Calreticulin: a multifunctional protein with potential therapeutic applications for chronic wounds
 Andrew P. Sawaya1*
Andrew P. Sawaya1*  Nicole M. Vecin1 Jamie L. Burgess1 Nkemcho Ojeh1,2 Gabrielle DiBartolomeo1
Nicole M. Vecin1 Jamie L. Burgess1 Nkemcho Ojeh1,2 Gabrielle DiBartolomeo1  Rivka C. Stone1
Rivka C. Stone1  Irena Pastar1
Irena Pastar1  Marjana Tomic-Canic1
Marjana Tomic-Canic1- 1Wound Healing and Regenerative Medicine Research Program, Dr Phillip Frost Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Faculty of Medical Sciences, The University of the West Indies, Bridgetown, Barbados
Calreticulin is recognized as a multifunctional protein that serves an essential role in diverse biological processes that include wound healing, modification and folding of proteins, regulation of the secretory pathway, cell motility, cellular metabolism, protein synthesis, regulation of gene expression, cell cycle regulation and apoptosis. Although the role of calreticulin as an endoplasmic reticulum-chaperone protein has been well described, several studies have demonstrated calreticulin to be a highly versatile protein with an essential role during wound healing. These features make it an ideal molecule for treating a complex, multifactorial diseases that require fine tuning, such as chronic wounds. Indeed, topical application of recombinant calreticulin to wounds in multiple models of wound healing has demonstrated remarkable pro-healing effects. Among them include enhanced keratinocyte and fibroblast migration and proliferation, induction of extracellular matrix proteins, recruitment of macrophages along with increased granulation tissue formation, all of which are important functions in promoting wound healing that are deregulated in chronic wounds. Given the high degree of diverse functions and pro-healing effects, application of exogenous calreticulin warrants further investigation as a potential novel therapeutic option for chronic wound patients. Here, we review and highlight the significant effects of topical application of calreticulin on enhancing wound healing and its potential as a novel therapeutic option to shift chronic wounds into healing, acute-like wounds.
The pathophysiology and clinical burden of chronic wounds
Chronic non-healing wounds represent a major healthcare burden for patients and healthcare professionals (1–3). The main types of chronic wounds include venous leg ulcers (VLU), diabetic foot ulcers (DFU) and pressure ulcers (PU), and they all share a serious clinical burden due to the high incidence and recurrence, and associated complications (4). They are frequently associated with underlying conditions that include vascular disease, diabetes, and aging and the severity of chronic wounds as a serious medical disease is generally overlooked, despite a high mortality rate of 50% (5, 6). Five-year mortality rates for complications of DFUs such as minor and major amputations are 46.2% and 56.6%, respectively, compared to a pooled five-year cancer mortality rate of 31% (7). In addition, the excessive costs for diabetes care surpassed the costs associated with cancer care in 2017 (7). In addition, chronic wounds have a significant impact on quality of life due to lifestyle modifications, reduced motility, and social isolation and are associated with increased risk of depression, anxiety, and suicide (8, 9). Despite the severity of the clinical problem associated with chronic wounds, treatments remain limited and often not efficacious in restoring healing. Over 50% of DFUs and over 70% of VLU fail to heal depending on the wound size and duration (10–12), emphasizing the essential need to better understand the mechanisms that lead to chronic wounds for the development of new therapeutic options.
The physiological wound healing response progresses through a series of overlapping phases including hemostasis, inflammation, proliferation, and remodeling (13–16). Chronic wounds fail to progress through the phases of healing in a timely manner and are considered to be “stuck in a chronic inflammatory state,” contributing to inhibition of healing (17). This paradigm has been the longstanding rationale to therapeutically target the inflammatory process to curb inflammation. However, this has proven to be clinically unsuccessful. Recent studies have demonstrated an imbalance between pro-inflammatory and anti-inflammatory responses along with impaired function and signaling of inflammatory cells in DFUs and VLUs that fail to reach levels of acute healing wounds, rendering them incapable of progressing through the healing process (18–21). These previously unrecognized findings provide the basis for why therapies aimed at suppressing inflammation in chronic wounds has been clinically unsuccessful.
Keratinocytes play a crucial role in mediating the wound healing response and are among the first cells to respond to injury. Keratinocytes release several cytokines and factors that include IL-1β, TNFα, IL-6 and EGF that act in paracrine fashion to alert neighboring cells and local immune cells that the barrier has been breached (16, 22). In addition, these factors stimulate keratinocytes to acquire an activated phenotype (23). Upon activation, keratinocytes become hyperproliferative and migratory and begin to close the wound through re-epithelialization. The activated phenotype of keratinocytes is characterized by upregulation of the wound-inducible keratin genes, Krt6, 16 and 17 along with upregulation of integrins and rearrangement of the actin cytoskeletal components to promote a migratory phenotype (22). Once keratinocytes have covered the wound, they begin the differentiation process to restore the epidermal barrier. In chronic wounds, keratinocyte activation is deregulated, in which they display a hyper-proliferative and non-migratory phenotype with hyperkeratosis and parakeratosis at the wound edge, further contributing to impaired barrier formation (16, 17, 24). The hyperproliferative, non-migratory epidermis is a hallmark of chronic wounds that is due to increased levels of the β-catenin/c-myc pathway (17, 25, 26). The overexpression of the β-catenin/c-myc pathway has profound effects on keratinocyte function that include downregulation of cytoskeletal components preventing a migratory phenotype along with (26) depletion of epidermal stem cells, contributing to inhibition of healing (25, 27).
Fibroblasts also play a crucial role in the wound healing process and are the main cell type responsible for the production of extracellular matrix (ECM) and granulation tissue as well as wound contraction, recruitment of immune cells, and angiogenesis (10, 28). Fibroblasts demonstrate phenotypic plasticity in response to wounding, which can contribute to proper healing or its pathogenesis (29–34). Fibroblasts regulate the wound healing response through secretion of signaling molecules and ECM proteins, adopt a transient contractile phenotype, and can serve as progenitors for specialized differentiated mesenchymal cells such as adipocytes (35, 36). Dysregulated fibroblasts in chronic wounds exhibit poor migration, proliferation, and ECM deposition, and increased senescence resulting in insufficient granulation tissue with increased fibrosis (17, 37–39). Aberrant Notch1 signaling has been shown to inhibit fibroblast growth and differentiation into myofibroblasts and diminished their potential for stimulating an angiogenic response (40). In addition, chronic wound fibroblasts exhibit dysregulated senescence, further contributing to impaired wound healing (41, 42). Furthermore, the hyperglycemic microenvironment of DFUs induces metabolic epigenetic memory that sustains DNA methylation patterns of genes critical to wound healing that contributes to impaired fibroblast migration as well as accelerates their senescence (37, 43, 44). In addition, chronic wound fibroblasts display dysfunctional lysosomal capacity and resultant impaired protein turnover, and aberrant TGF-β activity that causes impaired myofibroblast contraction and increased fibrosis (45, 46).
Immune cells play critical roles in all phases of the wound healing process (17). Neutrophils are among the first cell types to arrive at the site of injury that function to eliminate microbes to prevent infection and remove cellular debris (47, 48). Monocytes immediately follow and appear at the site of injury where they are activated in response to microbial products and inflammatory mediators that enable the transition to M1 macrophages, a pro-inflammatory phenotype that aids in preventing infection (49, 50). As the inflammatory response progresses, macrophages transition to an anti-inflammatory and pro-healing M2 phenotype that aids in repair of tissue (10, 50). In addition, the removal of apoptotic neutrophils by macrophages play an important role in augmenting the inflammatory response. They are also a rich source of pro-inflammatory cytokines (IL-1β and IL-6) and growth factors (FGF, EGF, and PDGF) that aid in initiating granulation tissue formation. We have previously demonstrated that immune cell recruitment of macrophages and neutrophils are deregulated in the wound edge of DFUs (19, 21). In addition, we identified transcriptional networks mediated by the FOXM1 transcription factor to be suppressed in DFUs that induce formation of neutrophil extracellular traps (NET), releasing cytotoxic proteins that can damage surrounding tissue, further contributing to inhibition of healing (19, 51). The inability of chronic wounds to clear the NETs along with decreased pro-healing M2 macrophages prevent chronic wounds from progressing through the wound healing process, causing them to be stalled in the inflammatory phase (52).
Effective treatments for chronic wounds remain limited due to the complexity of their pathophysiology, requiring a multifactorial approach that can target several aspects of the processes deregulated in chronic wounds. Here, we highlight the role of calreticulin (CALR) that functions as a multifunctional and versatile protein with remarkable pro-healing effects as a potential novel therapeutic option for chronic wounds.
Calreticulin: overview
Calreticulin is a 46 kDa, high-capacity calcium-binding protein that is recognized for its role in regulating cellular processes in response to stress (53–56). The significance of CALR is underscored by the fact that knockout of calreticulin is embryonically lethal (57). It was initially identified as an endoplasmic reticulum (ER)-resident protein that resides within the lumen of the ER with two major functions: directing the proper folding of proteins, and homeostatic control of calcium levels in the cytosol and ER (54, 55, 58). However, later studies have demonstrated CALR to be localized in multiple compartments within the cell as well as in the extracellular matrix in which it has been demonstrated to be a potent pro-healing factor during wound healing (58–63). The initial discovery of CALR as a pro-healing factor came from studies that demonstrated purified hyaluronic acid (HA) accelerated wound healing in rodent models (64). However, it was later found that the pro-healing effects were mitigated by protease treatment, but not by hyaluronidase treatment, indicating an active protein to be in complex with HA responsible for the pro-healing effects (65, 66). Subsequent studies revealed, through purification and amino-acid sequencing, the active protein to be CALR.
The structure of calreticulin consists of three distinct functional domains that include the N-terminal domain, the middle P-domain, and C-terminal domain (55, 58). The N-terminal domain is the most conserved domain of CALR among different species and contains the signal sequence required for localization to the ER, whereas the middle P and C-terminal domains contain multiple high and low affinity calcium binding sites, respectively (58). The middle P-domain is rich in proline residues and consists of a pair of three repeated sequences that function in lectin-like chaperone activity termed A and B repeats with amino acid sequence PXXIXDPDAXKPEDWDE, and GXWXPPXIXNPXYX, respectively (55, 58). The C-terminal domain contains the canonical KDEL sequence necessary for the retrieval and retention of CALR in the ER. Together with its structural and functional homolog, calnexin, CALR directs the proper folding of proteins primarily through its lectin-like chaperone activity (67). Unlike CALR, calnexin is integrated within the ER membrane to coordinate proper folding of nascent proteins. Quality control of protein folding is carried out by recognition of oligosaccharides added to nascent polypeptides (68). The coordination between CALR and calnexin ensure proper folding of proteins by recognizing aberrant oligosaccharides and retaining the newly formed glycoprotein within the ER until the protein has acquired the proper conformation to enter the secretory pathway. Prolonged interaction with calreticulin/calnexin can activate the ER-associated degradation pathway (ERAD) to prevent accumulation of improperly folded proteins (69). Although studies on ER stress in chronic wounds are limited, several factors are known to induce ER stress that include hypoxia, oxidative stress and infection that are all associated with the pathophysiology of chronic wounds (70–72). ER stress has been shown to be elevated in diabetic and pressure ulcer mouse models, contributing to impaired healing (71, 72). Moreover, ER stress related genes were found to be upregulated in venous leg ulcers (70). However, it remains to be determined the role of CALR in mediating ER stress in chronic wounds.
CALR also has a critical role in regulating calcium homeostasis (73, 74). Calcium is a widely known important signaling molecule with multiple effects that include regulation of gene expression, cell adhesion and promoting keratinocyte differentiation (16, 75–77). Interestingly, CALR expression is greater in the suprabasal layers of the epidermis where calcium concentrations are higher relative to basal keratinocytes (60, 78), suggesting a role for CALR in regulating the differentiation process. Although the role of CALR in regulating epidermal differentiation remains unknown, it is interesting to reason that given its high expression level in the epidermis, particularly suprabasal keratinocytes, and its capacity to modulate calcium levels, it may play a previously unrecognized role in regulating epidermal differentiation. Calcium concentrations are three-fold higher in the lumen of the ER relative to the cytosol. The calcium storage capacity in the ER-lumen is enhanced by calcium-binding proteins that include CALR, Grp94, immunoglobulin-heavy-chain-binding protein (55, 58). Depletion of calcium stores from the ER-lumen can have profound effects that include impaired chaperone function and accumulation of misfolded proteins and blockage of the secretory pathway. In addition, calcium levels have a role in regulating expression of CALR in which depletion of intracellular calcium stores activates its gene expression (75, 79).
CALR is unique from most other ER-resident proteins in that its localization is not restricted to the ER (80). It is also found localized throughout the cell in intracellular compartments (e.g., nucleus, cytosol), cell surface as well as in extracellular compartments where it can interact and activate various signaling molecules to promote wound healing. Although cell surface CALR lacks a transmembrane domain, it complexes with its co-receptor low-density lipoprotein receptor-related protein 1 (LRP1) (81, 82). The LRP1/Calreticulin complex acts as a cell surface receptor for thrombospondin-1 (TSP-1) that is necessary to activate focal adhesion disassembly to promote migration (81–84).
Despite having the canonical KDEL ER-retention signal sequence, the exact mechanisms and how CALR localizes to multiple compartments remains poorly understood. Nevertheless, its localization to multiple compartments has demonstrated specific roles beyond functioning as an ER-chaperone protein during wound healing that include regulation of cell migration, focal adhesion disassembly, cellular proliferation, promoting phagocytosis, resistance to cell death and regulation of transcription factors (81–87), all of which are deregulated in chronic wounds. The extra-ER functions and application as a pro-healing factor in various wound healing models have been well established, making it an attractive therapeutic option for patients with chronic wounds (Table 1).
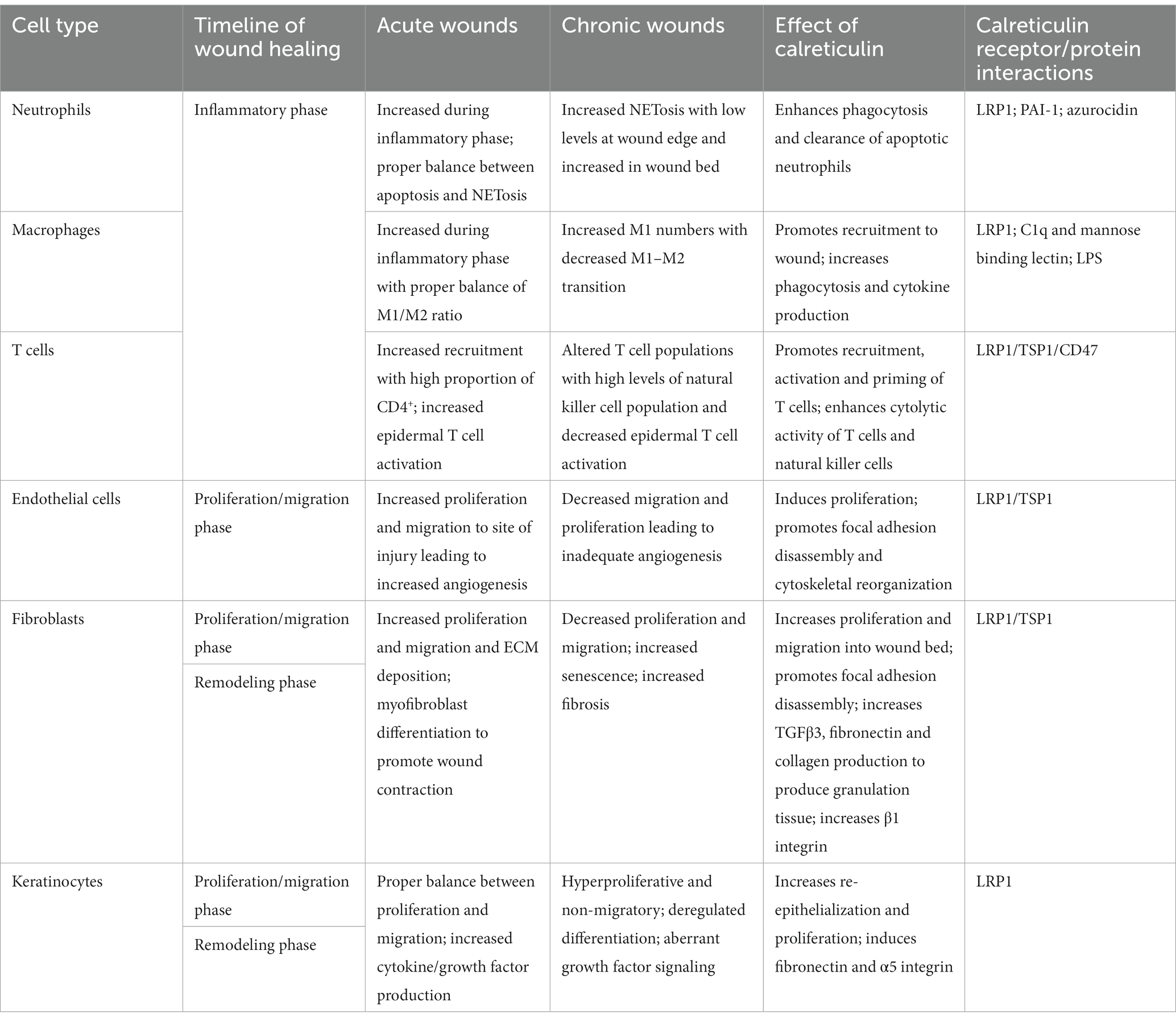
Table 1. Summary of the cellular components involved in acute and chronic wounds and the effects of calreticulin on each cell type in potentially reversing chronic wound pathophysiology.
Calreticulin effects on keratinocyte function
Exogenous CALR treatment has shown significant effects in promoting keratinocyte migration in preclinical models of wound healing. In vivo studies using impaired healing models that include diabetic (db/db) mice models and steroid-treated porcine models, demonstrated enhanced rate of re-epithelization in CALR-treated wounds compared to controls (59, 60, 62). This was further supported by in vitro studies that showed CALR enhanced keratinocyte migration in a dose-dependent manner by a twofold higher migration rate than treatment with epidermal growth factor (60, 61), a potent stimulator of keratinocyte migration. Moreover, in vivo studies demonstrated CALR to be more effective in promoting wound healing than PDGF-BB, the only FDA approved recombinant protein for chronic wounds.
Although the exact mechanisms by which CALR promotes keratinocyte migration are poorly understood, it is possible that CALR may stimulate actin cytoskeletal signaling through activation of the LRP1 receptor (88). In chronic wounds, actin cytoskeletal signaling is deregulated due to increased caveolin-1 (Cav1) expression, contributing to inhibition of migration (89). Increased Cav1 expression results in increased RhoA activity, preventing directional migration (89). In addition, studies have demonstrated that deletion of CALR increased caveolin expression and endocytosis (90). Although the effects of CALR on Cav1 levels have not been investigated in the context of wound healing, signaling through the LRP1/Calreticulin complex has been shown to inhibit RhoA activity (88), providing a potential mechanism of CALR action in reversing Cav1 negative effects on keratinocyte migration.
CALR has also been shown to regulate steroid receptor activity and block their ability to regulate gene expression (87, 91, 92). Activation of steroid receptors by their cognizant ligands are known to play an important role in regulating the wound healing response, particularly glucocorticoids (GCs). GCs are well known potent inhibitors of wound healing that exert their effects through the GR (93–96). GC synthesis is increased in chronic wounds, contributing to inhibition of healing (89). CALR has been demonstrated to antagonize GR and prevent its ability to regulate gene expression as well as function as a nuclear export for GR (91, 92, 97), suggesting an additional pro-healing mechanism for CALR. Activation of GR was shown to exert profound diverse effects including inhibition of apoptosis, interferon-y signaling pathway, promoted terminal differentiation while inhibiting early differentiation markers, along with inhibition of keratinocyte migration (17, 24, 96). In addition, GR interacts with a variety of signaling pathways and transcription factors important in wound healing. GR has been shown to antagonize the epidermal growth factor (EGF) pathway (98), one of the most potent stimulators of keratinocyte migration. Inhibition of EGF signaling by GR resulted in suppression of the early wound-healing marker, keratin 6 (K6), resulting in impaired healing. Furthermore, GR activation leads to activation of the β-catenin/c-myc pathway resulting in a hyperproliferative epidermal phenotype, a hallmark of chronic wounds (26, 99). Moreover, activation of c-myc leads to epidermal stem cell depletion, further contributing to the inability of the tissue to properly respond to injury (27). In addition, GCs have been shown to inhibit deposition of ECM proteins such as collagen and cytoskeletal components preventing a migratory phenotype (93, 96), all of which CALR has been demonstrated to promote and may potentially reverse in a chronic wound setting. Although the role of CALR in inhibiting GR has not been demonstrated in the context of wound healing, the potential to reverse the wide-spread effects of GR activity in chronic wounds with CALR would provide a highly advantageous effect to shift chronic wounds to an acute-like healing wound.
Furthermore, chronic wounds exhibit deregulation of keratinocyte differentiation in which the early markers of differentiation, keratins 1 and 10, at the non-healing edge of chronic wounds were found to be suppressed, whereas the late differentiation markers involucrin and transglutaminase were upregulated, contributing to inhibition of healing (24). In normal wound healing, keratinocytes undergo calcium-dependent differentiation as they migrate upward, eventually terminally differentiating to make up the cornified, outermost layer of skin (17, 100). A major role of CALR is maintaining calcium homeostasis (101) and given that keratinocyte differentiation is calcium-dependent (77), it may play a critical role in potentially reversing the impaired differentiation process in chronic wounds. However, further studies are needed to elucidate possible effects of CALR on keratinocyte differentiation.
The potent effects of CALR in enhancing keratinocyte and fibroblasts migration in preclinical models of wound healing underscore its efficacy as a potential therapy for wound healing disorders. Further investigations into the mechanistic aspects that CALR has on keratinocyte function, that include targeting the wound healing inhibitors GR and Cav1, are needed to further demonstrate its potency as a potential therapeutic to reverse the effects of these factors in chronic wounds to stimulate healing.
Calreticulin effects on fibroblasts
The effects of CALR studied in porcine and murine wound models demonstrated that CALR induced a tissue regenerative response that was evident by the presence of epidermal appendages and lack of scarring through regulation of fibroblast function (61, 63). In addition, CALR induced a dose-dependent increase in granulation tissue formation, producing granulation tissue with greater depth and cellularity and enhanced wound tensile strength (60, 62). Fibroblasts are responsible for granulation tissue formation for contraction of the wound bed and in chronic wounds this function is impaired (63, 102). Fibroblasts isolated from db/db mice and compared to fibroblasts isolated from nondiabetic wild-type mice demonstrated that CALR significantly enhanced migration and wound closure (59). This was further supported by in vitro wound healing assays with human fibroblasts cultured in hyperglycemic conditions that demonstrated significantly enhanced fibroblast migration after treatment with 50 ng/mL CALR (59). Moreover, CALR stimulated increased granulation tissue formation, induced fibroblast production of collagen I, fibronectin, α5β1 integrins and elastin through signaling mediated by TGF-β3 (63), all of which promotes fibroblast migration into the wound bed (61, 63). While TGF-β1 is known for its role in scar formation and fibrosis as well as its wound healing effects, TGF-β3 is antifibrotic and promotes reduced scarring, supporting anti-scaring effects of CALR (103, 104). Moreover, topical application of CALR showed increased proliferation of dermal fibroblasts with increased dermal cellularity and organized collagen fibrils (62). Interestingly, knockout of either CALR or LRP1 in fibroblast cell lines resulted in inhibition of migration in the presence of TSP-1 (85, 86), confirming the importance of this receptor complex in promoting migration.
Calreticulin effects on stem cell function
Epidermal stem cells of the skin are located in at least three distinct lineages, namely, the hair follicle bulge region, the epidermal basal layer and the base of sebaceous glands (10). These cells play an important role in epidermal homeostasis, regeneration and contribute to wound repair (105). Other adult stem cells such as hair follicle dermal papilla and dermal sheath cells, melanocyte progenitors, bone marrow-derived stem cells, mesenchymal stem cells and adipose progenitors have also been reported to contribute to wound healing (106). Adult stem cells promote healing by speeding-up re-epithelialization, exhibiting plasticity, accelerating angiogenesis, and releasing paracrine signaling molecules (106). Depletion of stem cells caused by overexpression of β-catenin/c-myc pathway has been shown to contribute to delayed and impaired wound healing (26). Although the effects of CALR on adult stem cells in the context of wound healing is poorly understood, its role in developmental processes and modulation of embryonic stem cell fate decisions has been reported (107, 108). Studies have shown that CALR knockout leads to embryonic lethality in mice whereas this phenotype is rescued when CALR is constitutively expressed (109) thus proposing CALR as a key regulator of cardiomyogenesis and vital for controlled cardiomyocyte development (109). The effects of CALR on cardiac development have been shown to be mediated through indirect transcriptional activity regulation of nuclear factor of activated T cell (NFAT3) (57) and myocyte-enhancer factor (MEF) 2C (110) by affecting calcineurin activity. Impaired nuclear translocation of these transcription factors in CALR-null cells were reestablished upon re-expression of CALR (57, 110). Furthermore, CALR plays an important role in central nervous system development (111), osteogenesis (107) and chondrogenesis (108) and hematopoiesis (112, 113). Pilquil et al. demonstrated that CALR regulated a switch between osteoblast and chondrocyte lineages derived from murine embryonic stem cells via calcineurin/NFATC1 axis and GSK3β-deactivation (108). CALR has been shown to affect β-catenin associated pathways and Notch and phosphoinositide 3 kinase signaling all of which are important in regulating stem cell fate decisions (114–117). It is interesting to note that several of these pathways overlap with stem cell regulation during wound healing that include NFATC1 (118) and GR/β-catenin/GSK3β/c-myc (26, 27, 99), suggesting a potential role for CALR in the regulation of progenitor cells during wound healing, however further studies are needed. The diverse effects of CALR in promoting development and wound healing as well as its ability to affect known stem cell associated pathways and antagonize activity of GR/β-catenin/c-myc pathway suggests CALR may participate in modulating stem cell regulation, preserve and/or protect local and systemic adult stem cell populations. Further research on the precise mechanisms underlying CALR’s role in stem cells in general and keratinocyte stem cells could provide valuable insights for potential therapeutic strategies in chronic wounds.
Calreticulin effects on immune cell response
CALR is predicted to play a role in the attraction and migration of many immune cells including monocytes (61), T lymphocytes (119), natural killer cells (120), and dendritic cells (121). A major component of the wound healing inflammatory response involves tight regulation of the type and number of immune cell infiltration (10). While increased immune cells are a normal and necessary part of the wound healing process, the location, appropriate activation, and duration are critical for proper wound repair. Chronic wounds are characterized by an extended, suboptimal inflammatory response, with significantly decreased levels of neutrophils and macrophages compared to normal healing wounds (19, 20). The role of CALR in macrophage recruitment was demonstrated in porcine studies which found that CALR treated porcine wounds showed increased wound healing and, unlike wounds treated with buffer or PDGF-BB, showed up to three times the number of macrophages in the granulation tissue that resolved once past the inflammatory phase of wound healing (60, 61). Interestingly, approximately 50% of the macrophages were sequestered to the microvasculature of the dermis rather than the extracellular matrix (62), though the role of CALR in this localization and its subsequent effect in wound healing are still unclear.
CALR has been found to be a critical signal for immune cell activation and cytokine production. Cell surface expression of CALR on monocytes increases during their lifetime while in neutrophils, CALR expression is diminished over time (122). Flow cytometry and ELISA analysis found CALR was released from the cell surface when neutrophils are activated (123). Neutrophils secrete azurocidin, which has been shown to bind to CALR and induce cytokine production in monocytes (124). A partial recombinant CALR fragment from mice was found to stimulate human monocytes and B cell cytokine secretion (125), as well as stimulate B cell differentiation into antibody secreting cells (126). Interestingly, stimulation of macrophages with recombinant CALR induced expression of TNFα and IL-6. These findings were further supported by utilizing an anti-CALR antibody that blocked their expression (124, 127). Moreover, treatment of mice with anti-CALR antibody reduced CXCL15, IL-6, IL-1β, TNF-α expression and recruitment of immune cells (127). In addition, CALR was reported to act as an opsonin for bacterial clearance by binding with LPS and other danger-associated molecular patterns (DAMPS) that triggered induction of cytokines production in macrophages, indicating CALR may function as a pathogen-associated molecular pattern (PAMPs) involved in the clearance of pathogens (128). As chronic wounds are characterized by deregulated immune cell response at the wound edge, it is of interest that CALR was found to increase immune cell activity, suggesting CALR may restore proper immune cell response in chronic wounds.
A major challenge with chronic wounds is deregulation of the immune cell response that traps chronic wounds in a prolonged and dampened inflammatory state. Neutrophils play a major role in wound healing and are deregulated in chronic wounds (19, 48). Chronic wounds are characterized by increased NET formation in which neutrophils decondense their chromatin and release their contents into the extracellular space that acts as nets to trap and maintain microbial infection from spreading (51, 129–131). However, excessive NETosis is detrimental to the wound healing process and diabetes has been shown to prime neutrophils to undergo NETosis and inhibit wound healing (129, 130). We found increased NET formation to result from suppression of the FOXM1 network in DFUs (51). Although CALR has shown some effects on regulating the immune cell response, its effects on neutrophils in the context of wound healing have not been fully explored. Given the pro-healing effects in preclinical models of diabetic wound healing, it would be interesting to establish the effects of CALR on regulating the neutrophil response and the potential to inhibit NET formation and restore neutrophil function in chronic wounds to promote a proper inflammatory response.
Calreticulin effects on apoptosis and phagocytosis
Considering wound debridement is standard of care in the treatment of DFUs, the role of CALR in skin as a naturally occurring component of wound debridement through phagocytosis of apoptotic or infected cells is of considerable interest (61, 132). CALR has been shown to be a major regulator of immunogenic cell death (ICD) and acts as a key discriminator against other forms of cell death (133–135). ICD is triggered by specific inducers that lead to ER stress, the release and membrane exposure of CALR, recognition of DAMPs, and activation of immune cells that elicits antigen-specific inflammatory responses (133–135). Studies in the cancer field have shown the translocation of intracellular CALR to the cell surface is required for phagocytosis of apoptotic tumor cells by dendritic cells and induction of an effective immune response (136, 137), including priming of cytotoxic T cells (138, 139). CALR has been constitutively found on several cell surfaces which has been shown to induce phagocytosis when CALR is uncoupled from other phagocytic inhibitor molecules, such as CD47 (140). The loss of CD47 association led to not only apoptotic, but even viable erythrocytes and leukocytes phagocytosed by macrophages and nonprofessional phagocytes (132).
Removal of apoptotic cells is divided into three steps namely recruitment, engagement and engulfing (141). Phagocytes are first recruited to the apoptotic cells by “find-me” signals released by the cells. Engagement follows which occurs through binding directly or via bridging molecules in response to ‘eat-me’ signals on the apoptotic outer cell membrane. Phagocytes then engulf the apoptotic cells with the help of engulfment-receptors which engage directly with ‘eat-me’ signals (141). Cell-surface CALR has been shown to be vital for cellular response to ICD, apoptosis and efferocytosis (132, 133). It plays a key role as a bridging molecule in recognizing and removing apoptotic cells via the LRP1/C1q collectin/ficolin pathway by direct binding to the complement protein C1q, collectin, and ficolin proteins that recognize apoptotic cells (123, 142, 143). Binding of CALR to LRP1 has been shown to induce phagocytosis and pro-inflammatory responses (144). Moreover, both endogenous CALR (132) and exogeneous CALR secreted by phagocytes serve as ‘eat-me’ signals (145, 146) required for clearance by phagocytic cells along with others such as phosphatidylserine (147). CALR in conjunction with LRP1 has been shown to mediate efferocytosis (132) where CALR is present on the surface of the apoptotic cell to be taken up by the phagocyte expressing LRP1 in a trans configuration. This requires CD47/integrin-associated protein (IAP), a known “do not eat me” signal described for viable cells, to be down-regulated on the apoptotic cell to block engagement of CD47 with the cell surface signal regulatory protein-α (SIRP-α) Src homology 2 domain-containing protein tyrosine phosphatase substrate on the phagocytic cell.
Overexpression of CALR has been shown to result in heightened cellular susceptibility to apoptosis, whereas CALR-deficient cells from CALR knockout mice exhibited significant resistance to staurosporine-induced apoptosis. This resistance was accompanied by reduced cytochrome c release from mitochondria and diminished caspase 3 enzyme activity (74). In Drosophila studies, cell surface CALR is required for apoptotic cell phagocytosis by hemocyte-derived phagocytes and is blocked by CALR antibodies (148). Moreover, dead CALR-null mouse embryo fibroblast cells were not phagocytosed unless rescued by exogenous addition of CRT (132). These studies highlight the crucial role CALR plays in cellular responses to apoptosis and efferocytosis. Given that CALR is involved in these processes, it is likely that CALR may contribute to the wound debridement process of nonviable tissue, damaged, dead or unwanted cells and bacterial clearance by phagocytosis to resolve inflammation and promote chronic wound healing (61). These findings implicate CALR to have beneficial pro-healing effects through regulating apoptosis and removal of non-viable tissue/cells that is of considerable interest and needs further investigation.
Conclusions and future directions
The various pro-healing functions of CALR makes it an attractive therapeutic option for patients with chronic wounds (Figure 1). Further studies are needed to determine the full effects of CALR action on additional processes of wound healing that include antimicrobial properties, angiogenesis, and effective topical delivery methods of CALR to patients.
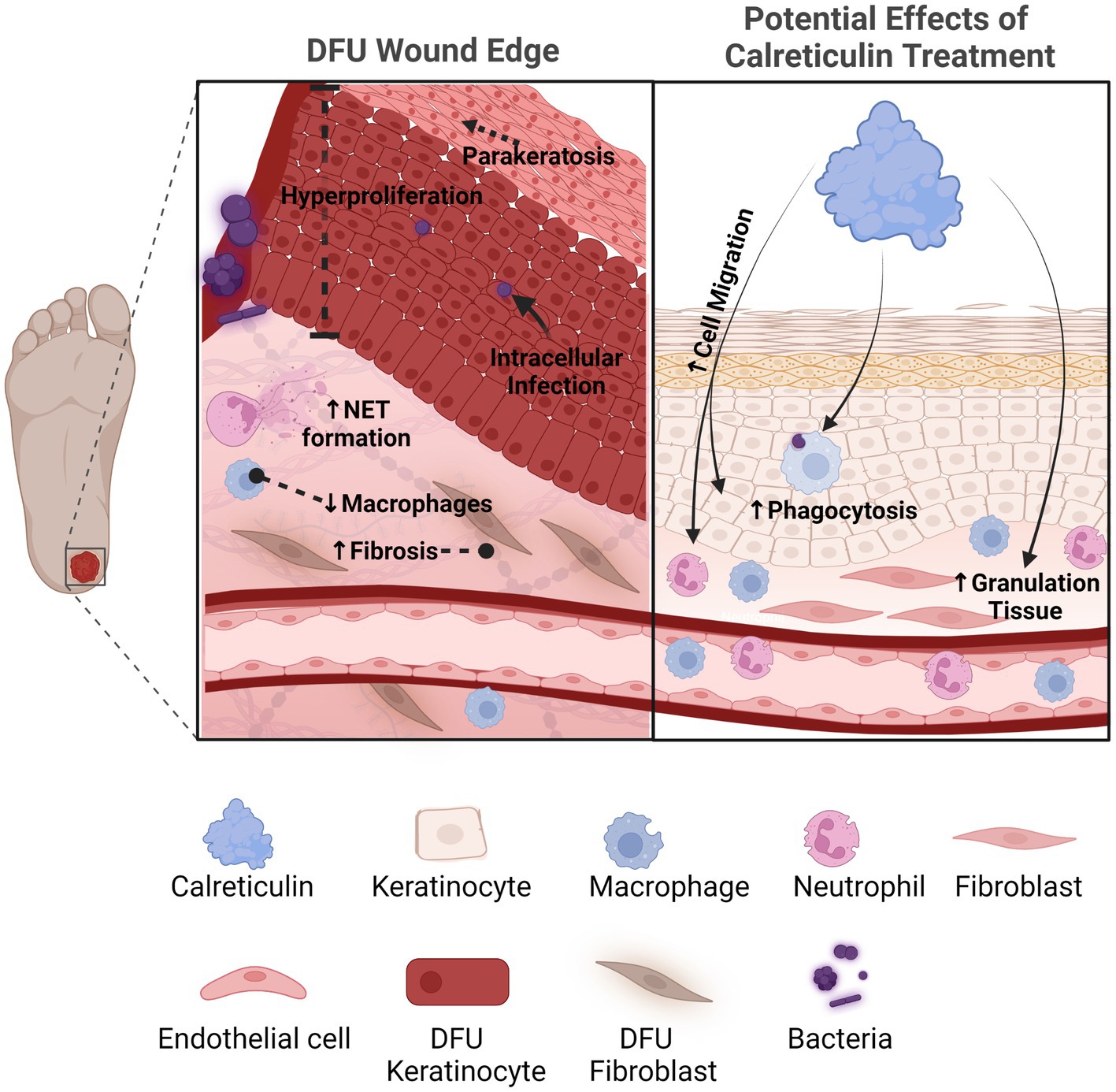
Figure 1. Calreticulin as a novel therapeutic with multiple pro-healing effects for the treatment of chronic wounds. Diabetic foot ulcers compared to acute healing wounds indicates the diverse effects calreticulin exerts on the wound healing process. Calreticulin enhances keratinocyte migration/proliferation, fibroblast function, granulation tissue and phagocytosis, all processes that are deregulated in DFUs that calreticulin may reverse to stimulate healing. Created with BioRender.com.
The microbial burden of chronic wounds remains a major clinical challenge (149–151). Unresolved infection in chronic wounds further delays healing and contributes to the prolonged inflammation of the disease (10). Chronic wounds are characterized by a perturbed microbiome with increased bacterial biofilm formation (152). Although any direct antimicrobial effects of CALR during wound healing have not been determined, CALR has been shown to play a role in mediating the immune cell response during infection. Phagocytosis of pathogen-induced dying cells by dendritic cells is induced by exposure of CALR on the cell surface (153). There has been speculation that intracellular pathogens have found ways to avoid phagocytosis induced by CALR surface exposure (153), a speculation that is particularly relevant in DFUs considering recent studies have found that even DFUs with no clinical sign of infection harbor intracellular Staphylococcus aureus in keratinocytes (154). In neutrophils, CALR was found to bind an anti-bacterial peptide shown to have therapeutic activity against methicillin resistant S. aureus MRSA infected mice and stimulate superoxide anion production (124, 155). Conjugation of a recombinant CALR fragment with laminarin, a β-1,3-glucan, activated B cells and induced antibodies that prevented Candida albicans growth in vitro (156). However, further studies are needed to further characterize CALR as an antimicrobial in the context of wound healing.
Angiogenesis is an important aspect of wound healing that is deregulated in chronic wounds (157–159). Vasculature abnormalities are the result of impaired recruitment and function of endothelial cells and response to growth factors (e.g., VEGF), which contributes to decreased blood flow leading to ischemia and impaired healing (160). Studies on the effects of CALR on angiogenesis has shown mixed results, suggesting CALR may have context-dependent effects on the angiogenesis process. In vitro studies have demonstrated CALR increased proliferation of microvascular endothelial cells and VEGF production (60, 161). However, the effects of CALR on angiogenesis in tumor models have demonstrated that CALR undergoes N-terminal cleavage to generate the CALR fragment, vasostatin, that acts as an inhibitor of angiogenesis by preventing endothelial cell adhesion (162, 163). Nevertheless, it was demonstrated that the vasostatin fragment does not impair wound healing at concentrations used in tumor models (164). These findings indicate the context- and concentration-dependent regulation of CALR, further supporting the need to characterize the effects of CALR on angiogenesis in the context of wound healing.
CALR has also been shown to regulate several metabolic processes that include glucose transport, insulin receptor expression and iron metabolism (165–168), all of which play important roles in wound healing that are deregulated in chronic wounds. It was demonstrated that CALR regulates expression of insulin receptor and the GLUT1 transporter to maintain proper glucose levels (165, 167, 168). High levels of glucose is known to result in increased GLUT1 and insulin insensitivity (169). Under high glucose conditions, CALR promoted the degradation of GLUT1 mRNA resulting in overall downregulation of glucose transport into endothelial cells (165). In addition, CALR has been shown to act as a receptor for heparin in complex with LRP1 to prevent intracellular responses to high glucose concentrations (170). Moreover, the synergistic effects of CALR and heparin stimulated extracellular hyaluronan synthesis (170). In addition to regulating glucose metabolism, CALR has been shown to regulate iron metabolism (166), a process deregulated in chronic wounds in which excessive iron load contributes to impaired macrophage polarization and inhibition of healing (49). It was demonstrated that increased iron levels resulted in increased CALR expression in response to iron-induced oxidative damage in Caco-2 cells (166), a human intestinal epithelial cell line. Although the effects of CALR on metabolic processes in the context of wound healing remains to be investigated, it is interesting to speculate that CALR can have significant effects in restoring proper metabolic processes during wound healing that are deregulated in chronic wounds.
Another important aspect of future investigations into implementing topical CALR as a therapy for chronic wounds would be to evaluate the best course and formulation of topical delivery to patients. Exciting advances in nanotechnology-based drug delivery systems have been utilized as a delivery vehicle for topical based therapies. These include multiple matrix-based delivery systems such as electro-spun collagen or hydrogel-based delivery systems that may facilitate sustained topical delivery of CALR at the wound site and protects its activity from the high protease activity present in chronic wounds (171–175). Although topical application of recombinant proteins such as EGF and TGFβ, have shown success in several preclinical models of wound healing, the majority have failed to show efficacy in clinical trials, in part, due to the high degree of protease activity in chronic wounds (10, 176, 177). In addition, the receptors for these growth factors were found to be downregulated and mis-localized in the cytoplasm in patients with chronic wounds (46, 178), preventing patients from responding to exogenously applied growth factors. Although no clinical trials have been conducted with CALR in chronic wounds to date, these studies provide valuable lessons to learn from that can improve recombinant protein therapy in the chronic wound setting. Future work will be necessary to further characterize the receptors and factors that CALR interacts with in chronic wound tissue to assure full therapeutic potential of CALR. In addition, strategies to prevent degradation of CALR in the wound environment need to be investigated to protect its activity and allow CALR to exert its multiple pro-healing functions. These approaches will need to be tested in various in vivo models for determining an effective delivery system for CALR that can further support FDA applications for CALR as a topical treatment for patients with chronic wounds.
It is evident that the pathophysiology of chronic wounds is extremely complex that demands a multifactorial therapeutic approach that can target the multiple processes that are deregulated. Despite the advances made in understanding CALR effects in wound healing, many questions remain unanswered and the impact it has on wound healing may extend even further that may reveal additional novel mechanisms of CALR action in promoting wound healing. Given the multiple pro-healing effects of CALR that have already been established, topical CALR may provide a powerful therapeutic approach targeting multiple aspects of the chronic wound pathophysiology with maximum efficacy achieving the needed shift from a non-healing wound into an acute wound with healing properties.
Author contributions
AS and MT-C: conceptualization, design, and review and editing. AS, NV, JB, GD, RS, NO, and IP: writing—original draft and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the startup funds from the Dr. Phillip Frost Department of Dermatology, University of Miami Miller School of Medicine (to AS).
Acknowledgments
We recognize the major contribution of Leslie I Gold (NYU School of Medicine) in the study of calreticulin function in general and in wound healing in particular. We are grateful to the Tissue Regeneration Sciences (TRS) and Philip Frost Department of Dermatology and Cutaneous Surgery for their generous support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Burgess, JL, Wyant, WA, Abdo Abujamra, B, Kirsner, RS, and Jozic, I. Diabetic wound-healing science. Medicina. (2021) 57:1072. doi: 10.3390/medicina57101072
2. Gould, L, Abadir, P, Brem, H, Carter, M, Conner-Kerr, T, Davidson, J, et al. Chronic wound repair and healing in older adults: current status and future research. Wound Repair Regen. (2015) 23:1–13. doi: 10.1111/wrr.12245
3. Schneider, C, Stratman, S, and Kirsner, RS. Lower extremity ulcers. Med Clin North Am. (2021) 105:663–79. doi: 10.1016/j.mcna.2021.04.006
4. Singer, AJ, Tassiopoulos, A, and Kirsner, RS. Evaluation and Management of Lower-Extremity Ulcers. N Engl J Med. (2017) 377:1559–67. doi: 10.1056/NEJMra1615243
5. Alavi, A, Sibbald, RG, Mayer, D, Goodman, L, Botros, M, Armstrong, DG, et al. Diabetic foot ulcers: part I. pathophysiology and prevention. J Am Acad Dermatol. (2014) 70:1.e1–1.e18. doi: 10.1016/j.jaad.2013.06.055
6. Robbins, JM, Strauss, G, Aron, D, Long, J, Kuba, J, and Kaplan, Y. Mortality rates and diabetic foot ulcers: is it time to communicate mortality risk to patients with diabetic foot ulceration? J Am Podiatr Med Assoc. (2008) 98:489–93. doi: 10.7547/0980489
7. Armstrong, DG, Swerdlow, MA, Armstrong, AA, Conte, MS, Padula, WV, and Bus, SA. Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J Foot Ankle Res. (2020) 13:16. doi: 10.1186/s13047-020-00383-2
8. Hoban, C, Sareen, J, Henriksen, CA, Kuzyk, L, Embil, JM, and Trepman, E. Mental health issues associated with foot complications of diabetes mellitus. Foot Ankle Surg. (2015) 21:49–55. doi: 10.1016/j.fas.2014.09.007
9. Zhou, K, and Jia, P. Depressive symptoms in patients with wounds: a cross-sectional study. Wound Repair Regen. (2016) 24:1059–65. doi: 10.1111/wrr.12484
10. Eming, SA, Martin, P, and Tomic-Canic, M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. (2014) 6:265sr6. doi: 10.1126/scitranslmed.3009337
11. Lebrun, E, Tomic-Canic, M, and Kirsner, RS. The role of surgical debridement in healing of diabetic foot ulcers. Wound Repair Regen. (2010) 18:433–8. doi: 10.1111/j.1524-475X.2010.00619.x
12. Margolis, DJ, Berlin, JA, and Strom, BL. Which venous leg ulcers will heal with limb compression bandages? Am J Med. (2000) 109:15–9. doi: 10.1016/S0002-9343(00)00379-X
13. Barrientos, S, Stojadinovic, O, Golinko, MS, Brem, H, and Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. (2008) 16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x
14. Martin, P, and Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br J Dermatol. (2015) 173:370–8. doi: 10.1111/bjd.13954
15. Morton, LM, and Phillips, TJ. Wound healing and treating wounds: differential diagnosis and evaluation of chronic wounds. J Am Acad Dermatol. (2016) 74:589–605. doi: 10.1016/j.jaad.2015.08.068
16. Pastar, I, Stojadinovic, O, Yin, NC, Ramirez, H, Nusbaum, AG, Sawaya, A, et al. Epithelialization in wound healing: a comprehensive review. Adv Wound Care. (2014) 3:445–64. doi: 10.1089/wound.2013.0473
17. Pastar, I, Balukoff, NC, Marjanovic, J, Chen, VY, Stone, RC, and Tomic-Canic, M. Molecular pathophysiology of chronic wounds: Current state and future directions. Cold Spring Harb Perspect Biol. (2023) 15:a041243. doi: 10.1101/cshperspect.a041243
18. Ramirez, HA, Pastar, I, Jozic, I, Stojadinovic, O, Stone, RC, Ojeh, N, et al. Staphylococcus aureus triggers induction of miR-15B-5P to diminish DNA repair and deregulate inflammatory response in diabetic foot ulcers. J Invest Dermatol. (2018) 138:1187–96. doi: 10.1016/j.jid.2017.11.038
19. Sawaya, AP, Stone, RC, Brooks, SR, Pastar, I, Jozic, I, Hasneen, K, et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat Commun. (2020) 11:4678. doi: 10.1038/s41467-020-18276-0
20. Stone, RC, Stojadinovic, O, Rosa, AM, Ramirez, HA, Badiavas, E, Blumenberg, M, et al. A bioengineered living cell construct activates an acute wound healing response in venous leg ulcers. Sci Transl Med. (2017) 9:eaaf8611. doi: 10.1126/scitranslmed.aaf8611
21. Theocharidis, G, Thomas, BE, Sarkar, D, Mumme, HL, Pilcher, WJR, Dwivedi, B, et al. Single cell transcriptomic landscape of diabetic foot ulcers. Nat Commun. (2022) 13:181. doi: 10.1038/s41467-021-27801-8
22. Freedberg, IM, Tomic-Canic, M, Komine, M, and Blumenberg, M. Keratins and the keratinocyte activation cycle. J Invest Dermatol. (2001) 116:633–40. doi: 10.1046/j.1523-1747.2001.01327.x
23. Tomic-Canic, M, Komine, M, Freedberg, IM, and Blumenberg, M. Epidermal signal transduction and transcription factor activation in activated keratinocytes. J Dermatol Sci. (1998) 17:167–81. doi: 10.1016/S0923-1811(98)00016-4
24. Stojadinovic, O, Pastar, I, Vukelic, S, Mahoney, MG, Brennan, D, Krzyzanowska, A, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med. (2008) 12:2675–90. doi: 10.1111/j.1582-4934.2008.00321.x
25. Marjanovic, J, Ramirez, HA, Jozic, I, Stone, RC, Wikramanayake, TC, Head, CR, et al. Dichotomous role of miR193b-3p in diabetic foot ulcers maintains inhibition of healing and suppression of tumor formation. Sci Transl Med. (2022) 14:eabg8397. doi: 10.1126/scitranslmed.abg8397
26. Stojadinovic, O, Brem, H, Vouthounis, C, Lee, B, Fallon, J, Stallcup, M, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. (2005) 167:59–69. doi: 10.1016/S0002-9440(10)62953-7
27. Stojadinovic, O, Pastar, I, Nusbaum, AG, Vukelic, S, Krzyzanowska, A, and Tomic-Canic, M. Deregulation of epidermal stem cell niche contributes to pathogenesis of nonhealing venous ulcers. Wound Repair Regen. (2014) 22:220–7. doi: 10.1111/wrr.12142
28. Talbott, HE, Mascharak, S, Griffin, M, Wan, DC, and Longaker, MT. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell. (2022) 29:1161–80. doi: 10.1016/j.stem.2022.07.006
29. Driskell, RR, Lichtenberger, BM, Hoste, E, Kretzschmar, K, Simons, BD, Charalambous, M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. (2013) 504:277–81. doi: 10.1038/nature12783
30. Foster, DS, Januszyk, M, Yost, KE, Chinta, MS, Gulati, GS, Nguyen, AT, et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc Natl Acad Sci U S A. (2021) 118:e2110025118. doi: 10.1073/pnas.2110025118
31. Mascharak, S, Talbott, HE, Januszyk, M, Griffin, M, Chen, K, Davitt, MF, et al. Multi-omic analysis reveals divergent molecular events in scarring and regenerative wound healing. Cell Stem Cell. (2022) 29:315. doi: 10.1016/j.stem.2021.12.011
32. Guerrero-Juarez, CF, Dedhia, PH, Jin, S, Ruiz-Vega, R, Ma, D, Liu, Y, et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat Commun. (2019) 10:650. doi: 10.1038/s41467-018-08247-x
33. Mascharak, S, Desjardins-Park, HE, Davitt, MF, Griffin, M, Borrelli, MR, Moore, AL, et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science. (2021) 372:eaba2374. doi: 10.1126/science.aba2374
34. Plikus, MV, Wang, X, Sinha, S, Forte, E, Thompson, SM, Herzog, EL, et al. Fibroblasts: origins, definitions, and functions in health and disease. Cells. (2021) 184:3852–72. doi: 10.1016/j.cell.2021.06.024
35. Plikus, MV, Guerrero-Juarez, CF, Ito, M, Li, YR, Dedhia, PH, Zheng, Y, et al. Regeneration of fat cells from myofibroblasts during wound healing. Science. (2017) 355:748–52. doi: 10.1126/science.aai8792
36. Shook, BA, Wasko, RR, Rivera-Gonzalez, GC, Salazar-Gatzimas, E, Lopez-Giraldez, F, Dash, BC, et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science. (2018) 362:eaar2971. doi: 10.1126/science.aar2971
37. Liang, L, Stone, RC, Stojadinovic, O, Ramirez, H, Pastar, I, Maione, AG, et al. Integrative analysis of miRNA and mRNA paired expression profiling of primary fibroblast derived from diabetic foot ulcers reveals multiple impaired cellular functions. Wound Repair Regen. (2016) 24:943–53. doi: 10.1111/wrr.12470
38. Pastar, I, Marjanovic, J, Liang, L, Stone, RC, Kashpur, O, Jozic, I, et al. Cellular reprogramming of diabetic foot ulcer fibroblasts triggers pro-healing miRNA-mediated epigenetic signature. Exp Dermatol. (2021) 30:1065–72. doi: 10.1111/exd.14405
39. Stone, RC, Stojadinovic, O, Sawaya, AP, Glinos, GD, Lindley, LE, Pastar, I, et al. A bioengineered living cell construct activates metallothionein/zinc/MMP8 and inhibits TGFbeta to stimulate remodeling of fibrotic venous leg ulcers. Wound Repair Regen. (2020) 28:164–76. doi: 10.1111/wrr.12778
40. Shao, H, Li, Y, Pastar, I, Xiao, M, Prokupets, R, Liu, S, et al. Notch1 signaling determines the plasticity and function of fibroblasts in diabetic wounds. Life Sci Alliance. (2020) 3:e202000769. doi: 10.26508/lsa.202000769
41. Wilkinson, HN, Clowes, C, Banyard, KL, Matteuci, P, Mace, KA, and Hardman, MJ. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J Invest Dermatol. (2019) 139:1171–81. doi: 10.1016/j.jid.2019.01.005
42. Wilkinson, HN, and Hardman, MJ. Senescence in wound repair: emerging strategies to target chronic healing wounds. Front Cell Dev Biol. (2020) 8:773. doi: 10.3389/fcell.2020.00773
43. Kashpur, O, Smith, A, Gerami-Naini, B, Maione, AG, Calabrese, R, Tellechea, A, et al. Differentiation of diabetic foot ulcer-derived induced pluripotent stem cells reveals distinct cellular and tissue phenotypes. FASEB J. (2019) 33:1262–77. doi: 10.1096/fj.201801059
44. Park, LK, Maione, AG, Smith, A, Gerami-Naini, B, Iyer, LK, Mooney, DJ, et al. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics. (2014) 9:1339–49. doi: 10.4161/15592294.2014.967584
45. Berberich, B, Thriene, K, Gretzmeier, C, Kuhl, T, Bayer, H, Athanasiou, I, et al. Proteomic profiling of fibroblasts isolated from chronic wounds identifies disease-relevant signaling pathways. J Invest Dermatol. (2020) 140:2280–90. doi: 10.1016/j.jid.2020.02.040
46. Pastar, I, Stojadinovic, O, Krzyzanowska, A, Barrientos, S, Stuelten, C, Zimmerman, K, et al. Attenuation of the transforming growth factor beta-signaling pathway in chronic venous ulcers. Mol Med. (2010) 16:92–101. doi: 10.2119/molmed.2009.00149
47. Kim, MH, Liu, W, Borjesson, DL, Curry, FR, Miller, LS, Cheung, AL, et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol. (2008) 128:1812–20. doi: 10.1038/sj.jid.5701223
48. Wilgus, TA, Roy, S, and McDaniel, JC. Neutrophils and wound repair: positive actions and negative reactions. Adv Wound Care. (2013) 2:379–88. doi: 10.1089/wound.2012.0383
49. Sindrilaru, A, Peters, T, Wieschalka, S, Baican, C, Baican, A, Peter, H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. (2011) 121:985–97. doi: 10.1172/JCI44490
50. Wynn, TA, and Vannella, KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
51. Sawaya, AP, Stone, RC, Mehdizadeh, S, Pastar, I, Worrell, S, Balukoff, NC, et al. FOXM1 network in association with TREM1 suppression regulates NET formation in diabetic foot ulcers. EMBO Rep. (2022) 23:e54558. doi: 10.15252/embr.202154558
52. DiPietro, LA, Wilgus, TA, and Koh, TJ. Macrophages in healing wounds: paradoxes and paradigms. Int J Mol Sci. (2021) 22:950. doi: 10.3390/ijms22020950
53. Groenendyk, J, Wang, WA, Robinson, A, and Michalak, M. Calreticulin and the heart. Cells. (2022) 11:1722. doi: 10.3390/cells11111722
54. Krause, KH, and Michalak, M. Calreticulin. Cells. (1997) 88:439–43. doi: 10.1016/S0092-8674(00)81884-X
55. Michalak, M, Corbett, EF, Mesaeli, N, Nakamura, K, and Opas, M. Calreticulin: one protein, one gene, many functions. Biochem J. (1999) 344:281–92. doi: 10.1042/bj3440281
56. Michalak, M, Milner, RE, and Burns, K. Opas M. Calreticulin. Biochem J. (1992) 285:681–92. doi: 10.1042/bj2850681
57. Mesaeli, N, Nakamura, K, Zvaritch, E, Dickie, P, Dziak, E, Krause, KH, et al. Calreticulin is essential for cardiac development. J Cell Biol. (1999) 144:857–68. doi: 10.1083/jcb.144.5.857
58. Michalak, M, Groenendyk, J, Szabo, E, Gold, LI, and Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. (2009) 417:651–66. doi: 10.1042/BJ20081847
59. Greives, MR, Samra, F, Pavlides, SC, Blechman, KM, Naylor, SM, Woodrell, CD, et al. Exogenous calreticulin improves diabetic wound healing. Wound Repair Regen. (2012) 20:715–30. doi: 10.1111/j.1524-475X.2012.00822.x
60. Nanney, LB, Woodrell, CD, Greives, MR, Cardwell, NL, Pollins, AC, Bancroft, TA, et al. Calreticulin enhances porcine wound repair by diverse biological effects. Am J Pathol. (2008) 173:610–30. doi: 10.2353/ajpath.2008.071027
61. Gold, LI, Eggleton, P, Sweetwyne, MT, Van Duyn, LB, Greives, MR, Naylor, SM, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. (2010) 24:665–83. doi: 10.1096/fj.09-145482
62. Gold, LI, Rahman, M, Blechman, KM, Greives, MR, Churgin, S, Michaels, J, et al. Overview of the role for calreticulin in the enhancement of wound healing through multiple biological effects. J Investig Dermatol Symp Proc. (2006) 11:57–65. doi: 10.1038/sj.jidsymp.5650011
63. Pandya, UM, Manzanares, MA, Tellechea, A, Egbuta, C, Daubriac, J, Jimenez-Jaramillo, C, et al. Calreticulin exploits TGF-beta for extracellular matrix induction engineering a tissue regenerative process. FASEB J. (2020) 34:15849–74. doi: 10.1096/fj.202001161R
64. Burd, DA, Greco, RM, Regauer, S, Longaker, MT, Siebert, JW, and Garg, HG. Hyaluronan and wound healing: a new perspective. Br J Plast Surg. (1991) 44:579–84. doi: 10.1016/0007-1226(91)90093-Y
65. Bakshandeh, N, Siebert, JW, Cabrera, RC, Eidelman, Y, Longaker, M, Freund, RW, et al. Isolation and partial characterization of hyaluronan-protein-collagen complex (HA-PC) from fetal sheep skin of different gestational ages. Biochem Int. (1992) 28:843–51.
66. Cabrera, RC, Siebert, JW, Eidelman, Y, Gold, LI, Longaker, MT, and Garg, HG. The in vivo effect of hyaluronan associated protein-collagen complex on wound repair. Biochem Mol Biol Int. (1995) 37:151–8.
67. Bedard, K, Szabo, E, Michalak, M, and Opas, M. Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int Rev Cytol. (2005) 245:91–121. doi: 10.1016/S0074-7696(05)45004-4
68. Hebert, DN, and Molinari, M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. (2007) 87:1377–408. doi: 10.1152/physrev.00050.2006
69. Gold, L, Williams, D, Groenendyk, J, Michalak, M, and Eggleton, P. Unfolding the complexities of ER chaperones in health and disease: report on the 11th international calreticulin workshop. Cell Stress Chaperones. (2015) 20:875–83. doi: 10.1007/s12192-015-0638-4
70. Bachar-Wikstrom, E, Manchanda, M, Bansal, R, Karlsson, M, Kelly-Pettersson, P, Skoldenberg, O, et al. Endoplasmic reticulum stress in human chronic wound healing: rescue by 4-phenylbutyrate. Int Wound J. (2021) 18:49–61. doi: 10.1111/iwj.13525
71. Cui, FF, Pan, YY, Xie, HH, Wang, XH, Shi, HX, Xiao, J, et al. Pressure combined with ischemia/reperfusion injury induces deep tissue injury via endoplasmic reticulum stress in a rat pressure ulcer model. Int J Mol Sci. (2016) 17:284. doi: 10.3390/ijms17030284
72. Schurmann, C, Goren, I, Linke, A, Pfeilschifter, J, and Frank, S. Deregulated unfolded protein response in chronic wounds of diabetic Ob/Ob mice: a potential connection to inflammatory and angiogenic disorders in diabetes-impaired wound healing. Biochem Biophys Res Commun. (2014) 446:195–200. doi: 10.1016/j.bbrc.2014.02.085
73. Michalak, M, Robert Parker, JM, and Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. (2002) 32:269–78. doi: 10.1016/S0143416002001884
74. Nakamura, K, Zuppini, A, Arnaudeau, S, Lynch, J, Ahsan, I, Krause, R, et al. Functional specialization of calreticulin domains. J Cell Biol. (2001) 154:961–72. doi: 10.1083/jcb.200102073
75. Burns, K, Atkinson, EA, Bleackley, RC, and Michalak, M. Calreticulin: from Ca2+ binding to control of gene expression. Trends Cell Biol. (1994) 4:152–4. doi: 10.1016/0962-8924(94)90190-2
76. Hwang, J, Kalinin, A, Hwang, M, Anderson, DE, Kim, MJ, Stojadinovic, O, et al. Role of scarf and its binding target proteins in epidermal calcium homeostasis. J Biol Chem. (2007) 282:18645–53. doi: 10.1074/jbc.M702035200
77. Morasso, MI, and Tomic-Canic, M. Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell. (2005) 97:173–83. doi: 10.1042/BC20040098
78. Elias, P, Ahn, S, Brown, B, Crumrine, D, and Feingold, KR. Origin of the epidermal calcium gradient: regulation by barrier status and role of active vs passive mechanisms. J Invest Dermatol. (2002) 119:1269–74. doi: 10.1046/j.1523-1747.2002.19622.x
79. Waser, M, Mesaeli, N, Spencer, C, and Michalak, M. Regulation of calreticulin gene expression by calcium. J Cell Biol. (1997) 138:547–57. doi: 10.1083/jcb.138.3.547
80. Johnson, S, Michalak, M, Opas, M, and Eggleton, P. The ins and outs of calreticulin: from the ER lumen to the extracellular space. Trends Cell Biol. (2001) 11:122–9. doi: 10.1016/S0962-8924(01)01926-2
81. Murphy-Ullrich, JE. Thrombospondin-1 signaling through the Calreticulin/LDL receptor related protein 1 Axis: functions and possible roles in Glaucoma. Front Cell Dev Biol. (2022) 10:898772. doi: 10.3389/fcell.2022.898772
82. Orr, AW, Pedraza, CE, Pallero, MA, Elzie, CA, Goicoechea, S, Strickland, DK, et al. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol. (2003) 161:1179–89. doi: 10.1083/jcb.200302069
83. Coppolino, MG, Woodside, MJ, Demaurex, N, Grinstein, S, St-Arnaud, R, and Dedhar, S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature. (1997) 386:843–7. doi: 10.1038/386843a0
84. Goicoechea, S, Orr, AW, Pallero, MA, Eggleton, P, and Murphy-Ullrich, JE. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J Biol Chem. (2000) 275:36358–68. doi: 10.1074/jbc.M005951200
85. Goicoechea, S, Pallero, MA, Eggleton, P, Michalak, M, and Murphy-Ullrich, JE. The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J Biol Chem. (2002) 277:37219–28. doi: 10.1074/jbc.M202200200
86. Pallero, MA, Elzie, CA, Chen, J, Mosher, DF, and Murphy-Ullrich, JE. Thrombospondin 1 binding to calreticulin-LRP1 signals resistance to anoikis. FASEB J. (2008) 22:3968–79. doi: 10.1096/fj.07-104802
87. Burns, K, Duggan, B, Atkinson, EA, Famulski, KS, Nemer, M, Bleackley, RC, et al. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature. (1994) 367:476–80. doi: 10.1038/367476a0
88. Orr, AW, Pallero, MA, Xiong, WC, and Murphy-Ullrich, JE. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J Biol Chem. (2004) 279:48983–92. doi: 10.1074/jbc.M404881200
89. Jozic, I, Sawaya, AP, Pastar, I, Head, CR, Wong, LL, Glinos, GD, et al. Pharmacological and genetic inhibition of Caveolin-1 promotes epithelialization and wound closure. Mol Ther. (2019) 27:1992–2004. doi: 10.1016/j.ymthe.2019.07.016
90. Massaeli, H, Viswanathan, D, Pillai, DG, and Mesaeli, N. Endoplasmic reticulum stress enhances endocytosis in calreticulin deficient cells. Biochim Biophys Acta, Mol Cell Res. (2019) 1866:727–36. doi: 10.1016/j.bbamcr.2018.12.003
91. Dedhar, S, Rennie, PS, Shago, M, Hagesteijn, CY, Yang, H, Filmus, J, et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nature. (1994) 367:480–3. doi: 10.1038/367480a0
92. Michalak, M, Burns, K, Andrin, C, Mesaeli, N, Jass, GH, Busaan, JL, et al. Endoplasmic reticulum form of calreticulin modulates glucocorticoid-sensitive gene expression. J Biol Chem. (1996) 271:29436–45. doi: 10.1074/jbc.271.46.29436
93. Jozic, I, Abujamra, BA, Elliott, MH, Wikramanayake, TC, Marjanovic, J, Stone, RC, et al. Glucocorticoid-mediated induction of caveolin-1 disrupts cytoskeletal organization, inhibits cell migration and re-epithelialization of non-healing wounds. Commun Biol. (2021) 4:757. doi: 10.1038/s42003-021-02298-5
94. Pastar, I, Stojadinovic, O, Sawaya, AP, Stone, RC, Lindley, LE, Ojeh, N, et al. Skin metabolite, farnesyl pyrophosphate, regulates epidermal response to inflammation, oxidative stress, and migration. J Cell Physiol. (2016) 231:2452–63. doi: 10.1002/jcp.25357
95. Sawaya, AP, Pastar, I, Stojadinovic, O, Lazovic, S, Davis, SC, Gil, J, et al. Topical mevastatin promotes wound healing by inhibiting the transcription factor c-Myc via the glucocorticoid receptor and the long non-coding RNA Gas5. J Biol Chem. (2018) 293:1439–49. doi: 10.1074/jbc.M117.811240
96. Stojadinovic, O, Lee, B, Vouthounis, C, Vukelic, S, Pastar, I, Blumenberg, M, et al. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem. (2007) 282:4021–34. doi: 10.1074/jbc.M606262200
97. Holaska, JM, Black, BE, Love, DC, Hanover, JA, Leszyk, J, and Paschal, BM. Calreticulin Is a receptor for nuclear export. J Cell Biol. (2001) 152:127–40. doi: 10.1083/jcb.152.1.127
98. Lee, B, Vouthounis, C, Stojadinovic, O, Brem, H, Im, M, and Tomic-Canic, M. From an enhanceosome to a repressosome: molecular antagonism between glucocorticoids and EGF leads to inhibition of wound healing. J Mol Biol. (2005) 345:1083–97. doi: 10.1016/j.jmb.2004.11.027
99. Jozic, I, Vukelic, S, Stojadinovic, O, Liang, L, Ramirez, HA, Pastar, I, et al. Stress signals, mediated by membranous glucocorticoid receptor, activate PLC/PKC/GSK-3beta/beta-catenin pathway to inhibit wound closure. J Invest Dermatol. (2017) 137:1144–54. doi: 10.1016/j.jid.2016.11.036
100. Candi, E, Schmidt, R, and Melino, G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. (2005) 6:328–40. doi: 10.1038/nrm1619
101. Arnaudeau, S, Frieden, M, Nakamura, K, Castelbou, C, Michalak, M, and Demaurex, N. Calreticulin differentially modulates calcium uptake and release in the endoplasmic reticulum and mitochondria. J Biol Chem. (2002) 277:46696–705. doi: 10.1074/jbc.M202395200
102. Cook, H, Davies, KJ, Harding, KG, and Thomas, DW. Defective extracellular matrix reorganization by chronic wound fibroblasts is associated with alterations in TIMP-1, TIMP-2, and MMP-2 activity. J Invest Dermatol. (2000) 115:225–33. doi: 10.1046/j.1523-1747.2000.00044.x
103. Lichtman, MK, Otero-Vinas, M, and Falanga, V. Transforming growth factor beta (TGF-beta) isoforms in wound healing and fibrosis. Wound Repair Regen. (2016) 24:215–22. doi: 10.1111/wrr.12398
104. Ramirez, H, Patel, SB, and Pastar, I. The role of TGFbeta signaling in wound epithelialization. Adv Wound Care. (2014) 3:482–91. doi: 10.1089/wound.2013.0466
105. Haensel, D, Jin, S, Sun, P, Cinco, R, Dragan, M, Nguyen, Q, et al. Defining epidermal basal cell states during skin homeostasis and wound healing using single-cell transcriptomics. Cell Rep. (2020) 30:3932–47 e6. doi: 10.1016/j.celrep.2020.02.091
106. Ojeh, N, Pastar, I, Tomic-Canic, M, and Stojadinovic, O. Stem cells in skin regeneration, wound healing, and their clinical applications. Int J Mol Sci. (2015) 16:25476–501. doi: 10.3390/ijms161025476
107. Alvandi, Z, Al-Mansoori, LJR, and Opas, M. Calreticulin regulates Src kinase in osteogenic differentiation from embryonic stem cells. Stem Cell Res. (2020) 48:101972. doi: 10.1016/j.scr.2020.101972
108. Pilquil, C, Alvandi, Z, and Opas, M. Calreticulin regulates a switch between osteoblast and chondrocyte lineages derived from murine embryonic stem cells. J Biol Chem. (2020) 295:6861–75. doi: 10.1074/jbc.RA119.011029
109. Papp, S, Dziak, E, and Opas, M. Embryonic stem cell-derived cardiomyogenesis: a novel role for calreticulin as a regulator. Stem Cells. (2009) 27:1507–15. doi: 10.1002/stem.85
110. Lynch, J, Guo, L, Gelebart, P, Chilibeck, K, Xu, J, Molkentin, JD, et al. Calreticulin signals upstream of calcineurin and MEF2C in a critical ca(2+)-dependent signaling cascade. J Cell Biol. (2005) 170:37–47. doi: 10.1083/jcb.200412156
111. Zhang, X, Szabo, E, Michalak, M, and Opas, M. Endoplasmic reticulum stress during the embryonic development of the central nervous system in the mouse. Int J Dev Neurosci. (2007) 25:455–63. doi: 10.1016/j.ijdevneu.2007.08.007
112. Galluzzi, L, and Kroemer, G. Calreticulin surface presentation can promote quality control of hematopoietic stem cells. Trends Immunol. (2022) 43:950–2. doi: 10.1016/j.it.2022.10.008
113. Salati, S, Prudente, Z, Genovese, E, Pennucci, V, Rontauroli, S, Bartalucci, N, et al. Calreticulin affects hematopoietic stem/progenitor cell fate by impacting erythroid and megakaryocytic differentiation. Stem Cells Dev. (2018) 27:225–36. doi: 10.1089/scd.2017.0137
114. Groenendyk, J, and Michalak, M. Disrupted WNT signaling in mouse embryonic stem cells in the absence of calreticulin. Stem Cell Rev Rep. (2014) 10:191–206. doi: 10.1007/s12015-013-9488-6
115. Fadel, MP, Szewczenko-Pawlikowski, M, Leclerc, P, Dziak, E, Symonds, JM, Blaschuk, O, et al. Calreticulin affects beta-catenin-associated pathways. J Biol Chem. (2001) 276:27083–9. doi: 10.1074/jbc.M101676200
116. Fan, X, Yao, Y, and Zhang, Y. Calreticulin promotes proliferation and extracellular matrix expression through notch pathway in cardiac fibroblasts. Adv Clin Exp Med. (2018) 27:887–92. doi: 10.17219/acem/74430
117. Orr, AW, Pallero, MA, and Murphy-Ullrich, JE. Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J Biol Chem. (2002) 277:20453–60. doi: 10.1074/jbc.M112091200
118. Horsley, V, Aliprantis, AO, Polak, L, Glimcher, LH, and Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cells. (2008) 132:299–310. doi: 10.1016/j.cell.2007.11.047
119. Forslow, A, Liu, Z, and Sundqvist, KG. Receptor communication within the lymphocyte plasma membrane: a role for the thrombospondin family of matricellular proteins. Cell Mol Life Sci. (2007) 64:66–76. doi: 10.1007/s00018-006-6255-8
120. Zheng, Y, Li, C, Xin, P, Peng, Q, Zhang, W, Liu, S, et al. Calreticulin increases growth and progression of natural killer/T-cell lymphoma. Aging. (2020) 12:23822–35. doi: 10.18632/aging.104030
121. Chen, R, Huang, M, Yang, X, Chen, XH, Shi, MY, Li, ZF, et al. CALR-TLR4 complex inhibits non-small cell lung Cancer progression by regulating the migration and maturation of dendritic cells. Front Oncol. (2021) 11:743050. doi: 10.3389/fonc.2021.697697
122. Donnelly, S, Roake, W, Brown, S, Young, P, Naik, H, Wordsworth, P, et al. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. (2006) 54:1543–56. doi: 10.1002/art.21783
123. Eggleton, P, Lieu, TS, Zappi, EG, Sastry, K, Coburn, J, Zaner, KS, et al. Calreticulin is released from activated neutrophils and binds to C1q and mannan-binding protein. Clin Immunol Immunopathol. (1994) 72:405–9. doi: 10.1006/clin.1994.1160
124. Okuyama, Y, Cho, JH, Nakajima, Y, Homma, K, Sekimizu, K, and Natori, S. Binding between azurocidin and calreticulin: its involvement in the activation of peripheral monocytes. J Biochem. (2004) 135:171–7. doi: 10.1093/jb/mvh020
125. Li, Y, Zeng, X, He, L, and Yuan, H. Dendritic cell activation and maturation induced by recombinant calreticulin fragment 39-272. Int J Clin Exp Med. (2015) 8:7288–96.
126. Zhang, T, Xia, Y, Zhang, L, Bao, W, Hong, C, and Gao, XM. CD1d(hi)CD5(+) B cells differentiate into antibody-secreting cells under the stimulation with calreticulin fragment. Protein Cell. (2013) 4:872–81. doi: 10.1007/s13238-013-3062-5
127. Jiang, Z, Chen, Z, Hu, L, Qiu, L, and Zhu, L. Calreticulin blockade attenuates murine acute lung injury by inducing polarization of M2 subtype macrophages. Front Immunol. (2020) 11:11. doi: 10.3389/fimmu.2020.00011
128. Pandya, UM, Egbuta, C, Abdullah Norman, TM, Chiang, CE, Wiersma, VR, Panchal, RG, et al. The biophysical interaction of the danger-associated molecular pattern (DAMP) Calreticulin with the pattern-associated molecular pattern (PAMP) lipopolysaccharide. Int J Mol Sci. (2019) 20:408. doi: 10.3390/ijms20020408
129. Fadini, GP, Menegazzo, L, Rigato, M, Scattolini, V, Poncina, N, Bruttocao, A, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. (2016) 65:1061–71. doi: 10.2337/db15-0863
130. Wong, SL, Demers, M, Martinod, K, Gallant, M, Wang, Y, Goldfine, AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. (2015) 21:815–9. doi: 10.1038/nm.3887
131. Jorch, SK, and Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
132. Gardai, SJ, McPhillips, KA, Frasch, SC, Janssen, WJ, Starefeldt, A, Murphy-Ullrich, JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cells. (2005) 123:321–34. doi: 10.1016/j.cell.2005.08.032
133. Kielbik, M, Szulc-Kielbik, I, and Klink, M. Calreticulin-multifunctional chaperone in immunogenic cell death: potential significance as a prognostic biomarker in ovarian Cancer patients. Cells. (2021) 10:130. doi: 10.3390/cells10010130
134. Obeid, M, Tesniere, A, Ghiringhelli, F, Fimia, GM, Apetoh, L, Perfettini, JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
135. Tang, D, Kang, R, Berghe, TV, Vandenabeele, P, and Kroemer, G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
136. Obeid, M, Tesniere, A, Panaretakis, T, Tufi, R, Joza, N, van Endert, P, et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev. (2007) 220:22–34. doi: 10.1111/j.1600-065X.2007.00567.x
137. Tesniere, A, Apetoh, L, Ghiringhelli, F, Joza, N, Panaretakis, T, Kepp, O, et al. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. (2008) 20:504–11. doi: 10.1016/j.coi.2008.05.007
138. Chaput, N, De Botton, S, Obeid, M, Apetoh, L, Ghiringhelli, F, Panaretakis, T, et al. Molecular determinants of immunogenic cell death: surface exposure of calreticulin makes the difference. J Mol Med. (2007) 85:1069–76. doi: 10.1007/s00109-007-0214-1
139. Sipione, S, Ewen, C, Shostak, I, Michalak, M, and Bleackley, RC. Impaired cytolytic activity in calreticulin-deficient CTLs. J Immunol. (2005) 174:3212–9. doi: 10.4049/jimmunol.174.6.3212
140. Subramanian, S, Tsai, R, and Discher, DE. The 'metabolon,' CD47, and the 'phagocytic synapse': molecular co-localization and species divergence. Transfus Clin Biol. (2006) 13:31–8. doi: 10.1016/j.tracli.2006.02.009
141. Atkin-Smith, GK. Phagocytic clearance of apoptotic, necrotic, necroptotic and pyroptotic cells. Biochem Soc Trans. (2021) 49:793–804. doi: 10.1042/BST20200696
142. Ogden, CA, deCathelineau, A, Hoffmann, PR, Bratton, D, Ghebrehiwet, B, Fadok, VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. (2001) 194:781–96. doi: 10.1084/jem.194.6.781
143. Honore, C, Hummelshoj, T, Hansen, BE, Madsen, HO, Eggleton, P, and Garred, P. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum. (2007) 56:1598–607. doi: 10.1002/art.22564
144. Gardai, SJ, Xiao, YQ, Dickinson, M, Nick, JA, Voelker, DR, Greene, KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cells. (2003) 115:13–23. doi: 10.1016/S0092-8674(03)00758-X
145. Feng, M, Marjon, KD, Zhu, F, Weissman-Tsukamoto, R, Levett, A, Sullivan, K, et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun. (2018) 9:3194. doi: 10.1038/s41467-018-05211-7
146. Feng, M, Chen, JY, Weissman-Tsukamoto, R, Volkmer, JP, Ho, PY, McKenna, KM, et al. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci U S A. (2015) 112:2145–50. doi: 10.1073/pnas.1424907112
147. Fadok, VA, de Cathelineau, A, Daleke, DL, Henson, PM, and Bratton, DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. (2001) 276:1071–7. doi: 10.1074/jbc.M003649200
148. Kuraishi, T, Manaka, J, Kono, M, Ishii, H, Yamamoto, N, Koizumi, K, et al. Identification of calreticulin as a marker for phagocytosis of apoptotic cells in Drosophila. Exp Cell Res. (2007) 313:500–10. doi: 10.1016/j.yexcr.2006.10.027
149. Pastar, I, Nusbaum, AG, Gil, J, Patel, SB, Chen, J, Valdes, J, et al. Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS One. (2013) 8:e56846. doi: 10.1371/journal.pone.0056846
150. Tomic-Canic, M, Burgess, JL, O'Neill, KE, Strbo, N, and Pastar, I. Skin microbiota and its interplay with wound healing. Am J Clin Dermatol. (2020) 21:36–43. doi: 10.1007/s40257-020-00536-w
151. White, EK, and Grice, EA. The wound microbiome. Cold Spring Harbor Perspectives in Biol. (2023) 15:a041218. doi: 10.1101/cshperspect.a041218
152. Pastar, I, Dinic, M, Verpile, R, Meng, J, Marjanovic, J, Burgess, J, et al. Chronic wound microenvironment mediates selection of biofilm-forming multi drug resistant Staphylococcus epidermidis with capability to impair healing. Res Sq. (2023) [Preprint]. doi: 10.21203/rs.3.rs-2562300/v1
153. Kepp, O, Senovilla, L, Galluzzi, L, Panaretakis, T, Tesniere, A, Schlemmer, F, et al. Viral subversion of immunogenic cell death. Cell Cycle. (2009) 8:860–9. doi: 10.4161/cc.8.6.7939
154. Pastar, I, Sawaya, AP, Marjanovic, J, Burgess, JL, Strbo, N, Rivas, KE, et al. Intracellular Staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest. (2021) 131:e133727. doi: 10.1172/JCI133727
155. Cho, JH, Homma, K, Kanegasaki, S, and Natori, S. Activation of human neutrophils by a synthetic anti-microbial peptide, KLKLLLLLKLK-NH2, via cell surface calreticulin. Eur J Biochem. (1999) 266:878–85. doi: 10.1046/j.1432-1327.1999.00920.x
156. Li, WJ, Long, K, Dong, HL, and Gao, XM. Adjuvanticity of a recombinant calreticulin fragment in assisting anti-beta-glucan IgG responses in T cell-deficient mice. Clin Vaccine Immunol. (2013) 20:582–9. doi: 10.1128/CVI.00689-12
157. Eming, SA, Brachvogel, B, Odorisio, T, and Koch, M. Regulation of angiogenesis: wound healing as a model. Prog Histochem Cytochem. (2007) 42:115–70. doi: 10.1016/j.proghi.2007.06.001
158. Okonkwo, UA, Chen, L, Ma, D, Haywood, VA, Barakat, M, Urao, N, et al. Compromised angiogenesis and vascular integrity in impaired diabetic wound healing. PLoS One. (2020) 15:e0231962. doi: 10.1371/journal.pone.0231962
159. Okonkwo, UA, and DiPietro, LA. Diabetes and Wound Angiogenesis. Int J Mol Sci. (2017) 18:1419. doi: 10.3390/ijms18071419
160. Lauer, G, Sollberg, S, Cole, M, Flamme, I, Sturzebecher, J, Mann, K, et al. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol. (2000) 115:12–8. doi: 10.1046/j.1523-1747.2000.00036.x
161. Ding, H, Hong, C, Wang, Y, Liu, J, Zhang, N, Shen, C, et al. Calreticulin promotes angiogenesis via activating nitric oxide signalling pathway in rheumatoid arthritis. Clin Exp Immunol. (2014) 178:236–44. doi: 10.1111/cei.12411
162. Pike, SE, Yao, L, Jones, KD, Cherney, B, Appella, E, Sakaguchi, K, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. (1998) 188:2349–56. doi: 10.1084/jem.188.12.2349
163. Pike, SE, Yao, L, Setsuda, J, Jones, KD, Cherney, B, Appella, E, et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood. (1999) 94:2461–8. doi: 10.1182/blood.V94.7.2461.419a26_2461_2468
164. Lange-Asschenfeldt, B, Velasco, P, Streit, M, Hawighorst, T, Pike, SE, Tosato, G, et al. The angiogenesis inhibitor vasostatin does not impair wound healing at tumor-inhibiting doses. J Invest Dermatol. (2001) 117:1036–41. doi: 10.1046/j.0022-202x.2001.01519.x
165. Totary-Jain, H, Naveh-Many, T, Riahi, Y, Kaiser, N, Eckel, J, and Sasson, S. Calreticulin destabilizes glucose transporter-1 mRNA in vascular endothelial and smooth muscle cells under high-glucose conditions. Circ Res. (2005) 97:1001–8. doi: 10.1161/01.RES.0000189260.46084.e5
166. Nunez, MT, Osorio, A, Tapia, V, Vergara, A, and Mura, CV. Iron-induced oxidative stress up-regulates calreticulin levels in intestinal epithelial (Caco-2) cells. J Cell Biochem. (2001) 82:660–5. doi: 10.1002/jcb.1194
167. Ramos, RR, Swanson, AJ, and Bass, J. Calreticulin and Hsp90 stabilize the human insulin receptor and promote its mobility in the endoplasmic reticulum. Proc Natl Acad Sci U S A. (2007) 104:10470–5. doi: 10.1073/pnas.0701114104
168. Jalali, S, Aghasi, M, Yeganeh, B, and Mesaeli, N. Calreticulin regulates insulin receptor expression and its downstream PI3 kinase/Akt signalling pathway. Biochim Biophys Acta. (2008) 1783:2344–51. doi: 10.1016/j.bbamcr.2008.08.014
169. Lu, L, Seidel, CP, Iwase, T, Stevens, RK, Gong, YY, Wang, X, et al. Suppression of GLUT1; a new strategy to prevent diabetic complications. J Cell Physiol. (2013) 228:251–7. doi: 10.1002/jcp.24133
170. Wang, AJ, Ren, J, Wang, A, and Hascall, VC. Heparin and calreticulin interact on the surface of early G0/G1 dividing rat mesangial cells to regulate hyperglycemic glucose-induced responses. J Biol Chem. (2023) 299:103074. doi: 10.1016/j.jbc.2023.103074
171. Ali, IH, Khalil, IA, and El-Sherbiny, IM. Single-dose electrospun nanoparticles-in-nanofibers wound dressings with enhanced epithelialization, collagen deposition, and granulation properties. ACS Appl Mater Interfaces. (2016) 8:14453–69. doi: 10.1021/acsami.6b04369
172. Patel, Z, Gharat, SA, Al-Tabakha, MM, Ashames, A, Boddu, SHS, and Momin, MM. Recent advancements in electrospun nanofibers for wound healing: polymers, clinical and regulatory perspective. Crit Rev Ther Drug Carrier Syst. (2022) 39:83–118. doi: 10.1615/CritRevTherDrugCarrierSyst.2022039840
173. Stack, ME, Mishra, S, Parimala Chelvi Ratnamani, M, Wang, H, Gold, LI, and Wang, H. Biomimetic extracellular matrix nanofibers electrospun with Calreticulin promote synergistic activity for tissue regeneration. ACS Appl Mater Interfaces. (2022) 14:51683–96. doi: 10.1021/acsami.2c13887
174. Soni, SS, D'Elia, AM, Alsasa, A, Cho, S, Tylek, T, O'Brien, EM, et al. Sustained release of drug-loaded nanoparticles from injectable hydrogels enables long-term control of macrophage phenotype. Biomater Sci. (2022) 10:6951–67. doi: 10.1039/D2BM01113A
175. Witherel, CE, Sao, K, Brisson, BK, Han, B, Volk, SW, Petrie, RJ, et al. Regulation of extracellular matrix assembly and structure by hybrid M1/M2 macrophages. Biomaterials. (2021) 269:120667. doi: 10.1016/j.biomaterials.2021.120667
176. Barrientos, S, Brem, H, Stojadinovic, O, and Tomic-Canic, M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen. (2014) 22:569–78. doi: 10.1111/wrr.12205
177. Yager, DR, Chen, SM, Ward, SI, Olutoye, OO, Diegelmann, RF, and Kelman, CI. Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen. (1997) 5:23–32. doi: 10.1046/j.1524-475X.1997.50108.x
Keywords: calreticulin (CALR), chronic wounds, keratinocytes, wound healing, topical therapies
Citation: Sawaya AP, Vecin NM, Burgess JL, Ojeh N, DiBartolomeo G, Stone RC, Pastar I and Tomic-Canic M (2023) Calreticulin: a multifunctional protein with potential therapeutic applications for chronic wounds. Front. Med. 10:1207538. doi: 10.3389/fmed.2023.1207538
Edited by:
Piul Rabbani, New York University, United StatesReviewed by:
Suneel Kumar, Rutgers, The State University of New Jersey, United StatesJordan Robin Yaron, Arizona State University, United States
Xiaoxing Kou, Sun Yat-sen University, China
Copyright © 2023 Sawaya, Vecin, Burgess, Ojeh, DiBartolomeo, Stone, Pastar and Tomic-Canic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew P. Sawaya, asawaya@med.miami.edu