 Eiseul Kim
Eiseul Kim Seung-Min Yang
Seung-Min Yang Dayoung Kim
Dayoung Kim Hae-Yeong Kim
Hae-Yeong Kim- Institute of Life Sciences & Resources and Department of Food Science and Biotechnology, Kyung Hee University, Yongin, South Korea
Lacticaseibacillus casei, Lacticaseibacillus chiayiensis, and Lacticaseibacillus zeae are very closely related Lacticaseibacillus species. L. casei has long been proposed as a probiotic, whereas studies on functional characterization for L. chiayiensis and L. zeae are some compared to L. casei. In this study, L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 were isolated from raw milk, and their probiotic properties were investigated. Genomic analysis demonstrated the role of L. chiayiensis and L. zeae as probiotic candidates. The three strains were tolerant to acid and bile salt, with inhibitory action against pathogenic bacterial strains and capacity of antioxidants. Complete genome sequences of the three strains were analyzed to highlight the probiotic properties at the genetic level, which results in the discovery of genes corresponding to phenotypic characterization. Moreover, genes known to confer probiotic characteristics were identified, including genes related to biosynthesis, defense machinery, adhesion, and stress adaptation. The comparative genomic analysis with other available genomes revealed 256, 214, and 32 unique genes for FBL6, FBL7, and FBL8, respectively. These genomes contained individual genes encoding proteins that are putatively involved in carbohydrate transport and metabolism, prokaryotic immune system for antiviral defense, and physiological control processes. In particular, L. casei FBL6 had a bacteriocin gene cluster that was not present in other genomes of L. casei, resulting in this strain may exhibit a wide range of antimicrobial activity compared to other L. casei strains. Our data can help us understand the probiotic functionalities of the three strains and suggest that L. chiayiensis and L. zeae species, which are closely related to L. casei, can also be considered as novel potential probiotic candidate strains.
Introduction
Probiotics are defined as live microorganisms that benefit host health when ingested in adequate amounts (Ye et al., 2020). Lactic acid bacteria can be utilized as probiotics provided that they have the desired properties, such as safety assurance for consumers, stability, and persistence in the gastrointestinal tract (Ye et al., 2020). Moreover, the ability to inhibit pathogens’ growth, improve microbial balance in the intestines, and regulate mucosal and systemic immunity is required. Many strains belonging to lactic acid bacteria function as probiotics, and they also play a significant role in improving the quality of fermented foods (Zhang et al., 2018).
Lacticaseibacillus casei, Lacticaseibacillus chiayiensis, and Lacticaseibacillus zeae belong to the Lacticaseibacillus species and are very closely related phenotypically and genotypically along with Lacticaseibacillus paracasei and Lacticaseibacillus rhamnosus (Kim et al., 2020b). L. casei is one of the most studied lactic acid bacterial strains associated with probiotics and used in industrial and applied health applications (Hill et al., 2018). Strains considered to be probiotics have been proven to exhibit beneficial properties by promoting health associated with the regulation of immune response and improvement of atopic dermatitis (Mazé et al., 2010; Kang et al., 2017). L. casei is often used in the fermentation of dairy products, resulting in improved flavor and texture (Hill et al., 2018). L. chiayiensis was first isolated in 2018 from cow manure (Huang et al., 2018), and since then, this species has not been reported anymore. L. zeae, designated as an independent species from L. casei in 2020, has been said to exhibit antimicrobial activity against two strains of enterotoxigenic Escherichia coli (Zhou et al., 2014; Huang et al., 2020; Chen et al., 2021).
Whole-genome sequencing of potential probiotic strains ensures accurate taxonomic assignment while also providing genetic data concerning the presence of probiotic traits, possible metabolic activities, and safety assessment such as virulence factors, antibiotic resistance genes, and genetics related to hazardous metabolites (Guinane et al., 2016; Li et al., 2017). However, despite its importance for predicting the critical abilities of putative probiotic strains, the current genomic information of L. zeae and L. chiayiensis is fairly limited, resulting in impossible genetic access. Currently, one complete genome of L. zeae was reported but none for L. chiayiensis. However, the draft genome of L. chiayiensis has been reported in the literature (Huang et al., 2018). Also, reports on probiotic properties and genomic evidence to support their functionality are not yet available for the two species. Genomic analysis, together with experimental studies, is important for constructing a probiotic profile of a novel strain. Therefore, in this study, we investigated a phenotypic assay for L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 and identified related genes that are responsible for their probiotic properties. Moreover, to gain a better insight into the probiotic effects, the comparative genomic analysis was conducted among the newly sequenced three strains and publicly available genomes, as well as a comparison of genetic characteristics of the three species.
Materials and Methods
Isolation of Strains and Identification
Raw milk samples were randomly collected from eight different ranches in Gyeonggi-do, Korea. Prior to the collection of raw milk samples, the udder was washed with distilled water and dried. The samples were obtained and directly placed into sterile tubes and immediately transferred to the laboratory. For the isolation of lactic acid bacterial strains, the serially diluted milk samples were spread on an MRS agar plate (Difco, Becton & Dickinson, Sparks, MD, United States) and incubated at 37°C for 48 h under anaerobic conditions. The different individual colonies were randomly selected according to morphological differences and purified on MRS broth (Difco). All isolates were stored at −80°C in glycerol until probiotic characterization.
Genomic DNA of the presumptive Lacticaseibacillus strain was extracted using the DNeasy Blood & Tissue Kits (Qiagen, Hilden, Germany) following the manufacturer’s protocol to identify isolates. These strains were identified via 16S rRNA gene sequencing with 27F/1492R primer pairs. The identification of L. casei, L. chiayiensis, and L. zeae was performed using a real-time PCR method targeting unique genes for each species (Kim et al., 2020b,2021). Three isolates demonstrating higher probiotic activities compared with other isolates were finally selected and designated respectively as FBL6, FBL7, and FBL8.
Genome Sequencing and Assembly
Cultured cells were harvested via centrifugation at 13,600 × g for 5 min. The total genomic DNA for genome sequencing was extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, United States) following the manufacturer’s protocols. Genomic DNA was sheared into 10-kb fragments, and then SMRTbell libraries were prepared using the SMRTbell Template Prep Kit v. 1.0 (Pacific Bioscience, Menlo Park, CA, United States). Prepared libraries were sequenced on a PacBio Sequel I sequencer (Pacific Bioscience) using the Sequel Sequencing Kit v. 3.0 on single-molecule real-time sequencing technology cell 1M v. 2.0. The generated raw reads were filtered to remove adapter sequences and short reads. Filtered subreads were assembled through the hierarchical genome assembly process.
Genomic Analysis
Genes of genomes were predicted using Prokka v. 1.12, and the corresponding function annotation was performed by Clusters of Orthologous Groups (COGs). The most important probiotic genes and stress-related genes were determined by the Rapid Annotations using Subsystem Technology and Prokka. Putative virulence factors were detected using VirulenceFinder v. 2.0, and antimicrobial resistance genes acquired within three genomes were identified using the Comprehensive Antibiotic Resistance Database (CARD) v. 3.1.1 and ResFinder v. 4.1. The pathogenicity of the three genomes was determined using PathogenFinder v. 1.1. Putative prophage insert regions were identified using the PHage Search Tool Enhanced Release (PHASTER) (Arndt et al., 2016). Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated genes (Cas) were detected using CRISPRCasFinder (Couvin et al., 2018). Secondary metabolite biosynthesis gene clusters were identified using antiSMASH bacterial v. 5.0 (Blin et al., 2019). Carbohydrate-active enzymes (CAZymes) within three genomes were identified using the CAZy database.1
Comparative Genomic Analysis
For the comparative genomic analysis, publicly available genome sequences of L. casei, L. chiayiensis, L. zeae, and other reported Lacticaseibacillus species, L. paracasei, and L. rhamnosus, were downloaded from the NCBI database (Table 1). The phylogenetic relatedness among the FBL6, FBL7, and FBL8 strains and other strains was investigated using 16S rRNA gene sequences, average nucleotide identity (ANI), and core and pan-genome sequences to infer their evolutionary relationships. 16S rRNA gene sequences were extracted from whole-genome sequences, and a phylogenetic tree was created using the neighbor-joining method. A phylogenetic identification by ANI analysis was conducted using orthologous average nucleotide identity (OrthoANI) (Lee et al., 2016). A phylogenetic tree based on pan-genome was constructed using the pan-genome workflow in the Anvi’o software v. 6.0 (Eren et al., 2015). Pan-genome and core-genome were analyzed using the Bacterial Pan-genome Analysis (BPGA) tool v. 1.3 with the default parameters. The assignment of COG functional categories for genes derived from core-genome and pan-genome was performed using USEARCH in the BPGA tool against the COG database.
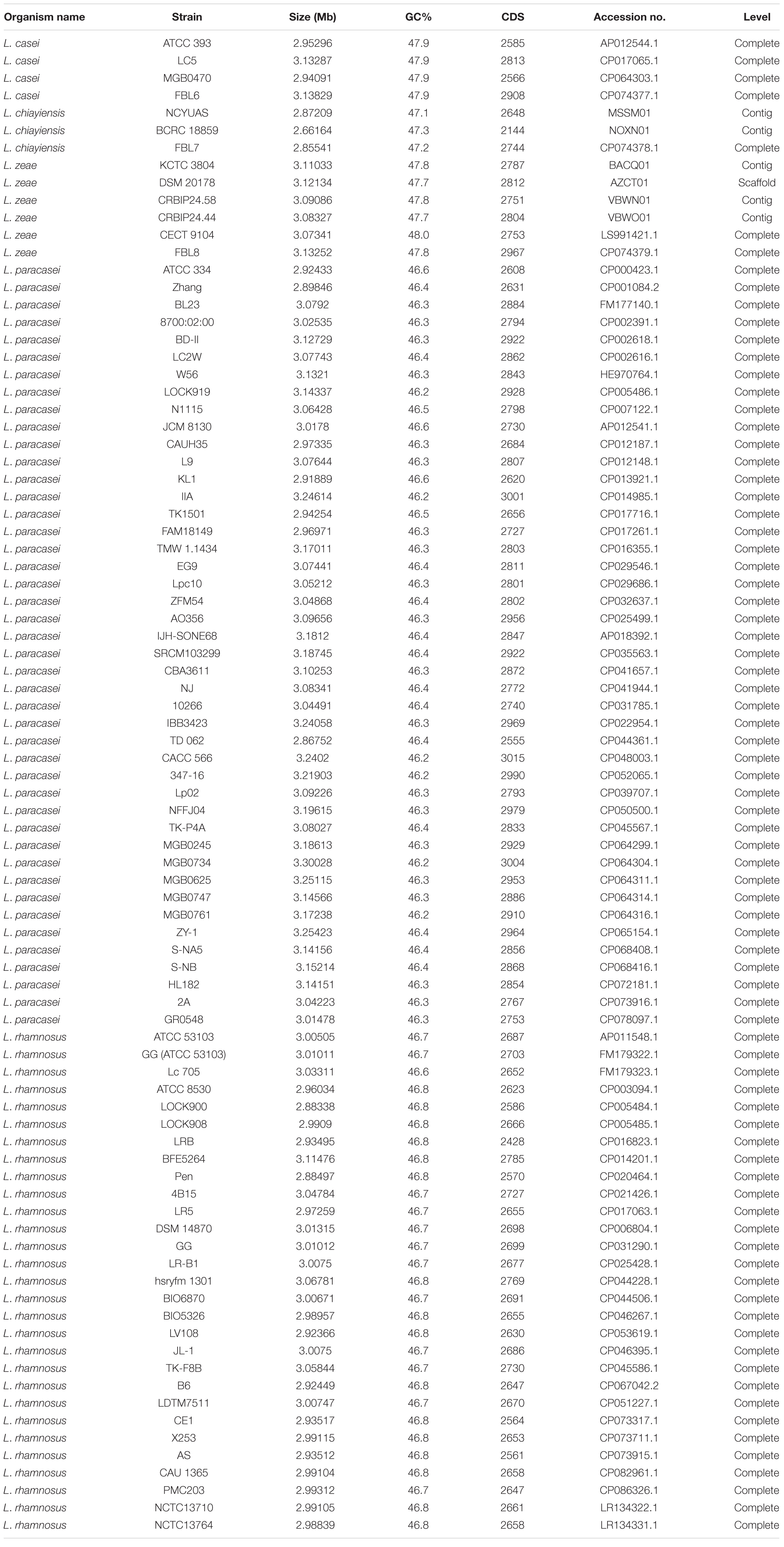
Table 1. Genome features of L. casei, L. chiayiensis, and L. zeae used in this study.
Evaluation of the Probiotic Properties of FBL6, FBL7, and FBL8
Acid and Bile Salt Tolerance
Tolerance to acid and bile salt was observed according to the previous studies (Yu et al., 2019; Jomehzadeh et al., 2020). Each strain was incubated in MRS broth at 37°C overnight. Aliquots (0.1 mL) of each active culture were inoculated in 10 mL MRS broth adjusted to pH 2.5 with 0.1 N hydrochloric acid (HCl) and then incubated for 3 h at 37°C. Each strain was inoculated in MRS broth containing 0.3% ox gall bile salt (Sigma-Aldrich, St. Louis, MO, United States) and incubated for 24 h at 37°C to confirm tolerance to bile salt. After incubation, cell suspension was spread on an MRS agar plate, and then viable cell count that survived at low pH and bile salt were enumerated by plate counting. L. rhamnosus American Type Culture Collection (ATCC) 53103 (LGG) was utilized as the control. The survival rate (%) of each strain was calculated using the following formula: (N/N0) × 100, where N and N0 denote viable cell numbers after culturing with low pH or bile salt and initial viable cell numbers, respectively.
Antimicrobial Activity
The antimicrobial activity of strains against pathogenic Gram-positive and Gram-negative bacterial strains was determined using the well-diffusion method (Jomehzadeh et al., 2020). Listeria monocytogenes ATCC 19111, L. ivanovii ATCC 19119, Bacillus cereus ATCC 21772, E. coli O157:H7 ATCC 43894, E. coli O1:K1:H7 ATCC 11775, and Salmonella Enteritidis ATCC 4931 were grown on nutrient broth (Difco) at 37°C overnight. The FBL6, FBL7, and FBL8 strains were grown in MRS broth at 37°C for 24 h and then centrifuged at 8,000 × g for 5 min to prepare a supernatant. The pathogens (106 CFU/mL) were inoculated on Mueller–Hinton agar (Difco) and then cultured by putting the supernatant of the three strains in 8-mm-diameter wells. After incubation for 24 h at 37°C, antimicrobial activity was determined by measuring the inhibition zone surrounding the well.
Antioxidant Activity by Radical Scavenging Ability
The 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging ability of the strains was analyzed according to the previous studies (Yu et al., 2019). The activated cell (200 μL) was mixed with 1 mL of 0.4 mM DPPH solution and reacted in a dark room for 30 min. After centrifugation at 8,000 × g for 5 min, the absorbance of each supernatant was measured at 517 nm. The control group used the same amount of distilled water as the sample, and 0.2 mM ascorbic acid was used as the positive control. Scavenging activity (%) was calculated using the following formula: [1 – (Asample/Acontrol)] × 100, where Acontrol and Asample indicate the absorbance of the control and absorbance of the sample, respectively.
Adhesion Ability
The adhesion ability was evaluated using the human intestinal epithelial adenocarcinoma cell line (HT-29) (Yu et al., 2019). The HT-29 cell was obtained from the ATCC (Manassas, VA, United States). The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing fetal bovine serum (FBS, 10%, v/v) at 37°C in a humidified atmosphere of 5% CO2 until a 70–80% confluent monolayer was obtained. The HT-29 cells (1 × 105 cells/well) were seeded into a 24-well culture plate and cultured until a confluent monolayer was observed. After replacing the FBS-free RPMI 1640 medium, the isolates (1 × 108 CFU/mL) were added to each well, with each strain being tested in triplicates and incubated for 2 h. Subsequently, each well was rinsed three times with PBS, and the attached cells were lysed with 0.1% Triton X-100 (Sigma-Aldrich). The number of adherent bacteria was counted by spreading the serial dilutions on MRS agar. The adhesin value (%) was calculated using the following formula: N1/N0 × 100, where N0 and N1 denote the initial viable cell numbers and viable cell numbers obtained from the HT-29 cells, respectively.
Autoaggregation and Coaggregation
Autoaggregation and coaggregation tests were conducted according to previous studies (Lee et al., 2014). Autoaggregation of strains was observed to indirectly confirm intestinal cell adhesion. The cell (4 mL) suspended in PBS (108 CFU/mL) was mixed by vortexing for 20 s and then incubated at room temperature for 5 h to determine autoaggregation. Each time, 0.1 mL of each supernatant was mixed with 0.9 mL PBS, and absorbance was measured at 600 nm. The following formula was used to calculate autoaggregation of strain (%): A0–(At/A0) × 100, where At and A0 denote the absorbance at 5 h and absorbance at 0 h, respectively.
For the measurement of coaggregation, the cell suspensions of each strain and pathogenic strain were mixed and then vortexed for 20 s. The control tubes were prepared using 4 mL of putative probiotic strain and pathogenic strain in each tube. After incubation for 5 h, the absorbance of the suspension was measured at 600 nm. The following formula was used to calculate the coaggregation of a strain: {[(Ax + Ay)/2] – Amix/(Ax + Ay)/2} × 100, where Ax and Ay denote the bacterial suspension in the control tube, and Amix indicates the mixed bacterial suspension at 5 h.
Statistical Analysis
All experiments for probiotic properties were conducted in triplicate, and the statistical analysis of values was performed using R version 4.1.0. Significant differences (p < 0.05) between the mean ± standard deviation values were determined by one-way analysis of variance and Duncan’s multiple range test.
Results and Discussion
General Genome Features
The complete genome of L. casei FBL6 contained a chromosome of 3,138,294 bp with a guanine-cytosine (GC) content of 47.9% (Supplementary Table 1). The complete genomes of L. chiayiensis FBL7 and L. zeae FBL8 contained a chromosome of 2,855,405 bp with a GC content of 47.2% and a chromosome of 3,132,522 bp with a GC content of 47.8%, respectively. Previous studies have reported that strains occurring in host-associated environments, such as the gut and oral, showed small genome size and gene number, whereas strains present in soil have the largest genome size (Bentkowski et al., 2015; Cobo-Simón and Tamames, 2017; Jia et al., 2022). They also reported a positive correlation between genome size and ubiquity, suggesting that microorganisms with larger genome size can adapt to more environmental conditions. FBL6, FBL7, and FBL8 had a larger genome size and higher GC content than L. acidophilus NCFM (1,993,564 bp, 34.7%), L. helveticus CAUH18 (2,160,583 bp, 36.8%), and L. johnsonii ZLJ010 (1,999,879 bp, 34.91%) but were similar to L. plantarum EM (3,649,371 bp, 44.14%), L. paracasei ZFM54 (3,048,677 bp, 46.35%), and L. rhamnosus GG (3,010,111 bp, 46.7%) (Altermann et al., 2005; Kankainen et al., 2009; Yang et al., 2016; Zhang et al., 2019; Kim et al., 2020a; Qureshi et al., 2020). Among these, L. plantarum, which is ecologically flexible, has a large genome size (Kim et al., 2020a), similar to L. casei, L. chiayiensis, and L. zeae, indicating that the latter can adapt well to various environments.
In each genome, 2,908 coding genes of FBL6, 2,744 genes of FBL7, and 2,967 genes of FBL8 were assigned to COG families comprising 20 functional categories. In the three genomes, most coding genes were classified for general function prediction only (R, 13.11–13.43%), carbohydrate transport and metabolism (G, 11.68–13.01%), transcription (K, 8.87–9.19%), and amino acid transport and metabolism (E, 8.78–8.97%) (Supplementary Figure 1). The high proportion of genes involved in carbohydrate and amino acid metabolism indicates that three strains might utilize and degrade a wide range of carbohydrates and proteins (Heo et al., 2021).
Phylogenetic Comparison of L. casei, L. chiayiensis, and L. zeae
Phylogenetic comparison based on the 16S rRNA gene sequences demonstrated that L. casei, L. chiayiensis, and L. zeae were clustered according to the species (Figure 1A). In a phylogenetic tree based on the 16S rRNA gene sequences, FBL6 demonstrated a 99.62% similarity to other L. casei strains. In addition, it showed a high degree of similarity to L. chiayiensis (99.43%) and L. zeae (99.36%). FBL7 exhibited a 99.81% similarity to other L. chiayiensis strains and a high degree of similarity to L. zeae (99.49%). In some L. zeae strains, the 16S rRNA gene was present as a short fragment; thus, the remaining strains except these were compared with the 16S rRNA gene of FBL8. As a result, FBL8 demonstrated a high similarity (99.87%) to the L. zeae CECT9104 and L. zeae CRBIP24.44 strains.
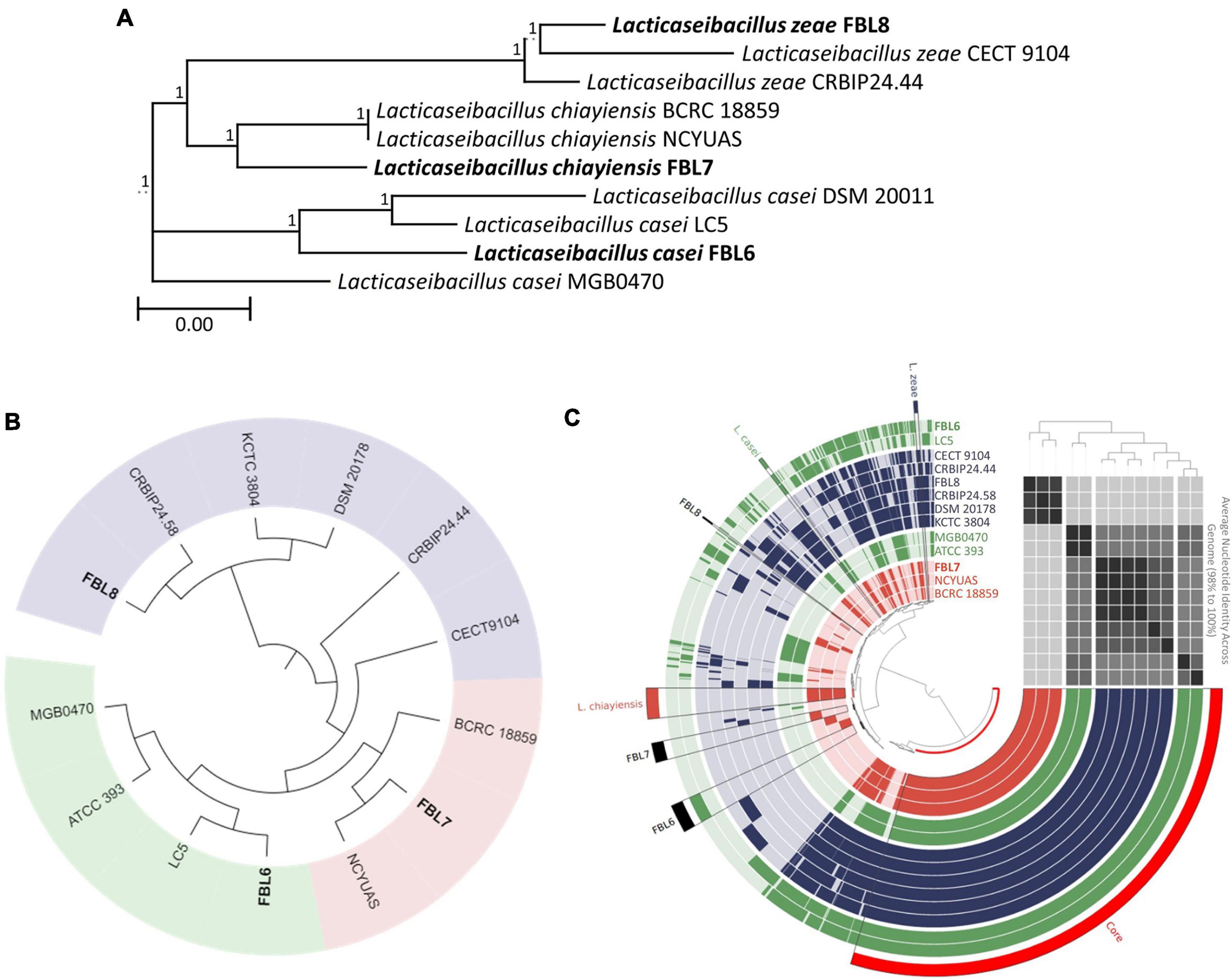
Figure 1. Phylogeny analysis based on (A) 16S rRNA gene sequences and (B) core-genome and (C) pan-genome. In tree based on pan-genome distribution, each ring represents one genome. The green, blue, and red rings indicate the L. casei, L. chiayiensis, and L. zeae, respectively. The tinted bright and dark color in ring represent gene absence and presence, respectively. ANI values were plotted as heatmap determined at high similarity (black) and low similarity (gray).
According to the orthoANI calculation, FBL6 shared 95.36–95.81% sequence similarity to other L. casei genomes and 88.09–93.75% to L. chiayiensis and L. zeae genomes (Supplementary Table 2). L. casei FBL6 demonstrated a closer relationship with LC5. L. casei LC5 is a probiotic strain isolated from fermented dairy products and used as commercial probiotics (Kang et al., 2017). This strain has a probiotic effect on atopic dermatitis. FBL7 demonstrated 99.24 and 98.67% similarity to the genomes of L. chiayiensis NCYUAS and BCRC 18859, respectively. FBL8 shared 96.65–99.56% sequence similarities to other L. zeae genomes and high sequence similarities to other L. casei and L. chiayiensis genomes (88.12–94.38%). Also, in the phylogenetic analysis based on core-genome and pan-genome, FBL6, FBL7, and FBL8 were classified as L. casei, L. chiayiensis, and L. zeae and demonstrated a closer relationship with L. casei LC5, L. chiayiensis CRBLP24.58, and L. zeae NCYUAS, respectively (Figures 1B,C). The three species showed a high similarity to the 16S rRNA gene, which is more than 99.17% of each other, and it was confirmed that this gene could not be distinguished. Conversely, the analysis based on the whole-genome sequence clearly distinguishes the three species, suggesting that the genome sequence can help in achieving better understanding of the evolutionary history than the 16S rRNA gene.
Pan-Genome and Core-Genome
Pan-genome and core-genome are used for investigating genomic diversity within closely related microorganisms or the same species (Zhang et al., 2019). Pan-genome is the entire set of genes of all strains that reflects the receptive capacity of the genetic determinants (Zhang et al., 2019). A core-genome is a set of genes shared by all strains in a species, including genetic determinants, for maintaining the characteristics of species (Bosi et al., 2016; Zhang et al., 2019). Four genomes of L. casei had a pan-genome of 3,423 orthologs, which were divided into a core-genome of 1,890 orthologs (55.21%), an accessory-genome of 853 orthologs (24.92%), and a unique genome of 680 orthologs (19.87%) (Figure 2). The pan-genome size was 1.24 times the average size of these four genomes, and the core-genome constituted approximately 68.84% of each L. casei genome. In the core-genome, 1,570 orthologs could be assigned in 20 COG categories. Some highly conserved functional categories, such as general function prediction only (R, 13.65%), carbohydrate transport and metabolism (G, 10.06%), and function unknown (S, 9.21%), were enriched among the core-genome of L. casei (Figure 2).
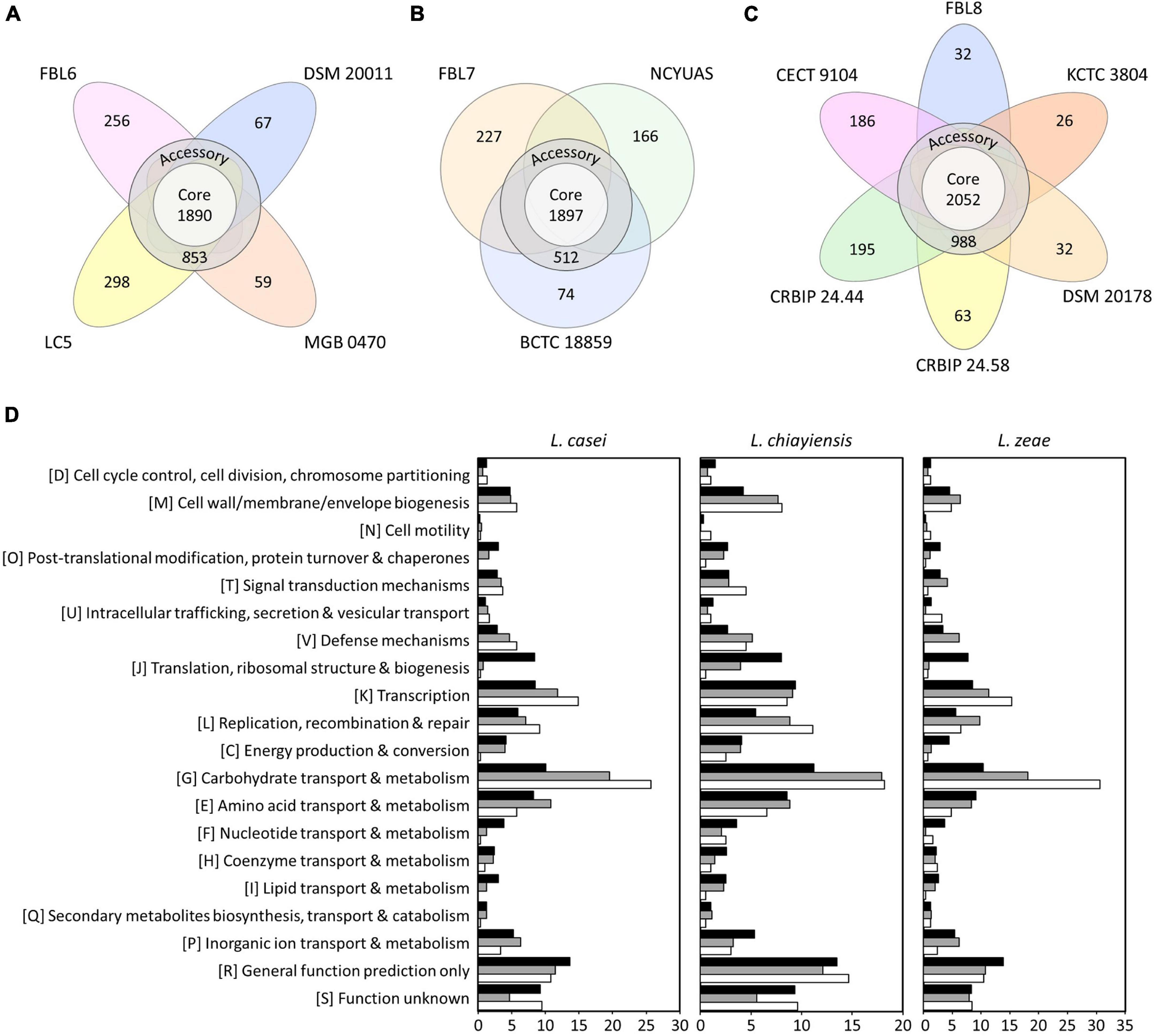
Figure 2. The number of core, accessory, and unique genes among (A) L. casei, (B) L. chiayiensis, and (C) L. zeae genomes, and (D) the number of genes assigned in cluster of orthologous group (COG) categories. Black, gray, and white bars represent COGs of the core, accessory, and unique genes, respectively.
Three genomes of L. chiayiensis had a pan-genome of 2,876 orthologs, divided into a core-genome of 1,897 orthologs (65.96%), an accessory-genome of 512 orthologs (17.80%), and a unique genome of 467 orthologs (16.24%) (Figure 2). In the L. chiayiensis core-genome, 1,523 ortholog gene clusters could be assigned in 20 COG categories. Some highly conserved functional categories, such as general function prediction only (R, 13.48%), carbohydrate transport and metabolism (G, 11.20%), and transcription (K, 9.39%), were enriched among the core-genome of L. zeae.
Six genomes of L. zeae had a pan-genome of 3,574 orthologs, which were divided into a core-genome of 2,052 orthologs (57.41%), an accessory-genome of 988 orthologs (27.64%), and a unique genome of 534 orthologs (14.94%) (Figure 2). In the core-genome, 1,687 orthologs could be assigned in 20 COG categories. Some highly conserved functional categories, such as general function prediction only (R, 13.84%), carbohydrate transport and metabolism (G, 10.32%), and amino acid transport and metabolism (E, 9.12%), were enriched among the core-genome of L. zeae. Other Lactobacillus species, such as L. johnsonii and L. ruminis, which have a relatively small genome size, had a high proportion of genes related to core functions, such as translation, replication, and cell cycle control (Kant et al., 2017; Zhang et al., 2019). Contrarily, owing to its large genome size, L. casei, L. chiayiensis, and L. zeae had a relatively high number of proteins for amino acids and carbohydrate metabolisms, similar to L. plantarum (Boekhorst et al., 2004; Zhang et al., 2019).
Unique Genome Characteristics
Unique Genome of L. casei FBL6
Lacticaseibacillus casei FBL6 had 256 unique genes, including 126 proteins with known functions and 130 hypothetical proteins of unknown function (Supplementary Table 3). To further investigate the function of the encoded protein of unique genes, they were assigned to the COG categories. Of the 256 individual genes in the FBL6 genomes, 100 were assigned to 13 COG categories (Supplementary Figure 2). In total, 24 genes were enriched in carbohydrate transport and metabolism (G, 24%). It contains genes such as PTS sugar transporter subunits IIB and IIA and PTS lactose/cellobiose transporter subunit IIA. These individual genes may endow FBL6 with probiotic strain properties that utilize various carbohydrates (Zhang et al., 2019). A total of 16 genes in the FBL6 genome were assigned to replication, recombination, and repair (L, 16%), containing genes associated with mobile elements. Furthermore, 16 genes were assigned in transcription (K, 16%), containing several transcriptional regulators. Previous study has reported that the presence of these transcriptional regulators can act on specific genes to regulate the expression and give them several benefits when they are present in an environment such as the gut (Zhang et al., 2019). However, further research is required to determine their association with genes and probiotics affected by transcriptional regulators.
Unique Genome of L. chiayiensis FBL7
Lacticaseibacillus chiayiensis FBL7 had 214 unique genes, including 140 proteins with known functions and 87 hypothetical proteins (Supplementary Table 4). Of the 214 unique genes, 73 were assigned to 17 COG categories (Supplementary Figure 2). A total of 13 genes were abundant in replication, recombination, and repair (L, 17.8%). In FBL7, CRISPR-associated protein Cas4 (locus_00047), which is not found in other L. chiayiensis strains, was present. This gene is one of the core CRISPR-associated (Cas) proteins involved in the prokaryotic immune system for antiviral defense (Zhang et al., 2012). Owing to the presence of this protein, FBL7 may be protected against mobile elements, including viruses, but other L. chiayiensis strains may not. Nine genes were assigned to carbohydrate transport and metabolism (G, 12.34%) and nine also to amino acid transport and metabolism (E, 12.34%). These unique genes endow FBL7 with the ability to obtain extra resources and protect itself against adverse stimuli, allowing better adaptation to various environments (Zhang et al., 2019).
Unique Genome of L. zeae FBL8
Lacticaseibacillus zeae FBL8 had 32 unique genes, including 26 proteins with known functions and 6 hypothetical proteins (Supplementary Table 5). Of the 32 unique genes, 17 were assigned to 11 COG functional categories (Supplementary Figure 2). Three genes were assigned to replication, recombination, and repair (L, 17.65%), containing several transposases and mobile element proteins. Two genes were assigned to the cell wall, membrane, and envelopment biogenesis (M, 11.76%). Two genes were assigned to signal transduction mechanisms (T, 11.76%), including two-component system sensors, histidine kinase, and response regulator. For lactic acid bacteria to survive while passing through the gastrointestinal tract, they need to have the ability to sense and respond to conditions in various changing environments (Zhang et al., 2019). They are generally detected by a two-component system (Cui and Qu, 2011), which consists of a membrane-associated histidine kinase that detects specific environmental signals and a cytoplasmic response regulator regulating gene expression (Cui and Qu, 2011). FBL8 carried a distinguished two-component system sensor histidine kinase. Thus, this unique gene might confer FBL8 to control various physiological processes, such as stress and toxicity.
Comparative Genomic Analysis of Lacticaseibacillus Strains
Comparative genomic analysis was performed to compare the genomic characteristics between these three strains and other reported 83 Lacticaseibacillus strains. Of the 237,017 coding genes, 896 genes were shared by all genomes, comprising the core-genome. Accessory-genome is composed of 5,467 coding genes, and 1,642 coding genes were found only in a single genome. The pan- and core-genome were annotated COG database and then assigned to functional categories. Some highly conserved functional categories, such as translation, ribosomal structure and biogenesis (J, 12.71%), general function prediction only (R, 12.35%), function unknown (S, 10.45%), and amino acid transport and metabolism (E, 7.4%), were enriched among the core-genome of Lacticaseibacillus strains (Supplementary Figure 3). Translation functions are important for maintaining physiological parameters during changes to which gut microorganisms are exposed, suggesting their adaptive and survival roles (Sharma et al., 2018). Similar to our study, genomic analysis of Bifidobacterium showed high conservation of “Translation, ribosomal structure, and biogenesis (J)” class in the core-genome, demonstrating the importance of information storage for these bacteria to adapt and survive in the gut (Sharma et al., 2018). In addition, for the accessory-genome and unique-genome, we observed enrichment of genes in the COG category “Carbohydrate transport and metabolism (G, 17.73–17.52%).” Among the 1,642 unique genes, 65, 70, and 8 coding genes were specific to L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8, respectively. These unique genes include MucBP domain-containing protein, LysM peptidoglycan-binding domain-containing protein, and hypothetical proteins.
Probiotic-Related Genes
Lifestyle Adaptation to Stress
Lactic acid bacteria, which are widely used in food fermentation, need to survive in various environmental conditions, including pH, salinity, and temperature (Ye et al., 2020). Resistance to environmental conditions is one of the desirable properties of lactic acid bacteria. Proteins of adaptation to stress regulate evolution tolerance and demonstrate genetic adaptation (Ye et al., 2020).
L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 contained several genes encoding stress-related proteins (Table 2). The FBL6 genome harbored the eight genes (locus_00683 to locus_00690), which encodes F0F1 ATP synthetase, and the gene locus_01410, which encodes the cholylglycine hydrolase; these proteins are associated with resistance to low pH and bile. Cholylglycine hydrolase is responsible for converting conjugated bile acid into free bile acid and confers probiotics in the gastrointestinal tract (Heo et al., 2021). FBL6 genome possessed genes encoding heat stress proteins, including molecular chaperone (locus_02010), dnaK (locus_00321), GroES (locus_02686), GroEL (locus_02687), and heat shock protein GrpE (locus_00320). Furthermore, this genome has an ABC transport system (locus_01791 to locus_01794) and glycine betaine ABC transport system (locus_02770 to locus_02772), which may have an osmotic pressure function (Heo et al., 2021). FBL6 may be involved in oxidative stress resistance by encoding proteins such as thiol peroxidase (locus_01096), thioredoxin reductase (locus_00895), and glutaredoxin (locus_00073), which catalyze glutathione-dependent disulfide reductions (Zhang et al., 2019). FBL6, FBL7, and FBL8 contained genes encoding stress-related proteins, such as bile, acid, and oxidative stress tolerance genes. Moreover, two genomes carried genes that encode proteases related to stress response, heat stress proteins. These results suggest that three strains might resist multiple stress conditions and thus reach the intestines through the gastrointestinal tract.
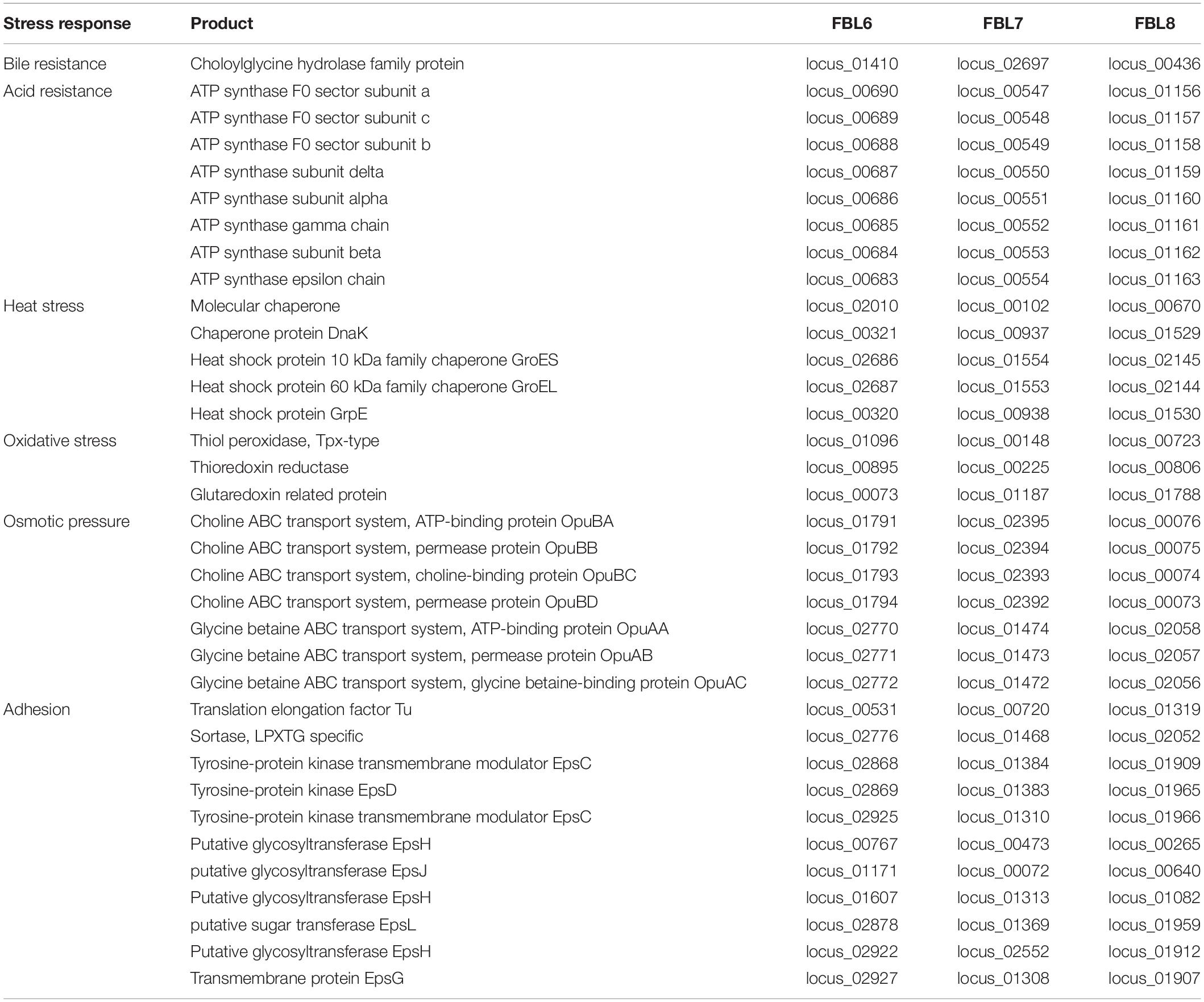
Table 2. Genes coding for proteins involved in stress resistance.
Regulatory proteins such as transcriptional regulators and sigma factors are important for microorganisms to adapt to different environments (Zhang et al., 2019). FBL6 had genes encoding several proteins involved in transcriptional regulation, including 110 transcriptional regulators and one sigma factor (RpoD). FBL7 and FBL8 had 100 and 105 transcriptional regulators, respectively, and one sigma factor in both strains. These genomes had fewer regulatory proteins than other probiotic species, L. johnsonii, and similar to L. plantarum (Zhang et al., 2018, 2019). A previous study has demonstrated that many regulatory proteins are contained in large genomes, contributing to various lifestyle differences (Zhang et al., 2019). Thus, L. johnsonii does not require a complex regulatory system, whereas L. casei, L. chiayiensis, and L. zeae, with their free-living lifestyle, appear to have regulatory machinery that can adapt to various environmental niches (Zhang et al., 2019).
Adhesion
Adhesion to the intestinal mucosa epithelium is a prerequisite for demonstrating a probiotic effect, and bacterial surface proteins are known to be involved in the colonization of the intestinal mucosa (Ye et al., 2020). The potential surface exposure protein of lactic acid bacteria plays a significant role in bacterial interactions with the environment or host (Ye et al., 2020). L. casei FBL6 encodes cell-surface proteins, such as lipoprotein signal peptide (locus_00412) and elongation factor Tu (locus_00531) (Table 2). In addition, in this genome, coding genes, such as LPXTG-specific sortase (locus_00278) and exopolysaccharides (eps) cluster, which are related to strain adhesion to the surrounding epithelial tissue, were contained within the FBL6 genome. Exopolysaccharide contributes strains and host interactions within the intestinal epithelial cells and mucosa and is involved in certain essential functions of probiotics, such as immunomodulation, antioxidation, aggregation, and biofilm formation (Alayande et al., 2020). Sortase-dependent proteins are cell-surface proteins in lactic acid bacteria and play a significant role in adhesion (Alayande et al., 2020). Therefore, the adhesion-related genes identified in the FBL6 genome may provide stability of strain and prolong its antagonistic effect on unwanted gut microorganisms, aiding in the effective colonization of the intestinal environment and inhibition of pathogens. Similarly, genes related to adhesion were also present in L. chiayiensis FBL7 and L. zeae FBL8.
Safety Assessment
One of the essential requirements for probiotics is their safety. Probiotics should be least resistant to critical antibiotics, as antibiotic resistance in bacteria can be inherent or acquired through mutation or horizontal gene transfer (Soni et al., 2021). The search using ResFinder and CARD returned no hits for antibiotic resistance genes in L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 genomes. As a result of the strain’s pathogenicity investigation, the calculated matched pathogenic families for FBL6 were 0, and the unmatched pathogenic families were 83. The probability that the strain is a human pathogen is 0.218; thus, this strain was predicted not to be a pathogen. These results suggest that this depicts the safety of the strain and reduces the risk of transferring antibiotic resistance genes to the host’s native gut microbiota. Also, FBL7 and FBL8 did not contain pathogenicity factors. The probability of being a pathogen in the three genomes was very low (FBL7, 0.164; FBL8, 0.213), and these strains were predicted to be non-human pathogens. Therefore, FBL6, FBL7, and FBL8 strains are safe.
Mobile Genetic Element Analysis
PHAST predicted one intact prophage element and questionable prophage element in L. casei FBL6 and L. chiayiensis FBL7, respectively, and two intact prophage elements in L. zeae FBL8. Prophages are often detected in the genomes of probiotic Lactobacillus species (Stefanovic and McAuliffe, 2018; Feyereisen et al., 2019; Zhang et al., 2019). The prophage region in FBL6 extended from 1,379,286 to 1,410,750 bp and contained 46 proteins with an intact prophage element from locus_01305 to locus_01350 (Supplementary Table 6); this was the part of the prophage BH1 detected in Lactobacillus (accession no. NC_048737). For FBL7, the prophage region contained 52 proteins with a questionable prophage element (Supplementary Table 7). The genes in the prophage region were homologous to the genes of bacteriophage T25 (NC_048625) observed in Lactobacillus. For FBL8, the prophage 1 region contained 50 proteins with an intact prophage element, and prophage 2 contained 19 proteins with an intact prophage element (Supplementary Table 8). The genes in prophage 1 and 2 regions were homologous to a gene of phage BH1 (NC_048737) found in Lactobacillus and a gene of phage StauST398 (NC_023499) found in Staphylococcus, respectively. In these bacteriophage regions, virulence factors or genes related to pathogenic properties could not be detected.
CRISPR refers to a family of DNA repeats providing acquired immunity to foreign genetic elements (Zhang et al., 2019). These are composed of short and highly conserved repeats, interspaced by spacers, which are variable sequences, and often exist adjacent to the cas genes. The presence of these CRISPR loci increases the genomic stability of strains, allowing them to adapt to various environments. The FBL6 genome contained two CRISPR loci (Supplementary Table 9). The genomes of FBL7 and FBL8 harbored CRISPR regions, with associated spacer and Cas-gene. The FBL7 genome contained three CRISPR loci. The detected CRISPR Cas system was of type IIA (cas1, cas9, and csn2) and type IC (cas1, cas2, cas3, cas4, cas5, cas7, and cas8). The FBL8 genome contained five CRISPR loci. The detected CRISPR/CRISPR-associated (Cas) system was of type IC (cas1, cas2, cas3, cas4, cas5, cas7, and cas8). The presence of these CRISPR loci will provide the strains with the ability to defend themselves against plasmids, phages, and insertion sequences (Alayande et al., 2020). Therefore, the CRISPR Cas system could prevent L. casei, L. chiayiensis, and L. zeae from acquiring virulence or antibiotic resistance genes through horizontal gene transfer.
Secondary Metabolic Product
The probiotic strain should survive in the intestine by competing with other microorganisms after adhesion or colonization in the gut (Heo et al., 2021). Strains with the ability to produce bacteriocin have the advantage of competing with other gut microorganisms (Heo et al., 2021). L. casei FBL6 encoded three secondary metabolic enzymes, ribosomally synthesized, and post-translationally modified peptide product (RiPP) cluster, all of which were predicted to produce bacteriocin. The first RiPP-like cluster demonstrated high similarity to bacteriocin IIc (Figure 3A). Some core biosynthetic genes and other genes in this cluster were specifically present only in FBL6 but not in different L. casei strains (Supplementary Table 10). These genes were helix–turn–helix transcriptional regulators, Blp family class II bacteriocin. In the second RiPP cluster, a gene encoding bacteriocin sakacin-P was only present in FBL6. Sakacins, class II bacteriocin, were first identified in L. sakei and demonstrated a bactericidal effect on food pathogens and spoilage species (Fallico et al., 2011). In particular, it has been reported that sakacin-P has relatively high activity against L. monocytogenes compared with other bacteriocins and is therefore promising for use in the food industry (Møretrø et al., 2000). Due to the presence of this gene, FBL6 may have antimicrobial activity against pathogens, but other L. casei strains may not. Moreover, the Rrf2 family transcriptional regulator, ABC transporter permease, and transporter-related genes in the third RiPP cluster were specifically present only in the FBL6 genome. L. chiayiensis FBL7 encoded five RiPP clusters, all of which were predicted to be bacteriocins, such as sakacin-P and bacteriocin IIc (Figure 3B). Unlike other L. chiayiensis strains, some transcriptional regulators, DNA-binding proteins, and hypothetical proteins were only present in the FBL7 genome (Supplementary Table 11). L. zeae FBL8 encoded four RiPP clusters, all of which were predicted to be bacteriocins (Figure 3C). The RiPP clusters in FBL8 were also present in other L. zeae genomes (Supplementary Table 12).
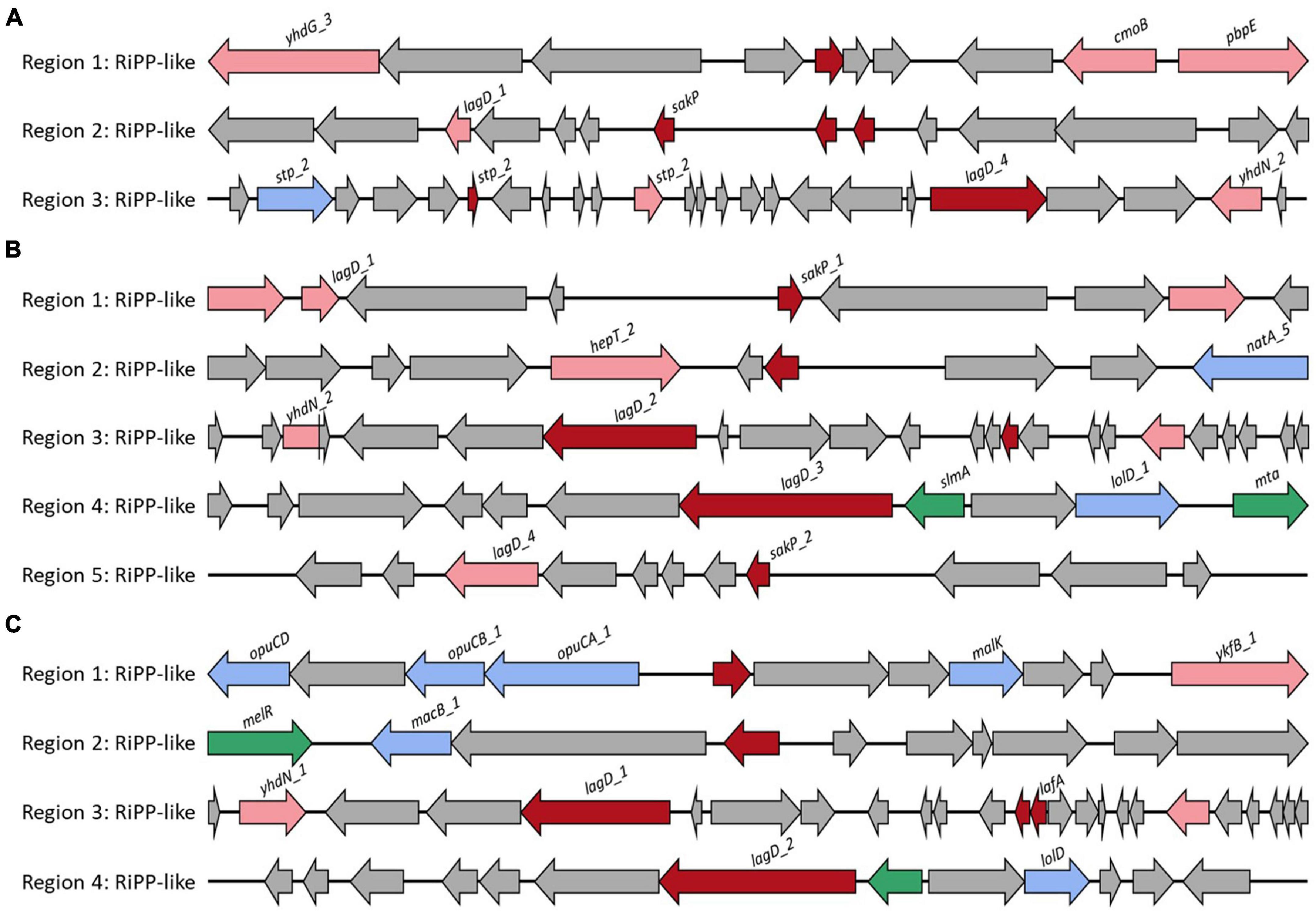
Figure 3. Biosynthetic gene cluster of secondary metabolites. Gene clusters for the biosynthesis of bacteriocin in (A) L. casei FBL6, (B) L. chiayiensis FBL7, and (C) L. zeae FBL8. The red, pink, blue, green, and gray arrows represent core biosynthetic genes, additional biosynthetic genes, transport-related genes, regulatory genes, and other genes, respectively.
Carbohydrate-Active Enzymes
The L. casei FBL6 genome contained 119 genes in the CAZymes gene families as follows: 61 glycoside hydrolases (GHs), 40 glycosyltransferases (GTs), 3 carbohydrate esterases (CEs), 1 auxiliary activity (AA), 13 carbohydrate-binding modules (CBMs), and 1 polysaccharide lyase (PL) (Figure 4). The L. chiayiensis FBL7 genome contained 105 genes in CAZymes gene families: 60 GHs, 29 GTs, 3 CEs, 1 AA. 11 CBMs, and 1 PL. Among them, GH78, GT0, and CBM48 were only predicted in FBL7, not in other L. chiayiensis strains. The L. zeae FBL8 genome contained 115 genes in CAZymes gene families: 56 GHs, 43 GTs, 3 CFs, 1 AA, 9 CBMs, and 3 PLs. The results demonstrated that the carbohydrate utilization patterns of FBL6, FBL7, and FBL8 were different. GH127 (β-L-arabinofuranosidase) and GT111 (β-1,3-galactofuranosyltransferase) were only predicted in the FBL6 strain, whereas GH154 (β-glucuronidase) and GH43-11 (β-xylosidase) were only predicted in the FBL7 strain. Also, GH15 (glucoamylase), GH105 (unsaturated rhamnogalacturonyl hydrolase), and PL8 (hyaluronate lyase) were only predicted in the FBL8 strain. A previous study has reported that the larger the number of CAZymes encoding genes in the genome, the higher the probiotic potential for immune stimulation and pathogen defense (Zhang et al., 2018). FBL6, FBL7, and FBL8 exhibited a higher diversity of carbohydrate-activation enzymes, which were relatively abundant than other strains.
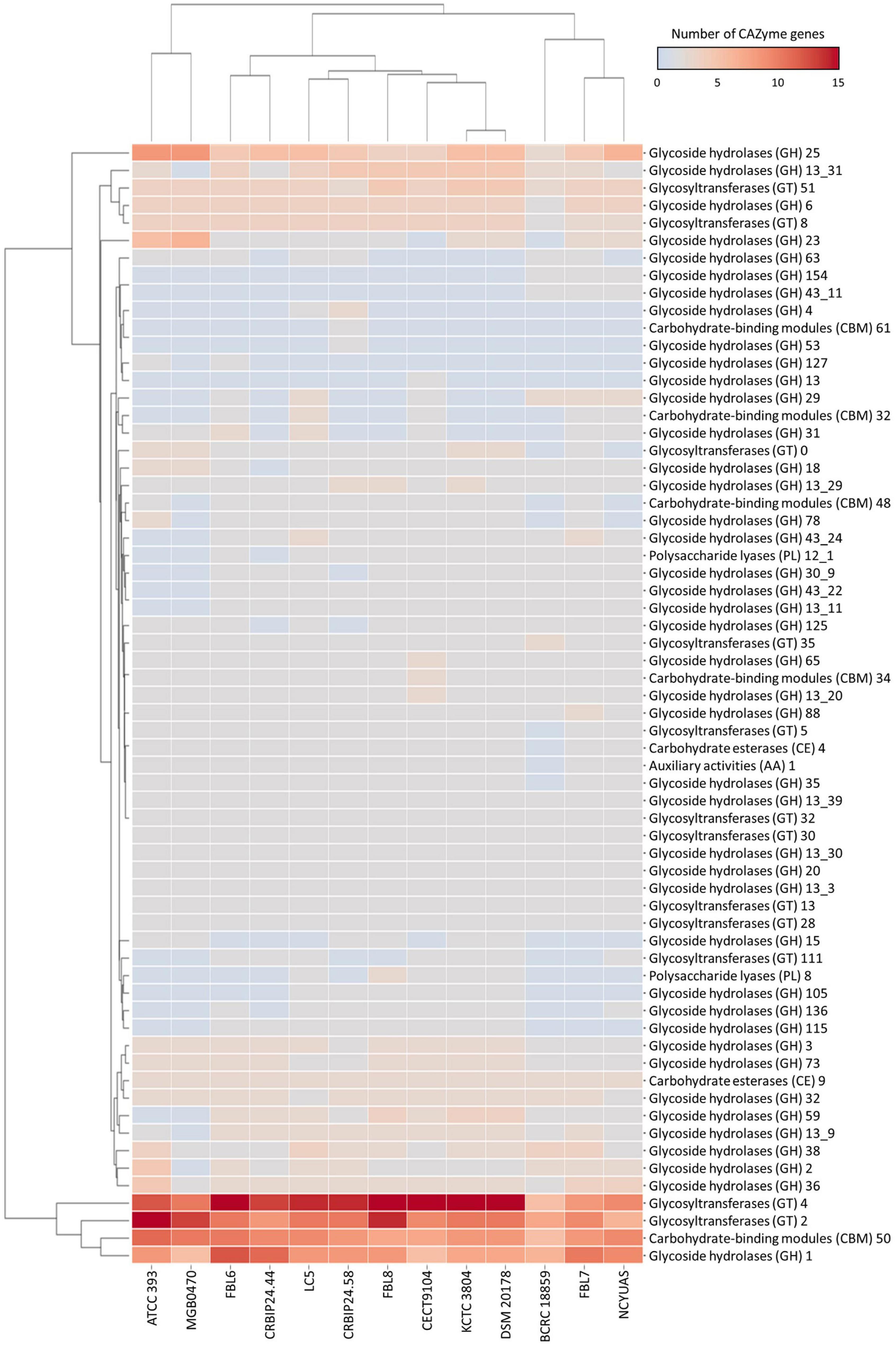
Figure 4. Heatmap for distribution of the number of genes of carbohydrate-active enzymes in L. casei FBL6, L. chiayiensis FBL7, L. zeae FBL8, and other strains. The right and bottom of the heatmap present carbohydrate-active enzymes and genomes, respectively.
Probiotic Properties Under in vitro Conditions
Acid and Bile Resistance
The effects of probiotic strains depend on their ability to multiply in the gastrointestinal tract to exhibit functionality and survive in natural host defenses (Ye et al., 2020). For the cells to reach the intestine and exhibit functionality, probiotic strains must be resistant to low pH and bile salt (Yu et al., 2019). The ability of the strains to survive in simulated gastric juice and bile salt was evaluated in vitro. The survival of FBL6, FBL7, and FBL8 was not affected at all by the low-acid environment, and these strains exhibited survival rates of 96.7 ± 12.75% to 120.3 ± 6.14%, confirming the high stability against acid (Figure 5A). Also, the three strains showed survival rates of 85.4 ± 8.97% to 108.8 ± 9.10% at a high concentration of bile salt, indicating that they were not affected by bile salt to survive (Figure 5B). Therefore, these results suggest that three strains possess the characteristics of probiotics with high survival rates at low pH and bile concentration.
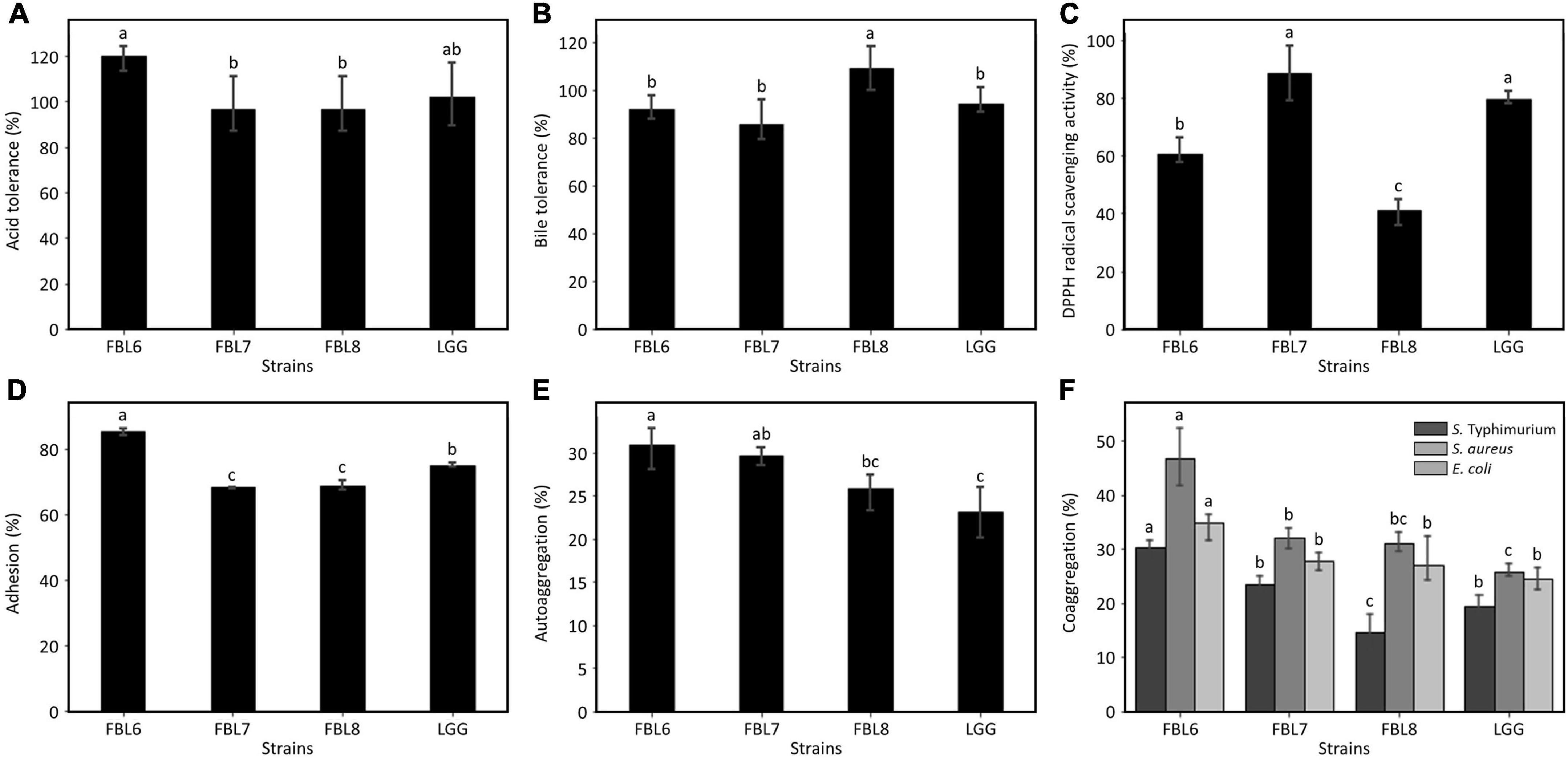
Figure 5. Evaluation of the probiotic properties of L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8. (A) The acid and (B) bile tolerance of three strains. (C) The antioxidant activity and (D) adhesion activity of three strains. (E) The autoaggregation and (F) coaggregation of three strains on S. Typhimurium ATCC 14028, S. aureus ATCC 6538, and E. coli ATCC 43894. Data value represents the mean ± standard deviation (n = 3). Different letters on each bar indicate significant differences (p < 0.05).
Antioxidant Activity
The DPPH radical scavenging activity of the strains was analyzed to observe the antioxidant effect, which is one of the probiotic properties. As a result, the three strains exhibited a DPPH scavenging potential (Figure 5C). There is no significant difference in DPPH radical scavenging activity between L. chiayiensis FBL7 strain and control strain, L. rhamnosus GG. The L. casei FBL6 and L. zeae FBL8 strains showed lower activities. The DPPH radical scavenging activities of the three strains ranged from 41.0 ± 4.62% to 88.6 ± 9.52%, and all strains exhibited antioxidant activity.
Antimicrobial Properties
The inhibitors produced by probiotic strains can be alternatives to antibiotics (Li et al., 2021). The antimicrobial properties of the three strains against the representative pathogenic strains were investigated. L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 had a broad antimicrobial spectrum (Table 3). These strains exhibited antimicrobial activity against Gram-negative bacteria and Gram-positive bacteria, such as Listeria, B. cereus, E. coli, and Salmonella. The antimicrobial activity of probiotic strains is mainly mediated by the production of some inhibitory substances, such as bacteriocins and lactic acid (Ye et al., 2020). These strains have genes involved in the production of antimicrobial peptides in the genomes, and it is presumed that these substances contributed to the inhibitory effect on pathogenic strains.
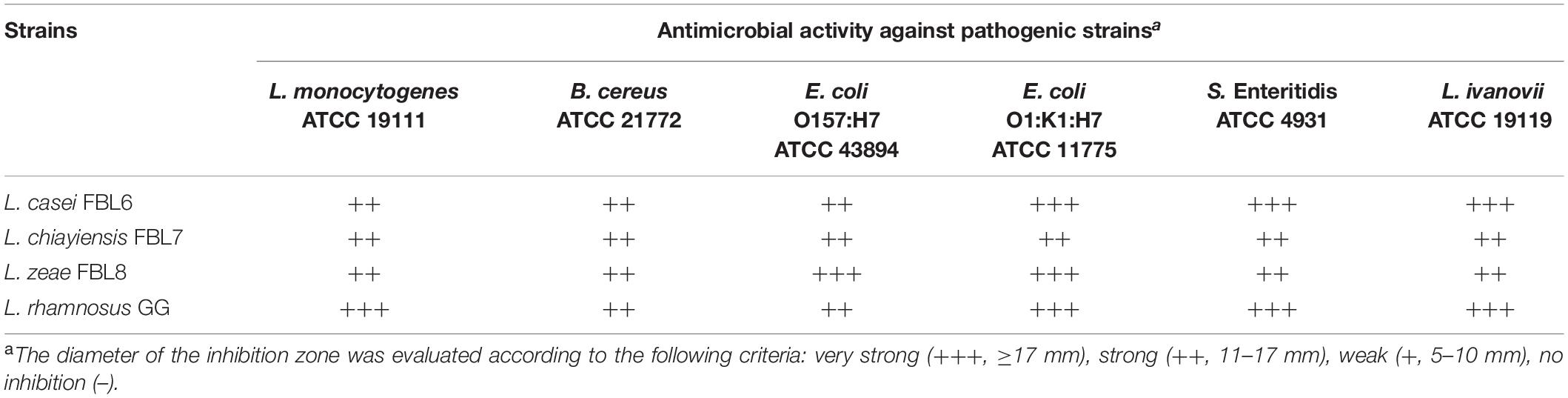
Table 3. Antimicrobial activity of L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8.
Adhesion Ability
The adhesion ability of probiotic strain has been proven to vary depending on their species or strain because the production of proteinaceous secretory adhesion and the properties of bacterial cell membranes contribute to intestinal cell attachments (Yu et al., 2019). L. casei FBL6 exhibited an adhesion capability of 85.4 ± 1.37%, which is higher than that of the control strain, L. rhamnosus GG (Figure 5D). This result is consistent with a previous study demonstrating the probiotic L. casei strain’s high adhesion ability (Yu et al., 2019). L. chiayiensis FBL6 and L. zeae FBL7 strains demonstrated lower adhesion capabilities than L. rhamnosus GG but were similar to other probiotic strains, such as L. plantarum subsp. plantarum SW03, L. rhamnosus KCTC 12202BP, and L. gasseri 5R01 (Oh and Jung, 2015; Son et al., 2017; Oh et al., 2018). These results suggest that three strains have potentially fulfilled the adhesion capacity requirements for probiotics.
Autoaggregation and Coaggregation
Autoaggregation is the phenomenon of clumping of bacteria of the same strain and is correlated with the adhesion ability of the probiotic strain to the intestinal epithelial cells (Lee et al., 2014). This ability is a precondition for colonization and improved persistence in the gastrointestinal tract (Lee et al., 2014). The autoaggregation rates of FBL6, FBL7, and FBL8 were measured. The results indicated that all strains possessed autoaggregation phenotypes (Figure 5E) and showed higher autoaggregation than the control strain, namely L. rhamnosus GG, except for L. zeae FBL8. There is no significant difference in autoaggregation between L. zeae FBL8 and L. rhamnosus GG.
Coaggregation refers to clumping between two different species. The coaggregation ability of the probiotic and pathogenic strains may hinder colonization by pathogenic strains in the gut, allowing a host defense mechanism against infection (Lee et al., 2014). Coaggregation of FBL6, FBL7, and FBL8 strains with pathogenic strains was measured. All strains demonstrated coaggregation with three pathogenic strains compared with the control strain, L. rhamnosus GG, except for the coaggregation of FBL8 with S. Typhimurium. FBL6 showed the best coaggregation rate with S. aureus (46.8 ± 5.41%) (Figure 5F). The genomic analysis and in vitro studies suggest that these strains may have adhesion ability in human intestinal epithelial cells.
Conclusion
This study was conducted to demonstrate the probiotic properties of L. casei FBL6, L. chiayiensis FBL7, and L. zeae FBL8 isolated from raw milk. The FBL7 and FBL8 strains are the first L. chiayiensis and L. zeae strains with phenotype and genotype studies. For the first time, this study revealed that L. chiayiensis and L. zeae, like L. casei, also exhibit probiotic properties. The genome sequencing and comparative genomic analysis data indicated that three strains play a role as a potential probiotic candidate with antibacterial activity and the ability to survive in environmental stresses, such as acid, bile, temperature, and oxidative stress. Comparative genomic analysis indicated the L. casei FBL6 carried a sakacin-P gene cluster that was not found in other L. casei genomes. Moreover, genes related to adaptation, such as carbohydrate-active enzymes and secondary metabolite gene clusters specific to the genomes of three strains, were also revealed. Our analysis results could provide a genetic basis for further investigation to show the three strains’ functional mechanism of probiotic characteristics. However, it is necessary to further elucidate its specific benefits for host health through in vitro and in vivo experiments.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI AND CP074377.1, CP074378.1, CP074379.1.
Author Contributions
EK and H-YK contributed to the conception and design of this study. EK and S-MY performed the genome analysis and comparative genomic analysis. S-MY and DK performed the in vitro probiotic test. EK prepared a draft manuscript. H-YK reviewed and edited the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant No. 2020R1A6A3A01100168).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.794315/full#supplementary-material
Footnotes
References
Alayande, K. A., Aiyegoro, O. A., Nengwekhulu, T. M., Katata-Seru, L., and Ateba, C. N. (2020). Integrated genome-based probiotic relevance and safety evaluation of Lactobacillus reuteri PNW1. PLoS One 15:e0235873. doi: 10.1371/journal.pone.0235873
Altermann, E., Russell, W. M., Azcarate-Peril, M. A., Barrangou, R., Buck, B. L., McAuliffe, O., et al. (2005). Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc. Natl. Acad. Sci. U. S. A. 102, 3906–3912. doi: 10.1073/pnas.0409188102
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bentkowski, P., Van Oosterhout, C., and Mock, T. (2015). A model of genome size evolution for prokaryotes in stable and fluctuating environments. Genome Biol. Evol. 7, 2344–2351. doi: 10.1093/gbe/evv148
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). AntiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Boekhorst, J., Siezen, R. J., Zwahlen, M. C., Vilanova, D., Pridmore, R. D., Mercenier, A., et al. (2004). The complete genomes of Lactobacillus plantarum and Lactobacillus johnsonii reveal extensive differences in chromosome organization and gene content. Microbiology 150, 3601–3611. doi: 10.1099/mic.0.27392-0
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., and Palsson, B. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 113, E3801–E3809. doi: 10.1073/pnas.1523199113
Chen, Y., Liu, S., Yu, C., Azevedo, P., Liu, S., O, K., et al. (2021). Evaluating the Effectiveness of Lactobacillus zeae against Enterotoxigenic Escherichia coli F4 Infection in an In Vitro Porcine Intestinal Epithelial Cell Model. ACS Food Sci. Technol. 1, 215–228. doi: 10.1021/acsfoodscitech.0c00069
Cobo-Simón, M., and Tamames, J. (2017). Relating genomic characteristics to environmental preferences and ubiquity in different microbial taxa. BMC Genomics 18:499. doi: 10.1186/s12864-017-3888-y
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Cui, Y., and Qu, X. (2011). Comparative analysis of two component signal transduction systems of the Lactobacillus acidophilus group. Brazilian J. Microbiol. 42, 147–157. doi: 10.1590/S1517-83822011000100019
Eren, A. M., Esen, ÖC., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Fallico, V., McAuliffe, O., Ross, R. P., Fitzgerald, G. F., and Hill, C. (2011). “The potential of lacticin 3147, enterocin AS-48, lacticin 481, variacin and sakacin P for food biopreservation,” in Protective Cultures, Antimicrobial Metabolites and Bacteriophages for Food and Beverage Biopreservation, ed <gnm>C.</gnm> <snm>Lacroix</snm> (Amsterdam: Elsevier), 100–128. doi: 10.1533/9780857090522.1.100
Feyereisen, M., Mahony, J., Kelleher, P., Roberts, R. J., O’Sullivan, T., Geertman, J. M. A., et al. (2019). Comparative genome analysis of the Lactobacillus brevis species. BMC Genomics 20:416. doi: 10.1186/s12864-019-5783-1
Guinane, C. M., Crispie, F., and Cotter, P. D. (2016). “Value of microbial genome sequencing for probiotic strain identification and characterization: promises and pitfalls,” in The Gut-Brain Axis Dietary, Probiotic, and Prebiotic Interventions on the Microbiota, eds N. Hyland and C. Stanton (Amsterdam: Elsevier), 45–60. doi: 10.1016/B978-0-12-802304-4.00004-9
Heo, S., Kim, J.-H., Kwak, M.-S., Sung, M.-H., and Jeong, D.-W. (2021). Functional annotation genome unravels potential probiotic Bacillus velezensis Strain KMU01 from traditional Korean fermented kimchi. Foods 10:563. doi: 10.3390/foods10030563
Hill, D., Sugrue, I., Tobin, C., Hill, C., Stanton, C., and Ross, R. P. (2018). The Lactobacillus casei group: history and health related applications. Front. Microbiol. 9:2107. doi: 10.3389/fmicb.2018.02107
Huang, C. H., Chen, C. C., Liou, J. S., Lee, A. Y., Blom, J., Lin, Y. C., et al. (2020). Genome-based reclassification of Lactobacillus casei: emended classification and description of the species Lactobacillus zeae. Int. J. Syst. Evol. Microbiol. 70, 3755–3762. doi: 10.1099/ijsem.0.003969
Huang, C. H., Liou, J. S., Lee, A. Y., Tseng, M., Miyashita, M., Huang, L., et al. (2018). Polyphasic characterization of a novel species in the Lactobacillus casei group from cow manure of Taiwan: description of L. chiayiensis sp. nov. Syst. Appl. Microbiol. 41, 270–278. doi: 10.1016/j.syapm.2018.01.008
Jia, J., Liu, M., Feng, L., and Wang, Z. (2022). Comparative genomic analysis reveals the evolution and environmental adaptation of Acinetobacter johnsonii. Gene 808:145985. doi: 10.1016/j.gene.2021.145985
Jomehzadeh, N., Javaherizadeh, H., Amin, M., Saki, M., Al-Ouqaili, M. T. S., Hamidi, H., et al. (2020). Isolation and identification of potential probiotic Lactobacillus species from feces of infants in southwest Iran. Int. J. Infect. Dis. 96, 524–530. doi: 10.1016/j.ijid.2020.05.034
Kang, J., Chung, W. H., Lim, T. J., Whon, T. W., Lim, S., and Nam, Y. D. (2017). Complete genome sequence of Lactobacillus casei LC5, a potential probiotics for atopic dermatitis. Front. Immunol. 8:413. doi: 10.3389/fimmu.2017.00413
Kankainen, M., Paulin, L., Tynkkynen, S., Von Ossowski, I., Reunanen, J., Partanen, P., et al. (2009). Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human-mucus binding protein. Proc. Natl. Acad. Sci. U. S. A. 106, 17193–17198. doi: 10.1073/pnas.0908876106
Kant, R., Palva, A., and Von Ossowski, I. (2017). An in silico pan-genomic probe for the molecular traits behind Lactobacillus ruminis gut autochthony. PLoS One 12:e0175541. doi: 10.1371/journal.pone.0175541
Kim, E., Yang, S. M., Cho, E. J., and Kim, H. Y. (2020b). Novel real-time PCR assay for Lactobacillus casei group species using comparative genomics. Food Microbiol. 90:103485. doi: 10.1016/j.fm.2020.103485
Kim, E., Chang, H. C., and Kim, H. Y. (2020a). Complete genome sequence of Lactobacillus plantarum EM, A putative probiotic strain with the cholesterol-lowering effect and antimicrobial activity. Curr. Microbiol. 77, 1871–1882. doi: 10.1007/s00284-020-02000-8
Kim, E., Yang, S.-M., and Kim, H.-Y. (2021). Differentiation of Lacticaseibacillus zeae Using Pan-Genome Analysis and Real-Time PCR Method Targeting a Unique Gene. Foods 10:2112. doi: 10.3390/foods10092112
Lee, I., Kim, Y. O., Park, S. C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Lee, K. W., Park, J. Y., Sa, H. D., Jeong, J. H., Jin, D. E., Heo, H. J., et al. (2014). Probiotic properties of Pediococcus strains isolated from jeotgals, salted and fermented Korean sea-food. Anaerobe 28, 199–206. doi: 10.1016/j.anaerobe.2014.06.013
Li, B., Jin, D., Evivie, S. E., Li, N., Yan, F., Zhao, L., et al. (2017). Safety assessment of Lactobacillus helveticus KLDS1.8701 based on whole genome sequencing and oral toxicity studies. Toxins (Basel). 9:301. doi: 10.3390/toxins9100301
Li, X. Y., Li, L. X., Li, Y., Zhou, R. C., Li, B., Gu, X., et al. (2021). Complete genome sequencing of Peyer’s patches-derived Lactobacillus taiwanensis CLG01, a potential probiotic with antibacterial and immunomodulatory activity. BMC Microbiol. 21:68. doi: 10.1186/s12866-021-02127-z
Mazé, A., Boël, G., Zúñiga, M., Bourand, A., Loux, V., Yebra, M. J., et al. (2010). Complete genome sequence of the probiotic Lactobacillus casei strain BL23. J. Bacteriol. 192, 2647–2648. doi: 10.1128/JB.00076-10
Møretrø, T., Aasen, I. M., Storrø, I., and Axelsson, L. (2000). Production of sakacin P by Lactobacillus sakei in a completely defined medium. J. Appl. Microbiol. 88, 536–545. doi: 10.1046/j.1365-2672.2000.00994.x
Oh, N. S., Joung, J. Y., Lee, J. Y., and Kim, Y. (2018). Probiotic and anti-inflammatory potential of Lactobacillus rhamnosus 4B15 and Lactobacillus gasseri 4M13 isolated from infant feces. PLoS One 13:e0192021. doi: 10.1371/journal.pone.0192021
Oh, Y. J., and Jung, D. S. (2015). Evaluation of probiotic properties of Lactobacillus and Pediococcus strains isolated from Omegisool, a traditionally fermented milletalcoholic beverage in Korea. LWT Food Sci. Technol. 63, 437–444. doi: 10.1016/j.lwt.2015.03.005
Qureshi, N., Gu, Q., and Li, P. (2020). Whole genome sequence analysis and in vitro probiotic characteristics of a Lactobacillus strain Lactobacillus paracasei ZFM54. J. Appl. Microbiol. 129, 422–433. doi: 10.1111/jam.14627
Sharma, V., Mobeen, F., and Prakash, T. (2018). Exploration of survival traits, probiotic determinants, host interactions, and functional evolution of bifidobacterial genomes using comparative genomics. Genes (Basel). 9:477. doi: 10.3390/genes9100477
Son, S. H., Jeon, H. L., Jeon, E. B., Lee, N. K., Park, Y. S., Kang, D. K., et al. (2017). Potential probiotic Lactobacillus plantarum Ln4 from kimchi: evaluation of β-galactosidase and antioxidant activities. LWT Food Sci. Technol. 85, 181–186. doi: 10.1016/j.lwt.2017.07.018
Soni, R., Nanjani, S., and Keharia, H. (2021). Genome analysis reveals probiotic propensities of Paenibacillus polymyxa HK4. Genomics 113, 861–873. doi: 10.1016/j.ygeno.2020.10.017
Stefanovic, E., and McAuliffe, O. (2018). Comparative genomic and metabolic analysis of three Lactobacillus paracasei cheese isolates reveals considerable genomic differences in strains from the same niche. BMC Genomics 19:205. doi: 10.1186/s12864-018-4586-0
Yang, Y., An, H., Zhai, Z., Wang, G., Li, J., and Hao, Y. (2016). Complete genome sequence of Lactobacillus helveticus CAUH18, A potential probiotic strain originated from koumiss. J. Biotechnol. 224, 18–19. doi: 10.1016/j.jbiotec.2016.03.004
Ye, K., Li, P., and Gu, Q. (2020). Complete genome sequence analysis of a strain Lactobacillus pentosus ZFM94 and its probiotic characteristics. Genomics 112, 3142–3149. doi: 10.1016/j.ygeno.2020.05.015
Yu, H. S., Jang, H. J., Lee, N. K., and Paik, H. D. (2019). Evaluation of the probiotic characteristics and prophylactic potential of Weissella cibaria strains isolated from kimchi. LWT 112:108229. doi: 10.1016/j.lwt.2019.05.127
Zhang, J., Kasciukovic, T., and White, M. F. (2012). The CRISPR Associated Protein Cas4 Is a 5′ to 3′ DNA Exonuclease with an Iron-Sulfur Cluster. PLoS One 7:e47232. doi: 10.1371/journal.pone.0047232
Zhang, W., Ji, H., Zhang, D., Liu, H., Wang, S., Wang, J., et al. (2018). Complete genome sequencing of Lactobacillus plantarum ZLP001, a potential probiotic that enhances intestinal epithelial barrier function and defense against pathogens in pigs. Front. Physiol. 9:1689. doi: 10.3389/fphys.2018.01689
Zhang, W., Wang, J., Zhang, D., Liu, H., Wang, S., Wang, Y., et al. (2019). Complete Genome sequencing and comparative genome characterization of Lactobacillus johnsonii ZLJ010, a potential probiotic with health-promoting properties. Front. Genet. 10:812. doi: 10.3389/fgene.2019.00812
Keywords: Lacticaseibacillus chiayiensis, Lacticaseibacillus zeae, complete genome sequencing, comparative genomics, probiotic, Lacticaseibacillus casei
Citation: Kim E, Yang S-M, Kim D and Kim H-Y (2022) Complete Genome Sequencing and Comparative Genomics of Three Potential Probiotic Strains, Lacticaseibacillus casei FBL6, Lacticaseibacillus chiayiensis FBL7, and Lacticaseibacillus zeae FBL8. Front. Microbiol. 12:794315. doi: 10.3389/fmicb.2021.794315
Received: 13 October 2021; Accepted: 10 December 2021;
Published: 07 January 2022.
Edited by:
Si Hong Park, Oregon State University, United StatesReviewed by:
Zhi-Qiang Xiong, University of Shanghai for Science and Technology, ChinaCorina-Diana Ceapa, Universidad Nacional Autónoma de México, Mexico
Copyright © 2022 Kim, Yang, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hae-Yeong Kim, hykim@khu.ac.kr