 1166–1179. doi:10.1111/j.1460-9568.2008.06426.x
Beirowski, B., Babetto, E., Gilley, J., Mazzola, F., Conforti, L., Janeckova, L., et al. (2009). Non-Nuclear WldS Determines its Neuroprotective Efficacy for Axons and Synapses In Vivo. J. Neurosci. 29, 653–668. doi:10.1523/JNEUROSCI.3814-08.2009
Beirowski, B., Berek, L., Adalbert, R., Wagner, D., Grumme, D. S., Addicks, K., et al. (2004). Quantitative and Qualitative Analysis of Wallerian Degeneration Using Restricted Axonal Labelling in YFP-H Mice. J. Neurosci. Methods 134, 23–35. doi:10.1016/j.jneumeth.2003.10.016
Belinda, L. W.-C., Wei, W. X., Hanh, B. T. H., Lei, L. X., Bow, H., and Ling, D. J. (2008). SARM: A Novel Toll-Like Receptor Adaptor, is Functionally Conserved from Arthropod to Human. Mol. Immunol. 45, 1732–1742. doi:10.1016/j.molimm.2007.09.030
Bloom, A. J., Mao, X., Strickland, A., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2021). Constitutively Active SARM1 Variants Found in ALS Patients Induce Neuropathy. bioRxiv. doi:10.1101/2021.04.16.439886
Boissonnas, A., Louboutin, F., Laviron, M., Loyher, P.-L., Reboussin, E., Barthelemy, S., et al. (2020). Imaging Resident and Recruited Macrophage Contribution to Wallerian Degeneration. J. Exp. Med. 217, 20200471. doi:10.1084/jem.20200471
Bosanac, T., Hughes, R. O., Engber, T., Devraj, R., Brearley, A., Danker, K., et al. (2021). Pharmacological SARM1 Inhibition Protects Axon Structure and Function in Paclitaxel-Induced Peripheral Neuropathy. Brain, awab184. doi:10.1093/brain/awab184
Bratkowski, M., Xie, T., Thayer, D. A., Lad, S., Mathur, P., Yang, Y.-S., et al. (2020). Structural and Mechanistic Regulation of the Pro-Degenerative NAD Hydrolase SARM1. Cell Rep. 32, 107999. doi:10.1016/j.celrep.2020.107999
Broz, P., and Dixit, V. M. (2016). Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 16, 407–420. doi:10.1038/nri.2016.58
Büki, A., Siman, R., Trojanowski, J. Q., and Povlishock, J. T. (1999). The Role of Calpail-Mediated Spectrin Proteolysis in Traumatically Induced Axonal Injury. J. Neuropathol. Exp. Neurol. 58, 365–375. doi:10.1097/00005072-199904000-00007
Burdett, H., Kobe, B., and Anderson, P. A. (2019). Animal NLRs Continue to Inform Plant NLR Structure and Function. Arch. Biochem. Biophys. 670, 58–68. doi:10.1016/j.abb.2019.05.001
Camacho-Pereira, J., Tarragó, M. G., Chini, C. C. S., Nin, V., Escande, C., Warner, G. M., et al. (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-dependent Mechanism. Cell Metab. 23, 1127–1139. doi:10.1016/j.cmet.2016.05.006
Cantó, C., Menzies, K. J., and Auwerx, J. (2015). NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act Between Mitochondria and the Nucleus. Cell Metab. 22, 31–53. doi:10.1016/j.cmet.2015.05.023
Carlsson, E., Ding, J. L., and Byrne, B. (2016). SARM Modulates MyD88-Mediated TLR Activation through BB-Loop Dependent TIR-TIR Interactions. Biochim. Biophys. Acta Mol. Cell Res. 1863, 244–253. doi:10.1016/j.bbamcr.2015.11.021
Carole, C., Nathalie, P., Jérôme, R., Laurence, S., Jean-François, G., Yuji, K., et al. (2004). TLR-independent Control of Innate Immunity in Caenorhabditis elegans by the TIR Domain Adaptor Protein TIR-1, an Ortholog of Human SARM. Nat. Immunol. 5, 488–494. doi:10.1038/ni1060
Carty, M., and Bowie, A. G. (2019). SARM: From Immune Regulator to Cell Executioner. Biochem. Pharmacol. 161, 52–62. doi:10.1016/j.bcp.2019.01.005
Carty, M., Goodbody, R., Schröder, M., Stack, J., Moynagh, P. N., and Bowie, A. G. (2006). The Human Adaptor SARM Negatively Regulates Adaptor Protein TRIF-dependent Toll-like Receptor Signaling. Nat. Immunol. 7, 1074–1081. doi:10.1038/ni1382
Carty, M., Kearney, J., Shanahan, K. A., Hams, E., Sugisawa, R., Connolly, D., et al. (2019). Cell Survival and Cytokine Release After Inflammasome Activation is Regulated by the Toll-IL-1R Protein SARM. Immunity 50, 1412–1424. doi:10.1016/j.immuni.2019.04.005
Cashman, C. R., and Höke, A. (2015). Mechanisms of Distal Axonal Degeneration in Peripheral Neuropathies. Neurosci. Lett. 596, 33–50. doi:10.1016/j.neulet.2015.01.048
Chen, B., Banton, M. C., Singh, L., Parkinson, D. B., and Dun, X.-P. (2021). Single Cell Transcriptome Data Analysis Defines the Heterogeneity of Peripheral Nerve Cells in Homeostasis and Regeneration. Front. Cell. Neurosci. 15, 624826. doi:10.3389/fncel.2021.624826
Cheng, H.-C., Ulane, C. M., and Burke, R. E. (2010). Clinical Progression in Parkinson Disease and the Neurobiology of Axons. Ann. Neurol. 67, 715–725. doi:10.1002/ana.21995
Chini, C. C. S., Peclat, T. R., Warner, G. M., Kashyap, S., Espindola-Netto, J. M., de Oliveira, G. C., et al. (2020). CD38 Ecto-Enzyme in Immune Cells is Induced During Aging and Regulates NAD+ and NMN Levels. Nat. Metab. 2, 1284–1304. doi:10.1038/s42255-020-00298-z
Chuang, C.-F., and Bargmann, C. I. (2005). A Toll-Interleukin 1 Repeat Protein at the Synapse Specifies Asymmetric Odorant Receptor Expression via ASK1 MAPKKK Signaling. Genes Develop. 19, 270–281. doi:10.1101/gad.1276505
Cohen, M. S., Ghosh, A. K., Kim, H. J., Jeon, N. L., and Jaffrey, S. R. (2012). Chemical Genetic-Mediated Spatial Regulation of Protein Expression in Neurons Reveals an Axonal Function for WldS. Chem. Biol. 19, 179–187. doi:10.1016/j.chembiol.2012.01.012
Coleman, M. (2005). Axon Degeneration Mechanisms: Commonality amid Diversity. Nat. Rev. Neurosci. 6, 889–898. doi:10.1038/nrn1788
Coleman, M. P., Conforti, L., Buckmaster, E. A., Tarlton, A., Ewing, R. M., Brown, M. C., et al. (1998). An 85-kb Tandem Triplication in the Slow Wallerian Degeneration (Wlds) Mouse. Proc. Natl. Acad. Sci. 95, 9985–9990. doi:10.1073/pnas.95.17.9985
Coleman, M. P., and Höke, A. (2020). Programmed Axon Degeneration: from Mouse to Mechanism to Medicine. Nat. Rev. Neurosci. 21, 183–196. doi:10.1038/s41583-020-0269-3
Conforti, L., Gilley, J., and Coleman, M. P. (2014). Wallerian Degeneration: An Emerging Axon Death Pathway Linking Injury and Disease. Nat. Rev. Neurosci. 15, 394–409. doi:10.1038/nrn3680
Conforti, L., Tarlton, A., Mack, T. G. A., Mi, W., Buckmaster, E. A., Wagner, D., et al. (2000). A Ufd2/D4Cole1e Chimeric Protein and Overexpression of Rbp7 in the Slow Wallerian Degeneration (WldS) Mouse. Proc. Natl. Acad. Sci. 97, 11377–11382. doi:10.1073/pnas.97.21.11377
Covarrubias, A. J., Kale, A., Perrone, R., Lopez-Dominguez, J. A., Pisco, A. O., Kasler, H. G., et al. (2020). Senescent Cells Promote Tissue NAD+ Decline during Ageing via the Activation of CD38+ Macrophages. Nat. Metab. 2, 1265–1283. doi:10.1038/s42255-020-00305-3
Dangl, J. L., Horvath, D. M., and Staskawicz, B. J. (2013). Pivoting the Plant Immune System from Dissection to Deployment. Science 341, 746–751. doi:10.1126/science.1236011
De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C. J. (2008). Role of Axonal Transport in Neurodegenerative Diseases. Annu. Rev. Neurosci. 31, 151–173. doi:10.1146/annurev.neuro.31.061307.090711
Demarest, T. G., Babbar, M., Okur, M. N., Dan, X., Croteau, D. L., Fakouri, N. B., et al. (2019). NAD+Metabolism in Aging and Cancer. Annu. Rev. Cancer Biol. 3, 105–130. doi:10.1146/annurev-cancerbio-030518-055905
Di Stefano, M., Loreto, A., Orsomando, G., Mori, V., Zamporlini, F., Hulse, R. P., et al. (2017). NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr. Biol. 27, 784–794. doi:10.1016/j.cub.2017.01.070
Di Stefano, M., Nascimento-Ferreira, I., Orsomando, G., Mori, V., Gilley, J., Brown, R., et al. (2015). A Rise in NAD Precursor Nicotinamide Mononucleotide (NMN) After Injury Promotes Axon Degeneration. Cell. Death. Differ. 22, 731–742. doi:10.1038/cdd.2014.164
Doron, S., Melamed, S., Ofir, G., Leavitt, A., Lopatina, A., Keren, M., et al. (2018). Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science 359, eaar4120. doi:10.1126/science.aar4120
Durães, F., Pinto, M., and Sousa, E. (2018). Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 11, 44. doi:10.3390/ph11020044
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD + Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 93, 1334–1343. doi:10.1016/j.neuron.2017.02.022
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., Yim, A. K. Y., DiAntonio, A., et al. (2018). TIR Domain Proteins are an Ancient Family of NAD+-consuming Enzymes. Curr. Biol. 28, 421–430. doi:10.1016/j.cub.2017.12.024
Feldman, E. L., Nave, K.-A., Jensen, T. S., and Bennett, D. L. H. (2017). New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 93, 1296–1313. doi:10.1016/j.neuron.2017.02.005
Fernandes, K. A., Mitchell, K. L., Patel, A., Marola, O. J., Shrager, P., Zack, D. J., et al. (2018). Role of SARM1 and DR6 in Retinal Ganglion Cell Axonal and Somal Degeneration Following Axonal Injury. Exp. Eye Res. 171, 54–61. doi:10.1016/j.exer.2018.03.007
Ferri, A., Sanes, J. R., Coleman, M. P., Cunningham, J. M., and Kato, A. C. (2003). Inhibiting Axon Degeneration and Synapse Loss Attenuates Apoptosis and Disease Progression in a Mouse Model of Motoneuron Disease. Curr. Biol. 13, 669–673. doi:10.1016/s0960-9822(03)00206-9
Figley, M. D., Gu, W., Nanson, J. D., Shi, Y., Sasaki, Y., Cunnea, K., et al. (2021). SARM1 is a Metabolic Sensor Activated by an Increased NMN/NAD+ Ratio to Trigger Axon Degeneration. Neuron 109, 1118–1136. doi:10.1016/j.neuron.2021.02.009
Fogh, I., Ratti, A., Gellera, C., Lin, K., Tiloca, C., Moskvina, V., et al. (2014). A Genome-Wide Association Meta-Analysis Identifies a Novel Locus at 17q11.2 Associated with Sporadic Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 23, 2220–2231. doi:10.1093/hmg/ddt587
Formentini, L., Moroni, F., and Chiarugi, A. (2009). Detection and Pharmacological Modulation of Nicotinamide Mononucleotide (NMN) In Vitro and In Vivo. Biochem. Pharmacol. 77, 1612–1620. doi:10.1016/j.bcp.2009.02.017
Geden, M. J., and Deshmukh, M. (2016). Axon Degeneration: Context Defines Distinct Pathways. Curr. Opin. Neurobiol. 39, 108–115. doi:10.1016/j.conb.2016.05.002
Geisler, S., Doan, R. A., Cheng, G. C., Cetinkaya-Fisgin, A., Huang, S. X., Höke, A., et al. (2019a). Vincristine and Bortezomib Use Distinct Upstream Mechanisms to Activate a Common SARM1-Dependent Axon Degeneration Program. JCI. Insight. 4, e129920. doi:10.1172/jci.insight.129920
Geisler, S., Doan, R. A., Strickland, A., Huang, X., Milbrandt, J., and DiAntonio, A. (2016). Prevention of Vincristine-Induced Peripheral Neuropathy by Genetic Deletion of SARM1 in Mice. Brain 139, 3092–3108. doi:10.1093/brain/aww251
Geisler, S., Huang, S. X., Strickland, A., Doan, R. A., Summers, D. W., Mao, X., et al. (2019b). Gene Therapy Targeting SARM1 Blocks Pathological Axon Degeneration in Mice. J. Exp. Med. 216, 294–303. doi:10.1084/jem.20181040
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 Activation Triggers Axon Degeneration Locally via NAD+ Destruction. Science 348, 453–457. doi:10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon Self-Destruction: New Links Among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. doi:10.1016/j.neuron.2015.12.023
Gerdts, J., Summers, D. W., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2013). Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J. Neurosci. 33, 13569–13580. doi:10.1523/JNEUROSCI.1197-13.2013
Gilley, J., Adalbert, R., Yu, G., and Coleman, M. P. (2013). Rescue of Peripheral and CNS Axon Defects in Mice Lacking NMNAT2. J. Neurosci. 33, 13410–13424. doi:10.1523/jneurosci.1534-13.2013
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an Essential Survival Factor for Maintenance of Healthy Axons. Plos. Biol. 8, e1000300. doi:10.1371/journal.pbio.1000300
Gilley, J., Jackson, O., Pipis, M., Estiar, M. A., Gan-Or, Z., Goutman, S. A., et al. (2021). Enrichment of SARM1 Alleles Encoding Variants with Constitutively Hyperactive NADase in Patients with ALS and Other Motor Nerve Disorders. medRxiv. doi:10.1101/2021.06.17.21258268
Gilley, J., Orsomando, G., Nascimento-Ferreira, I., and Coleman, M. P. (2015). Absence of SARM1 Rescues Development and Survival of NMNAT2-Deficient Axons. Cell Rep. 10, 1974–1981. doi:10.1016/j.celrep.2015.02.060
Gould, S. A., Gilley, J., Ling, K., Jaffar-Nejad, P., Ringo, F., and Coleman, M. (2021). Sarm1 Haploinsufficiency and Low Expression Levels After Antisense Oligonucleotides Delays Programmed Axon Degeneration. SSRN J. 45. doi:10.2139/ssrn.3806068
Gürtler, C., Carty, M., Kearney, J., Schattgen, S. A., Ding, A., Fitzgerald, K. A., et al. (2014). SARM Regulates CCL5 Production in Macrophages by Promoting the Recruitment of Transcription Factors and RNA Polymerase II to the Ccl5 Promoter. J. Immunol. 192 (10), 4821–4832. doi:10.4049/jimmunol.1302980
Guse, A. (2004). Biochemistry, Biology, and Pharmacology of Cyclic Adenosine Diphosphoribose (cADPR). Curr. Med. Chem. 11, 847–855. doi:10.2174/0929867043455602
Hasbani, D., and Omalley, K. (2006). WldS Mice are Protected Against the Parkinsonian Mimetic MPTP. Exp. Neurol. 202, 93–99. doi:10.1016/j.expneurol.2006.05.017
Henninger, N., Bouley, J., Sikoglu, E. M., An, J., Moore, C. M., King, J. A., et al. (2016). Attenuated Traumatic Axonal Injury and Improved Functional Outcome after Traumatic Brain Injury in Mice Lacking Sarm1. Brain 139, 1094–1105. doi:10.1093/brain/aww001
Horsefield, S., Burdett, H., Zhang, X., Manik, M. K., Shi, Y., Chen, J., et al. (2019). NAD+ Cleavage Activity by Animal and Plant TIR Domains in Cell Death Pathways. Science 365, 793–799. doi:10.1126/science.aax1911
Hou, Y.-J., Banerjee, R., Thomas, B., Nathan, C., García-Sastre, A., Ding, A., et al. (2013). SARM is Required for Neuronal Injury and Cytokine Production in Response to central Nervous System Viral Infection. J. Immunol. 191, 875–883. doi:10.4049/jimmunol.1300374
Howell, G. R., Libby, R. T., Jakobs, T. C., Smith, R. S., Phalan, F. C., Barter, J. W., et al. (2007). Axons of Retinal Ganglion Cells are Insulted in the Optic Nerve Early in DBA/2J Glaucoma. J. Cell. Biol. 179, 1523–1537. doi:10.1083/jcb.200706181
Hughes, R. O., Bosanac, T., Mao, X., Engber, T. M., DiAntonio, A., Milbrandt, J., et al. (2021). Small Molecule SARM1 Inhibitors Recapitulate the SARM1−/− Phenotype and Allow Recovery of a Metastable Pool of Axons Fated to Degenerate. Cell Rep. 34, 108588. doi:10.1016/j.celrep.2020.108588
Huppke, P., Wegener, E., Gilley, J., Angeletti, C., Kurth, I., Drenth, J. P. H., et al. (2019). Homozygous NMNAT2 Mutation in Sisters With Polyneuropathy and Erythromelalgia. Exp. Neurol. 320, 112958. doi:10.1016/j.expneurol.2019.112958
Jiang, Y., Liu, T., Lee, C.-H., Chang, Q., Yang, J., and Zhang, Z. (2020). The NAD+-Mediated Self-Inhibition Mechanism of Pro-Neurodegenerative Sarm1. Nature 588, 658–663. doi:10.1038/s41586-020-2862-z
Julian, V., and Byrne, A. B. (2020). TIR-1/SARM1 Inhibits Axon Regeneration. bioRxiv. doi:10.1101/2020.06.23.165852
Kawai, T., and Akira, S. (2010). The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat. Immunol. 11, 373–384. doi:10.1038/ni.1863
Kim, Y., Zhou, P., Qian, L., Chuang, J.-Z., Lee, J., Li, C., et al. (2007). MyD88-5 Links Mitochondria, Microtubules, and JNK3 in Neurons and Regulates Neuronal Survival. J. Exp. Med. 204, 2063–2074. doi:10.1084/jem.20070868
Klim, J. R., Williams, L. A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B. N., Mordes, D. A., et al. (2019). ALS-Implicated Protein TDP-43 Sustains Levels of STMN2, a Mediator of Motor Neuron Growth and Repair. Nat. Neurosci. 22, 167–179. doi:10.1038/s41593-018-0300-4
Ko, K. W., Milbrandt, J., and DiAntonio, A. (2020). SARM1 Acts Downstream of Neuroinflammatory and Necroptotic Signaling to Induce Axon Degeneration. J. Cell. Biol. 219, e201912047. doi:10.1083/jcb.201912047
Lapointe, N. E., Morfini, G., Brady, S. T., Feinstein, S. C., Wilson, L., and Jordan, M. A. (2013). Effects of Eribulin, Vincristine, Paclitaxel and Ixabepilone on Fast Axonal Transport and Kinesin-1 Driven Microtubule Gliding: Implications for Chemotherapy-Induced Peripheral Neuropathy. Neurotoxicology 37, 231–239. doi:10.1016/j.neuro.2013.05.008
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 30, 630–655. doi:10.1016/j.cmet.2019.09.001
Li, W. H., Huang, K., Cai, Y., Wang, Q. W., Zhu, W. J., Hou, Y. N., et al. (2021). Permeant Fluorescent Probes Visualize the Activation of SARM1 and Uncover an Anti-Neurodegenerative Drug Candidate. bioRxiv. doi:10.1101/2021.02.24.432704
Lindborg, J. A., Mack, M., and Zigmond, R. E. (2017). Neutrophils Are Critical for Myelin Removal in a Peripheral Nerve Injury Model of Wallerian Degeneration. J. Neurosci. 37, 10258–10277. doi:10.1523/jneurosci.2085-17.2017
Liu, K., Yang, L., Wang, G., Liu, J., Zhao, X., Wang, Y., et al. (2021). Metabolic Stress Drives Sympathetic Neuropathy within the Liver. Cell Metab. 33, 666–675. doi:10.1016/j.cmet.2021.01.012
Loreto, A., Angeletti, C., Gu, W., Osborne, A., Nieuwenhuis, B., Gilley, J., et al. (2020a). Potent Activation of SARM1 by NMN Analogue VMN Underlies Vacor Neurotoxicity. bioRxiv. doi:10.1101/2020.09.18.304261
Loreto, A., Di Stefano, M., Gering, M., and Conforti, L. (2015). Wallerian Degeneration is Executed by an NMN-SARM1-dependent Late Ca2+ Influx but Only Modestly Influenced by Mitochondria. Cell Rep. 13, 2539–2552. doi:10.1016/j.celrep.2015.11.032
Loreto, A., Hill, C. S., Hewitt, V. L., Orsomando, G., Angeletti, C., Gilley, J., et al. (2020b). Mitochondrial Impairment Activates the Wallerian Pathway through Depletion of NMNAT2 Leading to SARM1-dependent Axon Degeneration. Neurobiol. Dis. 134, 104678. doi:10.1016/j.nbd.2019.104678
Loring, H. S., and Thompson, P. R. (2020). Emergence of SARM1 as a Potential Therapeutic Target for Wallerian-Type Diseases. Cell Chem. Biol. 27, 1–13. doi:10.1016/j.chembiol.2019.11.002
Lubińska, L. (1977). Early Course of Wallerian Degeneration in Myelinated Fibres of the Rat Phrenic Nerve. Brain Res. 130, 47–63. doi:10.1016/0006-8993(77)90841-1
Lubińska, L. (1982). Patterns of Wallerian Degeneration of Myelinated Fibres in Short and Long Peripheral Stumps and in Isolated Segments of Rat Phrenic Nerve. Interpretation of the Role of Axoplasmic Flow of the Trophic Factor. Brain Res. 233, 227–240. doi:10.1016/0006-8993(82)91199-4
Lukacs, M., Gilley, J., Zhu, Y., Orsomando, G., Angeletti, C., Liu, J., et al. (2019). Severe Biallelic Loss-of-Function Mutations in Nicotinamide Mononucleotide Adenylyltransferase 2 (NMNAT2) in Two Fetuses with Fetal Akinesia Deformation Sequence. Exp. Neurol. 320, 112961. doi:10.1016/j.expneurol.2019.112961
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of Wallerian Degeneration Does Not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1, 27–33. doi:10.1111/j.1460-9568.1989.tb00771.x
Luo, X., and Kraus, W. L. (2012). On PAR with PARP: Cellular Stress Signaling Through poly(ADP-Ribose) and PARP-1. Genes Develop. 26, 417–432. doi:10.1101/gad.183509.111
Ma, S., Lapin, D., Liu, L., Sun, Y., Song, W., Zhang, X., et al. (2020). Direct Pathogen-Induced Assembly of an NLR Immune Receptor Complex to Form a Holoenzyme. Science 370, eabe3069. doi:10.1126/science.abe3069
Mabbitt, P. D., Loreto, A., DéryFletcher, M.-A. A. J., Fletcher, A. J., Stanley, M., Pao, K.-C., et al. (2020). Structural Basis for RING-Cys-Relay E3 Ligase Activity and its Role in Axon Integrity. Nat. Chem. Biol. 16, 1227–1236. doi:10.1038/s41589-020-0598-6
Mack, T. G. A., Reiner, M., Beirowski, B., Mi, W., Emanuelli, M., Wagner, D., et al. (2001). Wallerian Degeneration of Injured Axons and Synapses is Delayed by a Ube4b/Nmnat Chimeric Gene. Nat. Neurosci. 4, 1199–1206. doi:10.1038/nn770
Maekawa, T., Kufer, T. A., and Schulze-Lefert, P. (2011). NLR Functions in Plant and Animal Immune Systems: So Far and yet So Close. Nat. Immunol. 12, 817–826. doi:10.1038/ni.2083
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2013). Comparative Genomics of Defense Systems in Archaea and Bacteria. Nucleic Acids Res. 41, 4360–4377. doi:10.1093/nar/gkt157
Malavasi, F., Deaglio, S., Funaro, A., Ferrero, E., Horenstein, A. L., Ortolan, E., et al. (2008). Evolution and Function of the ADP Ribosyl cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 88, 841–886. doi:10.1152/physrev.00035.2007
Martin, R., Qi, T., Zhang, H., Liu, F., King, M., Toth, C., et al. (2020). Structure of the Activated ROQ1 Resistosome Directly Recognizing the Pathogen Effector XopQ. Science 370, eabd9993. doi:10.1126/science.abd9993
Melamed, Z. e., López-Erauskin, J., Baughn, M. W., Zhang, O., Drenner, K., Sun, Y., et al. (2019). Premature Polyadenylation-Mediated Loss of Stathmin-2 Is a Hallmark of TDP-43-Dependent Neurodegeneration. Nat. Neurosci. 22, 180–190. doi:10.1038/s41593-018-0293-z
Milde, S., Gilley, J., and Coleman, M. P. (2013). Subcellular Localization Determines the Stability and Axon Protective Capacity of Axon Survival Factor Nmnat2. PLoS Biol. 11, e1001539. doi:10.1371/journal.pbio.1001539
Mink, M., Fogelgren, B., Olszewski, K., Maroy, P., and Csiszar, K. (2001). A Novel Human Gene (SARM) at Chromosome 17q11 Encodes a Protein with a SAM Motif and Structural Similarity to Armadillo/β-Catenin that is Conserved in Mouse, Drosophila, and Caenorhabditis elegans. Genomics 74, 234–244. doi:10.1006/geno.2001.6548
Mori, V., Amici, A., Mazzola, F., Di Stefano, M., Conforti, L., Magni, G., et al. (2014). Metabolic Profiling of Alternative NAD Biosynthetic Routes in Mouse Tissues. PLoS One 9, e113939. doi:10.1371/journal.pone.011393910.1371/journal.pone.0113939
Nanson, J. D., Kobe, B., and Ve, T. (2019). Death, TIR, and RHIM: Self‐Assembling Domains Involved in Innate Immunity and Cell‐Death Signaling. J. Leukoc. Biol. 105, 363–375. doi:10.1002/jlb.mr0318-123R
Navarro, M. N., Gómez de las Heras, M. M., and Mittelbrunn, M. (2021). Nicotinamide Adenine Dinucleotide Metabolism in the Immune Response, Autoimmunity and Inflammageing. Br. J. Pharmacol. 1–18. doi:10.1111/bph.15477
Navas, L. E., and Carnero, A. (2021). NAD+ Metabolism, Stemness, the Immune Response, and Cancer. Signal Transduct. Target Ther. 6, 2. doi:10.1038/s41392-020-00354-w
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., et al. (2017). Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron 95, 78–91. doi:10.1016/j.neuron.2017.06.031
Nikiforov, A., Kulikova, V., and Ziegler, M. (2015). The Human NAD Metabolome: Functions, Metabolism and Compartmentalization. Crit. Rev. Biochem. Mol. Biol. 50, 284–297. doi:10.3109/10409238.2015.1028612
Nimma, S., Ve, T., Williams, S. J., and Kobe, B. (2017). Towards the Structure of the TIR-Domain Signalosome. Curr. Opin. Struct. Biol. 43, 122–130. doi:10.1016/j.sbi.2016.12.014
O’Neill, L. A. J., Fitzgerald, K. A., and Bowie, A. G. (2003). The Toll-IL-1 Receptor Adaptor Family Grows to Five Members. Trends Immunol. 24, 286–289. doi:10.1016/s1471-4906(03)00115-7
O'Neill, L. A. J., Golenbock, D., and Bowie, A. G. (2013). The History of Toll-Like Receptors - Redefining Innate Immunity. Nat. Rev. Immunol. 13, 453–460. doi:10.1038/nri3446
Ofir, G., Herbst, E., Baroz, M., Cohen, D., Millman, A., Doron, S., et al. (2021). Antiviral Activity of Bacterial TIR Domains via Signaling Molecules that Trigger Cell Death. bioRxiv. doi:10.1101/2021.01.06.425286
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is Required for Activation of an Injury-Induced Axon Death Pathway. Science 337, 481–484. doi:10.1126/science.1223899
Pan, Z.-G., and An, X.-S. (2018). SARM1 Deletion Restrains NAFLD Induced by High Fat Diet (HFD) through Reducing Inflammation, Oxidative Stress and Lipid Accumulation. Biochem. Biophys. Res. Commun. 498, 416–423. doi:10.1016/j.bbrc.2018.02.115
Panneerselvam, P., and Ding, J. L. (2015). Beyond TLR Signaling-The Role of SARM in Antiviral Immune Defense, Apoptosis & Development. Int. Rev. Immunol. 34, 432–444. doi:10.3109/08830185.2015.1065826
Peng, J., Yuan, Q., Lin, B., Panneerselvam, P., Wang, X., Luan, X. L., et al. (2010). SARM Inhibits Both TRIF- and MyD88-Mediated AP-1 Activation. Eur. J. Immunol. 40, 1738–1747. doi:10.1002/eji.200940034
Perkins, N. M., and Tracey, D. J. (2000). Hyperalgesia Due to Nerve Injury: Role of Neutrophils. Neuroscience 101, 745–757. doi:10.1016/S0306-4522(00)00396-1
Perraud, A.-L., Fleig, A., Dunn, C. A., Bagley, L. A., Launay, P., Schmitz, C., et al. (2001). ADP-Ribose Gating of the Calcium-Permeable LTRPC2 Channel Revealed by Nudix Motif Homology. Nature 411, 595–599. doi:10.1038/35079100
Peters, O. M., Lewis, E. A., Osterloh, J. M., Weiss, A., Salameh, J. S., Metterville, J., et al. (2018). Loss of Sarm1 Does Not Suppress Motor Neuron Degeneration in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 27, 3761–3771. doi:10.1093/hmg/ddy260
Press, C., and Milbrandt, J. (2008). Nmnat Delays Axonal Degeneration Caused by Mitochondrial and Oxidative Stress. J. Neurosci. 28, 4861–4871. doi:10.1523/JNEUROSCI.0525-08.2008
Pudla, M., Limposuwan, K., and Utaisincharoen, P. (2011). Burkholderia Pseudomallei-Induced Expression of a Negative Regulator, Sterile-α and Armadillo Motif-Containing Protein, in Mouse Macrophages: A Possible Mechanism for Suppression of the MyD88-Independent Pathway. Infect. Immun. 79, 2921–2927. doi:10.1128/IAI.01254-10
Ramón y Cajal, S. (1991). Cajal's Degeneration & Regeneration of the Nervous System. London, United Kingdom: Oxford University Press. ISBN: 0195065166
Saatman, K. E., Creed, J., and Raghupathi, R. (2010). Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 7, 31–42. doi:10.1016/j.nurt.2009.11.002
Sajadi, A., Schneider, B. L., and Aebischer, P. (2004). Wlds-Mediated protection of Dopaminergic Fibers in an Animal Model of Parkinson Disease. Curr. Biol. 14, 326–330. doi:10.1016/j.cub.2004.01.053
Sasaki, Y., Nakagawa, T., Mao, X., DiAntonio, A., and Milbrandt, J. (2016). NMNAT1 Inhibits Axon Degeneration via Blockade of SARM1-Mediated NAD+ Depletion. eLife 5, e19749. doi:10.7554/eLife.19749
Sasaki, Y., Vohra, B. P. S., Baloh, R. H., and Milbrandt, J. (2009a). Transgenic Mice Expressing the Nmnat1 Protein Manifest Robust Delay in Axonal Degeneration In Vivo. J. Neurosci. 29, 6526–6534. doi:10.1523/JNEUROSCI.1429-09.2009
Sasaki, Y., Vohra, B. P. S., Lund, F. E., and Milbrandt, J. (2009b). Nicotinamide Mononucleotide Adenylyl Transferase-Mediated Axonal Protection Requires Enzymatic Activity but Not Increased Levels of Neuronal Nicotinamide Adenine Dinucleotide. J. Neurosci. 29, 5525–5535. doi:10.1523/JNEUROSCI.5469-08.2009
Shen, C., Vohra, M., Zhang, P., Mao, X., Figley, M. D., Zhu, J., et al. (2021). Multiple Domain Interfaces Mediate SARM1 Autoinhibition. Proc. Natl. Acad. Sci. USA 118, e2023151118. doi:10.1073/pnas.2023151118
Simon, D. J., and Watkins, T. A. (2018). Therapeutic Opportunities and Pitfalls in the Treatment of Axon Degeneration. Curr. Opin. Neurol. 31, 693–701. doi:10.1097/WCO.0000000000000621
Singhal, A., and Cheng, C. Y. (2019). Host NAD+ Metabolism and Infections: Therapeutic Implications. Int. Immunol. 31, 59–67. doi:10.1093/intimm/dxy068
Sporny, M., Guez-Haddad, J., Khazma, T., Yaron, A., Dessau, M., Shkolnisky, Y., et al. (2020). Structural Basis for SARM1 Inhibition and Activation under Energetic Stress. Elife 9, e62021. doi:10.7554/eLife.62021
Sporny, M., Guez-Haddad, J., Lebendiker, M., Ulisse, V., Volf, A., Mim, C., et al. (2019). Structural Evidence for an Octameric Ring Arrangement of SARM1. J. Mol. Biol. 431, 3591–3605. doi:10.1016/j.jmb.2019.06.030
Sta, M., Cappaert, N. L. M., Ramekers, D., Baas, F., and Wadman, W. J. (2014). The Functional and Morphological Characteristics of Sciatic Nerve Degeneration and Regeneration after Crush Injury in Rats. J. Neurosci. Methods 222, 189–198. doi:10.1016/j.jneumeth.2013.11.012
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and Transport Deficits Early in the Pathogenesis of Alzheimer’s Disease. Science 307, 1282–1288. doi:10.1126/science.1105681
Summers, D. W., DiAntonio, A., and Milbrandt, J. (2014). Mitochondrial Dysfunction Induces Sarm1-Dependent Cell Death in Sensory Neurons. J. Neurosci. 34, 9338–9350. doi:10.1523/JNEUROSCI.0877-14.2014
Summers, D. W., Frey, E., Walker, L. J., Milbrandt, J., and DiAntonio, A. (2020). DLK Activation Synergizes with Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Mol. Neurobiol. 57, 1146–1158. doi:10.1007/s12035-019-01796-2
Summers, D. W., Milbrandt, J., and DiAntonio, A. (2018). Palmitoylation Enables MAPK-Dependent Proteostasis of Axon Survival Factors. Proc. Natl. Acad. Sci. USA 115, E8746–E8754. doi:10.1073/pnas.1806933115
Szretter, K. J., Samuel, M. A., Gilfillan, S., Fuchs, A., Colonna, M., and Diamond, M. S. (2009). The Immune Adaptor Molecule SARM Modulates Tumor Necrosis Factor Alpha Production and Microglia Activation in the Brainstem and Restricts West Nile Virus Pathogenesis. J. Virol. 83, 9329–9338. doi:10.1128/JVI.00836-09
Thompson, M. R., Kaminski, J. J., Kurt-Jones, E. A., and Fitzgerald, K. A. (2011). Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 3, 920–940. doi:10.3390/v3060920
Tsao, J. W., George, E. B., and Griffin, J. W. (1999). Temperature Modulation Reveals Three Distinct Stages of Wallerian Degeneration. J. Neurosci. 19, 4718–4726. doi:10.1523/JNEUROSCI.19-12-04718.1999
Turkiew, E., Falconer, D., Reed, N., and Höke, A. (2017). Deletion of Sarm1 Gene is Neuroprotective in Two Models of Peripheral Neuropathy. J. Peripher. Nerv. Syst. 22, 162–171. doi:10.1111/jns.12219
Uccellini, M. B., Bardina, S. V., Sánchez-Aparicio, M. T., White, K. M., Hou, Y.-J., Lim, J. K., et al. (2020). Passenger Mutations Confound Phenotypes of SARM1-Deficient Mice. Cell Rep. 31, 107498. doi:10.1016/j.celrep.2020.03.062
Vajjhala, P. R., Ve, T., Bentham, A., Stacey, K. J., and Kobe, B. (2017). The Molecular Mechanisms of Signaling by Cooperative Assembly Formation in Innate Immunity Pathways. Mol. Immunol. 86, 23–37. doi:10.1016/j.molimm.2017.02.012
van Rheenen, W., Shatunov, A., Dekker, A. M., Mclaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-Wide Association Analyses Identify New Risk Variants and the Genetic Architecture of Amyotrophic Lateral Sclerosis. Nat. Genet. 48, 1043–1048. doi:10.1038/ng.3622
Ve, T., Williams, S. J., and Kobe, B. (2015). Structure and Function of Toll/Interleukin-1 Receptor/Resistance Protein (TIR) Domains. Apoptosis 20, 250–261. doi:10.1007/s10495-014-1064-2
Vérièpe, J., Fossouo, L., and Parker, J. A. (2015). Neurodegeneration in C. elegans Models of ALS Requires TIR-1/Sarm1 Immune Pathway Activation in Neurons. Nat. Commun. 6, 7319. doi:10.1038/ncomms8319
Walker, L. J., Summers, D. W., Sasaki, Y., Brace, E., Milbrandt, J., and DiAntonio, A. (2017). MAPK Signaling Promotes Axonal Degeneration by Speeding the Turnover of the Axonal Maintenance Factor NMNAT2. Elife 6, e22540. doi:10.7554/eLife.22540
Waller, A. (1851). Experiments on the Section of the Glosso-Pharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres. Edinb. Med. Surg. J. 76, 369–376. doi:10.1098/rstl.1850.0021
Wan, L., Essuman, K., Anderson, R. G., Sasaki, Y., Monteiro, F., Chung, E.-H., et al. (2019). TIR Domains of Plant Immune Receptors Are NAD+-Cleaving Enzymes that Promote Cell Death. Science 365, 799–803. doi:10.1126/science.aax1771
Wang, J., Fox, M. A., and Povlishock, J. T. (2013). Diffuse Traumatic Axonal Injury in the Optic Nerve Does Not Elicit Retinal Ganglion Cell Loss. J. Neuropathol. Exp. Neurol. 72, 768–781. doi:10.1097/NEN.0b013e31829d8d9d
Wang, J. T., Medress, Z. A., Vargas, M. E., and Barres, B. A. (2015). Local Axonal Protection by WldS as Revealed by Conditional Regulation of Protein Stability. Proc. Natl. Acad. Sci. USA 112, 10093–10100. doi:10.1073/pnas.1508337112
Wang, J., Zhai, Q., Chen, Y., Lin, E., Gu, W., Mcburney, M. W., et al. (2005). A Local Mechanism Mediates NAD-Dependent Protection of Axon Degeneration. J. Cel. Biol. 170, 349–355. doi:10.1083/jcb.200504028
Wang, Q., Zhang, S., Liu, T., Wang, H., Liu, K., Wang, Q., et al. (2018). Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep. 23, 716–724. doi:10.1016/j.celrep.2018.03.071
Warburg, O., and Christian, W. (1936). Pyridin, der wasserstoffübertragende Bestandteil von Gärungsfermenten. Helv. Chim. Acta. 19, E79–E88. doi:10.1002/hlca.193601901199
White, M. A., Lin, Z., Kim, E., Henstridge, C. M., Pena Altamira, E., Hunt, C. K., et al. (2019). Sarm1 Deletion Suppresses TDP-43-Linked Motor Neuron Degeneration and Cortical Spine Loss. Acta Neuropathol. Commun. 7, 166. doi:10.1186/s40478-019-0800-9
Williams, P. A., Harder, J. M., Foxworth, N. E., Cardozo, B. H., Cochran, K. E., and John, S. W. M. (2017a). Nicotinamide and WLDS Act Together to Prevent Neurodegeneration in Glaucoma. Front. Neurosci. 11, 232. doi:10.3389/fnins.2017.00232
Williams, P. A., Harder, J. M., Foxworth, N. E., Cochran, K. E., Philip, V. M., Porciatti, V., et al. (2017b). Vitamin B3 modulates Mitochondrial Vulnerability and Prevents Glaucoma in Aged Mice. Science 355, 756–760. doi:10.1126/science.aal0092
Xiong, X., Hao, Y., Sun, K., Li, J., Li, X., Mishra, B., et al. (2012). The Highwire Ubiquitin Ligase Promotes Axonal Degeneration by Tuning Levels of Nmnat Protein. PLoS Biol. 10, e1001440. doi:10.1371/journal.pbio.1001440
Yahata, N., Yuasa, S., and Araki, T. (2009). Nicotinamide Mononucleotide Adenylyltransferase Expression in Mitochondrial Matrix Delays Wallerian Degeneration. J. Neurosci. 29, 6276–6284. doi:10.1523/JNEUROSCI.4304-08.2009
Yan, T., Feng, Y., Zheng, J., Ge, X., Zhang, Y., Wu, D., et al. (2010). Nmnat2 Delays Axon Degeneration in Superior Cervical Ganglia Dependent on its NAD Synthesis Activity. Neurochem. Int. 56, 101–106. doi:10.1016/j.neuint.2009.09.007
Yang, J., Weimer, R. M., Kallop, D., Olsen, O., Wu, Z., Renier, N., et al. (2013). Regulation of Axon Degeneration after Injury and in Development by the Endogenous Calpain Inhibitor Calpastatin. Neuron 80, 1175–1189. doi:10.1016/j.neuron.2013.08.034
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological Axonal Death through a MAPK Cascade that Triggers a Local Energy Deficit. Cell 160, 161–176. doi:10.1016/j.cell.2014.11.053
Ydens, E., Amann, L., Asselbergh, B., Scott, C. L., Martens, L., Sichien, D., et al. (2020). Profiling Peripheral Nerve Macrophages Reveals Two Macrophage Subsets with Distinct Localization, Transcriptome and Response to Injury. Nat. Neurosci. 23, 676–689. doi:10.1038/s41593-020-0618-6
Yi, S., Tang, X., Yu, J., Liu, J., Ding, F., and Gu, X. (2017). Microarray and qPCR Analyses of Wallerian Degeneration in Rat Sciatic Nerves. Front. Cell. Neurosci. 11, 22. doi:10.3389/fncel.2017.00022
Ying, W. (2008). NAD+/NADH and NADP+/NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 10, 179–206. doi:10.1089/ars.2007.1672
Zhao, Z. Y., Xie, X. J., Li, W. H., Liu, J., Chen, Z., Zhang, B., et al. (2019). A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. Science 15, 452–466. doi:10.1016/j.isci.2019.05.001
Zhou, X., Jiang, T., Du, X., Zhou, P., Jiang, Z., Michal, J. J., et al. (2013). Molecular Characterization of Porcine SARM1 and its Role in Regulating TLRs Signaling during Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus Infection In Vivo. Develop. Comp. Immunol. 39, 117–126. doi:10.1016/j.dci.2012.02.001
Zhu, Y., Zhang, L., Sasaki, Y., Milbrandt, J., and Gidday, J. M. (2013). Protection of Mouse Retinal Ganglion Cell Axons and Soma from Glaucomatous and Ischemic Injury by Cytoplasmic Overexpression of Nmnat1. Invest. Ophthalmol. Vis. Sci. 54, 25–36. doi:10.1167/iovs.12-10861
Ziegler, M., and Oei, S. L. (2001). A Cellular Survival Switch: Poly(ADP-Ribosyl)ation Stimulates DNA Repair and Silences Transcription. Bioessays 23, 543–548. doi:10.1002/bies.1074
Zigmond, R. E., and Echevarria, F. D. (2019). Macrophage Biology in the Peripheral Nervous System after Injury. Prog. Neurobiol. 173, 102–121. doi:10.1016/j.pneurobio.2018.12.001
Keywords: NAD, NMNAT2, Sarm1, axon degeneration, innate immunity
Citation: Hopkins EL, Gu W, Kobe B and Coleman MP (2021) A Novel NAD Signaling Mechanism in Axon Degeneration and its Relationship to Innate Immunity. Front. Mol. Biosci. 8:703532. doi: 10.3389/fmolb.2021.703532
Received: 30 April 2021; Accepted: 28 June 2021;
Published: 08 July 2021.
Edited by:
Enrico Balducci, University of Camerino, ItalyReviewed by:
Janos Groh, University Hospital Würzburg, GermanyAntonio Orlacchio, Santa Lucia Foundation (IRCCS), Italy
Copyright © 2021 Hopkins, Gu, Kobe and Coleman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael P. Coleman, mc469@cam.ac.uk
†These authors have contributed equally to this work