Adult-onset neuronal intranuclear inclusion disease related retinal degeneration: a Chinese case series
 Chaoyi Feng1†
Chaoyi Feng1†  Qian Chen1†
Qian Chen1†  Xinghua Luan2
Xinghua Luan2  Ping Sun1
Ping Sun1  Yuwen Cao2
Yuwen Cao2  Jingying Wu2 Shige Wang2
Jingying Wu2 Shige Wang2  Xinghuai Sun1,3
Xinghuai Sun1,3  Li Cao2,4*
Li Cao2,4*  Guohong Tian1,3*
Guohong Tian1,3*- 1Department of Ophthalmology, Eye Ear Nose and Throat Hospital of Fudan University, Shanghai, China
- 2Department of Neurology, Shanghai Jiaotong University Affiliated Sixth People’s Hospital, Shanghai, China
- 3State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Institutes of Brain Science, Fudan University, Shanghai, China
- 4Shanghai Neurological Rare Disease Biobank and Precision Diagnostic Technical Service Platform, Shanghai, China
Purpose: To evaluate adult-onset neuronal intranuclear inclusion disease (NIID)-related retinopathy with guanine-guanine-cytosine repeat expansions in NOTCH2NLC.
Materials and methods: Neuro-ophthalmic evaluations, including best-corrected visual acuity, slit-lamp biomicroscopy, intraocular pressure (IOP), ultrasound biomicroscopy, pupillometry, fundus photography, fundus autofluorescence (FAF), optical coherence tomography (OCT), Humphrey visual field, full-field electroretinography (ERG), and multifocal ERG (mf-ERG) were performed in patients with gene-proven NIID.
Results: Nine patients (18 eyes) were evaluated, with a median age of 62 years (55–68) and only one man was included in our study. Six patients presented with decreased visual acuity or night blindness, whereas the other three were asymptomatic. The visual acuity was measured from 20/200 to 20/20. Miosis was present in eight patients, four of whom had ciliary process hypertrophy and pronation, and three of whom had shallow anterior chambers. Fundus photography, FAF, and OCT showed consistent structural abnormalities mainly started from peripapillary areas and localized in the outer layer of photoreceptors and inner ganglion cell layer. ERG and mf-ERG also revealed retinal dysfunction in the corresponding regions.
Conclusion: Patients with NIID showed both structural and functional retinopathies which were unique and different from common cone-rod dystrophy or retinitis pigmentosa. Patients with miosis may have a potential risk of an angle-closure glaucoma attack. Neuro-ophthalmic evaluations is essential for evaluating patients with NIID, even without visual symptom.
1 Introduction
Neuronal intranuclear inclusion disease (NIID) is a rare progressive neurodegenerative disorder characterized by the deposition of eosinophilic hyaline intranuclear inclusions in neuronal and somatic cells, as well as various organs (1–9). The manifestation of neurological symptoms is highly variable, and the clinical subtypes can be divided into infantile, juvenile, and adult-onset forms according to the age of onset (10). Adult-onset NIID is observed in sporadic and familial cases (1, 10, 11). Skin biopsy and gene testing for repeat expansions of guanine-guanine-cytosine (GGC) in the NOTCH2NLC gene can differentiate between NIID and other neurodegenerative disorders (12–15).
Ocular dysfunction in patients with NIID has been reported in many studies, mainly focusing on pupillary abnormalities such as miosis, which were more frequently observed in sporadic cases (94.4%) than in familial cases (63.6%) (1). Tai et al. (16) evaluated 223 Chinese patients with NIID and found that only 14.3% had visual loss.
As early as 1991, patients with biopsy-proven adult-onset NIID demonstrated a marked reduction in the dark-adapted b-wave response (17). Recently, Omoto et al. described a patient with sporadic adult-onset NIID who developed retinal dystrophy and abnormal electroretinography (ERG) results, presenting with childhood-onset night blindness (3). Nakamura et al. (6) reported on the ocular characteristics of NIID disease-related retinopathy in seven Asian patients, revealing similar features, including rod-cone dysfunction with progressive retinal degeneration in the peripapillary and midperipheral regions. This was a detailed study on retinopathy including patients with GGC repeat expansion in NIID disease.
Meanwhile, ubiquitin-positive neuronal intranuclear inclusions, which were previously considered to be unique pathologic evidence of NIID, have also been detected in patients with fragile X-associated tremor or ataxia syndrome (18), frontotemporal lobar degeneration, and motor neuron disease or amyotrophic lateral sclerosis (19). Neuro-ophthalmic evaluations, such as OCT and ERG are very useful techniques to differentiate the spectrum disorders. Therefore, thoroughly neuro-ophthalmic examination in neurodegenerative disease is very helpful for differentiate diagnosis, due to some distinctive pattern of retinopathies.
However, pupil light reflexes, structural and functional retinal changes in NIID have not been investigated in detail. In the present study, we utilized various techniques including pupillometry, fundus autofluorescence (FAF), optical coherence tomography (OCT), and electrophysiology to evaluate nine sporadic cases of adult-onset NIID with GGC repeat expansions in NOTCH2NLC.
2 Materials and methods
2.1 Participants
The study participants were patients diagnosed with adult-onset NIID by neurologists at the Shanghai Jiaotong University Affiliated Sixth People’s Hospital between July 2020 and March 2021, and then referred to the Department of Ophthalmology, Eye, Ear, Nose, and Throat Hospital of Fudan University to undergo screening for visual involvement.
All patients with NIID were diagnosed based on their clinical symptoms, brain magnetic resonance imaging, skin biopsy showing eosinophilic and ubiquitin-positive intranuclear inclusions, and gene testing confirming GGC repeat expansions in the NOTCH2NLC gene. The study protocol adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committee. Written informed consent was obtained from each participant after explaining the nature and purpose of the study.
2.2 Clinical investigations
Demographic data, including gender, age, and visual symptoms, were recorded. A detailed medical history was obtained from the patients. Findings on routine ophthalmologic examinations in the form of best-corrected visual acuity were recorded and converted to an early treatment diabetic retinopathy study letter score. Intraocular pressure (IOP) and depth of the anterior chamber were also evaluated by ultrasound biomicroscopy (UBM).
Pupillary sizes and light reflexes were tested using a pupilometer (NeurOptics DP2000; NeurOptics Inc., Irvine, CA, United States), a binocular system that stimulates the eyes directly, consensually, or bilaterally simultaneously and features automatic tracking and pupil detection. Spectral-domain OCT (SD-OCT) scans were performed using the Heidelberg Spectralis (Heidelberg Engineering Inc., Heidelberg, Germany). The macular ganglion cell-inner plexiform layer (GCIPL) and peripapillary retinal nerve fiber layer (RNFL) were measured using the Cirrus HD-OCT Model 4000 (Carl Zeiss Meditec AG, Jena, Germany). Other ophthalmic examinations included the Humphrey visual field (HVF) (Carl Zeiss Meditec AG) examination, ultrasound biomicroscopy (UBM; SUOER), fundus photography (Optos 200Tx), FAF (Optos 200Tx; Optos Plc, Dunfermline, Scotland), ERG (UTAS visual electrodiagnostic test system; LKC Technologies, Gaithersburg, MD, United States), and mf-ERG (VERIS; Electro-Diagnostic Imaging Inc., CA, United States).
3 Results
3.1 Demographics and ophthalmic tests
Nine patients diagnosed with gene proven NIID were evaluated. The median age of the patients was 62 years (range, 55–68). Among them, eight patients (88.9%) were women. Six patients had ocular symptoms such as blurred vision, decreased visual acuity, and night blindness. Furthermore, slit-lamp examination found six patients had mild to moderate age-related cataracts, and one patient had stellar vitreous degeneration. No patients had a high IOP level (more than 20 mm Hg) and UBM revealed that four patients with miosis had ciliary process hypertrophy and pronation, with shallow anterior chambers (anterior chamber deep less than 2.3 mm, Supplementary Content 1). One patient who underwent intraocular lens implantation had normal anterior chamber depth and angle, however, a hypertrophic ciliary process was still present. Detailed ophthalmic characteristics are shown in Table 1.
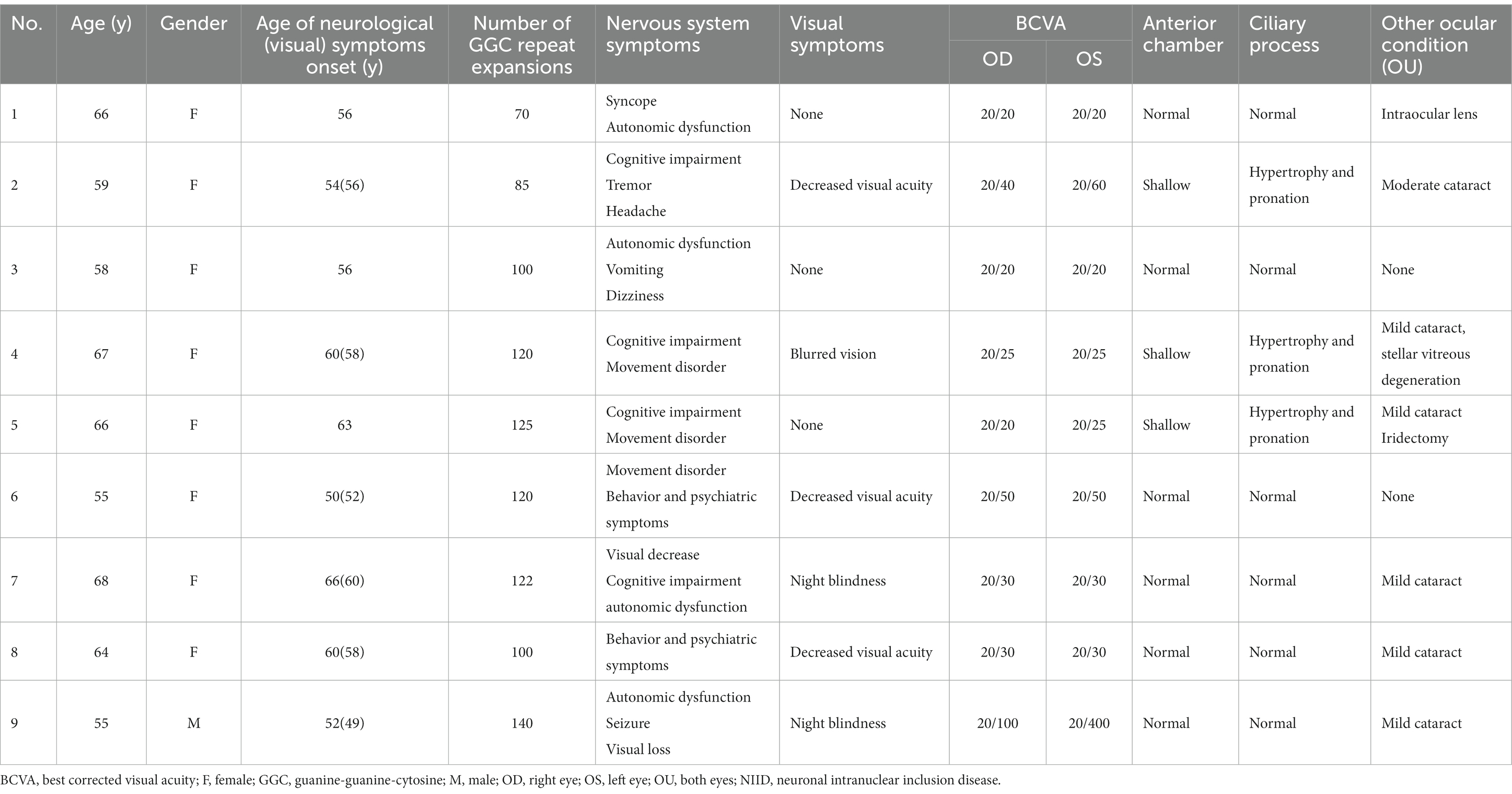
Table 1. Demographics and clinical characteristics of the nine patients with NIID.
3.2 Pupillary sizes and light reflexes
Eight individuals (all except patient 9) presented with miosis. The patients had a mean mesopic pupil diameter of 4.02 mm. The pupillary light reflexes of six patients were sluggish and presented an increased latency of constriction. Among them, patient 2 was unable to complete the pupilometer test (Figure 1).

Figure 1. Pupil sizes and light reflexes in patients with NIID. The mean pupil sizes and latency of the pupillary light reflexes in the normal population (red lines), and their standard deviation (red blocks). Patient 2 was unable to complete the pupilometer tests. NIID, neuronal intranuclear inclusion disease.
3.3 Structural retinal changes
All patients showed structural retinal abnormalities in both eyes, including those without visual symptoms. Fundus photographs showed peripapillary and posterior pole area retinal pigmentary changes encroaching the fovea. Additionally, wide-field FAF showed that the patients had different degrees of hypoautofluorescent areas in the peripapillary regions or posterior pole areas, indicating outer retinal dystrophy. Among them, patients 4, 5, 7, 8, and 9 had larger photoreceptor dystrophy regions that extended to the posterior poles, and hyperautofluorescent rings surrounding the hypoautofluorescent regions, indicating the border of the lesion (Figure 2). SD-OCT revealed structural changes in the outer retina throughout the patients, such as an ellipsoid zone (EZ) thinning, blurred interdigitation zone (IZ), and thinning of the outer nuclear layer. Hyperreflective fine dots were scattered throughout several of the eye layers in each patient, as shown in Figure 2. The areas of EZ abnormality shown on SD-OCT corresponded to the hyperautofluorescent rings on the wide-field FAF. Interestingly, patient 7 presented with night blindness 6 years prior to the diagnosis of NIID. The follow-up SD-OCT revealed progressive outer retinal dystrophy showing disruption of the EZ in the peripapillary regions initially, followed by the whole layer structure disappearing. The outer retinal abnormalities manifested as hypoautofluorescent patches in the FAF within the peripapillary and parafoveal regions and dramatically enlarged during the 2 years period (Figure 3). The nerve fiber layer of the peripapillary optic nerve (RNFL) and GCIPL of macular thickness were measured in six patients showing a decline in the peripapillary RNFL thickness in patients 4, 7, and 8 (Supplementary Content 1). All six patients had reduced macular GCIPL thickness; however, patients 4, 7, 8, and 9 had a significantly thinner macular GCIPL.
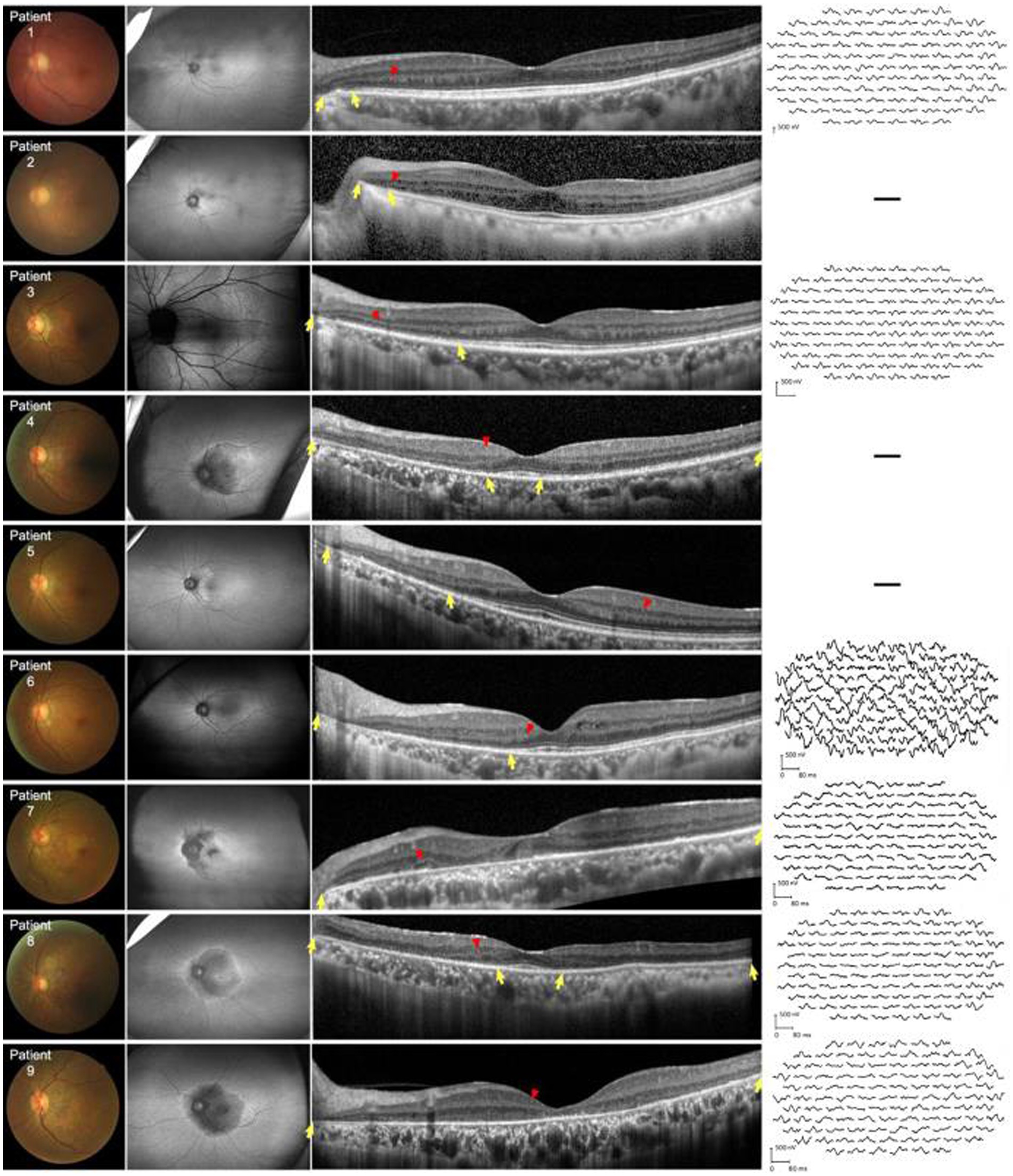
Figure 2. Fundus color, FAF, SD-OCT, and mf-ERG trace of nine patients with NIID. First left column: Fundus photographs show peripheral retinal pigmentary changes encroaching the fovea. Second left column: FAF images show constriction of the hyperautofluorescent ring corresponding to the border of EZ abnormalities. All patients had outer retinal dystrophy in the peripapillary regions. Patient 3 was assessed via Heidelberg OCT. In patients 4, 7, 8, and 9, dystrophy extended to the posterior poles which appeared as hypoautofluorescence. Third left column: horizontal SD-OCT cross-sections passing through the fovea for each patient showed EZ dystrophy (yellow arrows) and blurred IZ extending from the peripapillary regions to the posterior poles. Thinning of the outer nuclear layer can be seen in the corresponding regions. Hyperreflective fine dots (red arrows) scattered within the retina in all patients. Right column: mf-ERG of patients 1, 3, 6, 7, 8, and 9 revealed decreased b-wave amplitudes corresponding to the region of EZ and IZ abnormalities. EZ, ellipsoid zone; FAF, fundus autofluorescence; IZ, interdigitation zone; mf-ERG, multifocal full-field electroretinography; OCT, optical coherence tomography; SD-OCT, spectral-domain OCT.
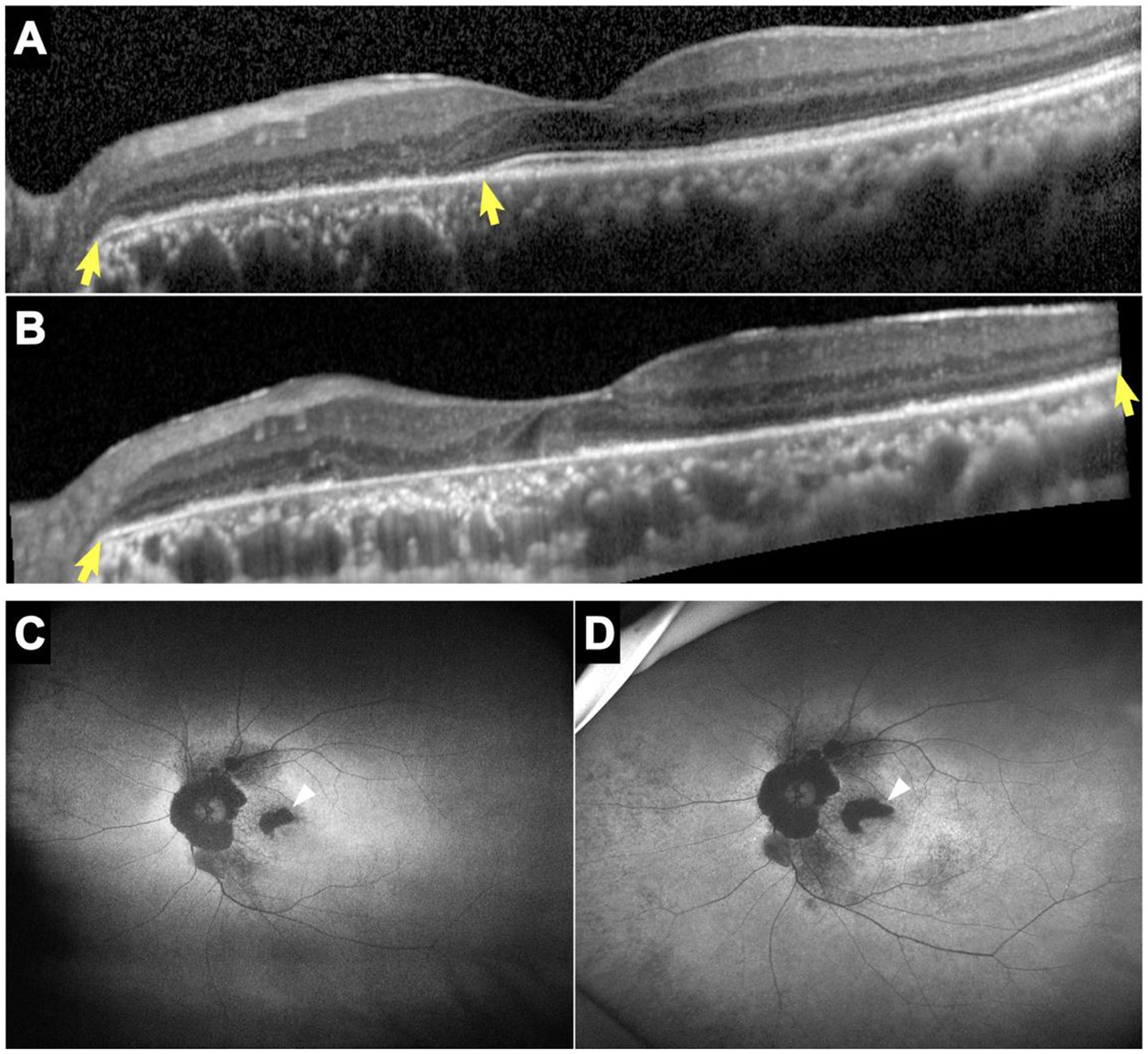
Figure 3. SD-OCT and FAF images of patient 7. Horizontal SD-OCT cross-sections passing through the fovea showed EZ dystrophy extending from the peripapillary regions to the entire posterior pole (yellow arrows) with loss of the IZ and outer nuclear layer (A: year 2014, B: year 2020). FAF showed hypoautofluorescent patches that enlarged noticeably over 2 years, particularly in the parafoveal regions (white arrows) (C: year 2018, D: year 2020). EZ, ellipsoid zone; FAF, fundus autofluorescence; IZ, interdigitation zone; SD-OCT, spectral-domain optical coherence tomography.
3.4 Functional retinal changes
Five patients underwent HVF tests, revealing physiological blind spot enlargement, bilateral temporal field defects that did not respect the vertical meridian, and visual field constriction (Supplementary Content 1). Furthermore, ERG and mf-ERG were performed on six of the patients without shallow anterior chambers, cases in which it was safe to dilate the pupils. ERG showed abnormalities in both retinal dark adaptation and light adaptation, including decreased amplitudes and increased latencies in all six patients (Figure 4). mf-ERG revealed various degrees of diffusely and asymmetrically decreased b-wave amplitudes and increased latencies in the posterior pole areas, indicating photoreceptor dysfunction. These functional abnormalities corresponded to the structural disruptions identified on FAF and OCT examinations. Discussion.
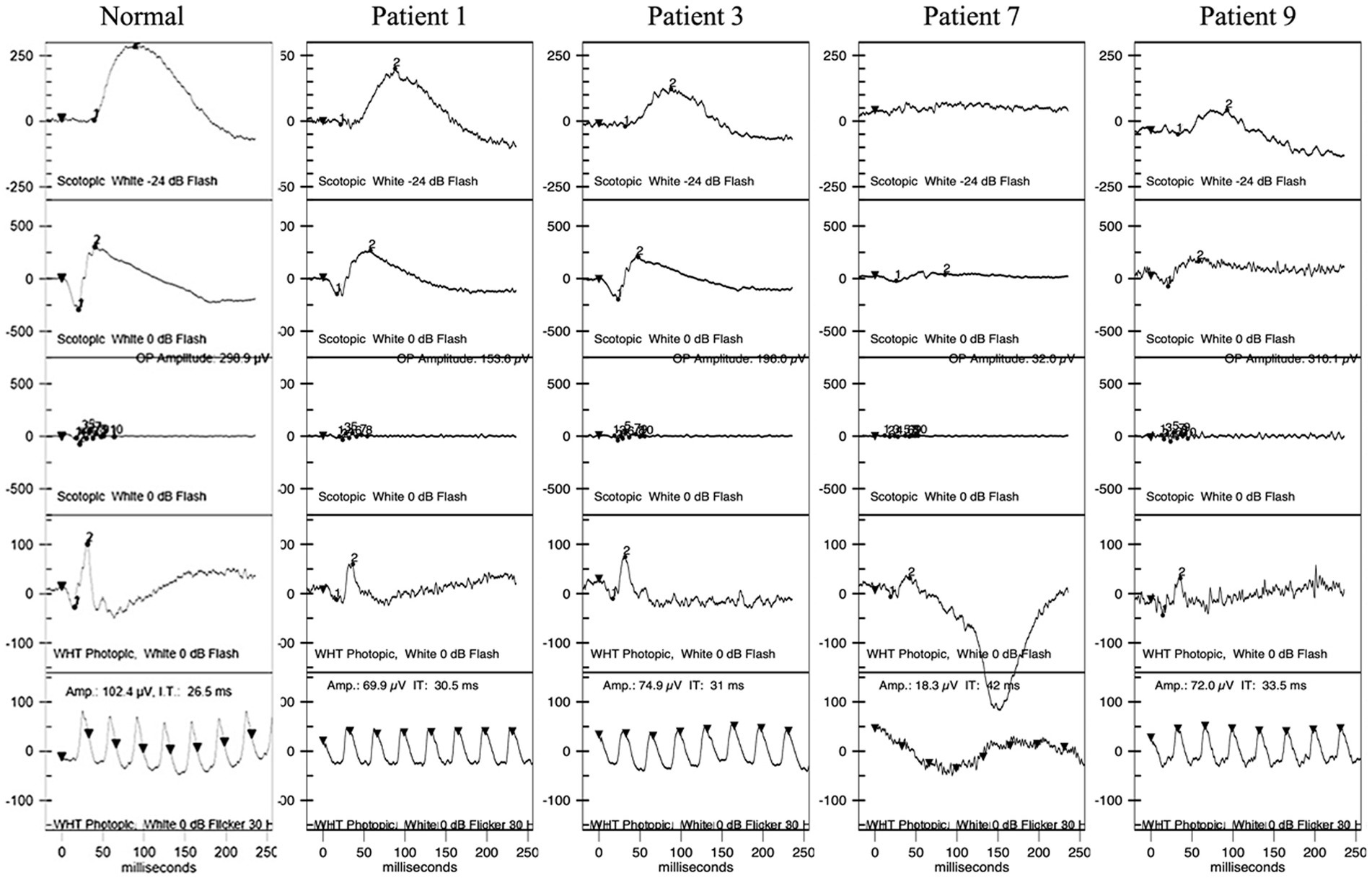
Figure 4. ERG of patients 1, 3, 7, and 9. Patient 1 had normal waves in dark adaptation. Patients 3, 7, and 9 had decreased amplitudes and increased latency of the b wave in dark adaptation with different degrees. All four patients had decreased amplitudes and increased latency in light adaptation. ERG, full-field electroretinography.
4 Discussion
4.1 Key findings
In 2019, Chinese and Japanese scholars revealed the causative gene of NIID. Due to relatively rare data, the prevalence of NIID is unclear; however, it is known to be relatively high in Japan and China, where a large percentage of the cases are reported (1–3, 13, 15, 16, 20).
Autonomic dysfunction of the pupils is frequently seen in patients with NIID (1–3, 6, 10, 11, 17, 21). However, previous studies have not quantified pupil sizes and light reflexes using pupillometry, a method which can reveal more information regarding the condition. We observed that the mean pupil diameters in patients with NIID ranged from 3.09 to 6.06 mm, dramatically smaller than the normal diameter range of 5.63 to 7.27 mm (22). The average latency of the pupillary light reflexes was 0.31 ± 0.05 s (23). The rate of miosis in our study was 88.9% (8/9) and the pupillary light reflexes were sluggish, as reported previously (17, 24). The unique feature of pupil size and light response is very helpful to ophthalmologists in identifying parasympathetic dysfunction among patients with NIID, which is usually diagnosed by a neurologist. One-third of the patients had shallow anterior chambers; however, as the pupillary sphincter and ciliary muscle are both parasympathetically innervated, we hypothesized that autonomic dysfunction may lead to relative hyperactivity of the parasympathetic nerve, causing pupil constriction and excessive contraction of the ciliary muscle, thereby increasing the risk of an angle-closure glaucoma attack. Notably, patient 5 underwent an iridectomy after evaluation as there were signs of possible acute attack of the glaucoma. Our findings suggest that patient’s with very shallow anterior chambers should be evaluated for prophylactic treatment to ensure the best clinical outcomes.
OCT and visual electrophysiology techniques verified that retinopathies, especially involving the outer layer of photoreceptors, were the initial and main sites of involvement in adult-onset NIID with GGC repeat expansions in NOTCH2NLC, a finding consistent with previous reports (2, 3, 6). Based on the follow-up OCT data of patient 7, we hypothesized that retinal dystrophy began in the peripapillary region in the early-stage and progressed centrifugally toward the fovea areas.
In addition, thinning of the macular GCIPL was present in all patients who underwent measurement, even with mild outer retinal dysfunction, which has not been mentioned in previous reports. This suggests that ganglion cells may also be involved in NIID-related retinal lesions. It has been reported that neuronal intranuclear inclusion bodies have been found in retinal ganglion cells. Moreover, ganglion cells and retinal nerve fiber loss of myelinated axons of the optic nerve were observed as histopathological findings of monozygotic twins with NIID (21), providing evidence for the thinner macular GCIPL instances in our study. Additional evidence of RGC pathological lesions were the hyperreflective fine dots observed in ganglion cell layer as well as other layers in retina using SD-OCT. Little is known about the pathogenesis of retinal involvement in patients with NIID. Photoreceptor cells, ganglion cells and retinal nerve fiber are parts of the visual nervous system. Since NIID shows extensive neurologic involvement, it is hypothesized that retinopathy associated with NIID has a similar mechanism.
Modes of functional visual electrophysiology such as ERG and mf-ERG can be very helpful in differentiating between specific NIID-related retinopathy and well-known cone-rod dystrophy or retinitis pigmentosa, which are caused by different gene mutations. The pattern of retinopathies within the patients with NIID in our study could not be simply classified as cone-rod dystrophy or rod-cone dystrophy, especially as only six patients fulfilled ERG and the degree of the lesions were diverse. At early stage (without visual loss, case 1 and case 5), the lesion began from the peripapillary areas and gradually involving the macular, which the lesion is not centered by macular. At late stage, the lesions were very subtle and focus in cone dysfunction, develop all over the retina, which the pattern was as well as retinitis pigmentosa. In classic cone-rod dystrophy, the lesions are usually centered within the macular region and a bulls-eye-like sign can be seen under ophthalmoscopic examination. This developed pattern of lesions from posterior pole to peripheral retina is also different from typical rod-cone dystrophy. Therefore, we addressed the pattern of retinopathies in NIID is unique as displayed by the findings.
Due to the heterogenous features and phenotypes of NIID, it is very challenging to diagnose without biopsy or gene testing for GGC repeat expansions in the NOTCH2NLC among atypical patients. According to the recently large sample study, Tai et al. reported less than 15% of patients with NIID had symptoms of visual loss (16). According to our data, some of the retinopathies were subtle and started from the peripapillary areas rather than the macular region. Therefore, patients often may not present with visual symptoms unless at the late-stage of the disease. As the eye is a visualized window of the central nervous system, detecting early structural and functional deterioration changes in the retina via ophthalmic techniques is an effective and less invasive diagnostic method. We found that comprehensive ophthalmic examinations provided valuable information to aid a physician’s ability to evaluate the diagnosis and prognosis of patients with NIID.
4.2 Limitations
The present study was a small sample observational clinical study. To solve this issue we will continue to recruit patients with NIID spectra. Additionally, a prospective follow-up study is needed to provide the long-term results.
5 Conclusion
In conclusion, adult-onset NIID with GGC repeat expansions in NOTCH2NLC is associated with a unique pattern of retinopathies, mainly originating from the peripapillary areas and involving the outer layer of photoreceptors and inner ganglion cells layer. Additionally, miosis and sluggish pupillary reflexes are often accompanied by NIID. Our findings provide evidence that neuro-ophthalmic evaluations, especially OCT, is essential for evaluating patients with NIID, even without visual symptom.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Institutional Ethics Review Board of the Eye, Ear, Nose, and Throat Hospital of the Fudan University Shanghai. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
GT and LC designed and conceptualized the study and revised the manuscript and discussions. CF, PS, QC, and YC was the major role in data acquisition. JW, XL, and SW interpreted the data. CF and XS performed statistical analysis. CF and QC drafted the manuscript for intellectual content. XS was supported by the grant. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Key Research and Development Program of China (2020YFA0112700 to XS) and Shanghai Science and Technology Innovation Action Plan (23DZ2291500 to LC). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Acknowledgments
The authors would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1188193/full#supplementary-material
References
1. Sone, J, Mori, K, Inagaki, T, Katsumata, R, Takagi, S, Yokoi, S, et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain. (2016) 139:3170–86. doi: 10.1093/brain/aww249
2. Yamada, W, Takekoshi, A, Ishida, K, Mochizuki, K, Sone, J, Sobue, G, et al. Case of adult-onset neuronal intranuclear hyaline inclusion disease with negative electroretinogram. Doc Ophthalmol. (2017) 134:221–6. doi: 10.1007/s10633-017-9584-z
3. Omoto, S, Hayashi, T, Matsuno, H, Higa, H, Kameya, S, Sengoku, R, et al. Neuronal intranuclear hyaline inclusion disease presenting with childhood-onset night blindness associated with progressive retinal dystrophy. J Neurol Sci. (2018) 388:84–6. doi: 10.1016/j.jns.2018.03.010
4. Hirose, B, Hisahara, S, Uesugi, H, Sone, J, Sobue, G, and Shimohama, S. Sporadic adult-onset neuronal intranuclear inclusion disease with abnormal electroretinogram, nerve conduction studies and somatosensory evoked potential. Rinsho Shinkeigaku. (2018) 58:407–10. doi: 10.5692/clinicalneurol.cn-001154
5. Hayashi, T, Katagiri, S, Mizobuchi, K, Yoshitake, K, Kameya, S, Matsuura, T, et al. Heterozygous GGC repeat expansion of NOTCH2NLC in a patient with neuronal intranuclear inclusion disease and progressive retinal dystrophy. Ophthalmic Genet. (2020) 41:93–5. doi: 10.1080/13816810.2020.1723119
6. Nakamura, N, Tsunoda, K, Mitsutake, A, Shibata, S, Mano, T, Nagashima, Y, et al. Clinical characteristics of neuronal intranuclear inclusion disease-related retinopathy with CGG repeat expansions in the NOTCH2NLC gene. Invest Ophthalmol Vis Sci. (2020) 61:27. doi: 10.1167/iovs.61.11.27
7. Weidenheim, KM, and Dickson, DW. Intranuclear inclusion bodies in an elderly demented woman: a form of intranuclear inclusion body disease. Clin Neuropathol. (1995) 14:93–9.
8. Sone, J, Tanaka, F, Koike, H, Inukai, A, Katsuno, M, Yoshida, M, et al. Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease. Neurology. (2011) 76:1372–6. doi: 10.1212/WNL.0b013e3182166e13
9. Sone, J, Kitagawa, N, Sugawara, E, Iguchi, M, Nakamura, R, Koike, H, et al. Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy. J Neurol Neurosurg Psychiatry. (2014) 85:354–6. doi: 10.1136/jnnp-2013-306084
10. Takahashi-Fujigasaki, J. Neuronal intranuclear hyaline inclusion disease. Neuropathology. (2003) 23:351–9. doi: 10.1046/j.1440-1789.2003.00524.x
11. Haltia, M, Tarkkanen, A, Somer, H, Palo, J, and Karli, H. Neuronal intranuclear inclusion disease. Clinical ophthalmological features and ophthalmic pathology. Acta Ophthalmol Suppl. (1986) 64:637–43. doi: 10.1111/j.1755-3768.1986.tb00680.x
12. Tian, Y, Wang, JL, Huang, W, Zeng, S, Jiao, B, Liu, Z, et al. Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am J Hum Genet. (2019) 105:166–76. doi: 10.1016/j.ajhg.2019.05.013
13. Ishiura, H, Shibata, S, Yoshimura, J, Suzuki, Y, Qu, W, Doi, K, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. (2019) 51:1222. doi: 10.1038/s41588-019-0458-z
14. Sun, QY, Xu, Q, Tian, Y, Hu, ZM, Qin, LX, Yang, JX, et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain. (2020) 143:222–33. doi: 10.1093/brain/awz372
15. Sone, J, Mitsuhashi, S, Fujita, A, Mizuguchi, T, Hamanaka, K, Mori, K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. (2019) 51:1215–21. doi: 10.1038/s41588-019-0459-y
16. Tai, H, Wang, A, Zhang, Y, Liu, S, Pan, Y, Li, K, et al. Clinical features and classification of neuronal Intranuclear inclusion disease. Neurol Genet. (2023) 9:e200057. doi: 10.1212/NXG.0000000000200057
17. Arrindell, EL, Trobe, JD, Sieving, PA, and Barnett, JL. Pupillary and electroretinographic abnormalities in a family with neuronal intranuclear hyaline inclusion disease. Arch Ophthalmol. (1991) 109:373–8. doi: 10.1001/archopht.1991.01080030075043
18. Gelpi, E, Botta-Orfila, T, Bodi, L, Marti, S, Kovacs, G, Grau-Rivera, O, et al. Neuronal intranuclear (hyaline) inclusion disease and fragile X-associated tremor/ataxia syndrome: a morphological and molecular dilemma. Brain. (2017) 140:e51. doi: 10.1093/brain/awx156
19. Al-Sarraj, S, King, A, Troakes, C, Smith, B, Maekawa, S, Bodi, I, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. (2011) 122:691–702. doi: 10.1007/s00401-011-0911-2
20. Deng, J, Gu, M, Miao, Y, Yao, S, Zhu, M, Fang, P, et al. Long-read sequencing identified repeat expansions in the 5′UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet. (2019) 56:758–64. doi: 10.1136/jmedgenet-2019-106268
21. Haltia, M, Somer, H, Palo, J, and Johnson, WG. Neuronal intranuclear inclusion disease in identical twins. Ann Neurol. (1984) 15:316–21. doi: 10.1002/ana.410150403
22. Linke, SJ, Baviera, J, Munzer, G, Fricke, OH, Richard, G, and Katz, T. Mesopic pupil size in a refractive surgery population (13,959 eyes). Optom Vis Sci. (2012) 89:1156–64. doi: 10.1097/OPX.0b013e318263c165
23. Kasthurirangan, S, and Glasser, A. Characteristics of pupil responses during far-to-near and near-to-far accommodation. Ophthalmic Physiol Opt. (2005) 25:328–39. doi: 10.1111/j.1475-1313.2005.00293.x
Keywords: neuronal intranuclear inclusion disease, NOTCH2NLC gene, retinal dystrophy, optical coherence tomography, fundus autofluorescence, pupilometer
Citation: Feng C, Chen Q, Luan X, Sun P, Cao Y, Wu J, Wang S, Sun X, Cao L and Tian G (2024) Adult-onset neuronal intranuclear inclusion disease related retinal degeneration: a Chinese case series. Front. Med. 11:1188193. doi: 10.3389/fmed.2024.1188193
Edited by:
Wensheng Li, Shanghai Aier Eye Hospital, ChinaReviewed by:
Yan Yan, Shanghai Jiao Tong University, ChinaChuntao Lai, Capital Medical University, China
Copyright © 2024 Feng, Chen, Luan, Sun, Cao, Wu, Wang, Sun, Cao and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Cao, caoli2000@yeah.net; Guohong Tian, valentian99@hotmail.com
†These authors have contributed equally to this work and share first authorship