Sarcoidosis: Progression to the chronic stage and pathogenic based treatment (narrative review)
 Anna Malkova1
Anna Malkova1  Yulia Zinchenko2
Yulia Zinchenko2  Anna Starshinova3*†
Anna Starshinova3*†  Dmitriy Kudlay4,5
Dmitriy Kudlay4,5  Igor Kudryavtsev6
Igor Kudryavtsev6  Anzhela Glushkova7
Anzhela Glushkova7  Piotr Yablonskiy1,2
Piotr Yablonskiy1,2  Yehuda Shoenfeld1,8,9
Yehuda Shoenfeld1,8,9- 1Laboratory of the Mosaic of Autoimmunity, St. Petersburg State University, Saint Petersburg, Russia
- 2Phthisiopulmonology Department, St. Petersburg Research Institute of Phthisiopulmonology, Saint Petersburg, Russia
- 3Almazov National Medical Research Centre, Saint Petersburg, Russia
- 4Department of Pharmacology, I.M. Sechenov First Moscow State Medical University, Moscow, Russia
- 5Laboratory of Personalized Medicine and Molecular Immunology, NRC Institute of Immunology FMBA of Russia, Moscow, Russia
- 6Department of Immunology, Institution of Experimental Medicine, Saint Petersburg, Russia
- 7V.M. Bekhterev National Research Medical Center for Psychiatry and Neurology, Saint Petersburg, Russia
- 8Sackler Faculty of Medicine, Ariel University, Ariel, Israel
- 9Zabludowicz Center for Autoimmune Diseases, Sheba Medical Center, Tel-Hashomer, Israel
Many factors confirm the autoimmune nature of sarcoidosis and help in determining the strategy of patient management and treatment initiation. However, the causes and the mechanisms of disease progression that result in fibrosis and insufficiency of the affected organ remain unclear. This narrative review aims to analyse the mechanisms and biomarkers of sarcoidosis progression, as well as the pathogenetic basis of sarcoidosis therapy. The following characteristics of progressive chronic sarcoidosis were revealed: the disease develops in patients with a genetic predisposition (SNP in genes GREM1, CARD15, TGF-β3, HLA-DQB1*06:02, HLA-DRB1*07/14/15), which contributes either the decreased ability of antigen elimination or autoimmune inflammation. Various prognostic biomarkers of disease progression (decreased levels of neopterin, elastase, sIL-2R, chitotriosidase, glycoprotein Krebs von den Lungen, Th17 cell count, reduced quantity of TNF-α in peripheral blood or bronchoalveolar lavage fluid) have been described and can potentially be used to determine the group of patients who will benefit from the use of corticosteroids/cytostatic drugs/biologics.
Introduction
According to the current concept, sarcoidosis occurs as a result of an exposure to various exogenous or endogenous antigens in subjects with a genetic predisposition to autoimmune disorders and is associated with the development of non-necrotizing granulomas in different organs (1).
The search for etiologic factors of sarcoidosis has revealed various infectious agents (from bacteria, viruses to fungi) as well as non-organic factors (e.g., silicone, silicates etc.) that could be associated with its development (2, 3). Long-term exposure to these factors is believed to cause a chronic over-stimulation of the immune response and chronic inflammation. Since no specific etiologic factor has been established, the pathogenesis of the disease remains poorly understood; furthermore, it appears impossible to create a disease model, develop a unified approach to therapy, and conduct non-clinical studies for the assessment of treatment efficacy (4).
Based on the pathologic findings, sarcoidosis is currently believed to be a granulomatous disease that can be either acute, subacute or chronic and is most commonly (in >90% of cases) associated with the involvement of the lungs and mediastinal lymph nodes as well as other organs and tissues and the development of non-necrotizing granulomas (1, 5, 6). It is known that a severe progressive course of sarcoidosis can lead to a failure of the affected organ, which requires enhanced therapy or even transplantation of healthy tissues (7). For this reason, it is important to understand the disease pathogenesis to inform patient management.
The aim of this review is to analyse the mechanisms and biomarkers of sarcoidosis progression, as well as the pathogenetic basis of sarcoidosis therapy.
Search strategy and selection criteria
The data for the narrative review were collected from the studies published in international databases MEDLINE, Current Contents, PubMed, Elsevier between 2001 and 2021 using the search words of “sarcoidosis, progression, pathogenesis, genetic markers, laboratory markers, treatment.” Inclusion criteria were original research with observation of patients with progressive sarcoidosis and comparison group (group without progression or the studied group before the progression), review articles and research articles on pathogenesis of sarcoidosis.
Granuloma formation
The key feature of the pathogenesis of sarcoidosis is the formation of granulomas in the lungs, mediastinal lymph nodes, skin and other organs. In patients who are genetically predisposed to this disease, a contact of antigen-presenting cells (macrophages, dendritic cells, epithelial cells) with an unknown foreign antigen results in the dysregulated immune response that is manifested as granulomatous inflammation (8). The main characteristic of sarcoidosis is the formation of non-caseating epithelioid granulomas in various organs, represented by lymphocytes, epithelioid and giant cells (asteroid and conchoidal bodies) (Figure 1). Unlike infectious-mediated granulomas in sarcoidosis, necrotic masses are not formed and ACE hyperproduction occurs (9, 10). The central part of a granuloma is composed of macrophages, modified macrophages, epithelioid cells and giant cells, with CD4+ T-lymphocytes between them (11). The peripheral part of granuloma is predominantly occupied with CD8+ T lymphocytes, fibroblasts, macrophages and fibrocytes. B lymphocytes are very rare in a granuloma usually (12). Central fibrinoid might be found (10).
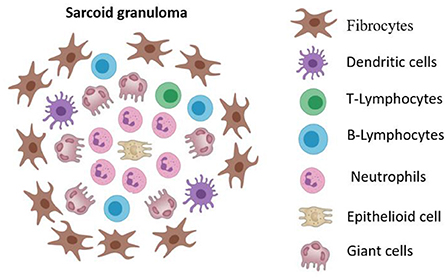
Figure 1. The cellular structure of sarcoid granuloma. Non-caseating epithelioid granuloma represented by lymphocytes, epithelioid and giant cells (asteroid and conchoidal bodies). The central part of a granuloma is composed of macrophages, modified macrophages, epithelioid cells and giant cells, with CD4+ T-lymphocytes between them. The peripheral part of granuloma is predominantly occupied with CD8+ T lymphocytes, fibroblasts, macrophages and fibrocytes.
Macrophages, dendritic cells and epithelial cells are the first cells to react with the antigens due to the presence of Toll-like receptors. Long-term exposure of Toll-like receptors to foreign antigens results in activation and epithelioid differentiation of macrophages, which then start producing pro-inflammatory cytokines (TNF-a, IL-1). Having bound to the antigen, dendritic cells migrate to lymph nodes, where they present the antigen to T-lymphocytes. Further activation of immune cells involves various chemokines such as CCL5 (for Th1) and CCL2 (for Th2). Much attention has been recently paid to the role of Th17 lymphocytes in the pathogenesis of sarcoidosis. These cells are a CD4+ lymphocyte subset that express IL-17A and have pro-inflammatory or anti-inflammatory properties as a result of the local inflammatory environment. Granuloma macrophages in patients with sarcoidosis were shown to express CCR20 thus attracting Th17 cells, while IL-23 expression causes a significant increase in IL-17A concentrations (13, 14). Since there is growing evidence of high plasticity of Th-cells (including Th17), the hypothesis of specific differentiation of T helper cells that are responsible for the initiation of granuloma formation and/or the development of chronic granulomas should be withdrawn.
The predominant phenotype among macrophages in sarcoidosis granulomas is M2, which have anti-inflammatory function and contribute to the disease progression. Moreover, in vitro and in vivo models have also demonstrated the presence of M2 at the initial stages of granuloma formation. There is growing evidence of the role of mTOR signaling pathway in the granuloma formation demonstrated in studies of molecular mechanisms of macrophage dysfunction. The activation of mTORC1 in murine macrophages was shown to result in the disease progression and formation of granulomas just as in humans with progressive sarcoidosis (14, 15). Metabolic adaptation to the environment and inflammatory conditions profoundly affects the regulation of autophagy, leading to the impaired antigen clearance and contributing to the persistence/progression of the granulomas.
More recent data also support the concept that sarcoidosis is a disorder associated not only with Th1 immune response but also with a potential dysfunction of regulatory immune cells and immune system impairment related to the incapability to eliminate the antigen, which is evidenced by the absence of necrotic tissue (16). The functional cell properties such as T-cell depletion may be as relevant cause of the progression of sarcoidosis as the profile of cytokine secretion.
The interaction of macrophages and T-cells and their relationship with the microenvironment (rather than individual mediators) should be the main objectives of disease profiling determining future personalized therapeutic approaches (17).
Genetic predisposing factors
The development of a chronic disease is associated with individual characteristics of the immune system indicating the existence of some genetic predisposing factors. Several candidate genes have been described in literature:
GREM1 gene
The human gene GREM1 encodes gremlin, a highly conserved glycoprotein, a member of the BMP antagonist family (Members of the TGF-β superfamily), including TGF-βs and bone morphogenetic proteins (BMPs). Heron et al. (18) found that sarcoidosis patients carrying the CC genotype of GREM1 had 6.4 times increased risk of fibrosis development.
CARD15 gene
The caspase recruitment domain (CARD) 15 gene (nucleotide oligomerisation domain (NOD)2) encodes an intracellular protein CARD15 of the NOD family. These proteins are involved in innate immunity through recognition of muramyl dipeptide, a component of bacterial cell wall peptidoglycan (19). It was shown that patients carrying the 2104T (702W) polymorphism of CARD15 gene were more likely to have radiographic stage IV disease at 4-yr follow-up. All patients possessing both CARD15 2104T and the C-C chemokine receptor (CCR)5 gene HHC haplotype had stage IV disease at presentation (20).
TGF-β genes
In the study of Kruit et al. the genes encoding isoforms of the transforming growth factor β were analyzed in patients with different stages of sarcoidosis. The frequency of TGF-beta3 4875 A, 17369 C alleles, the TGF-beta2 59941 G allele were significantly higher in sarcoidosis patients with pulmonary fibrosis developing than in patients with acute or /self-limiting and chronic sarcoidosis (p = 0.04; corrected p = 0.2; OR, 2.9) (21).
HLA-genes
The literature search showed that HLA-DQB1*06:02, HLA-DRB1*07/14/15 genotypes were associated with chronic sarcoidosis (22).
The predisposing genes are summarized in the Table 1.
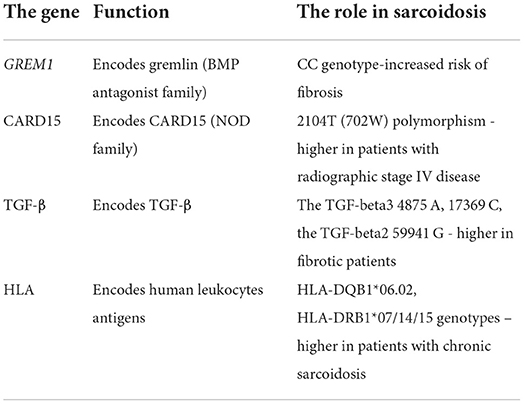
Table 1. Genetic predisposing factors of sarcoidosis progression.
In the research conducted by Lockstone et al. it was revealed that genes involved in leukocyte activation and differentiation, and cytokine production were overexpressed in patients with a progressive, fibrotic (P-F) pulmonary disease compared with the self-limiting group. Among these processes most significant changes were revealed in intracellular signaling (NF-kB and JAK-STAT cascades) and cell life (apoptosis, cell cycle, cell proliferation, and homeostasis) (23).
The pathogenesis of sarcoidosis progression
In some cases when it is impossible to eliminate the pathogen or chronic autoimmune inflammation develops (6, 24–26), collagen overproduction in the area of granulomas is induced, which eventually causes fibrosis and substitution of lung tissue with connective tissue. Fibrotic changes start at the periphery of granulomas and extend centrally (27).
The causes of fibrosis are unknown. However, some characteristics have been described in patients with chronic sarcoidosis.
Alterations of granuloma cell differentiation
Biopsy specimens obtained in patients with a progressive disorder demonstrated Treg with BTNL2 and ANXA11 mutations whose gene products have anti-inflammatory and immune regulatory properties. It is possible that Treg downregulate the effective inflammation and contribute to the development of chronic sarcoidosis (28, 29).
The role of Th2 in the progression of sarcoidosis is being actively studied. It has been suggested that a transition from a Th1 to a Th2 cytokine signature may occur in chronic sarcoidosis, perhaps as a response to persistent inflammation (30). The patients also showed an increase in IL-13 mRNA expression, which is one of the key cytokines of Th2 cells in peripheral blood (31). Animal studies (32) and tests of tissue samples obtained from sarcoidosis patients (33) demonstrated that the overproduction of Th2 cytokines was associated with an activation and differentiation of tissue macrophages with predominant formation of M2 cells. These cells contribute to the development and maintenance of the sites of chronic tissue inflammation, granuloma and fibrosis formation (34). The cytokines produced by Th2 cells - IL-4 and IL-13 - activate the STAT6 pathway in macrophages, with subsequent induction of numerous downstream mediators (34).
M2 macrophage polarization, as reflected by an increased CD163 expression, is associated with an enhanced antigen presentation within a select subset of regulatory T cells, leading to lower concentrations of IFN-γ and higher concentrations of IL-10 upon stimulation. Moreover, IL-13 has anti-inflammatory properties, including suppression of TNF-α release.
Cytokine production
IL-13 can stimulate macrophages to release TGF-β (35). In patients with a new-onset sarcoidosis, differences in lung lavage granulocyte counts and TGF-β release have been detected between those who eventually undergo clinical remission and those who develop a chronic disease course (36).
IL-13 and CCL2 secreted by Th2 were implicated in the pathogenesis of lung fibrosis models, including in idiopathic pulmonary fibrosis (37, 38) (Table 2). CCL2 promotes fibroblast survival, and also has been demonstrated to be elevated in sarcoid lung samples (39, 40).
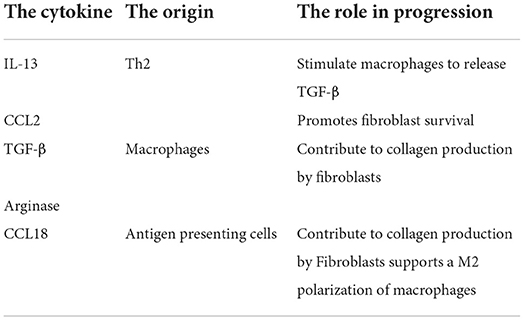
Table 2. The cytokines and its role in sarcoidosis progression.
CCL18 has been directly implicated in fibrosis (41). CCL18 production is associated with fibroblast stimulation and augmented collagen production (42). In sarcoidosis, increased CCL18 was associated with sarcoidosis-associated pulmonary fibrosis but not with other phenotypes of pulmonary disease. CCL18 supports a M2 polarization of macrophages in sarcoidosis-associated pulmonary fibrosis and suggests a pathogenic role of these macrophages in the fibrotic response.
M2 polarized macrophages preferentially express Arg1, which results is an increased arginase. Arginase metabolizes arginine to ornithine, a precursor for the collagen substrate proline (43). Th2 cytokine signature is thought to promote M2 polarization and was shown to correlate with the expression of Arg1 and with the development of fibrosis (44).
Cytokines released during the granuloma formation and macrophage transformation directly activate JAK-STAT pathway, which supports chronic inflammation (45). High production of IFN-γ induces a STAT1 in granuloma macrophages (46) while IL-6 – STAT3 in surrounding lymphocytes, which was confirmed by immunohistochemistry analysis of tissues of patients with cutaneous sarcoidosis (47).
The current understanding of sarcoidosis progression is shown in Figure 2.
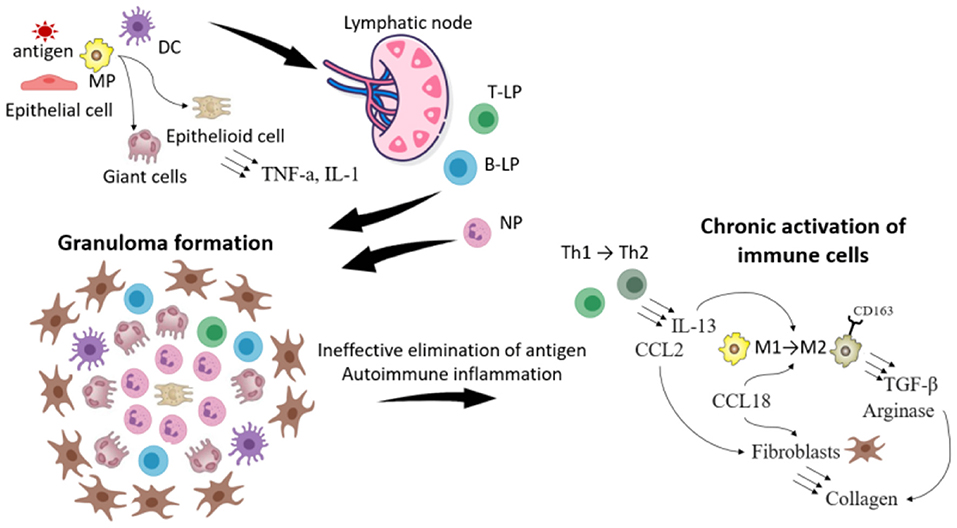
Figure 2. The scheme illustrating the progression of sarcoidosis. The foreign antigen is captured by macrophages (MP) and dendritic cells (DC). DC migrate to lymphoid nodes, where they present the antigen to T- and B-lymphocytes (T-, B-L). Meanwhile in the location of the inflammation MP because of the disability to eliminate the antigen transform to giant and epithelioid cells. T, B-L and neutrophils (NP) migrate in the location of the inflammation and the granuloma is formatted. Because of the impairment of the immune response (weak functions of leukocytes or in opposite the development of autoimmune reactions) immune cells are chronically activated, which leads to the change of their phenotype. Thus T-helper cells (Th1) of type 1 transform to type 2, which secrete IL-13 and CCL2. IL-13, CCL18 contributes to the transformation of MP type 1 to type 2 (expressing CD163), which produce TGF- β and arginase. All these processes stimulate synthesis of collagen by fibroblasts, which leads to the sarcoidosis progression and fibrosis.
Animal models
Animal models of sarcoidosis are difficult to induce, the previous attempts to evaluate the causes of the disease progression and the etiology-based treatment were unsuccessful (48). Animal studies have mainly demonstrated the development of pulmonary granulomas without fibrosis following exposure to various antigens (M. Tuberculosis peptides, P. Acne, carbon nanoparticles). In various studies, mice have developed multiple areas of granulomatosis (lungs, skin, liver, stomach, ganglia), which may indicate the features of the immune response in healthy mice, allowing for more effective elimination of pathogens. The granulomas observed in the experiment were shown to exist only if there was a continuous exposure to the adjuvant. We believe that sarcoidosis patients are chronically exposed by one or several triggers that contribute development and persistence of granulomas (48) or autoimmune inflammation (6).
Some models have shown similar tissue abnormalities as in patients with a chronic sarcoidosis. The authors of a study in a murine model with tuberous sclerosis complex 2 (TSC2) knockout gene described the development of granulomas showing epithelioid-like macrophages, M2 macrophage polarization, and mTORC1 pathway activation (15). TSC2 downregulates mTOR complex 1 (mTORC1) by inhibiting inflammatory immune responses in innate immune cells, such as monocytes, macrophages and dendritic cells. Genetic manipulation of the mTOR pathway in mice alters macrophage polarization and the production of inflammatory and immunomodulatory cytokines (49). Analysis of biopsy material showed an activated mTORC1 pathway significantly enriched (FDR < 0.001) and a significantly decreased mRNA expression of TSC1 in patients with the progressive relative to the self-limiting form of the disease (15).
The predictors of the progression of sarcoidosis
Some pathogenesis-related changes in the pulmonary tissue are accompanied by alterations in peripheral blood and bronchoalveolar lavage fluid in patients with progressive sarcoidosis. Accordingly, the observed correlation between some parameters and the development of granuloma inflammation in the tissues has revealed markers for sarcoidosis progression, namely increased levels of neopterin molecule synthesized from macrophages, sIL-2R marker of T-cell activation, chitotriosidase secreted by active macrophages, neutrophil-derived collagenase and elastase (50), and a low level of TNF-α synthesized from alveolar macrophages and T-cells (51).
Increased number of Th17 in peripheral blood (52) and higher bronchoalveolar (BAL) neutrophil elastase concentrations (36) were more characteristic for patients with progressive sarcoidosis. In addition, a number of studies have shown higher levels of neutrophils in BAL in patients with more advanced stages of the disease (53). Sarcoid granulomas are known to demonstrate a high glucose uptake (54), therefore in these loci increased glycolysis is observed (15). An increase in the concentration of Krebs von den Lungen-6 (KL-6) glycoprotein was observed and was often associated with fibrosis (51).
Some clinical characteristics such as extrapulmonary manifestations (especially cardiac sarcoidosis or neurosarcoidosis), composite physiologic index (CPI) - the index of lung function variables, which correlates with the degree of interstitial disease and CT measurements - the degree of fibrosis, enlargement of the pulmonary artery and the measures of diffuse lung carbon monoxide (DLCO) percentages were shown as markers of severity of pulmonary sarcoidosis (36, 48, 55, 56).
The use of Scadding radiographic stages can also predict the prognosis, the stage I disease was associated with the spontaneous resolution in 60 to 90% patients, while with an increase of stages the percentage of resolution possibility decreased (up to 0% in stage IV fibrotic disease) (57).
The whole list of possible laboratory and clinical markers predicting the progression of sarcoidosis is presented in Table 3.
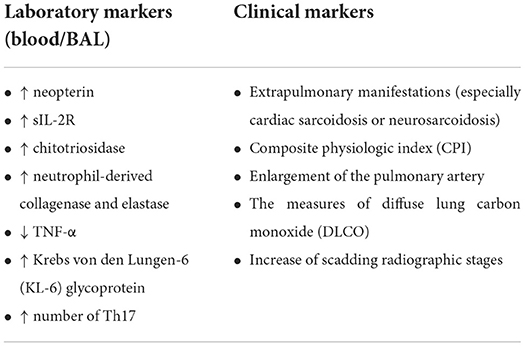
Table 3. The predictor markers of the progression of sarcoidosis.
Pathogenesis-based rationale for sarcoidosis therapy
In some patients, sarcoidosis may be associated with a failure in the affected organ, which requires treatment. However, a substantial proportion of patients are likely to subsequently develop a chronic or progressive sarcoidosis, an estimated percentage of 10% to 20% of patients will have sequelae, and a fatal outcome is observed in 6–7% of cases (58). The aim of the sarcoidosis therapy is the prevention of progression, fibrosis and the organ failure. Predicting the course of sarcoidosis during the initial assessment is a difficult task in clinical practice (59). A more detailed understanding of the pathogenesis of disease progression can help to formulate the criteria for prescribing therapy and improve understanding the mechanism of its effects.
Regulation of the immune response, as well as prevention of fibrosis due to suppression of the overactive macrophages, T-cells and cytokine (TNF-a, IL-1, IFN-γ and IL−6) production can be the main approaches of the sarcoidosis treatment (60, 61).
Drugs directed against autoimmune component
Current first-line therapy for sarcoidosis include corticosteroids (prednisolone, methylprednisolone) (62). The mechanism of action of this drug group is based on their rapid and potent anti-inflammatory activity as a result of suppression of cytokines (including TNF-α, INF-γ) that contribute to the development of granulomas (63, 64). The most suitable dosage and treatment regimen (from 15 mg in cases of lung involvement to 20–40 mg or more in patients with generalized disease and life-threatening conditions) have not been determined (62).
Recent studies have showed evidence for a role for humoral immunity in the sarcoidosis development indicating the possible use of anti-B-cell therapy. Therapy with anti-CD20 monoclonal antibody rituximab was associated with clinical improvement in sarcoidosis patients, which is most likely due to the role of “naive” and/or B- memory cells in the development of sarcoidosis (65–67). The exact role of rituximab as a third- or fourth-line therapy agent remains unclear, and further studies of B-cell immune response are required.
The prescribing factor for these therapy could be the finding of autoantibodies on BAL or blood serum, such as autoantibodies to mutated citrullinated vimentin (anti-MCV) (68, 69), and imbalance in B-cell population with the decrease in memory cells and increase in naïve and regulatory B-cells, which is common for autoimmune diseases (62, 63).
Drugs directed against immune cell differentiation
Second-line therapy includes cytotoxic agents (methotrexate, azathioprine, leflunomide, mycophenolate, as well as their combinations, mainly a combination of methotrexate and leflunomide). The main objective of the use of this group of drugs is to overcome the adverse effects of corticosteroids, taper their dose up to complete discontinuation (steroid-sparing effect). Methotrexate is the most commonly used second-line therapy agent. Methotrexate is a folic acid antagonist, which suppresses the production of TNF-α through adenosine A2A receptors, simultaneously inducing IL-4 and IL-13 that are up-regulators of M2 polarization. MTX can also polarize M0 to M2 via IL4-independent pathways. Thus, the desirable anti-inflammatory effects of MTX are leveled by an increased risk of fibrosis (70, 71).
Leflunomide, a dihydroorotase inhibitor, can be used alone or in combination with methotrexate, including in patients with generalized, progressive disease or with incomplete effect from other therapy. Leflunomide affects the inflammatory process by inhibiting Th17 cells and activating Tregs (72). Azathioprine affects RNA and DNA synthesis by inhibiting purine metabolism, suppresses lymphocyte proliferation, decreases the production of circulating T- and B-cells, and increases apoptosis in circulating lymphocytes. However, the exact mechanism of azathioprine effects on sarcoidosis has not been elucidated so far (73). Another second-line therapy option is mycophenolate mofetil (MMF), which inhibits the synthesis of purine nucleotides in lymphocytes and reduces the production of autoantibodies by B cells. MMF has been proven to be effective in small number of patients with pulmonary sarcoidosis, which manifested in pulmonary function improvement and nervous system effects. It has the lowest toxicity profile in all existing second-line therapy agents (74–76).
The third line includes the use of biological targeted therapy (77). Targeted TNF therapy was the first biological therapy used in patients with sarcoidosis. This is due to the fact that TNF-a is a product of macrophage activation that is a key pro-inflammatory cytokine that contributes to the spread of granulomatous inflammation. The role of TNF in disease progression has been demonstrated above. TNF-targeted therapy includes monoclonal antibodies (infliximab, adalimumab, golimumab), a recombinant protein that connects the TNF receptor to the permanent end of the IgG1 antibody (etanercept) and a pegylated Fab fragment of an anti-TNF-α humanized monoclonal antibody (certolizumab) (78). The drugs demonstrate various degree of effectiveness in different types of sarcoidosis; however, the available data are insufficient to develop a clear algorithm of their use. Infliximab is the most studied third-line drug. The recommended dose of infliximab together with maintenance therapy is 3–5 mg/kg administered every 4–8 weeks following the use of an initial loading dose. At the same time, the results of infliximab studies are somewhat contradictory and range from the evidence of improvement of lung lesions in 58.5% of patients to the lack of clinical significance (79, 80). The drug of choice for patients with infliximab resistance is adalimumab. Therapy with adalimumab was associated with an improvement during damage of skin and eye involvement, better pulmonary function, as well as reduced disease activity (81). At the same time, therapy with etanercept, a TNF receptor antagonist, has not been proven effective in sarcoidosis and therefore is not recommended. Neither golimumab [an anti-TNF-a human monoclonal antibody (IgG1)] nor ustekinumab (an IL-12 and IL-23 inhibitor) have been effective in respect of pulmonary function improvement, but have been shown to be partially effective in patients with sarcoid skin lesions (82).
Based on the hyper-expression of JAK-STAT pathway in granuloma cells the JAK-inhibitors were used in the treatment of patients with different types of sarcoidosis. The cases of efficient use of tofacitinib and ruxolitinib were described in pateints with cutaneous (47, 83), multi-organ (84), pulmonary sarcoidosis (85). The clinical studies of tofacitinib/ruxolitinib in sarcoidosis with/without corticosteroids (NCT03793439 and NCT03910543) are being conducted.
The described therapies can be beneficial for patients with weakened immune response, which can be diagnosed with the BAL or blood biomarkers, such as increased levels of neopterin, sIL-2R, chitotriosidase, neutrophil-derived collagenase and elastase (50), increase of cytokines (IL-13, CCL2, CCL18, TGF-β etc.) and a low level of TNF-α (51).
The described algorithm of the pathogenesis-based therapy of sarcoidosis is presented on Figure 3.
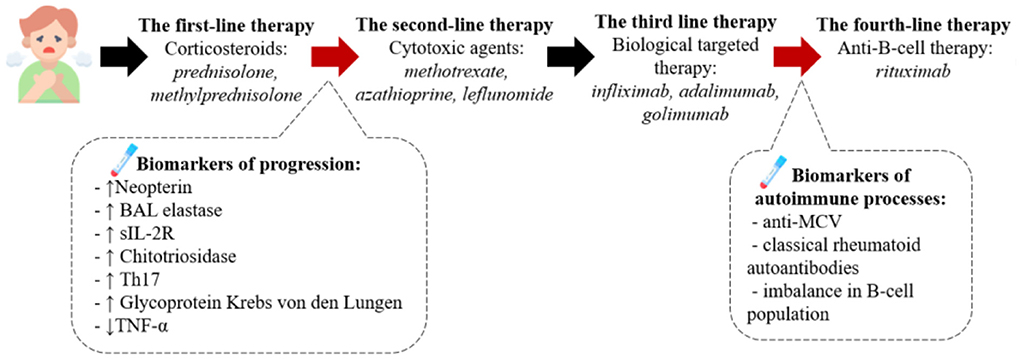
Figure 3. The pathogenesis-based tactic for sarcoidosis treatment. According to the clinical recommendations the first line therapy is corticosteroids, such as prednisolone or methylprednisolone. We assume the presence of biomarkers of the progression in blood or BALF can serve as criteria to start the second-line therapy presented by cytotoxic drugs (methotrexate, azathioprine etc.) and after – biological targeted therapy with anti-TNFα monoclonal antibodies. If the therapy has no beneficial effect we recommend to diagnose the presence of autoimmune processes (anti-MCV antibodies in the blood, the imbalance of B-cell population) and then consult with rheumatologist about the following treatment.
Conclusion
Based on the results of this analysis of published data, the following characteristics of progressive chronic sarcoidosis were revealed: the disease develops in patients with genetic predisposition (SNP in genes GREM1, CARD15, TGF-β3, HLA-DQB1*06.02, HLA-DRB1*07/14/15) as a result of granulomatous inflammation following an antigen presentation in the lungs. It either causes immune system impairment or activates autoimmune disorders. Thus, the antigen elimination becomes impossible, and immune cells change their differentiation (Th1 to Th2, MP1 to MP2) during the chronic inflammation process and start producing cytokines (IL-13, CCL2, CCL18, TGF-β etc.) and enzymes (Arginase) resulting in fibroblast activation and the development of fibrosis.
Various prognostic biomarkers of disease progression (increased levels of Neopterin, elastase, sIL-2R, Chitotriosidase, Glycoprotein Krebs von den Lungen, Th17 cell count, reduced quantity of TNF-α in peripheral blood or bronchoalveolar lavage fluid) have been described and can potentially be used to determine the group of patients who will benefit from the use of corticosteroids/ cytostatic drugs/biologics (see Figure 4).
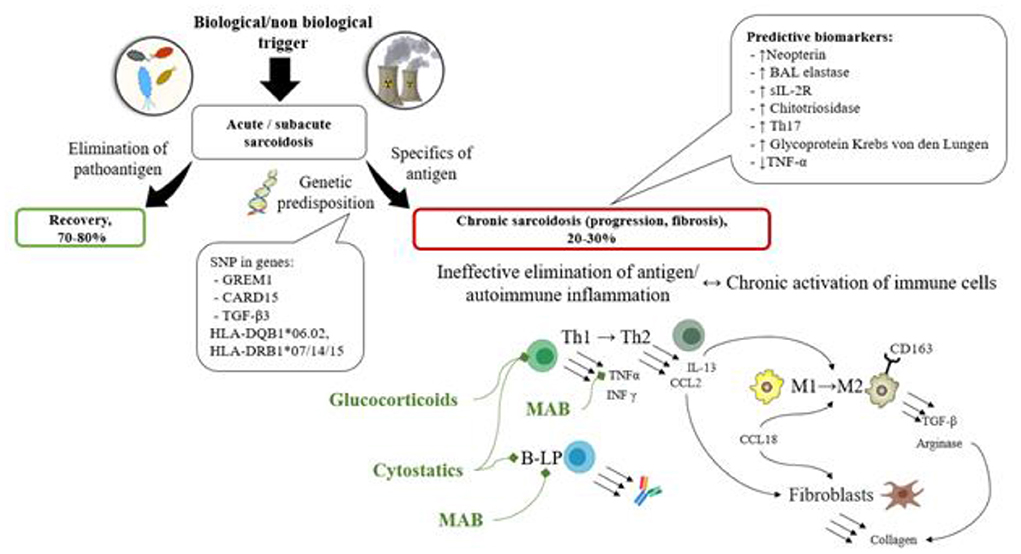
Figure 4. The scheme of the pathogenesis of sarcoidosis along with prognostic biomarkers, and treatment options based on the pathogenic processes. In patients with genetic predisposition (SNP in genes GREM1, CARD15, TGF-β3, HLA-DQB1*06.02, HLA-DRB1*07/14/15) immune system impairment is observed, characterized by immune response inefficacy or autoimmune potential. The antigen elimination becomes impossible, and immune cells change their differentiation (Th1 to Th2, MP1 to MP2) during the chronic inflammation process and start producing cytokines (IL-13, CCL2, CCL18, TGF-β etc.) and enzymes (arginase) resulting in fibroblast activation and the development of fibrosis. These processes can be represented by various prognostic biomarkers of disease progression (decreased levels of Neopterin, elastase, sIL-2R, Chitotriosidase, Glycoprotein Krebs von den Lungen, Th17 cell count, reduced quantity of TNF-α in peripheral blood or bronchoalveolar lavage fluid).
However, currently it is difficult to determine which prognostic markers can be considered indicators for the use of corticosteroids/cytostatic agents/biologics, therefore clinical studies are needed to evaluate the diagnostic significance of determining the described genetic and laboratory markers for selecting a management strategy.
Outstanding questions
At the moment, one of the problems of studying sarcoidosis is the lack of a clear connection of pathogenesis, a specific biomarker and the choice of treatment option. The solution of this practical issue can be aimed at: determining a group of patients with a low probability of self-limitation; pathogenetic justification for choosing a group of the 2nd and 3rd line therapy, based on the specific laboratory markers; determining patients at risk of relapse after drug withdrawal. For progress in these areas, it is necessary to continue research in the field of animal models of sarcoidosis, the connection of laboratory markers with variants of the course of the disease and their changes against the various therapy options, conducting clinical trials. This may make it possible to form clearer criteria for prescribing therapy, which in turn will increase its effectiveness.
Author contributions
AM and IK conducted analysis of the materials and wrote the manuscript. AS conducted analysis of the materials, wrote the manuscript, and was a coordinator of the project. DK, YZ, and AG wrote the manuscript. PY was a coordinator the project and wrote the manuscript. YS was a coordinator of the project. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grant of the Government of the Russian Federation for the state support of scientific research carried out under the supervision of leading scientists, agreement 14.W03.31.0009.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Crouser ED, Maier LA, Baughman RP, Abston E, Bernstein RC, Blankstein R, et al. Diagnosis and detection of sarcoidosis an official american thoracic society clinical practice guideline. Am J Respir Crit Care Med. (2020) 201:E26–51. doi: 10.1164/rccm.202002-0251ST
2. Judson MA. Environmental risk factors for sarcoidosis. Front Immunol. (2020) 11:1340. doi: 10.3389/fimmu.2020.01340
3. Kreider ME, Christie JD, Thompson B, Newman L, Rose C, Barnard J, et al. Relationship of environmental exposures to the clinical phenotype of sarcoidosis. Chest. (2005) 128:207–15. doi: 10.1016/S0012-3692(15)37948-4
4. Sellares J, Strambu I, Crouser ED, Freudenberg MA, Gulati M, Hart S, et al. New advances in the development of sarcoidosis models: a synopsis of a symposium sponsored by the foundation for sarcoidosis research. Sarcoidosis Vasc Diffus Lung Dis. (2018) 35:2–4. doi: 10.36141/svdld.v35i1.7032
5. Hunninghake GW, Costabel U, Ando M, Baughman R, Cordier JF, Du Bois R, et al. Statement on sarcoidosis. Am J Respir Crit Care Med. (1999) 160:736–55. doi: 10.1164/ajrccm.160.2.ats4-99
6. Starshinova AA, Malkova AM, Basantsova NY, Zinchenko YS, Kudryavtsev I V, Ershov GA, et al. Sarcoidosis as an autoimmune disease. Front Immunol. (2020) 10:2933. doi: 10.3389/fimmu.2019.02933
7. Puttgen KB. Diagnosis and management of. Pediatr Clin North Am. (2014) 61:383–402. doi: 10.1016/j.pcl.2013.11.010
8. Berge BT, KleinJan A, Muskens F, Hammad H, Hoogsteden HC, Hendriks RW, et al. Evidence for local dendritic cell activation in pulmonary sarcoidosis. Respir Res. (2012) 13:33. doi: 10.1186/1465-9921-13-33
9. Sakthivel P, Bruder D. Mechanism of granuloma formation in sarcoidosis. Curr Opin Hematol. (2017) 24:59–65. doi: 10.1097/MOH.0000000000000301
10. Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, Van den Blink B. Granuloma formation in pulmonary sarcoidosis. Front Immunol. (2013) 4:437. doi: 10.3389/fimmu.2013.00437
11. Oliver SJ, Kikuchi T, Krueger JG, Kaplan G. Thalidomide induces granuloma differentiation in sarcoid skin lesions associated with disease improvement. Clin Immunol. (2002) 102:225–36. doi: 10.1006/clim.2001.5173
12. Kita S, Tsuda T, Sugisaki K, Miyazaki E, Matsumoto T. Characterization of distribution of T lymphocyte subsets and activated T lymphocytes infiltrating into sarcoid lesions. Intern Med. (1995) 34:847–55. doi: 10.2169/internalmedicine.34.847
13. Crouser E. Role of imbalance between Th17 and regulatory T-cells in sarcoidosis. Curr Opin Pulm Med. (2018) 24:521–6. doi: 10.1097/MCP.0000000000000498
14. Wilson JL, Mayr HK, Weichhart T. Metabolic programming of macrophages: implications in the pathogenesis of granulomatous disease. Front Immunol. (2019) 10:2265. doi: 10.3389/fimmu.2019.02265
15. Linke M, Pham HTT, Katholnig K, Schnöller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol. (2017) 18:293–302. doi: 10.1038/ni.3655
16. Braun NA, Celada LJ, Herazo-Maya JD, Abraham S, Shaginurova G, Sevin CM, et al. Blockade of the programmed death-1 pathway restores sarcoidosis CD4+ T-cell proliferative capacity. Am J Respir Crit Care Med. (2014) 190:560–71. doi: 10.1164/rccm.201401-0188OC
17. Cinetto F, Scarpa R, Dell'Edera A, Jones MG. Immunology of sarcoidosis: old companions, new relationships. Curr Opin Pulm Med. (2020) 26:535–43. doi: 10.1097/MCP.0000000000000711
18. Heron M, Van Moorsel CHM, Grutters JC, Huizinga TWJ, Van Der Helm-Van Mil AHM, Nagtegaal MM, et al. Genetic variation in GREM1 is a risk factor for fibrosis in pulmonary sarcoidosis. Tissue Antigens. (2011) 77:112–7. doi: 10.1111/j.1399-0039.2010.01590.x
19. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Núñez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem. (2001) 276:4812–8. doi: 10.1074/jbc.M008072200
20. Sato H, Williams HRT, Spagnolo P, Abdallah A, Ahmad T, Orchard TR, et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur Respir J. (2010) 35:324–30. doi: 10.1183/09031936.00010209
21. Kruit A, Grutters JC, Ruven HJT, Van Moorsel CHM, Weiskirchen R, Mengsteab S, et al. Transforming growth factor-β gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest. (2006) 129:1584–91. doi: 10.1378/chest.129.6.1584
22. Malkova A, Starshinova A, Zinchenko Y, Basantsova N, Mayevskaya V, Yablonskiy P, et al. The opposite effect of human leukocyte antigen genotypes in sarcoidosis and tuberculosis: a narrative review of the literature. ERJ Open Res. (2020) 6:00155–2020. doi: 10.1183/23120541.00155-2020
23. Lockstone HE, Sanderson S, Kulakova N, Baban D, Leonard A, Kok WL, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med. (2010) 181:1367–75. doi: 10.1164/rccm.200912-1855OC
24. Kozlov VA, Savchenko AA, Kudryavtsev IV, Kozlov IG, Kudlai DA, Prodeus AP. BAG. Clinical immunology. Krasnoyarsk: Polikor (2020). p. 386.
25. Gavrilova N, Starshinova A, Zinchenko Y, Kudlay D, Shapkina V, Malkova A, et al. Small fiber neuropathy in sarcoidosis. Pathophysiol. (2021) 28:544–50. doi: 10.3390/pathophysiology28040035
26. Starshinova AA, Malkova AM, Zinchenko YS, Basantsova NY, Kudlai DA. An autoimmune component in the etiology of sarcoidosis. ?uberculosis Lung Dis. (2020) 98:54–62. doi: 10.21292/2075-1230-2020-98-5-54-62
27. Criado E, Sánchez M, Ramírez J, Arguis P, de Caralt TM, Perea RJ, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high- resolution CT with pathologic correlation. Radiographics. (2010) 30:1567–86. doi: 10.1148/rg.306105512
28. Valentonyte R, Hampe J, Huse K, Rosenstiel P, Albrecht M, Stenzel A, et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet. (2005) 37:357–64. doi: 10.1038/ng1519
29. Hofmann S, Franke A, Fischer A, Jacobs G, Nothnagel M, Gaede KI, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet. (2008) 40:1103–6. doi: 10.1038/ng.198
30. Teirstein AT, Morgenthau AS. “End-stage” pulmonary fibrosis in sarcoidosis. Mt Sinai J Med. (2009) 76:30–6. doi: 10.1002/msj.20090
31. Hauber HP, Gholami D, Meyer A, Pforte A. Increased interleukin-13 expression in patients with sarcoidosis. Thorax. (2003) 58:519–24. doi: 10.1136/thorax.58.6.519
32. Locke LW, Crouser ED, White P, Julian MW, Caceres EG, Papp AC, et al. IL-13–regulated macrophage polarization during granuloma formation in an in vitro human sarcoidosis model. Am J Respir Cell Mol Biol. (2019) 60:84–95. doi: 10.1165/rcmb.2018-0053OC
33. Shamaei M, Mortaz E, Pourabdollah M, Garssen J, Tabarsi P, Velayati A, et al. Evidence for M2 macrophages in granulomas from pulmonary sarcoidosis: a new aspect of macrophage heterogeneity. Hum Immunol. (2018) 79:63–9. doi: 10.1016/j.humimm.2017.10.009
34. Pechkovsky D V, Prasse A, Kollert F, Engel KMY, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. (2010) 137:89–101. doi: 10.1016/j.clim.2010.06.017
35. MacDonald TT. Decoy receptor springs to life and eases fibrosis. Nat Med. (2006) 12:13–4. doi: 10.1038/nm0106-13
36. Drent M, Jacobs JA, de Vries J, Lamers RJS, Liem IH, Wouters EFM. Does the cellular bronchoalveolar lavage fluid profile reflect the severity of sarcoidosis? Eur Respir J. (1999) 13:1338–44. doi: 10.1183/09031936.99.13613459
37. Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor β1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. (1996) 271:17779–84. doi: 10.1074/jbc.271.30.17779
38. Wen FQ, Kohyama T, Liu X, Yun KZ, Wang H, Hui JK, et al. Interleukin-4- and interleukin-13-enhanced transforming growth factor-β2 production in cultured human bronchial epithelial cells is attenuated by interferon-γ. Am J Respir Cell Mol Biol. (2002) 26:484–90. doi: 10.1165/ajrcmb.26.4.4784
39. Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T, et al. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol. (2007) 37:121–8. doi: 10.1165/rcmb.2005-0253OC
40. Palchevskiy V, Hashemi N, Weigt SS, Xue YY, Derhovanessian A, Keane MP, et al. Immune response CC chemokines CCL2 and CCL5 are associated with pulmonary sarcoidosis. Fibrogenes Tissue Repair. (2011) 4:1–12. doi: 10.1186/1755-1536-4-10
41. Prasse A, Pechkovsky D V, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. (2006) 173:781–92. doi: 10.1164/rccm.200509-1518OC
42. Luzina IG, Tsymbalyuk N, Choi J, Hasday JD, Atamas SP. CCL18-stimulated upregulation of collagen production in lung fibroblasts requires Sp1 signaling and basal Smad3 activity. J Cell Physiol. (2006) 206:221–8. doi: 10.1002/jcp.20452
43. Benson RC, Hardy KA, Morris CR. Arginase and arginine dysregulation in asthma. J Allergy. (2011) 2011:1–12. doi: 10.1155/2011/736319
44. Munder M, Eichmann K MM. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. (1998) 160:5347–54.
45. Wang A, Singh K, Ibrahim W, King B, Damsky W. The Promise of JAK Inhibitors for Treatment of Sarcoidosis and Other Inflammatory Disorders with Macrophage Activation: A Review of the Literature. Yale J Biol Med. (2020) 93:187–95.
46. Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene. (2000) 19:2619–27. doi: 10.1038/sj.onc.1203525
47. Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med. (2018) 379:2540–6. doi: 10.1056/NEJMoa1805958
48. Besnard V, Jeny F. Models contribution to the understanding of sarcoidosis pathogenesis: “are there good models of sarcoidosis?” J Clin Med. (2020) 9:2445. doi: 10.3390/jcm9082445
49. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. (2015) 15:599–614. doi: 10.1038/nri3901
50. Peroš-Golubičić T, Ivičević A, Bekić A, Alilović M, Tekavec-Trkanjec J, i Smojver-Ježek S. Lung lavage neutrophils, neutrophil elastase and albumin in the prognosis of pulmonary sarcoidosis. Preuzeto S, editor. Coll Antropol. (2001) 25:349–55. Available online at: https://hrcak.srce.hr/28345
51. Bergantini L, Bianchi F, Cameli P, Mazzei MA, Fui A, Sestini P, et al. Prognostic biomarkers of sarcoidosis: a comparative study of serum chitotriosidase, ACE, lysozyme, and KL-6. Dis Markers. (2019) 2019:8565423. doi: 10.1155/2019/8565423
52. Facco M, Cabrelle A, Teramo A, Olivieri V, Gnoato M, Teolato S, et al. Sarcoidosis is a Th1/Th17 multisystem disorder. Thorax. (2011) 66:144–50. doi: 10.1136/thx.2010.140319
53. Feng H, Yan L, Zhao Y, Li Z, Kang J. Neutrophils in bronchoalveolar lavage fluid indicating the severity and relapse of pulmonary sarcoidosis. Front Med. (2022) 8:3125. doi: 10.3389/fmed.2021.787681
54. Teirstein AS, Machac J, Almeida O, Lu P, Padilla ML, Iannuzzi MC. Results of 188 whole-body fluorodeoxyglucose positron emission tomography scans in 137 patients with sarcoidosis. Chest. (2007) 132:1949–53. doi: 10.1378/chest.07-1178
55. Kraaijvanger R, Janssen Bonás M, Vorselaars ADM, Veltkamp M. Biomarkers in the diagnosis and prognosis of sarcoidosis: current use and future prospects. Front Immunol. (2020) 11:1443. doi: 10.3389/fimmu.2020.01443
56. Walsh SLF, Wells AU, Sverzellati N, Keir GJ, Calandriello L, Antoniou KM, et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. Lancet Respir Med. (2014) 2:123–30. doi: 10.1016/S2213-2600(13)70276-5
57. Silva M, Nunes H, Valeyre D, Sverzellati N. Imaging of sarcoidosis. Clin Rev Allergy Immunol. (2015) 49:45–53. doi: 10.1007/s12016-015-8478-7
58. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. (2014) 383:1155–67. doi: 10.1016/S0140-6736(13)60680-7
59. Kobak S. Catch the rainbow: prognostic factor of sarcoidosis. Lung India. (2020) 37:425–32. doi: 10.4103/lungindia.lungindia_380_19
60. Gerke AK. Treatment of sarcoidosis: a multidisciplinary approach. Front Immunol. (2020) 11:3008. doi: 10.3389/fimmu.2020.545413
61. Le V, Crouser ED. Potential immunotherapies for sarcoidosis. Expert Opin Biol Ther. (2018) 18:399–407. doi: 10.1080/14712598.2018.1427727
62. Schutt AC, Bullington WM, Judson MA. Pharmacotherapy for pulmonary sarcoidosis: a delphi consensus study. Respir Med. (2010) 104:717–23. doi: 10.1016/j.rmed.2009.12.009
63. Gerke AK, Hunninghake G. The immunology of sarcoidosis. Clin Chest Med. (2008) 29:379–90. doi: 10.1016/j.ccm.2008.03.014
64. Grutters JC, van den Bosch JMM. Corticosteroid treatment in sarcoidosis. Eur Respir J. (2006) 28:627–36. doi: 10.1183/09031936.06.00105805
65. Ando M, Goto A, Takeno Y, Yamasue M, Komiya K, Umeki K, et al. Significant elevation of the levels of B-cell activating factor (BAFF) in patients with sarcoidosis. Clin Rheumatol. (2018) 37:2833–8. doi: 10.1007/s10067-018-4183-2
66. Bomprezzi R, Pati S, Chansakul C, Vollmer T. A case of neurosarcoidosis successfully treated with rituximab. Neurology. (2010) 75:568–70. doi: 10.1212/WNL.0b013e3181ec7ff9
67. Belkhou A, Younsi R, El Bouchti I, El Hassani S. Rituximab as a treatment alternative in sarcoidosis. Joint Bone Spine. (2008) 75:511–2. doi: 10.1016/j.jbspin.2008.01.025
68. Malkova A, Starshinova A, Zinchenko Y, Gavrilova N, Kudryavtsev I, Lapin S, et al. New laboratory criteria of the autoimmune inflammation in pulmonary sarcoidosis and tuberculosis. Clin Immunol. (2021) 227:108724. doi: 10.1016/j.clim.2021.108724
69. Kudryavtsev I, Serebriakova M, Starshinova A, Zinchenko Y, Basantsova N, Malkova A, et al. Imbalance in B cell and T follicular helper cell subsets in pulmonary sarcoidosis. Sci Rep. (2020) 10:1–10. doi: 10.1038/s41598-020-57741-0
70. Isshiki T, Yamaguchi T, Yamada Y, Maemura K, Makita K, Takeshima H, et al. Usefulness of low-dose methotrexate monotherapy for treating sarcoidosis. Intern Med. (2013) 52:2727–32. doi: 10.2169/internalmedicine.52.0976
71. Gerards AH, de Lathouder S, de Groot ER, Dijkmans BAC, Aarden LA. Inhibition of cytokine production by methotrexate. studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology. (2003) 42:1189–96. doi: 10.1093/rheumatology/keg323
72. Sahoo DH, Bandyopadhyay D, Xu M, Pearson K, Parambil JG, Lazar CA, et al. Effectiveness and safety of leflunomide for pulmonary and extrapulmonary sarcoidosis. Eur Respir J. (2011) 38:1145–50. doi: 10.1183/09031936.00195010
73. Vorselaars ADM, Wuyts WA, Vorselaars VMM, Zanen P, Deneer VHM, Veltkamp M, et al. Methotrexate vs azathioprine in second-line therapy of sarcoidosis. Chest. (2013) 144:805–12. doi: 10.1378/chest.12-1728
74. Brill AK, Ott SR, Geiser T. Effect and safety of mycophenolate mofetil in chronic pulmonary sarcoidosis: a retrospective study. Respiration. (2013) 86:376–83. doi: 10.1159/000345596
75. Androdias G, Maillet D, Marignier R, Pinède L, Confavreux C, Broussolle C, et al. Mycophenolate mofetil may be effective in CNS sarcoidosis but not in sarcoid myopathy. Neurology. (2011) 76:1168–72. doi: 10.1212/WNL.0b013e318212aafb
76. Hamzeh N, Voelker A, Forssén A, Gottschall EB, Rose C, Mroz P, et al. Efficacy of mycophenolate mofetil in sarcoidosis. Respir Med. (2014) 108:1663–9. doi: 10.1016/j.rmed.2014.09.013
77. Sweiss NJ, Welsch MJ, Curran JJ, Ellman MH. Tumor necrosis factor inhibition as a novel treatment for refractory sarcoidosis. Arthritis Care Res. (2005) 53:788–91. doi: 10.1002/art.21468
78. James WE, Baughman R. Treatment of sarcoidosis: grading the evidence. Expert Expert Rev Clin Pharmacol. (2018) 11:677–87. doi: 10.1080/17512433.2018.1486706
79. Kullberg S, Rivera N V, Abo Al Hayja M, Grunewald J, Eklund A. Changes in lung immune cells related to clinical outcome during treatment with infliximab for sarcoidosis. Clin Exp Immunol. (2020) 201:85–93. doi: 10.1111/cei.13438
80. Hostettler KE, Studler U, Tamm M, Brutsche MH. Long-term treatment with infliximab in patients with sarcoidosis. Respiration. (2012) 83:218–24. doi: 10.1159/000328738
81. Crommelin HA, Van Der Burg LM, Vorselaars ADM, Drent M, Van Moorsel CHM, Rijkers GT, et al. Efficacy of adalimumab in sarcoidosis patients who developed intolerance to infliximab. Respir Med. (2016) 115:72–7. doi: 10.1016/j.rmed.2016.04.011
82. Judson MA, Baughman RP, Costabel U, Drent M, Gibson KF, Raghu G, et al. Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur Respir J. (2014) 44:1296–307. doi: 10.1183/09031936.00000914
83. Damsky W, Thakral D, McGeary MK, Leventhal J, Galan A, King B. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. (2020) 82:612–21. doi: 10.1016/j.jaad.2019.05.098
84. Damsky W, Young BD, Sloan B, Miller EJ, Obando JA, King B. Treatment of multiorgan sarcoidosis with tofacitinib. ACR Open Rheumatol. (2020) 2:106–9. doi: 10.1002/acr2.11112
Keywords: sarcoidosis, pathogenesis, fibrosis, autoimmunity, immune therapy, adjuvant, autoimmune diseases
Citation: Malkova A, Zinchenko Y, Starshinova A, Kudlay D, Kudryavtsev I, Glushkova A, Yablonskiy P and Shoenfeld Y (2022) Sarcoidosis: Progression to the chronic stage and pathogenic based treatment (narrative review). Front. Med. 9:963435. doi: 10.3389/fmed.2022.963435
Received: 07 June 2022; Accepted: 03 August 2022;
Published: 06 September 2022.
Edited by:
Miriana d'Alessandro, University of Siena, ItalyReviewed by:
Prasanta Padhan, Kalinga Institute of Medical Sciences (KIMS), IndiaJavier Loricera, Hospital Universitario Marqués de Valdecilla, Spain
Copyright © 2022 Malkova, Zinchenko, Starshinova, Kudlay, Kudryavtsev, Glushkova, Yablonskiy and Shoenfeld. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Starshinova, starshinova_aa@almazovcentre.ru; starshinova_777@mail.ru
†ORCID: Anna Starshinova orcid.org/0000-0002-9023-6986