 Federica De Lise1
Federica De Lise1 Andrea Strazzulli2,3
Andrea Strazzulli2,3 Roberta Iacono1,2
Roberta Iacono1,2 Nicola Curci1,2Mauro Di Fenza1Luisa Maurelli1
Nicola Curci1,2Mauro Di Fenza1Luisa Maurelli1 Marco Moracci1,2,3
Marco Moracci1,2,3 Beatrice Cobucci-Ponzano1*
Beatrice Cobucci-Ponzano1*- 1Institute of Biosciences and BioResources – National Research Council of Italy, Naples, Italy
- 2Department of Biology, University of Naples Federico II, Complesso Universitario di Monte S. Angelo, Naples, Italy
- 3Task Force on Microbiome Studies, University of Naples Federico II, Naples, Italy
Genetic code decoding, initially considered to be universal and immutable, is now known to be flexible. In fact, in specific genes, ribosomes deviate from the standard translational rules in a programmed way, a phenomenon globally termed recoding. Translational recoding, which has been found in all domains of life, includes a group of events occurring during gene translation, namely stop codon readthrough, programmed ± 1 frameshifting, and ribosome bypassing. These events regulate protein expression at translational level and their mechanisms are well known and characterized in viruses, bacteria and eukaryotes. In this review we summarize the current state-of-the-art of recoding in the third domain of life. In Archaea, it was demonstrated and extensively studied that translational recoding regulates the decoding of the 21st and the 22nd amino acids selenocysteine and pyrrolysine, respectively, and only one case of programmed –1 frameshifting has been reported so far in Saccharolobus solfataricus P2. However, further putative events of translational recoding have been hypothesized in other archaeal species, but not extensively studied and confirmed yet. Although this phenomenon could have some implication for the physiology and adaptation of life in extreme environments, this field is still underexplored and genes whose expression could be regulated by recoding are still poorly characterized. The study of these recoding episodes in Archaea is urgently needed.
Introduction
Translation, in its basic mechanism, is universally conserved and is performed by one of the most complex and sophisticated cell machineries, the ribosomes, in which the majority of protein components are highly conserved in all of the domains of life. However, both the genetic code and its decoding are neither universal nor immutable due to the complex nature of translation. The genetic code is not quite universal; in fact, it is well established that the meaning of some codons in certain organelles and organisms has been reassigned (codon reassignment) for all the mRNAs belonging to that organelle or organism. Unlike codon reassignment, in non-canonical translation mechanisms the alteration of the translation rules does not occur for the whole organism but is limited only to specific genes and often correlated to particular physiological conditions that regulate their translation. The discovery of these gene expression regulatory mechanisms has completely changed our view of the disrupted genes that are often found during genome sequencing. In fact, sequenced genomes often reveal interrupted coding sequences and they are generally considered sequencing errors or pseudogenes. It is now well known that the majority of these interrupted genes are functional and encode proteins whose expression is regulated. Non-canonical translation mechanisms have been identified in all steps of the translation: initiation, elongation and termination. Well known strategies related to the initiation of translation are internal ribosome entry, leaky scanning, non-AUG initiation, ribosome shunting and reinitiation. These strategies are used extensively by viruses, presumably providing alternative ways to express different proteins from a single mRNA, facilitating the access to overlapping ORFs and overcoming the structural differences present in viral transcripts in comparison with cellular mRNAs. Furthermore, it has been shown that cancer cells exploit these alternative modes of translation initiation for their survival and proliferation under stressful conditions (for comprehensive reviews see Firth and Brierley, 2012; Pooggin and Ryabova, 2018; Sriram et al., 2018; Yang and Wang, 2019; Cao and Slavoff, 2020).
Programmed deviations from the standard translational rules occuring during translational elongation or termination steps are termed recoding (Gesteland and Atkins, 1996; Firth and Brierley, 2012) and, often in competition with standard decoding, have crucial roles in the regulation of gene expression (Baranov et al., 2002). These universal mechanisms are +1 or –1 programmed frameshifting (PRF) and ribosome hopping, which occur during the elongation step, and stop codon readthrough/redefinition occurring during the termination step (Farabaugh, 1996; Gesteland and Atkins, 1996; Baranov et al., 2002; Namy et al., 2004; Ling et al., 2015; Atkins et al., 2016; Rodnina et al., 2020).
In stop codon readthrough (Figure 1), the termination codon is decoded by a tRNA rather than a release factor, allowing ribosomes to synthesize an extended polypeptide. In specific genes, this tRNA carries the unusual amino acids selenocysteine (Hatfield and Gladyshev, 2002) or pyrrolysine (Namy et al., 2004), and specific stimulatory elements downstream to the stop codon regulate this process (Bertram et al., 2001). In PRF (Figure 1), ribosomes are induced to switch, upward or backward for +1 and –1 PRF, respectively, to an alternative, overlapping reading frame at a specific shift site (Farabaugh, 1996; Atkins et al., 2016). This is a regulated process and its frequency depends by genes and on the presence of stimulatory signals in the mRNA. PRF has been detected in organisms from all three domains of life, but it is very common in viruses (Baranov et al., 2006; Firth and Brierley, 2012), in which several recoding events have been described and characterized. Ribosome hopping (Figure 1) is a rarer recoding event in which the ribosome stops in a precise site of the mRNA and re-starts translation downstream bypassing few nucleotides. This mechanism has been discovered and studied in detail in the gene 60 of bacteriophage T4 (Herr et al., 2004). Ribosomal bypass occurs at hop elements where the ribosome block at the “take-off codon,” immediately upstream of a stop codon followed by a hairpin, determining the dissociation of the peptidyl-tRNA which re-associates at the “landing triplet,” 50 nt downstream, where the translation resumes. More recently, several bypassing elements (byps) have been reported in Magnusiomyces capitatus mitochondria, suggesting that hopping is more frequent than previously thought (Lang et al., 2014; Nosek et al., 2015). An updated list of genes regulated by recoding can be found in the Recode2 database (1 Bekaert et al., 2010).
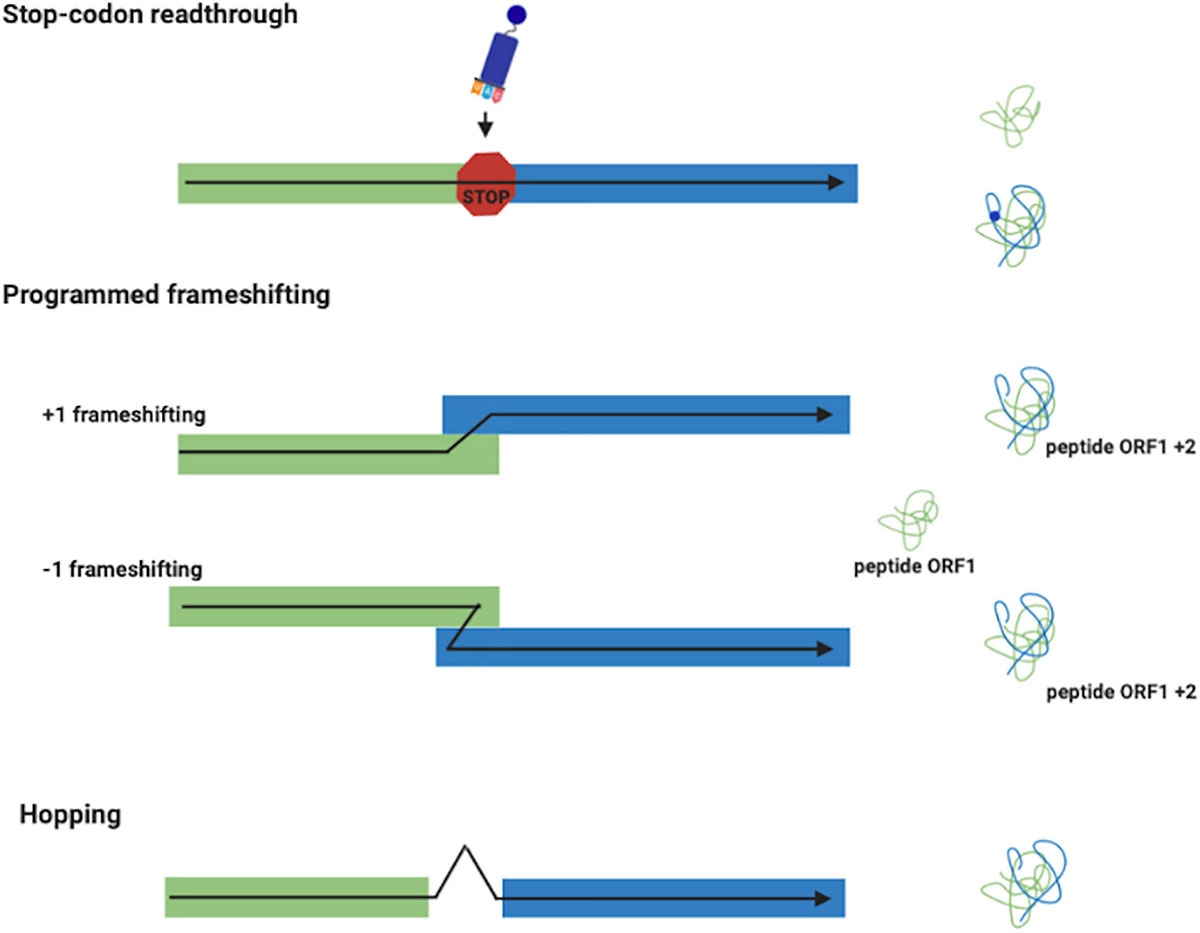
Figure 1. Recoding events. Stop-codon readthrough: a different meaning is assigned to a stop codon with the insertion of the unusual amino acids selenocysteine and pyrrolysine. Frameshifting (+1 and –1): produces two polypeptides from different reading frames of the same mRNA. Ribosome Hopping: synthesizes one protein from two open discontinuous reading frames.
In recent years, bioinformatic analyses of sequenced genomes available in databases have allowed the identification of numerous interrupted genes that could be potential candidates for genes whose expression is regulated by recoding (van Passel et al., 2007; Cobucci-Ponzano et al., 2010; Sharma et al., 2011). However, to date, most of these have been identified serendipitously. A huge boost to the study of non-canonical translation mechanisms came from the development of ribosome profiling, or ribo-seq, a technique that provides genome-wide information on protein synthesis (GWIPS) in vivo (Ingolia et al., 2009). Ribosome profiling is based on the deep sequencing of ribosome-protected mRNA fragments and the high resolution of this technique allows the determination of ribosome density along individual cellular mRNA molecules. The real power of ribosome profiling is in its ability to obtain position-specific information regarding ribosome locations on mRNAs, allowing the identification of unpredictable non-canonical translation events. Since its invention, the ribosome profiling technique has been applied in a range of studies in both prokaryotic and eukaryotic organisms, but only one analysis is reported in Archaea (Michel and Baranov, 2013; Brar and Weissman, 2015; Gelsinger et al., 2020).
In Archaea, non-canonical translation events have been demonstrated only during the elongation and the termination steps. In particular, termination codon readthrough events regulating the incorporation of the amino acids selenocysteine and pyrrolysine (Nicholas et al., 2018; Rother and Quitzke, 2018), and –1 PRF allowing the expression of a functional α-L-fucosidase (Cobucci-Ponzano et al., 2003a,b, 2005a, 2005b, 2006, 2012). More recently, –1 PRF was also reported in siphoviruses tailed virus 1 (HVTV-1) and three viruses (HCTV-1,2 and 5) that infect halophilic archaea (Pietila et al., 2013; Sencilo et al., 2013). Increasing evidence suggests that the flexibility of genetic code decoding is a trait selected during evolution to benefit microorganisms under certain physiological conditions, increasing their fitness (Ling et al., 2015). This could be particularly relevant for Archaea, often inhabiting extreme environments in which changes in nutrients, pH, temperatures, etc. are rather common and occur rapidly and reversibly, and may expose microbes to the necessity to modify reversibly gene expression through quick mechanisms (Iacono et al., 2020; Onofri et al., 2020; Strazzulli et al., 2020). Here, we summarize the current state of the art on the studies on the mechanisms of translational recoding found in Archaea, often living in extreme conditions, to provide an update of this interesting and relatively unknown mechanism of regulation of gene expression in the third domain of life.
Stop Codon Readthrough
In stop codon readthrough it is important to distinguish between two different mechanisms: ‘reassignment’ and ‘recoding’ (Atkins and Baranov, 2010). In codon reassignment, occurring for example in certain mitochondria (Barrell et al., 1979; Osawa et al., 1992), the meaning of particular codons is always reassigned. That codon has only the new meaning and this redefinition is context-independent. These reassignments mainly involve UAG or UGA codons encoding an amino acid instead of a termination signal. Instead, in context-dependent codon redefinition, such event only applies to particular stop codons. Stop codon readthrough is dynamic, with the new definition competing with the standard one, so only a part of the product reflects the new meaning. When it occurs, this redefinition mechanism is a recoding event (Figure 1) in which UAG or UGA specify for the amino acids selenocysteine and pyrrolysine, respectively.
The 21st Amino Acid: Selenocysteine
The twenty-first amino acid selenocysteine (Sec) contains selenium, an essential micronutrient for many organisms, and is translationally incorporated into proteins in Bacteria, Eukarya and Archaea (for a comprehensive review see Ambrogelly et al., 2007). Sec does not have a fully dedicated codon, but it is inserted in response to the UGA stop codons that are recoded in the presence of specific regulation signals in cis. When translating ribosomes encounter an UGA stop codon in the presence of regulative signals, they are loaded with a specific Sec-tRNA, promoting the insertion of a Sec residue in this location. In fact, in response to those signals, a Sec-specific elongation factor (SelB) replaces the standard EF-Tu uniquely for the translation of Sec UGA codons and recruits the specific Sec-tRNA (see below for the description of the mechanism of insertion). In bacteria, bSelB is homologous in the N-terminal part to the standard elongation factor ET-Tu, while it has a C-terminal extension responsible for binding to SECIS elements. In contrast to that, the C-terminal extension of the archaeal aSelB is shorter and unrelated to that of bacteria and these structural features are conserved in the eukaryotic homolog eSelB (Kromayer et al., 1996; Fagegaltier et al., 2000; Tujebajeva et al., 2000; Yoshizawa et al., 2005).
This structural difference is most likely the cause of the lack of binding of aSelB to a cognate SECIS element in vitro (Rother et al., 2000).
The presence of Sec as selenium carrier in natural proteins, called selenoproteins, was first demonstrated in clostridial glycine reductase (Cone et al., 1976). Sec was then found in enzymes maintaining cell redox balance defending the cell against reactive oxygen species. In humans, the selenoproteome comprises 25 members, whose biological functions have been implicated in diverse human diseases ranging from cardiovascular and endocrine disorders to abnormalities in immune responses and cancer (Bellinger et al., 2009).
Selenoproteins are often enzymes with oxidoreductase function in which Sec is the catalytic redox active site. Homologs proteins in which Sec is replaced with cysteine (Cys) exist for the great majority of selenoproteins, although they perform the same reaction less efficiently (Fomenko and Gladyshev, 2012). It is generally accepted that Sec is used in place of Cys due to its higher reactivity, which leads to improved catalytic efficiency, although the exchangeability of Sec and Cys is debated (Gromer et al., 2003; Castellano, 2009; Hondal and Ruggles, 2011; Hondal et al., 2013). The fact that the Sec-containing proteins are more active if compared to the Cys-containing versions was elegantly demonstrated by inactivating the Sec-specific elongation factor SelB in M. maripaludis JJ and observing that this led to overexpression of Cys-containing versions of selenoproteins (Rother et al., 2003). Selenoproteins are not present in all organisms but their distribution is scattered among all the three domains of life in which, however, they perform different functions (Mariotti et al., 2015). In Bacteria, selenoproteins are involved in redox homeostasis, electron transport/energy metabolism, compound detoxification, and oxidative protein folding. In contrast, in Archaea they are involved in methanogenesis, with the only exception of selenophosphate synthetase (SPS), involved in Sec biosynthesis (Stock and Rother, 2009; Rother and Krzycki, 2010). In Eukarya, selenoproteins are mainly involved in redox regulation, antioxidant defense, protein repair, and oxidative protein folding, with very few examples involved in compound detoxification, electron transport, and energy metabolism (Labunskyy et al., 2014). However, Bacteria and Archaea share a larger number of selenoprotein families if compared to Eukarya (Mariotti et al., 2016).
In Archaea, Bacteria and Eukarya, Sec is synthesized in a tRNA-bound fashion, although the mechanisms of Sec synthesis and insertion show differences in the three domains of life (Figure 2). While archaea and eukaryotes first catalyze the synthesis of phospho-Ser with the protein phosphoseryl-tRNASec kinase (PSTK), and then convert it to Sec, bacteria directly synthesize Sec from Ser (For a review see Rother and Quitzke, 2018).
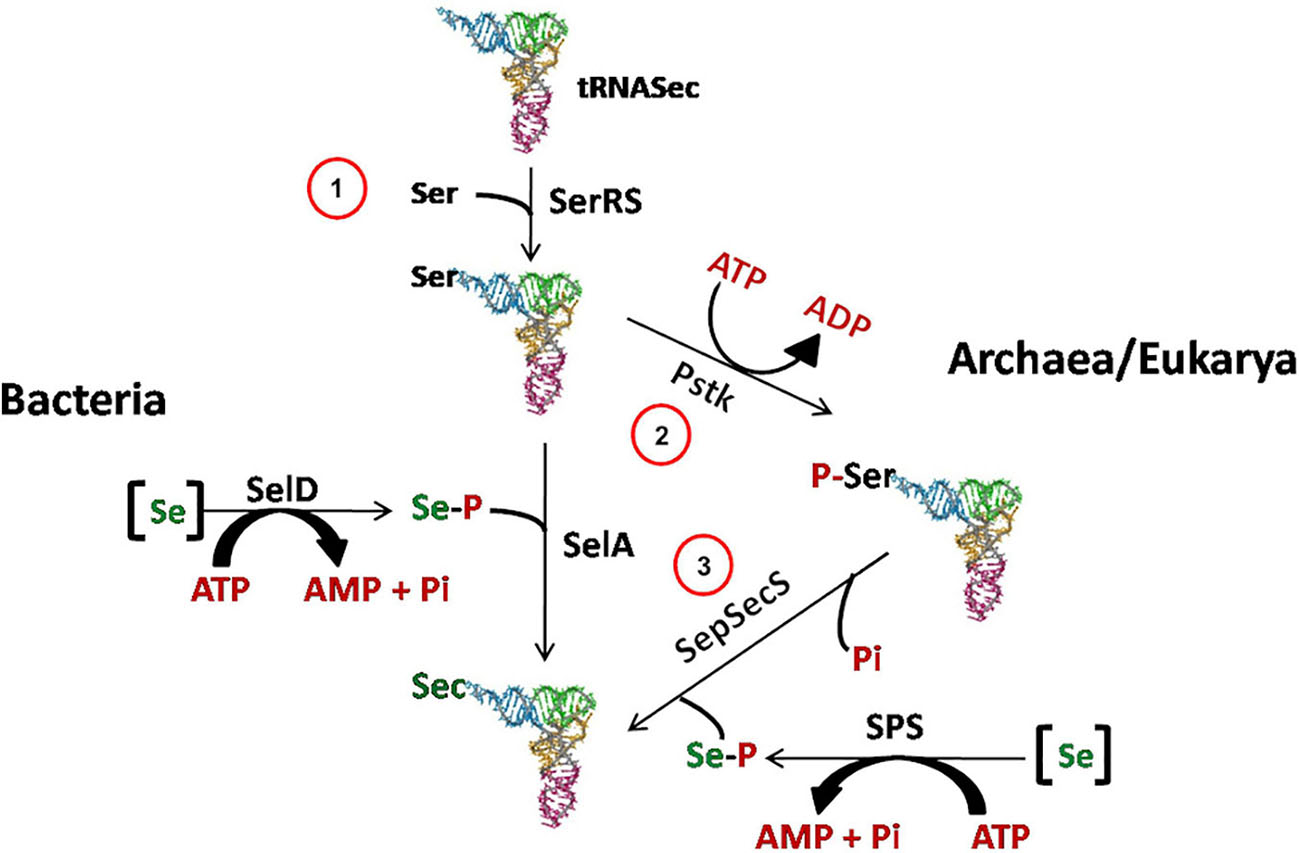
Figure 2. Sec biosynthesis in the three domains of life. In Archaea, as well as in Eukarya, Sec is synthesized in three steps. First (1), SerRS acylates tRNASec with serine to generate Ser-tRNASec. Then (2) PSTK forms Sep-tRNASec, which is converted to Sec-tRNASec by SepSecS in the presence of selenophosphate produced by selenophosphate synthetase (SPS) (3). -[Se]: reduced Se species; -SerRS: seryl-tRNA synthetase; -SelD/SPS: selenophosphate synthetase; -SelA: bacterial Sec synthase; -PstK: seryl-tRNASec kinase; -SepSecS: O-phosphoseryl-tRNA:selenocysteyl-tRNA synthase.
The insertion of Sec is driven by specific signals found in the selenoprotein gene transcripts in cis. These signals are RNA structures, named SECIS (SElenoCysteine Insertion Sequence) elements (Berry et al., 1991; Figure 3). In response to those signals, the specific elongation factor SelB replaces the standard EF-Tu and recruits the Sec-tRNA, promoting the insertion of Sec residues in a specific UGA (Hatfield and Gladyshev, 2002; Mariotti et al., 2016). Interestingly, SECIS elements do not share similarity in sequence or structure between the three domains of life (Krol, 2002). In bacteria, the SECIS element (bSECIS) is a stem–loop structure located within the coding sequence, immediately downstream of the recoded UGA. The bSECIS is bound directly by the elongation factor bSelB through its C-terminal extension (see above) (Figure 3). The eukaryotic SECIS elements are, instead, located in the 3′ UTR of selenoprotein transcripts and they do not interact directly with eSelB, but though the SECIS Binding Protein 2 SBP2 (Tujebajeva et al., 2000; Fletcher et al., 2001). In addition, it has been found that other factors are involved in eukaryal Sec insertion, as the ribosomal protein L30 (Chavatte et al., 2005).
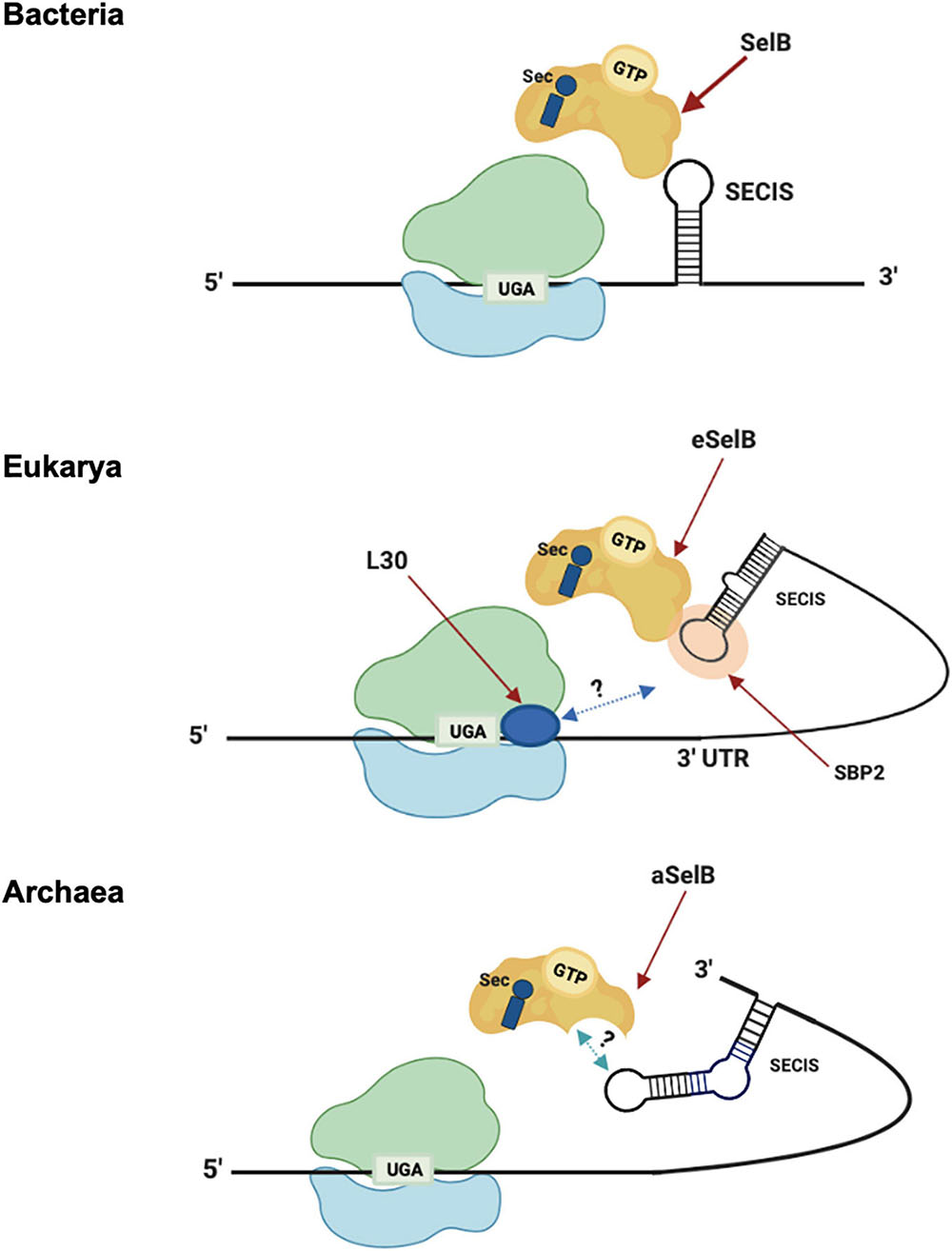
Figure 3. Sec translation in the three domains. Model of Sec incorporation in Bacteria (top), Eukarya (middle), and Archaea (bottom). -3′-UTR: 3′-untranslated region; -L30: ribosomal protein L30; -SBP2: SECIS-binding protein 2; -SECIS: Sec insertion sequence; -SelB/aSelB/eSelB: Sec-specific elongation factor.
The archaeal versions of SECIS (aSECIS) are characterized by two stems separated by an invariant asymmetric bulge (Krol, 2002; Kryukov and Gladyshev, 2004; Stock and Rother, 2009) and are normally located in the 3′ UTR of selenoprotein coding mRNA, with a single documented exception (Wilting et al., 1997). To date, no aSECIS binding factors have been identified. The SBP2 homolog has never been observed in archaea, and it has been shown that the archaeal SelB does not bind aSECIS elements (Mariotti et al., 2016). Thus, it has been proposed that the eukaryal Sec decoding mechanism, in which SBP2 is a key factor, evolved after the transition from archaeal to eukaryotic-like SECIS elements (Stock and Rother, 2009). From an evolutionary point of view, the distribution of selenoproteins in living organisms is consistent with the phylogenetic relationship between the organisms in the three domains of life (Mariotti et al., 2015). In addition, considering the clear homology between the key factors involved in the Sec pathway (tRNAsec, SelB, and the selenophosphate synthetase SPS/SelD) (Santesmasses et al., 2017), it was highlighted that it is very likely that this pathway originated only once in the history of life and was already present in the Last Universal Common Ancestor (LUCA) (Mariotti et al., 2016). However, the presence of this pathway in different living organisms appears to be very dynamic, showing both clear events of horizontal gene transfer and independent loss in many lineages (Zhang et al., 2006; Lobanov et al., 2008; Mariotti et al., 2015).
Selenoproteins are a quite rare feature among the Archaea. Sec was found in formate dehydrogenase, formylmethanofuran dehydrogenase, F420 reducing and non-reducing hydrogenases, HesB-like protein and heterodisulfide reductases (Kryukov and Gladyshev, 2004; Stock et al., 2010). For a detailed list of putative and known archaeal selenoproteins and their properties see Rother and Quitzke (2018). Interestingly, genes encoding selenoproteins, belonging to different families, and the full set of genes encoding for the key factors involved in the Sec pathway, have been found in Lokiarchaeota (Spang et al., 2015), considered the closest cultured archaeal relative of eukaryotes (Mariotti et al., 2016). The selenoprotein families identified in Lokiarchaeota were previously reported in other archaeal lineages (Stock and Rother, 2009), with the exception of the thioredoxin-like superfamily, found by bioinformatic analysis, both in bacteria (Zhang and Gladyshev, 2008) and eukaryotes (Jiang et al., 2012; Mariotti et al., 2013). Moreover, although the selenoprotein genes in Lokiarchaeota are typical of archaea, they possess conserved RNA structures similar to eukaryotic SECIS elements. This finding is the basis of a new theory proposing that eukaryotes have not reinvented the mechanism of insertion of the Sec as previously proposed, but rather that the Sec pathway has passed vertically from Archaea to Eukarya (Rother and Quitzke, 2018).
The 22nd Amino Acid: Pyrrolysine
Pyrrolysine (Pyl) was identified in 2002 as the 22nd proteinogenic amino acid (Hao et al., 2002; Srinivasan et al., 2002). From a biochemical perspective, Pyl is a typical L-lysine amino acid to which a pyrrole ring is branched on the lateral chain through an amide bond. This chemical modification is different from those present in other L-lysine derivatives found in some proteins from archaea like hypusine or methyllysine (Eichler and Adams, 2005). In fact, while in hypusine and methyllysine the modifications originate from post-translational events, Pyl is translationally incorporated (for a review see Brugère et al., 2018). This unusual and highly specialized amino acid is found in a small number of archaea able to metabolize methylamine as well as a few bacteria. The first hint of the presence of pyrrolysine (Pyl) has been reported in several Methanosarcina species with a total of 21 genes of mono, di-, and trimethylamine methyltransferases (MtmB, MtbB, and MttB, respectively) showing an in-frame amber UAG codon (James et al., 2001). Initially, the amino acid inserted into the UAG codon was identified as a lysine. Later, the three-dimensional structure resolution of the enzyme MtmB allowed to demonstrate that the amino acid was a Pyl. Furthermore, the identification of a specific tRNA for Pyl confirmed the hypothesis that Pyl is inserted into proteins during translation by a mechanism of recoding (Hao et al., 2002; Srinivasan et al., 2002). From these preliminary discoveries, several new pieces of information have been collected that have allowed to define the key factors involved in the biosynthesis and insertion of Pyl, the molecular mechanism underlying this recoding mechanism, its distribution and evolution, and the catalytic role of this amino acid.
The five Pyl genes involved in the biosynthesis and genetic encoding of Pyl are pylTSBCD (Srinivasan et al., 2002; Krzycki, 2004; Zhang et al., 2005; Longstaff et al., 2007) and, in most cases, they are organized in an operon-like structure as shown in Figure 4A. The Pyl genes have been found in bacterial and archaeal genomes and are usually clustered near the genes encoding the methylamine methyltransferases and other genes involved in methylamine metabolism (for a detailed description of the genomic contexts of Pyl-related genes see Gaston et al., 2011).
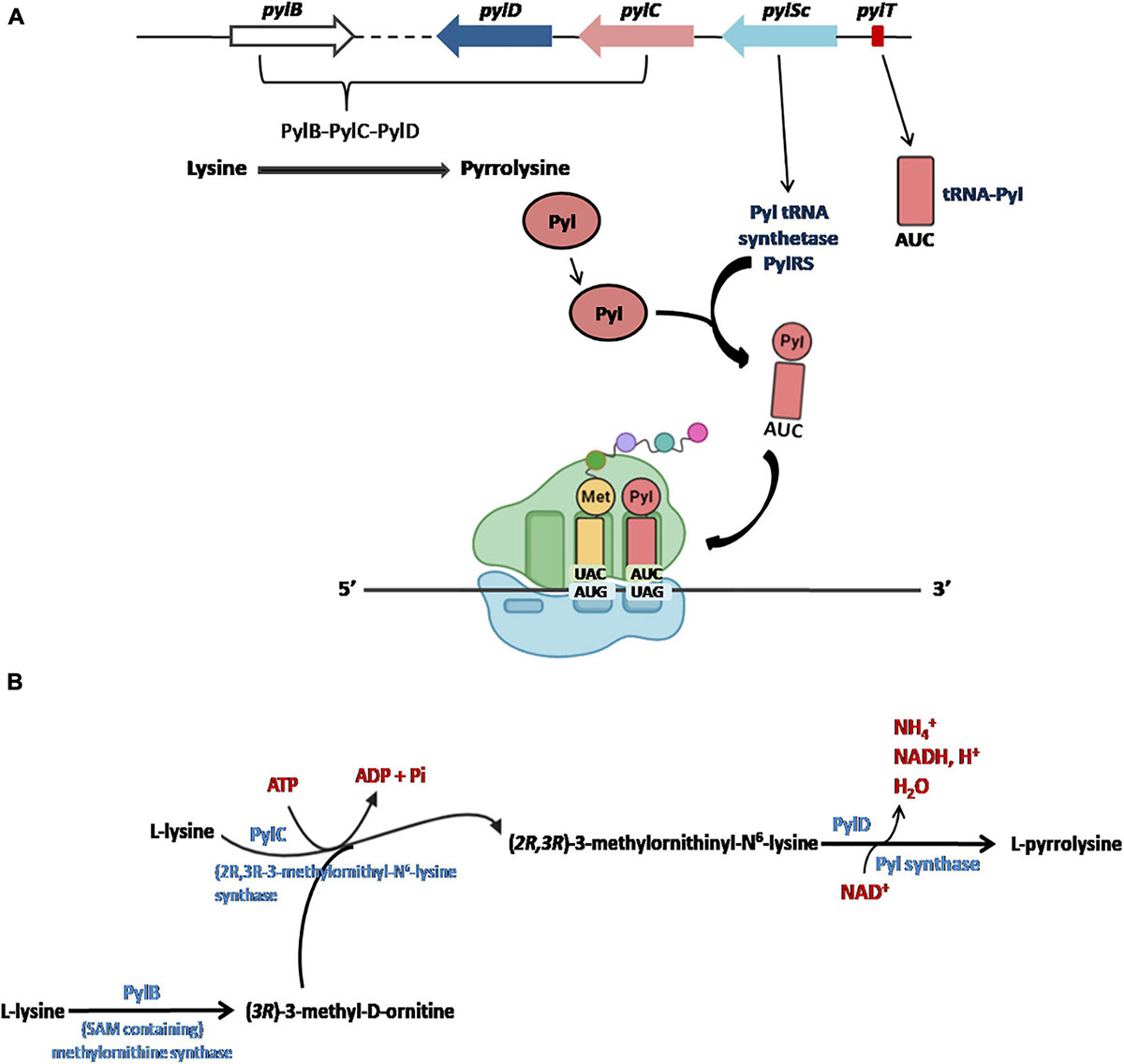
Figure 4. The Pyl insertion system (A). Pyl, synthesized by pylB, pylD, pylC, is charged on a specific tRNA (encoded by pylT) whose anticodon AUC recognizes UAG codons in a specific reaction catalyzed by PylRS (encoded PylSc). See text for details. Figures arranged from Brugère et al. (2018). Biosynthesis of Pyl (B). The complete biosynthesis pathway of L-pyrrolysine from two lysines catalyzed by PylB, PylC and PylD.
Initially, it had been proposed that the synthesis of Pyl took place from the lysyl-tRNA-Pyl (Srinivasan et al., 2002; Polycarpo et al., 2003), similarly to how it occurs for the synthesis of the Sec starting from the seryl-tRNA (Figure 2; Yoshizawa and Bock, 2009; Rother and Krzycki, 2010). However, it is now well documented that Pyl is synthesized by the enzymes PylB, PylC, and PylD from two equivalents of lysine. The two other genes of the Pyl system, pylT and pylS, encode, respectively, for the tRNAPyl, whose anticodon is complementary to the UAG codon, and the subunit of the tRNAPyl synthetase which directly esterified Pyl to the 3′-hydroxyl of tRNAPyl, clearly demonstrating that Pyl is made independently of tRNAPyl (Figure 4A; Blight et al., 2004; Polycarpo et al., 2004; Nozawa et al., 2009; Gaston et al., 2011; Tharp et al., 2018). The complete pathway of biosynthesis of Pyl is reported in Figure 4B (for a review see Brugère et al., 2018).
Although possible sequences that regulate Pyl (named PYLIS by analogy to the SECIS sequences, see above) were initially postulated (Namy et al., 2007), bioinformatic (Zhang et al., 2005) and biochemical studies have shown that no cis element is found or required in E. coli for the recoding of the UAG stop codon into Pyl (Longstaff et al., 2007; Namy et al., 2007). It follows that there is no specific context in the mRNA driving the recoding event, therefore it was proposed that Pyl insertion relies only on the competition between release factors and the tRNA-Pyl during translation. However, how the cell prevents all stop codons from being recoded is still to be elucidated, especially considering that cis signals have not been found. Interestingly, it has been reported that in the clostridial Acetohalobium arabaticum, UAG specifies Pyl only when the cells are grown in trimethylamine, while, when the cells are grown on pyruvate as a carbon source, UAG only specifies termination (Prat et al., 2012). Thus, this result indicates that Pyl insertion is regulated in specific physiological conditions and could suggest the presence of a trans-acting regulation factor expressed only in particular conditions which must still be identified.
Pyl is found in all methanogen methylamine methyltransferase genes and in some cases the readthrough efficiency of the UAG codon is as high as 97%. In these enzymes Pyl is always present in the active site, capturing methylamines before transferring one methyl group to a Co(I)-corrinoid cofactor of an associated protein (MtmC/MtbC/MttC) (Hao et al., 2002), suggesting that its role is fundamental for methylamine metabolism. More recently, it has been reported that natural MttB analogs without Pyl found in Desulfitobacterium hafniense has a glycine betaine methyltransferase activity (Ticak et al., 2014), confirming that methyltransferases containing Pyl are related to methylamines metabolism. The only other known Pyl-containing proteins are some transposases (Zhang et al., 2005), and a tRNAHis-guanylyltransferase Thg1 (Heinemann et al., 2009) both present in a subset of Methanosarcinales.
In archaea, pyl genes were initially identified in anaerobic methanogens living in environments where methylamines are available, namely, several Methanosarcinales (Deppenmeier et al., 2002; Galagan et al., 2002; Maeder et al., 2006), in Methanococcus burtonii (psychrophile) (Goodchild et al., 2004), and in Methanoalophilus mahii and Methanohalobium evestigatum (halophiles) (Rother and Krzycki, 2010; Gaston et al., 2011). More recently, the genes for the synthesis and encoding of Pyl were identified in several new lineages of methanogens, discovered by metagenomic approaches and distantly related to those mentioned above, in which the methanogenesis is dependent on methyl-compounds (Borrel et al., 2013; Evans et al., 2015; Petitjean et al., 2015; Nobu et al., 2016; Vanwonterghem et al., 2016; Sorokin et al., 2017). Pyl-containing methyltransferases needed for methylamine utilization are always present in these new lineages of methanogens that contain the Pyl system, strengthening the hypothesis that the Pyl system is dedicated to the incorporation of Pyl in these methyltransferases, and thus associated to methylamine utilization. Methanohalophilus, in which the Pyl-containing methyltransferases are absent (Fricke et al., 2006) are also lacking the Pyl system, suggesting that this recoding mechanism is linked to methylamine methyltransferases rather than to archaea performing methanogenesis based on methyl-compounds. In addition, it has been found that uncultured sugar-fermenters of the candidate division of Persephonarchaea, thriving in a hypersaline environment, harbor a complete set of genes for Pyl synthesis and mtmB, mtbB, and mttB genes (Guan et al., 2017). The components of the Pyl system in these archaea are phylogenetically related to those found in the bacteria Acetohalobium arabaticum who lives in the same environment, suggesting an event of horizontal gene transfer between these organisms (Guan et al., 2017). From its first discovery great advances have been made in understanding the role of this recoding event in archaea and allowing us to reveal that Pyl-system has a wide distribution and is not necessarily associated with methanogenesis in this domain of life (Brugère et al., 2018).
There are several hypotheses for the emergence of the Pyl system in living organisms. Among the others, one of the most recent, and strongly supported by current data, postulated that the Pyl trait is very ancient and probably only emerged once after LUCA and was linked to methanogenesis. The trait could have then evolved and preserved in organisms for which methylamine metabolism was fundamental to survive and could have been further spread across the bacterial and archaeal domains by horizontal gene transfer (Brugère et al., 2018).
Programmed Ribosomal Frameshifting
During standard mRNA translation the ribosome initiates protein synthesis at a start codon and moves by decoding three nucleotides at a time until it reaches a stop codon where translation is terminated. However, in some cases the ribosomes switch to an alternative reading frame on the mRNA by determining a translational slippage in the +1 or –1 direction (Farabaugh, 1996; Gesteland and Atkins, 1996; Figure 1). In contrast to spontaneous frameshifting, which produces non-functional polypeptides, PRF is generally in competition with standard decoding and typically leads to the synthesis of a functional polypeptide from an alternative frame with efficiencies varying from very low to as high as 80% (Tsuchihashi and Brown, 1992; Atkins et al., 2009). At the functional level there are two more common classes of regulation of PRF. In a first class, often termed ‘set ratio’ frameshifting, the proportion of ribosomes that shift frame is constant, thereby generating an extra N-terminally coincident product. In a second class, frameshift efficiency is dependent by the level of translation initiation or responsive to a trans-acting factor. Here, frameshifting, acting as a sensor and/or effector has a regulatory function, allows the synthesis of a functional trans-frame encoded product or alters mRNA half-life (Atkins et al., 2016). It has been well demonstrated that in eukaryotes, PRF can regulate the stability of an mRNA. In fact, it has been seen that following a PRF event, the ribosomes encounter a stop codon in the new reading frame that activates the nonsense-mediated decay pathway (Belew et al., 2014).
The PRF has been studied extensively in viruses, where –1 PRF plays an important role in viral propagation by modulating synthesis of viral proteins in specific stoichiometric ratios (Jacks and Varmus. 1985; Plant et al., 2010). The use of a –1 PRF mechanism for the expression of a viral gene was first identified in the Rous sarcoma virus (Jacks and Varmus, 1985). To date, it is well known that, for example, all coronaviruses utilize –1 PRF to control the relative expression of their proteins. In general, the early translated viral proteins are involved in neutralizing the host cellular immune response (ORF1a) and in genome replication and RNA synthesis (ORF1b). ORF1b is in the –1 reading frame with respect to ORF1a, and all coronaviruses, as well as SARS-CoV-2, utilize –1 PRF as a mean to synthesize the ORF2 encoded proteins (Kelly et al., 2020). PRF is well documented in retrotransposons and insertion elements too, while it is less common in cellular genes. Among the chromosomal genes, the best studied examples are the Antizyme (Matsufuji et al., 1995) in which + 1 PRF frameshifting functions both as a sensor of the polyamine levels and as an effector of a self-regulating circuit from yeasts to mammals. In the bacterial DNA polymerase, γ and τ subunits are produced in 1:1 molar ratio by –1 PRF from dnaX gene (Tsuchihashi and Kornberg, 1990; Mangold, 2005; Chen et al., 2014). For a comprehensive review on the genes expressed by PRF in Bacteria, Eukarya and viruses see Atkins et al. (2016); Rodnina et al. (2020). Among PRF, –1 frameshifting is more widespread with examples in all three domains of life (Luthi et al., 1990; Tsuchihashi and Kornberg, 1990; Cobucci-Ponzano et al., 2006; Wills et al., 2006; Belew et al., 2014), many of which are phylogenetically conserved.
As stated above, –1 PRF is generally in competition with standard decoding but it is facilitated by two regulatory elements in the mRNA sequence, a slippery site, where the transition to the –1 frame takes place, and a secondary structure element (a pseudoknot, a steam and loop or a kissing loop) at a defined distance of 5 to 9 nucleotides from the slippery site (Brierley et al., 1992, 2010; Atkinson et al., 1997; Lin et al., 2012; Choi et al., 2020). The slippery site, usually in the form of a heptanucleotide sequence X-XXY-YYZ, in which X can be any base, Y is usually A or U, and Z is any base but G (codons are shown in the 0 reading frame), allows for base pairing between the tRNA anticodon and the mRNA codon after shifting into the –1 reading frame. Prokaryotic frameshifting sites may contain additional stimulatory elements, such as an internal Shine-Dalgarno (SD)-like sequence upstream of the slippery site (Larsen et al., 1997; Choi et al., 2020) or tandem rare codons (Caliskan et al., 2017) both with the function of slowing down the translating ribosome and increasing the frameshifting efficiency. –1 PRF can be also facilitated by miRNAs binding as reported in the human mRNA encoding the HIV-1 co-receptor CCR5 (Belew et al., 2014), or proteins, as reported in some viruses (Kobayashi et al., 2010; Napthine et al., 2017; Wang et al., 2019) to the sequence following the slippery site.
Detailed studies on the molecular mechanism by which –1 PRF occurs have only recently been reported. These studies suggest that the molecular mechanisms are mainly two and depend on the availability of the aa-tRNAs of the codons in the slippery sequence (Namy et al., 2006; Chen et al., 2013, 2014; Caliskan et al., 2014, 2017; Kim et al., 2014; Yan et al., 2015; Korniy et al., 2019a, b). When the tRNAs reading the slippery sequence codons are abundant, –1 PRF occurs at the late stage of translocation, with two tRNAs moving through the ribosome, and requires the presence of the stimulatory element within the mRNA sequence. By contrast, in conditions in which aa-tRNAs are limited, the –1 PRF occurs via one-tRNA slippage of the P-site tRNA, when the A site is vacant, and its efficiency is independent of the stimulatory element within the mRNA sequence. This latter mechanism is often called “hungry” frameshifting, because it can be triggered by aa-tRNA limitation due to starvation (Gallant and Lindsley, 1992; Olubajo and Taylor, 2005; Temperley et al., 2010) (see below).
In Archaea only one case of –1 PRF has been reported (Cobucci-Ponzano et al., 2006). In the thermoacidophilic archaeon Saccharolobus solfataricus (formerly Sulfolobus solfataricus) (Sakai and Kurosawa, 2018) strain P2 the fucA1 gene was found to be organized in two open reading frames (ORFs) SSO11867 and SSO3060 of 81 and 426 amino acids, respectively, which are separated by a –1 frameshifting in a 40 bases overlap. These ORFs encode, respectively, for the N- and C-terminal part of a α-L-fucosidase. The overlap region between the two ORFs had the characteristic features of the genes expressed by –1 PRF, including a heptanucleotide A-AAA-AAT (codons are shown in the zero frame), flanked by a putative stem and loop and the tandem rare codons CAC (Figure 5). To test if these gene fragments could lead to a functional enzyme, a full-length gene, named framefucA, was produced by inserting specific site-directed mutations in the fucA1 gene, exactly in the position predicted by –1 PRF. In this way, the poly-A sequence of the slippery site was disrupted and a T nucleotide was inserted to restore a single reading frame between the two ORFs (Cobucci-Ponzano et al., 2003a). The framefucA mutant encoded for a polypeptide of 495 amino acids, that, remarkably, in recombinant form produced a fully functional α-L-fucosidase, named Ssα-fuc, which was thermophilic, thermostable and had an unusual non-americ structure (Cobucci-Ponzano et al., 2003b, 2005a; Rosano et al., 2004). The full-length protein FucA was expressed by –1 PRF in both E. coli and S. solfataricus showing for the first time that this kind of recoding is present in Archaea (Cobucci-Ponzano et al., 2006). The observation that the fucA1 interrupted gene directed the expression of low α-L-fucosidase activity in E. coli led to the isolation and characterization of the polypeptides expressed in the recombinant form demonstrating that the fucA1 gene produced in E. coli a mixture of two full-length polypeptides, both functional, with a total efficiency of about 5% (Xu et al., 2004; Cobucci-Ponzano et al., 2006). The identification of these polypeptides indicated that the translational recoding of fucA1 might occur in two ways, at least in E. coli (Figure 6): a simultaneous backward slippage of the ribosome when both the P- and the A-site tRNAs are occupied (Figure 6A) and/or the repositioning of the ribosome in the –1 frame when only the P-site tRNA is bound (Figure 6B) (Cobucci-Ponzano et al., 2006). The analysis of fucA1 –1 PRF in S. solfataricus by in vitro translation revealed that only the wild type slippery sequence led to the translation of a full-length product with good efficiency (about 10%), demonstrating that this process occurred in archaea (Cobucci-Ponzano et al., 2006). In vivo, full-length polypeptides from fucA1 were identified in S. solfataricus extracts, and reverse real-time PCR experiments and specific enzymatic assays confirmed that this enzyme was functionally expressed though at very low levels.
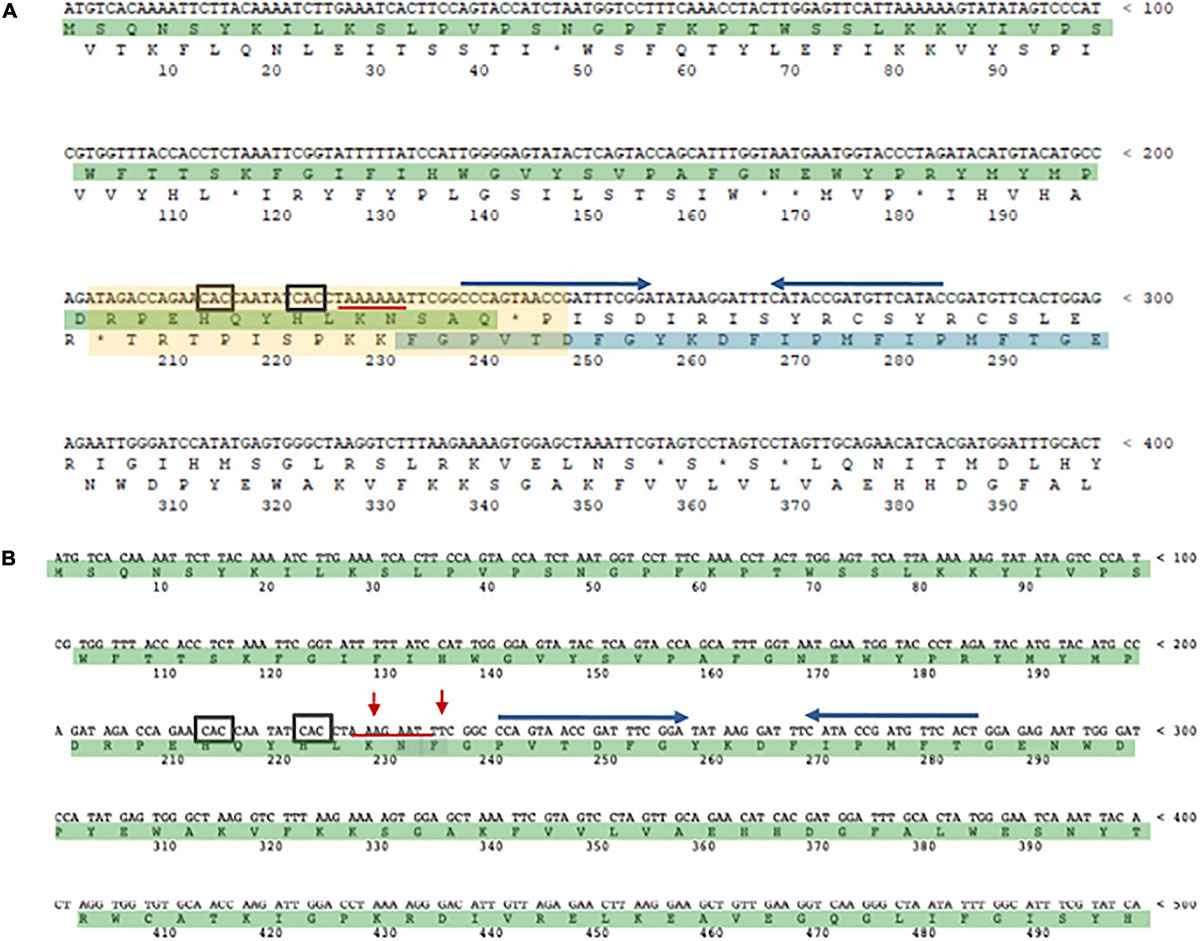
Figure 5. The α-L-fucosidase gene. (A) The N-terminal SSO11867 ORF (highlighted in green) is in the zero frame, the C-terminal SSO3060 ORF (highlighted in blue), for which only a fragment is shown, is in the –1 frame. The 40 bp region of overlap bertween the two ORFs is indicated with a light yellow rectangle. The slippery heptameric sequence is underlined with a red line. The rare codons CAC are indicated with a black square. The putative stem and loop region is indicated with blu arrows. (B) framefucA mutant gene (only a fragment is shown). The red arrows indicate the mutated nucleotides in the slippery sequence.
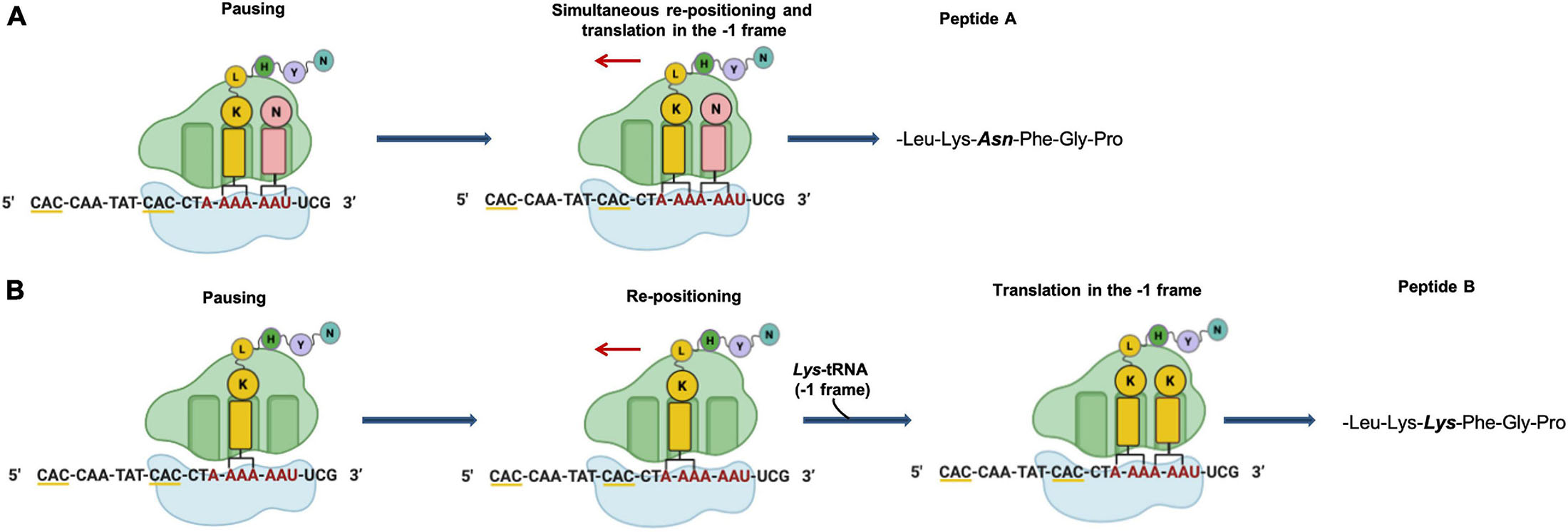
Figure 6. Putative mechanism of programmed –1 frameshifting. (A) Simultaneous P- and A-site slippage; (B) P-site slippage. The slippery heptameric sequence is indicated in red. Rare codons are underlined with yellow line.
Although these studies produced evidence that –1 PRF is present in archaea, several questions remain unanswered: to date, it is still not known why the translational of fucA1 in S. solfataricus is regulated by recoding, and if other genes are expressed by this mechanism in this or other archaea. However, since there are no α-L-fucosidase genes regulated by PRF in Bacteria and Eukarya, it has been suggested that this sophisticated mechanism of translational regulation preexisted in S. solfataricus and it was applied to the α-L-fucosidase gene for physiological reasons. Very recently, it has been found that fucA1 mRNA increases by 10 fold after S. solfataricus undergoes cold shock and in S. solfataricus cells grown in minimal medium containing the oligosaccharides of the hemicellulose xyloglucan (De Lise et al., 2021). Furthermore, this α-L-fucosidase has been shown to cooperate with other glycoside hydrolases from S. solfataricus for the hydrolysis of fucosylated xyloglucan oligosaccharides by removing the fucose moieties from this substrate with high efficiency in vitro (Curci et al., 2021). These new results will certainly need to be explored and could be of great help in understanding what the function of this enzyme is in vivo, and why its expression is regulated by 1- PRF.
Genomic sequencing showed that the fucA1 gene was also present in other archaea, all belonging to Crenarchaeota (for the compilation of these genes, see the Carbohydrate Active enZyme database2). The α-L-fucosidases from Sulfolobales showed 96% amino acid sequence identity and are all full length with the exception of the S. solfataricus, strain 98/2 which presented the frameshifting in the same position as the gene from strain P2. However, all Sulfolobales genes showed 100% DNA sequence identity in the region of the frameshifting, maintaining the rare codon, the slippery sequence, in which the stretch of A is shortened by one nucleotide in full-length genes, and the putative stem loop. On the contrary, the slippery sequence is not conserved in full-length α-L-fucosidase homologs from I. aggregans and C. maquilingensis. Remarkably, full-length α-L-fucosidases, in the region of the slippery sequence, have the same Lys or Asn amino acids observed in the full- length product of the wild-type interrupted fucA (Cobucci-Ponzano et al., 2006).
More recently, PRF events have been reported in some archaeal viruses. In particular, –1 PRF seems to be used by the siphoviruses tailed virus 1 (HVTV-1) and three viruses (HCTV-1,2 and 5) that infect halophilic archaea, while an event of +1 PRF appears to be present in the haloarchaeal myovirus tailed virus 2 (HSTV-2) (Pietila et al., 2013; Sencilo et al., 2013). In addition, it has been suggested that a frameshifting is presumably involved in the synthesis of magnesium chelatase from the archaea Methanocaldococcus and Methanococcus (Antonov et al., 2013b). Unfortunately, genes with frameshifts could be difficult to annotate by standard procedures and often might be annotated as two separate adjacent hypothetical genes (Antonov et al., 2013b). In recent years some bioinformatic tools have been developed with the aim of identifying possible genes regulated by frameshifting (Antonov and Borodovsky, 2010; Antonov et al., 2013a). However, none of these have been systematically tested on Archaea and it would be very useful to know whether the parameters used allow to identify possible genes regulated by frameshifting in this domain of life.
Conclusion
The identification of novel genes whose expression could be regulated translational recoding is not easy, either because disrupted genes are commonly considered non-functional pseudogenes or because technical limitations, and this is particularly true for Archaea for which molecular biology tools are still to be completely developed. Non-functional pseudogenes are present in organisms from all the living domains, though in some cases they have been demonstrated to be useful for an organism’s survival and adaptation to particular environmental changes (Harrison and Gerstein, 2002; Balakirev and Ayala, 2003; Hirotsune et al., 2003). In Archaea, 15 different species have been bioinformatically analyzed revealing a high number of predicted pseudogenes, the highest of which (8.6% of the annotated protein coding sequences) being in S. solfataricus. The expression of these genes has not been tested but, remarkably, all the frameshifts occurred in A/T rich DNA tracts resembling the slippery sequences regulating –1 PRF in cis (van Passel et al., 2007). In addition, a different bioinformatic analysis of other 16 Archaea genomes, allowed to identify a large number of disrupted genes, some of which resulted to be functional, as demonstrated by a high throughput proteomic analysis and functional characterization of some of them from S. solfataricus strain P2 (Cobucci-Ponzano et al., 2010). Interestingly one of the interrupted gene whose expression could be regulated by –1 PRF is the putative universal translation initiation factor SUI-1/aIF1 (Cobucci-Ponzano et al., 2010). This protein is essential in yeast forming the translation initiation complex and monitoring the maintenance of the correct translational reading frame in eukaryotes, such as it was suggested that it might govern programmed –1 frameshifting as a trans-acting factor (Cui et al., 1998; Kyrpides and Woese, 1998). Similarly, in vitro experiments performed with S. solfataricus cell fractions showed that aIF1 promotes translation complex binding to the ribosome, promoting discrimination against non-canonical start codons and enhancing translation efficiency (Hasenöhrl et al., 2006 RNA.; 12: 674–682.; Hasenöhrl et al., 2009 RNA.; 15: 2288–2298.) In the genome annotation of S. solfataricus, P2 strain, this gene is reported as interrupted by –1 frameshfting, but, once re-sequenced, it was found to be full-length, suggesting a possible sequencing error (Hasenöhrl et al., 2006; Cobucci-Ponzano et al., 2010). However, a high-throughput proteomic analysis revealed the presence of two peptides, one deriving from the full-length gene and the other one deriving from the translation of the annotated interrupted gene by –1 PRF (Cobucci-Ponzano et al., 2010). These data merit further investigation and could be of some help to elucidate the possible mechanism of expression of this gene in S. solfataricus and to shed some light of its role in vivo.
It has been suggested that the flexibility of the genetic code decoding, typical of recoding mechanisms, is a trait selected during evolution that may increase microbial fitness under certain conditions (Ling et al., 2015). The majority of Archaea populate extreme environments, which are often spots (e.g., hydrothermal vents, solfataras, etc.) surrounded by environments with milder conditions and frequently subjected to sudden changes that greatly, and temporarily, modify the chemical-physical parameters to which microorganisms must adapt. It is tempting to speculate that in these extreme environments translational recoding could be a way to maintain in a latent state the expression of certain genes, and up- or down-regulate them under specific conditions. Another important aspect to be considered is related to the understanding of the molecular mechanisms that lead to the improved fitness as a result of genetic code variation (Ling et al., 2015). This fostered a new research area in engineering synthetic organisms with new genetic codes and non-canonical amino acids (for a review see Hoffman et al., 2018). These engineered synthetic organisms will be very important to study the physiological effect of genetic code evolution (Ling et al., 2015). Thus, the study of translational recoding in Archaea is particularly important for its possible implications in the evolution of the genetic code and the correlation between the flexibility of the genetic code decoding and improved fitness in extreme environments.
Author Contributions
FDL and BC-P wrote the manuscript. AS, RI, NC, LM, MDF, and MM contributed to the article, edited English style, and approved the submitted version. MDF, MM, FDL, and BC-P edited the manuscript into its final format. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Italian Space Agency: “Life In Space (OPPS)” project (ASI N. 2019-3-U.O).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Giovanni del Monaco, Chiara Nobile, and Marco Petruzziello (IBBR-CNR) for administrative assistance.
Footnotes
References
Ambrogelly, A., Palioura, S., and Söll, D. (2007). Natural expansion of the genetic code. Nat. Chem. Biol. 3, 29–35. doi: 10.1038/nchembio847
Antonov, I., and Borodovsky, M. (2010). Genetack: frameshift identification in protein-coding sequences by the Viterbi algorithm. J. Bioinform Comput. Biol. 8, 535–551. doi: 10.1142/s0219720010004847
Antonov, I., Coakley, A., Atkins, J. F., Baranov, P. V., and Borodovsky, M. (2013b). Identification of the nature of reading frame transitions observed in prokaryotic genomes. Nucleic Acids Res. 41, 6514–6530. doi: 10.1093/nar/gkt274
Antonov, I., Baranov, P., and Borodovsky, M. (2013a). GeneTack database: genes with frameshifts in prokaryotic genomes and eukaryotic mRNA sequences. Nucleic Acids Res. 41, D152–D156. doi: 10.1093/nar/gks1062
Atkins, J. F., and Baranov, P. V. (2010). The distinction between recoding and codon reassignment. Genetics 185, 1535–1536. doi: 10.1534/genetics.110.11901
Atkins, J. F., Loughran, G., Bhatt, P. R., Firth, A. E., and Baranov, P. V. (2016). Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44, 7007–7078. doi: 10.1093/nar/gkw530
Atkinson, J., Dodge, M., and Gallant, J. (1997). Secondary structures and starvation-induced frameshifting. Mol. Microbiol. 26, 747–753. doi: 10.1046/j.1365-2958.1997.6101959.x
Balakirev, E. S., and Ayala, F. J. (2003). Pseudogenes: are they “Junk” or functional DNA? Annu. Rev. Genet. 37, 123–151. doi: 10.1146/annurev.genet.37.040103.103949
Baranov, P. V., Fayet, O., Hendrix, R. W., and Atkins, J. F. (2006). Recoding in bacteriophages and bacterial IS elements. Trends Genet. 22, 174–181. doi: 10.1016/j.tig.2006.01.005
Baranov, P. V., Gesteland, R. F., and Atkins, J. F. (2002). Recoding: translational bifurcations in gene expression. Gene 286, 187–201. doi: 10.1016/s0378-1119(02)00423-7
Barrell, B. G., Bankier, A. T., and Drouin, J. (1979). A different genetic code in human mitochondria. Nature 282, 189–194. doi: 10.1038/282189a0
Bekaert, M., Firth, A. E., Zhang, Y., Gladyshev, V. N., Atkins, J. F., and Baranov, P. V. (2010). Recode-2: new design, new search tools, and many more genes. Nucleic Acids Res. 38, 69–74. doi: 10.1093/nar/gkp788
Belew, A., Meskauskas, A., Musalgaonkar, S., Advani, V., Sulima, S., Kasprzak, W., et al. (2014). Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512, 265–269. doi: 10.1038/nature13429
Bellinger, F. P., Raman, A. V., Reeves, M. A., and Berry, M. J. (2009). Regulation and function of selenoproteins in human disease. Biochem. J. 422, 11–22. doi: 10.1042/BJ20090219
Berry, M. J., Banu, L., Chen, Y. Y., Mandel, S. J., Kieffer, J. D., Harney, J. W., et al. (1991). Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3′ untranslated region. Nature 353, 273–276. doi: 10.1038/353273a0
Bertram, G., Innes, S., Minella, O., Richardson, J., and Stansfield, I. (2001). Endless possibilities: translation termination and stop codon recognition. Microbiology 147, 255–269. doi: 10.1099/00221287-147-2-255
Blight, S. K., Larue, R. C., Mahapatra, A., Longstaff, D. G., Chang, E., Zhao, G., et al. (2004). Direct charging of tRNA(CUA) with pyrrolysine in vitro and in vivo. Nature 431, 333–335. doi: 10.1038/nature02895
Borrel, G., O’Toole, P. W., Harris, H. M., Peyret, P., Brugere, J. F., and Gribaldo, S. (2013). Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol. Evol. 5, 1769–1780. doi: 10.1093/gbe/evt128
Brar, G., and Weissman, J. (2015). Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 16, 651–664. doi: 10.1038/nrm4069
Brierley, I., Gilbert, R., Pennell, S., Atkins, J. F., and Gesteland, R. F. (2010). Recoding: expansion of decoding rules enriches gene expression. Nucleic Acids and Mol. Biol. 24, 149–174.
Brierley, I., Jenner, A., and Inglis, S. (1992). Mutational analysis of the “slippery-sequence” component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 227, 463–479. doi: 10.1016/0022-2836(92)90901-U
Brugère, J. F., Atkins, J. F., and Paul, W. O. (2018). Toole, guillaume borrel; pyrrolysine in archaea: a 22nd amino acid encoded through a genetic code expansion. Emerg. Top. Life Sci. 2, 607–618. doi: 10.1021/ja2054034
Caliskan, N., Katunin, V. I., Belardinelli, R., Peske, F., and Rodnina, M. V. (2014). Programmed –1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631. doi: 10.1016/j.cell.2014.04.041
Caliskan, N., Wohlgemuth, I., Korniy, N., Pearson, M., Peske, F., and Rodnina, M. V. (2017). Conditional switch between frameshifting regimes upon translation of dnaX mRNA. Mol. Cell. 66, 558–567. doi: 10.1016/j.molcel.2017.04.023
Cao, X., and Slavoff, S. A. (2020). Non-AUG start codons: expanding and regulating the small and alternative ORFeome. Exp. Cell. Res. 391:111973. doi: 10.1016/j.yexcr.2020.111973
Castellano, S. (2009). On the unique function of selenocysteine—insights from the evolution of selenoproteins. Biochim. Biophys. Acta 1790, 1463–1470. doi: 10.1016/j.bbagen.2009.03.027
Chavatte, L., Brown, B. A., and Driscoll, D. M. (2005). Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 12, 408–416. doi: 10.1038/nsmb922
Chen, C., Zhang, H., Broitman, S., Reiche, M., Farrell, I., Cooperman, B., et al. (2013). Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol. 20, 582–588. doi: 10.1038/nsmb.2544
Chen, J., Petrov, A., Johansson, M., Tsai, A., O’Leary, S., and Puglisi, J. (2014). Dynamic pathways of -1 translational frameshifting. Nature 512, 328–332. doi: 10.1038/nature13428
Choi, J., O’Loughlin, S., Atkins, J. F., and Puglisi, J. D. (2020). The energy landscape of –1 ribosomal frameshifting. Sci. Advan. 6:eaax6969. doi: 10.1126/sciadv.aax6969
Cobucci-Ponzano, B., Conte, F., Benelli, D., Londei, P., Flagiello, A., Monti, M., et al. (2006). The gene of an archaeal alpha-L-fucosidase is expressed by translational frameshifting. Nucleic Acids Res. 34, 4258–4268. doi: 10.1093/nar/gkl574
Cobucci-Ponzano, B., Guzzini, L., Benelli, D., Londei, P., Perrodou, E., Lecompte, O., et al. (2010). Functional characterization and high- throughput proteomic analysis of interrupted genes in the archaeon Sulfolobus solfataricus. J. Prot. Res. 9, 2496–2507. doi: 10.1021/pr901166q
Cobucci-Ponzano, B., Mazzone, M., Rossi, M., and Moracci, M. (2005a). Probing the catalytically essential residues of the α-L-fucosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. Biochemistry 44, 6331–6342. doi: 10.1021/bi047495f
Cobucci-Ponzano, B., Rossi, M., and Moracci, M. (2005b). Recoding in archaea. Mol. Microbiol. 55, 339–348. doi: 10.1111/j.1365-2958.2004.04400.x
Cobucci-Ponzano, B., Rossi, M., and Moracci, M. (2012). Translational recoding in archaea. Extremophiles 16, 793–803. doi: 10.1007/s00792-012-0482-8
Cobucci-Ponzano, B., Trincone, A., Giordano, A., Rossi, M., and Moracci, M. (2003a). Identification of an archaeal α-L-fucosidase encoded by an interrupted gene. Production of a functional enzyme by mutations mimicking programmed -1 frameshifting. J. Biol. Chem. 278, 14622–14631. doi: 10.1074/jbc.M211834200
Cobucci-Ponzano, B., Trincone, A., Giordano, A., Rossi, M., and Moracci, M. (2003b). Identification of the catalytic nucleophile of the family 29 α-L-fucosidase from Sulfolobus solfataricus via chemical rescue of an inactive mutant. Biochemistry 42, 9525–9531. doi: 10.1021/bi035036t
Cone, J. E., Del Río, R. M., Davis, J. N., and Stadtman, T. C. (1976). Chemical characterization of the selenoprotein component of clostridial glycine reductase: identification of selenocysteine as the organoselenium moiety. Proc. Natl. Acad. Sci. U.S.A. 73, 2659–2663. doi: 10.1073/pnas.73.8.2659
Cui, Y., Dinman, J. D., Kinzy, T. G., and Peltz, S. W. (1998). The Mof2/Sui1 protein is a general monitor of translational accuracy. Mol. Cell. Biol. 18, 1506–1516. doi: 10.1128/mcb.18.3.1506
Curci, N., Strazzulli, A., Iacono, R., De Lise, F., Maurelli, L., Di Fenza, M., et al. (2021). Xyloglucan oligosaccharides hydrolysis by exo-acting glycoside hydrolases from hyperthermophilic microorganism Saccharolobus solfataricus. Int. J. Mol. Sci. 22:3325. doi: 10.3390/ijms22073325
De Lise, F., Iacono, R., Strazzulli, A., Giglio, R., Curci, N., Maurelli, L., et al. (2021). Transcript regulation of the recoded archaeal α-L-fucosidase in vivo. Molecules 26:1861. doi: 10.3390/molecules26071861
Deppenmeier, U., Johann, A., Hartsch, T., Merkl, R., Schmitz, R. A., Martinez-Arias, R., et al. (2002). The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microb. Biotech. 4, 453–461.
Eichler, J., and Adams, M. W. (2005). Posttranslational protein modification in Archaea. Microbiol. Mol. Biol. Rev. 69, 393–425. doi: 10.1128/MMBR.69.3.393-425.2005
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., Golding, S. D., et al. (2015). Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438. doi: 10.1126/science.aac7745
Fagegaltier, D., Hubert, N., Yamada, K., Mizutani, T., Carbon, P., and Krol, A. (2000). Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 19, 4796–4805. doi: 10.1093/emboj/19.17.4796
Firth, A. E., and Brierley, I. (2012). Non-canonical translation in RNA viruses. J. Gen. Virol. 93, 1385–1409. doi: 10.1099/vir.0.042499-0
Fletcher, J. E., Copeland, P. R., Driscoll, D. M., and Krol, A. (2001). The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2. RNA 7, 1442–1453.
Fomenko, D. E., and Gladyshev, V. N. (2012). Comparative genomics of thiol oxidoreductases reveals widespread and essential functions of thiol based redox control of cellular processes. Antiox Redox Sign. 16, 193–201. doi: 10.1089/ars.2011.3980
Fricke, W. F., Seedorf, H., Henne, A., Krüer, M., Liesegang, H., Hedderich, R., et al. (2006). The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 188, 642–658. doi: 10.1128/JB.188.2.642-658.2006
Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., et al. (2002). The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12, 532–542. doi: 10.1101/gr.223902
Gallant, J., and Lindsley, D. (1992). Leftward ribosome frameshifting at a hungry codon. J Mo. Biol. 223, 31–40. doi: 10.1016/0022-2836(92)90713-t
Gaston, M. A., Zhang, L., Green-Church, K. B., and Krzycki, J. A. (2011). The complete biosynthesis of the genetically encoded amino acid pyrrolysine fromlysine. Nature 471, 647–650. doi: 10.1038/nature09918
Gelsinger, D. R., Dallon, E., Reddy, R., Mohammad, F., Buskirk, A. R., and DiRuggiero, J. (2020). Ribosome profiling in archaea reveals leaderless translation, novel translational initiation sites, and ribosome pausing at single codon resolution. Nucleic Acids Res. 48, 5201–5216. doi: 10.1093/nar/gkaa304
Gesteland, R. F., and Atkins, J. F. (1996). Recoding: dynamic reprogramming of translation. Annu. Rev. Biochem. 65, 741–768. doi: 10.1146/annurev.bi.65.070196.003521
Goodchild, A., Saunders, N. F., Ertan, H., Raftery, M., Guilhaus, M., Curmi, P. M. G., et al. (2004). A proteomic determination of cold adaptation in the Antarctic archaeon, Methanococcoides burtonii. Mol. Microbiol. 53, 309–321. doi: 10.1111/j.1365-2958.2004.04130.x
Gromer, S., Johansson, L., Bauer, H., Arscott, L. D., Rauch, S., Ballou, D. P., et al. (2003). Active sites of thioredoxin reductases: why selenoproteins? Proc. Natl. Acad. Sci. U.S.A. 100, 12618–12623. doi: 10.1073/pnas.2134510100
Guan, Y., Haroon, M. F., Alam, I., Ferry, J. G., and Stingl, U. (2017). Single-cell genomics reveals pyrrolysine-encoding potential in members of uncultivated archaeal candidate division MSBL-1. Environ. Microbiol. Rep. 9, 404–410. doi: 10.1111/1758-2229.12545
Hao, B., Gong, W., Ferguson, T. K., James, C. M., Krzycki, J. A., and Chan, M. K. (2002). A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 296, 1462–1466. doi: 10.1126/science.1069556
Harrison, P. M., and Gerstein, M. (2002). Studying Genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol. 318, 1155–1174. doi: 10.1016/s0022-2836(02)00109-2
Hasenöhrl, D., Benelli, D., Barbazza, A., Londei, P., and Bläsi, U. (2006). Sulfolobus solfataricus translation initiation factor 1 stimulates translation initiation complex formation. RNA 12, 674–682. doi: 10.1261/rna.2289306
Hasenöhrl, D., Fabbretti, A., Londei, P., Gualerzi, C. O., and Bläsi, U. (2009). Translation initiation complex formation in the crenarchaeon Sulfolobus solfataricus. RNA 15, 2288–2298. doi: 10.1261/rna.1662609
Hatfield, D. L., and Gladyshev, V. N. (2002). How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 22, 3565–3576. doi: 10.1128/mcb.22.11.3565-3576.2002
Heinemann, I. U., O’Donoghue, P., Madinger, C., Benner, J., Randau, L., Noren, C. J., et al. (2009). The appearance of pyrrolysine in tRNAHisguanylyltransferase by neutral evolution. Proc. Natl Acad. Sci. U.S.A. 106, 21103–21108. doi: 10.1073/pnas.0912072106
Herr, A. J., Wills, N. M., Nelson, C. C., Gesteland, R. F., and Atkins, J. F. (2004). Factors that influence selection of coding resumption sites in translational bypassing: minimal conventional peptidyl-tRNA: mRNA pairing can suffice. J. Biol. Chem. 279, 11081–11087. doi: 10.1074/jbc.M311491200
Hirotsune, S., Yoshida, N., Chen, A., Garrett, L., Sugiyama, F., Takahashi, S., et al. (2003). An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 423, 91–96. doi: 10.1038/nature01535
Hoffman, K. S., Crnković, A., and Söll, D. (2018). Versatility of synthetic tRNAs in genetic code expansion. Genes 9:537. doi: 10.3390/genes9110537
Hondal, R. J., Marino, S. M., and Gladyshev, V. N. (2013). Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid Redox Signal. 18, 1675–1689. doi: 10.1089/ars.2012.5013
Hondal, R. J., and Ruggles, E. L. (2011). Differing views of the role of selenium in thioredoxin reductase. Amino Acids 41, 73–89. doi: 10.1007/s00726-010-0494-6
Iacono, R., Cobucci-Ponzano, B., De Lise, F., Curci, N., Maurelli, L., Moracci, M., et al. (2020). Spatial metagenomics of three geothermal sites in Pisciarelli hot Spring focusing on the biochemical resources of the microbial consortia. Molecules 25:4023. doi: 10.3390/molecules25174023
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S., and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi: 10.1126/science.1168978
Jacks, T., and Varmus, H. (1985). Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science 230, 1237–1242. doi: 10.1126/science.2416054
James, C. M., Ferguson, T. K., Leykam, J. F., and Krzycki, J. A. (2001). The amber codon in the gene encoding the monomethylamine methyltransferase isolated from Methanosarcina barkeri is translated as a sense codon. J. Biol. Chem. 276, 34252–34258. doi: 10.1074/jbc.M102929200
Jiang, L., Ni, J., and Liu, Q. (2012). Evolution of selenoproteins in the metazoan. BMC Genomics 13:446. doi: 10.1186/1471-2164-13-446
Kelly, J. A., Olson, A. N., Neupane, K., Munshi, S., Emeterio, J. S., Pollack, L., et al. (2020). Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS-CoV-2. bioRxiv [preprint] doi: 10.1101/2020.03.13.991083
Kim, H., Liu, F., Fei, J., Bustamante, C., Gonzalez, R. Jr., and Tinoco, I. Jr. (2014). A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U.S.A. 111, 5538–5543. doi: 10.1073/pnas.1403457111
Kobayashi, Y., Zhuang, J., Peltz, S., and Dougherty, J. (2010). Identification of a cellular factor that modulates HIV-1 programmed ribosomal frameshifting. J. Biol. Chem. 285, 19776–19784. doi: 10.1074/jbc.M109.085621
Korniy, N., Goyal, A., Hoffmann, M., Samatova, E., Peske, F., Pohlmann, S., et al. (2019a). Modulation of HIV-1 Gag/Gag-Pol frameshifting by tRNA abundance. Nucleic Acids Res. 47, 5210–5222. doi: 10.1093/nar/gkz202
Korniy, N., Samatova, E., Anokhina, M., Peske, F., and Rodnina, M. (2019b). Mechanisms and biomedical implications of –1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 593, 1468–1482. doi: 10.1002/1873-3468.13478
Krol, A. (2002). Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie 84, 765–774. doi: 10.1016/s0300-9084(02)01405-0
Kromayer, M., Wilting, R., Tormay, P., and Böck, A. (1996). Domain structure of the prokaryotic selenocysteine-specific elongation factor SelB. J. Mol. Biol. 262, 413–420. doi: 10.1006/jmbi.1996.0525
Kryukov, G. V., and Gladyshev, V. N. (2004). The prokaryotic selenoproteome. EMBO Rep. 5, 538–543. doi: 10.1038/sj.embor.7400126
Krzycki, J. A. (2004). Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases. Curr. Opin. Chem. Biol. 8, 484–491. doi: 10.1016/j.cbpa.2004.08.012
Kyrpides, N. C., and Woese, C. R. (1998). Universally conserved translation initiation factors. Proc. Natl. Acad. Sci. U.S.A. 95, 224–228. doi: 10.1073/pnas.95.1.224
Labunskyy, V. M., Hatfield, D. L., and Gladyshev, V. N. (2014). Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 94, 739–777. doi: 10.1152/physrev.00039.2013
Lang, B. F., Jakubkova, M., Hegedusova, E., Daoud, R., Forget, L., Brejova, B., et al. (2014). Massive programmed translational jumping in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 111, 5926–5931. doi: 10.1073/pnas.1322190111
Larsen, B., Gesteland, R. F., and Atkins, J. F. (1997). Structural probing and mutagenic analysis of the stem-loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50%. J. Mol. Biol. 271, 47–60. doi: 10.1006/jmbi.1997.1162
Lin, Z., Gilbert, R., and Brierley, I. (2012). Spacer-length dependence of programmed -1 or -2 ribosomal frameshifting on a U6A heptamer supports a role for messenger RNA (mRNA) tension in frameshifting. Nucleic Acids Res. 40, 8674–8689. doi: 10.1093/nar/gks629
Ling, J., O’Donoghue, P., and Söll, D. (2015). Genetic code flexibility in microorganisms: novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 13, 707–772. doi: 10.1038/nrmicro3568
Lobanov, A. V., Hatfield, D. L., and Gladyshev, V. N. (2008). Reduced reliance on the trace element selenium during evolution of mammals. Genome Biol. 9:R62. doi: 10.1186/gb-2008-9-3-r62
Longstaff, D. G., Larue, R. C., Faust, J. E., Mahapatra, A., Zhang, L., Green- Church, K. B., et al. (2007). A natural genetic code expansion cassette enables transmissible biosynthesis and genetic encoding of pyrrolysine. Proc. Natl. Acad. Sci. U.S.A. 104, 1021–1026. doi: 10.1073/pnas.0610294104
Luthi, K., Moser, M., Ryser, J., and Weber, H. (1990). Evidence for a role of translational frameshifting in the expression of transposition activity of the bacterial insertion element IS1. Gene 88, 15–20. doi: 10.1016/0378-1119(90)90054-u
Maeder, D. L., Anderson, I., Brettin, T. S., Bruce, D. C., Gilna, P., Han, C. S., et al. (2006). The Methanosarcina barkeri genome: comparative analysis with Methanosarcina acetivorans and Methanosarcina mazei reveals extensive rearrangement within methanosarcinal genomes. J. Bacteriol. 188, 7922–7931. doi: 10.1128/JB.00810-06
Mangold, U. (2005). The antizyme family: polyamines and beyond. IUBMB Life 57, 671–676. doi: 10.1080/15216540500307031
Mariotti, M., Lobanov, A. V., Guigo, R., and Gladyshev, V. N. (2013). SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins. Nucleic Acids Res. 41:e149. doi: 10.1093/nar/gkt550
Mariotti, M., Lobanov, A. V., Manta, B., Santesmasses, D., Bofill, A., Guigó, R., et al. (2016). Lokiarchaeota marks the transition between the archaeal and eukaryotic selenocysteine encoding systems. Mol. Biol. Evol. 33, 2441–2453. doi: 10.1093/molbev/msw122
Mariotti, M., Santesmasses, D., Capella-Gutierrez, S., Mateo, A., Arnan, C., Johnson, R., et al. (2015). Evolution of selenophosphate synthetases: emergence and relocation of function through independent duplications and recurrent subfunctionalization. Genome Res. 25, 1256–1267. doi: 10.1101/gr.190538.115
Matsufuji, S., Matsufuji, T., Miyazaki, Y., Murakami, Y., Atkins, J. F., Gesteland, R. F., et al. (1995). Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 80, 51–60. doi: 10.1016/0092-8674(95)90450-6
Michel, A. M., and Baranov, P. V. (2013). Ribosome profiling: a Hi-Def monitor for protein synthesis at the genome-wide scale. WIREs RNA 4, 473–490. doi: 10.1002/wrna.1172
Namy, O., Moran, S., Stuart, D., Gilbert, R., and Brierley, I. (2006). A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441, 244–247. doi: 10.1038/nature04735
Namy, O., Rousset, J. P., Sawsan, N., and Brierley, I. (2004). Reprogrammed genetic decoding review in cellular gene expression. Mol. Cell. 13, 157–168. doi: 10.1016/s1097-2765(04)00031-0
Namy, O., Zhou, Y., Gundllapalli, S., Polycarpo, C. R., Denise, A., Rousset, J. P., et al. (2007). Adding pyrrolysine to the Escherichia coli genetic code. FEBS Lett. 581, 5282–5288. doi: 10.1016/j.febslet.2007.10.022
Napthine, S., Ling, R., Finch, L., Jones, J., Bell, S., Brierley, I., et al. (2017). Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun. 8:15582. doi: 10.1038/ncomms15582
Nicholas, P. R., Jean-François Brugère, J. F., Atkins, P. W., and O’Toole, G. B. (2018). Pyrrolysine in archaea: a 22nd amino acid encoded through a genetic code expansion. Emerg. Top. Life Sci. 2, 607–618. doi: 10.1042/ETLS20180094
Nobu, M. K., Narihiro, T., Kuroda, K., Mei, R., and Liu, W. T. (2016). Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 10, 2478–2487. doi: 10.1038/ismej.2016.33
Nosek, J., Tomaska, L., Burger, G., and Lang, B. F. (2015). Programmed translational bypassing elements in mitochondria: structure, mobility, and evolutionary origin. Trends Genet. 31, 187–194. doi: 10.1016/j.tig.2015.02.010
Nozawa, K., O’Donoghue, P., Gundllapalli, S., Araiso, Y., Ishitani, R., Umehara, T., et al. (2009). Pyrrolysyl-tRNA synthetase- tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 457, 1163–1167. doi: 10.1038/nature07611
Olubajo, B., and Taylor, E. (2005). A -1 frameshift in the HIV-1 env gene is enhanced by arginine deficiency via a hungry codon mechanism. Mutat. Res. 579, 125–132. doi: 10.1016/j.mrfmmm.2005.02.018
Onofri, S., Balucani, N., and Barone, V. (2020). The Italian National project of astrobiology-life in space-origin, presence, persistence of life in space, from molecules to extremophiles. Astrobiology 20, 580–582. doi: 10.1089/ast.2020.2247
Osawa, S., Jukes, T. H., Watanabe, K., and Muto, A. (1992). Recent evidence for evolution of the genetic code. Microbiol. Rev. 56, 229–264.
Petitjean, C., Deschamps, P., López-García, P., Moreira, D., and Brochier-Armanet, C. (2015). Extending the conserved phylogenetic core of archaea disentangles the evolution of the third domain of life. Mol. Biol. Evol. 32, 1242–1254. doi: 10.1093/molbev/msv015
Pietila, M. K., Laurinmaki, P., Russell, D. A., Ko, C. C., Jacobs-Sera, D., Butcher, S. J., et al. (2013). Insights into head-tailed viruses infecting extremely halophilic archaea. J. Virol. 87, 3248–3260. doi: 10.1128/JVI.03397-12
Plant, E., Rakauskaite, R., Taylor, D., and Dinman, J. (2010). Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J. Virol. 84, 4330–4340. doi: 10.1128/JVI.02480-09
Polycarpo, C., Ambrogelly, A., Bérubé, A., Winbush, S. M., McCloskey, J. A., Crain, P. F., et al. (2004). An aminoacyl-tRNA synthetase that specifically activates pyrrolysine. Proc. Natl. Acad. Sci. U. S. A. 101, 12450–12454. doi: 10.1073/pnas.0405362101
Polycarpo, C., Ambrogelly, A., Ruan, B., Tumbula-Hansen, D., Ataide, S. F., Ishitani, R., et al. (2003). Activation of the pyrrolysine suppressor tRNA requires formation of a ternary complex with class I and class II lysyl- tRNA synthetases. Mol. Cell. 12, 287–294. doi: 10.1016/S1097-2765(03)00280-6
Pooggin, M. M., and Ryabova, L. A. (2018). Ribosome shunting, polycistronic translation, and evasion of antiviral defenses in plant pararetroviruses and beyond. Front. Microbiol. 9:644. doi: 10.3389/fmicb.2018.00644
Prat, L., Heinemann, I. U., Aerni, H. R., Rinehart, J., O’Donoghue, P., and Söll, D. (2012). Carbon source-dependent expansion of the genetic code in bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, 21070–21075. doi: 10.1073/pnas.121861311
Rodnina, M. V., Korniy, N., and Klimova, M. (2020). Translational recoding: canonical translation mechanisms reinterpreted. Nucleic Acids Res. 48, 1056–1067. doi: 10.1093/nar/gkz783
Rosano, C., Zuccotti, S., Cobucci-Ponzano, B., Mazzone, M., Rossi, M., Moracci, M., et al. (2004). Structural characterization of the nonameric assembly of an Archaeal alpha-L-fucosidase by synchrotron small angle X-ray scattering. Biochem. Biophys. Res. Commun. 320, 176–182. doi: 10.1016/j.bbrc.2004.05.149
Rother, M., and Krzycki, J. A. (2010). Selenocysteine, pyrrolysine, and the unique energy metabolism of methanogenic archaea. Archaea 2010:453642. doi: 10.1155/2010/453642
Rother, M., Mathes, I., Lottspeich, F., and Böck, A. (2003). Inactivation of the selB gene in Methanococcus maripaludis: effect on synthesis of selenoproteins and their sulfur-containing homologs. J. Bacteriol. 185, 107–114. doi: 10.1128/JB.185.1.107-114.2003
Rother, M., and Quitzke, V. (2018). Selenoprotein synthesis and regulation in Archaea. Biochimt. Biophys. Acta (BBA) - Gen Subj. 1862, 2451–2462. doi: 10.1016/j.bbagen.2018.04.008
Rother, M., Wilting, R., Commans, S., and Böck, A. (2000). Identification and characterisation of the selenocysteine-specific translation factor SelB from the archaeon Methanococcus jannaschii. J. Mol. Biol. 299, 351–358. doi: 10.1006/jmbi.2000.3756
Sakai, H. D., and Kurosawa, N. (2018). Saccharolobus caldissimus gen. nov., sp. nov., a facultatively anaerobic iron-reducing hyperthermophilic archaeon isolated from an acidic terrestrial hot spring, and reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. nov. and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Microbiol. 68, 1271–1278. doi: 10.1099/ijsem.0.002665
Santesmasses, D., Mariotti, M., and Guigo, R. (2017). Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLoS Comput. Biol. 13:e1005383. doi: 10.1371/journal.pcbi.1005383
Sencilo, A., Jacobs-Sera, D., Russell, D. A., Ko, C. C., Bowman, C. A., Atanasova, N. S., et al. (2013). Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 10, 803–816. doi: 10.4161/rna.24045
Sharma, V., Firth, A. E., Antonov, I., Fayet, O., Atkins, J. F., Borodovsky, M., et al. (2011). A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol. Biol. Evol. 28, 3195–3211. doi: 10.1093/molbev/msr155
Sorokin, D. Y., Makarova, K. S., Abbas, B., Ferrer, M., Golyshin, P. N., Galinski, E. A., et al. (2017). Discovery of extremely halophilic, methyl-reducing Euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2:17081. doi: 10.1038/nmicrobiol.2017.81
Spang, A., Saw, J. H., Jørgensen, S. L., Zaremba-Niedzwiedzka, K., Martijn, J., Lind, A. E., et al. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179. doi: 10.1038/nature14447
Srinivasan, G., James, C. M., and Krzycki, J. A. (2002). Pyrrolysine encoded by UAG in Archaea: charging of a UAG- decoding specialized tRNA. Science 296, 1459–1462. doi: 10.1126/science.1069588
Sriram, A., Bohlen, J., and Teleman, A. A. (2018). Translation acrobatics: how cancer cells exploit alternate modes of translational initiation. EMBO Rep. 19:e45947. doi: 10.15252/embr.201845947
Stock, T., and Rother, M. (2009). Selenoproteins in archaea and Gram-positive bacteria. Biochim. Biophys. Acta 1790, 1520–1532. doi: 10.1016/j.bbagen.2009.03.022
Stock, T., Selzer, M., and Rother, M. (2010). In vivo requirement of selenophosphate for selenoprotein synthesis in archaea. Mol. Microbiol. 75, 149–160. doi: 10.1111/j.1365-2958.2009.06970.x
Strazzulli, A., Cobucci-Ponzano, B., Iacono, R., Giglio, R., Maurelli, L., Curci, N., et al. (2020). Discovery of hyperstable carbohydrate-active enzymes through metagenomics of extreme environments. FEBS J. 287, 1116–1137. doi: 10.1111/febs.15080
Temperley, R., Richter, R., Dennerlein, S., Lightowlers, R., and Chrzanowska-Lightowlers, Z. (2010). Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 327:301. doi: 10.1126/science.1180674
Tharp, J. M., Ehnbom, A., and Liu, W. R. (2018). tRNAPyl: structure, function, and applications. RNA Biol. 15, 441–452. doi: 10.1080/15476286.2017.1356561
Ticak, T., Kountz, D. J., Girosky, K. E., Krzycki, J. A., and Ferguson, D. J. (2014). A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 111, E4668–E4676. doi: 10.1073/pnas
Tsuchihashi, Z., and Brown, P. O. (1992). Sequence requirements for efficient translational frameshifting in the Escherichia coli dnaX gene and the role of an unstable interaction between tRNA(Lys) and an AAG lysine codon. Genes Dev. 6, 511–519. doi: 10.1101/gad.6.3.511
Tsuchihashi, Z., and Kornberg, A. (1990). Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 87, 2516–2520. doi: 10.1073/pnas.87.7.2516
Tujebajeva, R. M., Copeland, P. R., Xu, X. M., Carlson, B. A., Harney, J. W., Driscoll, D. M., et al. (2000). Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 1, 158–163. doi: 10.1093/embo-reports/kvd033
van Passel, M. W., Smillie, C. S., and Ochman, H. (2007). Gene decay in archaea. Archaea 2, 137–143. doi: 10.1155/2007/165723
Vanwonterghem, I., Evans, P. N., Parks, D. H., Jensen, P. D., Woodcroft, B. J., Hugenholtz, P., et al. (2016). Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1:16170. doi: 10.1038/nmicrobiol.2016.170
Wang, X., Xuan, Y., Han, Y., Ding, X., Ye, K., Yang, F., et al. (2019). Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 176, 625–635. doi: 10.1016/j.cell.2018.12.030
Wills, N., Moore, B., Hammer, A., Gesteland, R., and Atkins, J. (2006). A functional -1 ribosomal frameshift signal in the human paraneoplastic Ma3 gene. J/Biol. Chem. 281, 7082–7088. doi: 10.1074/jbc.M511629200
Wilting, R., Schorling, S., Persson, B. C., and Bock, A. (1997). Selenoprotein synthesis in archaea: identification of an mRNA element of Methanococcus jannaschii probably directing selenocysteine insertion. J Mol Biol. 266, 637–641. doi: 10.1006/jmbi.1996.0812
Xu, J., Hendrix, R. W., and Duda, R. L. (2004). Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell. 16, 11–21. doi: 10.1016/j.molcel.2004.09.006
Yan, S., Wen, J., Bustamante, C., and Tinoco, I. Jr. (2015). Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160, 870–881. doi: 10.1016/j.cell.2015.02.003
Yang, Y., and Wang, Z. (2019). IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 11, 911–919. doi: 10.1093/jmcb/mjz091
Yoshizawa, S., and Bock, A. (2009). The many levels of control on bacterial selenoprotein synthesis. Biochim. Biophys. Acta 1790, 1404–1414. doi: 10.1016/j.bbagen.2009.03.010
Yoshizawa, S., Rasubala, L., Ose, T., Kohda, D., Fourmy, D., and Maenaka, K. (2005). Structural basis for mRNA recognition by elongation factor SelB. Nat. Struct. Mol. Biol. 12, 198–203. doi: 10.1038/nsmb890
Zhang, Y., and Gladyshev, V. N. (2008). Trends in selenium utilization in marine microbial world revealed through the analysis of the global ocean sampling (GOS) project. PLoS Genet. 13,4:e1000095. doi: 10.1371/journal.pgen.1000095
Zhang, Y., Baranov, P. V., Atkins, J. F., and Gladyshev, V. N. (2005). Pyrrolysine and selenocysteine use dissimilar decoding strategies. J. Biol. Chem. 280, 20740–20751. doi: 10.1074/jbc.M501458
Keywords: alpha-fucosidase, recoding, frameshifting, pyrrolysine, selenocysteine, archaea
Citation: De Lise F, Strazzulli A, Iacono R, Curci N, Di Fenza M, Maurelli L, Moracci M and Cobucci-Ponzano B (2021) Programmed Deviations of Ribosomes From Standard Decoding in Archaea. Front. Microbiol. 12:688061. doi: 10.3389/fmicb.2021.688061
Received: 30 March 2021; Accepted: 04 May 2021;
Published: 04 June 2021.
Edited by:
Paola Londei, Sapienza University of Rome, ItalyReviewed by:
Emmanuelle Schmitt, UMR 7654 Bases moléculaires et régulation de la biosynthèse protéique, FranceDario Benelli, Sapienza University of Rome, Italy
Copyright © 2021 De Lise, Strazzulli, Iacono, Curci, Di Fenza, Maurelli, Moracci and Cobucci-Ponzano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatrice Cobucci-Ponzano, beatrice.cobucciponzano@ibbr.cnr.it