The Contribution of COL4A5 Splicing Variants to the Pathogenesis of X-Linked Alport Syndrome
 Tomohiko Yamamura
Tomohiko Yamamura Tomoko Horinouchi
Tomoko Horinouchi Yuya Aoto1
Yuya Aoto1  Rachel Lennon
Rachel Lennon Kandai Nozu
Kandai Nozu- 1Department of Pediatrics, Kobe University Graduate School of Medicine, Kobe, Japan
- 2Wellcome Centre for Cell-Matrix Research, Faculty of Biology Medicine and Health, University of Manchester, Manchester, United Kingdom
- 3Department of Paediatric Nephrology, Royal Manchester Children's Hospital, Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester, United Kingdom
X-linked Alport syndrome (XLAS) is caused by pathogenic variants in COL4A5 and is characterized by progressive kidney disease, hearing loss, and ocular abnormalities. Recent advances in genetic analysis and further understanding of genotype-phenotype correlations in affected male patients raises the importance of detecting splicing variants in COL4A5. Aberrant splicing of COL4A5 is caused not only by canonical splice site variants but also non-canonical splice site variants such as deep intronic changes or even substitutions in exons. Patients with splicing variants account for ~15% of all cases in XLAS. In addition, it has been shown that there is a significant difference in kidney survival depending on the aberrant splicing patterns of transcripts- in particular in-frame or out-of-frame nucleotide changes in transcripts. Therefore, cDNA analysis of patient mRNA is necessary to determine the impact of splice site variants and to confirm a diagnosis of XLAS and to predict the kidney prognosis. However, it is usually difficult to amplify COL4A5 transcripts extracted from peripheral blood leukocytes. For these cases, in vitro minigene assays or RNA sequence extracted from urine derived cells can confirm aberrant splicing patterns. Moreover, controlling aberrant splicing by nucleic acids or small molecular compounds in genetic diseases are attracting attention as a potential therapeutic strategy. Here, we review the frequency of splicing variants in COL4A5, the latest diagnostic strategies, and the prospects for new therapeutic approaches.
Introduction
Alport syndrome (AS) is an inherited disorder with progressive kidney disease, frequently accompanied by sensorineural hearing loss and specific ocular abnormalities (1–4). AS is caused by defects of in the type IV collagen network, a major structural component of basement membranes in the kidney, inner ear, and eye. Six distinct type IV collagen α-chains (α1–α6) have been identified and are encoded by the genes COL4A1–COL4A6. Pathogenic variants in the COL4A5, which encodes the type IV collagen α5 chain, are known to cause X-linked Alport syndrome (XLAS); XLAS is the most common inherited form of AS with ~80% of all AS patients (5). Our group confirmed this distribution in our cohort of all genetically diagnosed Japanese AS families (n = 397) where we found 74% (n = 295) had XLAS (6).
Regarding the genotype of XLAS, all types of variant category have been registered as causative variants to clinical genetic databases similar to other human inherited diseases. In addition, it is already known that strong genotype and kidney phenotype correlation exists in affected male patients with XLAS; patients with missense or small in-frame variants show less severe phenotypes compared to patients with truncating variants (e.g., nonsense, a small insertion/deletion leading to a premature stop codon) (7–10). In addition, we recently focused on the difference in transcripts of splicing variants in COL4A5 based on whether the abnormal transcript has in-frame deletion (the total number of nucleotides is multiple of 3) or out-of-frame deletion (not multiple of 3). According to this analysis, we revealed that male patients with splicing variants leading to in-frame transcripts had less severe phenotypes than those with out of frame transcripts (10, 11).
Recent advances in genetic analysis have enabled comprehensive and efficient screening of multiple genes including COL4A3, COL4A4, and COL4A5 for patients suspected as having Alport syndrome. However, specific variants causing abnormal splicing such as deep intronic variants cannot be detected by (targeted) exome sequencing and a consensus approach for detecting deep intronic variants has not been established (12–15). Moreover, transcript analysis targeting genomic DNA variants, which are suspected to causing aberrant splicing is challenging because of the stability of mRNA, and the extremely low expression level of COL4A5 transcripts in accessible cells such as peripheral blood leukocytes.
In this review, we provide a comprehensive overview of the investigation and functional analysis of splicing variants in the COL4A5 gene; including the frequency of these variants, the latest diagnostic strategies, and the prospects for new therapeutic approaches to regulate splicing patterns.
Splicing Abnormalities and Human Genetic Diseases
RNA splicing is a form of RNA processing in which precursor messenger RNA (pre-mRNA) is transformed into mature messenger RNA (mRNA) in the sequence of protein biosynthesis. In higher eukaryotes, the nucleotide sequence of genomic DNA (gDNA) is divided into the protein coding region (exon) and non-coding region (intron). Pre-mRNA newly transferred from gDNA include both exons and introns. The process of pre-mRNA splicing removes introns from pre-mRNA, and the remaining exons are combined to form mature mRNA (Figure 1).
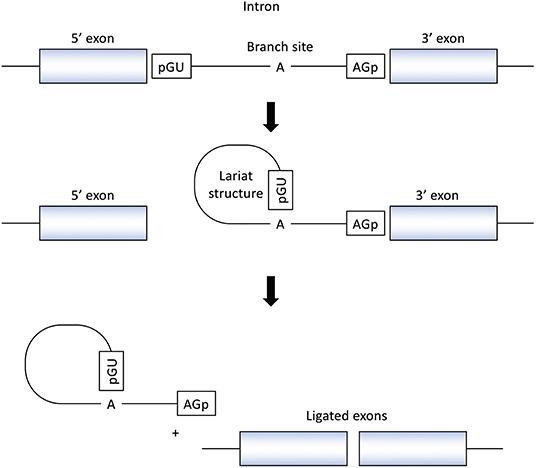
Figure 1. Splicing reactions and important splicing elements. There are several essential splicing motifs located in boundary region between exons and introns. In addition to highly conserved dinucleotides of both 5′ and 3′ side of intron (GU/AG), adenosine (A) at the branch site and polypyrimidine tract (not shown in figure) are located in intron. pre-mRNA splicing process take place in two transesterification steps. In the first step, the 2′-OH group of the adenosine (A) at the branch site performs a nucleophilic attack on a phosphate (p) at the 5′ splice site. This leads to cleavage of the 5′ exon from the intron and the formation of a lariat structure. In the following step, a second transesterification reaction, which involves the phosphate at the 3′ end of the intron, detach the intron from exon and ligates the two exons.
In the process of pre-mRNA splicing, specific sequences located in intron play an important role. Dinucleotides of both the 5' and 3' side of intron are highly conserved (GU/AG) and called the splice donor site and the splice acceptor site, respectively. In addition, other conserved motifs such as the branch site and the polypyrimidine tract, located upstream of the 3′ splice site is also important to determine the location of splicing. Pre-mRNA splicing takes place in two transesterification steps. In the first step, the 2′-OH group of the adenosine (A) at the branch site performs a nucleophilic attack on a phosphate (p) at the 5' splice site. This leads to cleavage of the 5′ exon from the intron and the formation of a lariat structure. In the following step, a second transesterification reaction, which involves the phosphate at the 3′ end of the intron, detaches the intron from exon and ligates the two exons (16) (Figure 1).
In addition to above essential splicing motifs, additional sequence elements known as enhancers or silencers are needed for accurate splicing. These regulatory sequences are located both in exons and introns and called exonic/intronic splicing enhancers/silencers. Furthermore, the splicing process, including the precise recognition of the splice site, is catalyzed by the huge complex of proteins and enzymes termed the spliceosome (17).
These splicing motifs or elements are known as a target of variation in genetic diseases. Variants in this region often cause aberrant splicing and result in pathology. The importance of splicing variants is illustrated by the fact that nearly 15% of human genetic diseases are estimated to be caused by variants located in the 5' or 3' consensus splice sites (18). In addition, comprehensive studies of cDNA analysis for all detected pathogenic variants in patients with ataxia-telangiectasia (OMIM#208900) and neurofibromatosis type I (OMIM#162200) revealed that nearly 50% of variants resulted in aberrant splicing patterns (19, 20). Surprisingly, among the splicing variants in these reports, a minority were detected in the conserved dinucleotide of 5' and 3' side (GU/AG) and most of other variants located in exons or other intronic regions and caused abnormal splicing. The mechanism of how variants located in exon region causes aberrant splicing is mainly explained by the disruption of splicing regulatory elements such as exonic splicing enhancer (ESE) or silencer (ESS) (21). If a nucleotide substitution is in the sequence of these important motifs, the signal to be recognized as an exon is weakened and this can cause exon skipping in process of splicing.
Splicing Variants in COL4A5 Gene
Frequency of Splicing Variants in COL4A5
To date, several retrospective studies reported the proportion of patients with splicing variants in XLAS. Jais et al. (7) investigated genetic and clinical characteristics in 195 families with XLAS and revealed that 29 families (14.9%) had splice site variants. Bekheirnia et al. (8) also reported the proportion of each variant in 175 families with male XLAS patient and it revealed 13.7% of all families possessed splice site variants. Recently, our group investigated 269 Japanese families with XLAS and we found that splicing variants were detected in 18.2% of all families (Table 1) (10). The higher proportion of splicing variants in our study was thought to be the effect of active transcriptional analysis to detect deep intronic variants and exonic splicing variants other than canonical dinucleotides splice site (GU/AG) variants. Interestingly, among 71 male XLAS patients from 49 families with splicing variants in our study, only 35 patients (26 families) had variants in canonical dinucleotides splice site of COL4A5 gene. These findings highlight the importance of splicing variants in COL4A5 and demonstrate similarity to the other inherited diseases mentioned in previous section.

Table 1. Mutational characteristics of reported cohort of X-linked Alport syndrome.
Genotype-Phenotype Correlation in Splicing Variants With XLAS
As described above there are strong genotype-phenotype correlations in males with XLAS; in particular, patients with missense or small in-frame variants (so-called non-truncating variants) have less severe phenotypes compared to patients with truncating variants (e.g., nonsense, small insertion, or deletion leading to out-of-frame sequences) (7–9). In addition, patients with splice site variants have shown intermediate severity, between the phenotypes associated with non-truncating and truncating variants.
Although splicing variants can be classified depending on their transcript pattern (i.e., in-frame or out-of-frame transcript), genotype-phenotype correlation analysis based on transcriptional analysis had not been conducted. Recently, we focused on this transcriptional difference in splicing variants and analyzed the kidney survival of patients with splicing variants. We found a significant difference between patients with in-frame splicing variants and those with out-of-frame splicing variants; the median kidney survival of patients with the in-frame splicing variants (n = 33) was 28 years, whereas it was 23 years for patients with the out-of-frame splicing variants (n = 32; P < 0.05) (10). This result demonstrates the value of transcriptional analysis for splicing variants in XLAS to estimate their kidney prognosis and enable genetic counseling.
Exonic Variants Causing Abnormal Splicing in COL4A5
Some exonic variants, which can be considered as missense or nonsense if transcriptional analysis is not conducted, affect splicing and may cause disease. In particular, single-base substitutions at the last nucleotide position in each exon are reported to likely affect splicing patterns (19, 20). However, no studies have addressed the characteristic of exonic variants in the COL4A5 gene which are likely to affect splicing. Therefore, we focused on the variants affecting the last nucleotide of exons in COL4A5 and conducted a comprehensive in vitro transcript analysis (22). We found 14 reported variants located in last nucleotide of any COL4A5 exon from the Human Gene Mutation Database (HGMD) and six novel variants from our cohort. All 14 variants in HGMD are reported as missense and most of them are glycine substitution, which is most common type of missense variant in COL4A5 (3). Furthermore, using an in vitro functional splicing analysis, 17 out of the total 20 variants showed aberrant splicing. In this study, we also conducted the genotype-phenotype correlation analysis of the splicing variants caused by substitution of last nucleotide in exons comparing to our previous report of missense variants. We found that the median age of developing end-stage kidney disease (ESKD) in cases with splicing variants was significantly worse than those with missense variants (27 vs. 40 years old, P < 0.01). From this result, we concluded that variants located in last nucleotide position of exons in COL4A5, even if they are glycine missense substitution, should be considered likely splicing variants and examined by transcriptional analysis.
In addition, the importance of synonymous or silent variants in abnormal splicing in COL4A5 should be considered. We analyzed COL4A5 transcripts in three patients clinically diagnosed XLAS with synonymous variants by using the in vitro functional splicing assay and analysis of patient mRNA. This revealed that all three cases showed aberrant splicing patterns (23, 24). Interestingly, among these three cases, one patient had both aberrant and normal transcripts by mRNA analysis, and they exhibited a milder phenotype. This finding suggested that synonymous variants in COL4A5 can affect splicing pattern and might show milder phenotype via producing mixture of both normal transcripts and aberrant splicing.
Intronic Variants Outside the Canonical Splice Site Causing Abnormal Splicing in COL4A5
Although the canonical dinucleotides splice site (GU/AG) is important for correct splicing and variants in this region cause aberrant splicing, intronic variants outside this region may also influence splicing. Indeed, there are several non-canonical intronic variants around exon-intron boundaries that have been reported in genomic databases such as HGMD (14). However, the pathogenicity of those intronic variants were not all proven by transcript analysis. Recently, our group reported the results of transcript analysis for seven non-canonical intronic variants in COL4A5 (6 reported variants and one from our cohort) by using in vitro splicing analysis with or without in vivo RNA sequencing. Consequently, five variants were expected to cause aberrant splicing (by skipping the respective exon) while one variant was found less likely to alter the splicing pattern (15). From the above, we should carefully judge the pathogenicity of non-canonical intronic variants and transcript analysis is recommended to assess their influence on splicing.
It has been known that deep intronic variants in COL4A5 also can cause aberrant splicing. Although this type of variant can be confirmed by only mRNA analysis because vast majority of intronic substitutions are polymorphisms, several pathogenic variants in deep introns of COL4A5 have been reported (14, 25). While variants close to exon-intron boundary frequently cause exon skipping, deep intronic variants in COL4A5 show the pathogenicity by the creation of cryptic exon (26, 27).
Diagnostic Strategy of COL4A5 Splicing Variants
Genetic Analysis for XLAS
Previously, Sanger sequencing was widely used for the genetic diagnosis of Alport syndrome. However, screening of all three Alport genes (COL4A3/COL4A4/COL4A5) by conventional Sanger sequencing is time-consuming because each gene contains ~50 exons with no hotspots. Therefore, targeted exome sequencing with Next Generation Sequencing (NGS) has become the first line screening method for the genetic analysis. However, it should be noted that targeted exome sequencing, which screen exons and exon–intron boundaries, cannot detect all types of variant.
For example, large deletion across exons and copy number variations (CNVs) could not be screened by direct sequence and multiplex ligation-dependent probe amplification (MLPA) is the only way to detect this type of variant. However, a paired analysis approach, comparing NGS data of patients and normal controls, recently enabled us to screen CNVs and we detected COL4A5 CNVs successfully with this method (28). Similarly, deep intronic variants causing aberrant splicing also could not be detected by standard sequencing for exons and exon-intron boundaries. Although whole genome sequencing (WGS) approach can screen deep intronic variants, it is difficult to detect pathogenic variants located in introns among the vast majority of non-pathogenic polymorphisms. Therefore, it is essential to carry out transcriptional analysis such as RNA sequencing to detect deep intronic variants.
Transcriptional analysis is important to assess the pathogenicity of intronic variants located outside canonical dinucleotides splice site (GU/AG). Variants located in canonical dinucleotide splice site can be diagnosed as pathogenic because of its critical effect on splicing (29), other intronic variants need to be assessed by transcriptional analysis to determine whether they cause aberrant splicing or not. In addition, it should be noted that there is a rare exception (<1%) of the type of canonical dinucleotide splice site (i.e., GC/AG etc.) and variants in this site do not always lead to exon skipping but sometimes partial deletion of exons or exonization of introns, which is important information to estimate renal prognosis for the evaluation of in-frame or out-of-frame deletions at the transcript levels (11, 14).
To detect pathogenic variants including splicing variants effectively, we conduct the genetic analysis for patients suspected as having XLAS in a stepwise manner, as shown in Figure 2. Briefly, targeted exome sequencing including all three Alport genes is performed first and then screening for copy number variations (CNVs) in the COL4A3/ COL4A4/COL4A5 genes is performed using paired analysis for patients in whom no pathogenic variants are detected by targeted exome sequencing. When paired analysis detects the possibility of CNVs in cases, MLPA is used to confirm CNVs. In addition, for patients with no obvious pathogenic variants, RNA sequencing using RT-PCR will be performed to detect aberrant splicing by intronic variants.
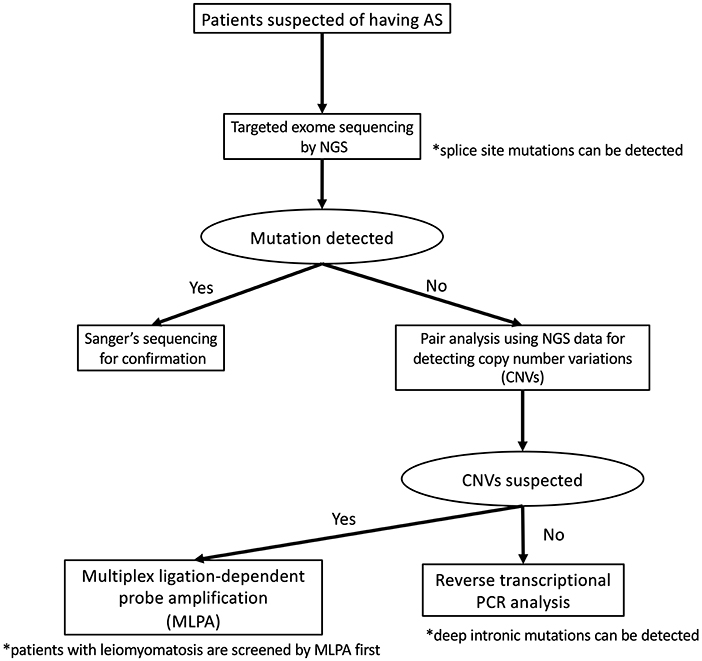
Figure 2. Mutational analysis approach for patients suspected as having Alport syndrome in our laboratory. In our laboratory, patients suspected as having Alport syndrome are first screened targeted exome sequencing by using NGS. If this first screening does not detect any pathogenic variants, screening for copy number variations (CNVs) in the COL4A4/COL4A4/COL4A5 genes is performed using paired analysis. When paired analysis detects the possibility of CNVs in cases, multiplex ligation-dependent probe amplification (MLPA) is used to confirm. In addition, for patients with no obvious pathogenic variants, RNA sequencing using reverse transcription PCR is performed.
Although the frequency of patients with CNVs is lower than those with splicing variants, our screening for CNVs can be conducted using the data of NGS analysis therefore this step is placed earlier than RNA sequencing. In addition, as the screening for CNVs using NGS data may not sufficiently detect CNVs of small size (smaller than 1,000 bp), we add MLPA analysis for the patients who are strongly suspected of having Alport syndrome from their clinical findings even if any pathogenic variants in COL4A3/COL4A4/COL4A5 genes were not detected by NGS and RNA sequencing.
Source of Transcriptional Analysis (Blood, Kidney, Hair Root, and Urine Derived Cell)
Peripheral leukocytes or hair roots have been traditionally used as a common source of mRNA for COL4A5 transcriptional analysis because of its accessibility. However, the expression level of COL4A5 mRNA in these samples is low and the nested PCR technique is required to amplify the targeted regions (14, 30). Although mRNA from patient kidney biopsies has abundant COL4A5 expression, the cDNA analysis for COL4A5 by using mRNA from kidney is not a common procedure since we now conduct a genetic analysis first approach for the diagnosis of Alport syndrome without performing a kidney biopsy. In contrast, patient urine samples have good accessibility and mRNA directly extracted from urine sediments is also abundant in COL4A5 expression and can be used for RT-PCR (10, 11, 14, 23).
Urine-derived cells have been shown to express podocyte markers, and podocytes are the main source of type IV collagen α3α4α5 in the glomerular basement membrane (31, 32). Sergio et al. reported that urine derived “podocyte-lineage” cells from a patient with Alport syndrome expressed all three Alport gene mRNA and could be used for RT PCR analysis (32). Our group also use this cultured urine derived cells as a main source of mRNA for RNA sequencing of Alport genes. Comparing to direct extraction of mRNA from urine sediment, mRNA from urine derived cultured cells is easy to handle because of its larger and stable amount of RNA.
In vitro Splicing Assay (Minigene Analysis)
In addition to transcript analysis using mRNA from patient derived samples, an in vitro functional splicing assay (minigene analysis) using an expression vector is also useful and can be applied for possible splicing variants in COL4A5. We routinely examine novel intronic variants or variants suspected of causing aberrant splicing by using a minigene assay, which is constructed to encode two cassette exons (A and B), an intervening sequence containing a multiple cloning site and a promoter region (Figure 3).
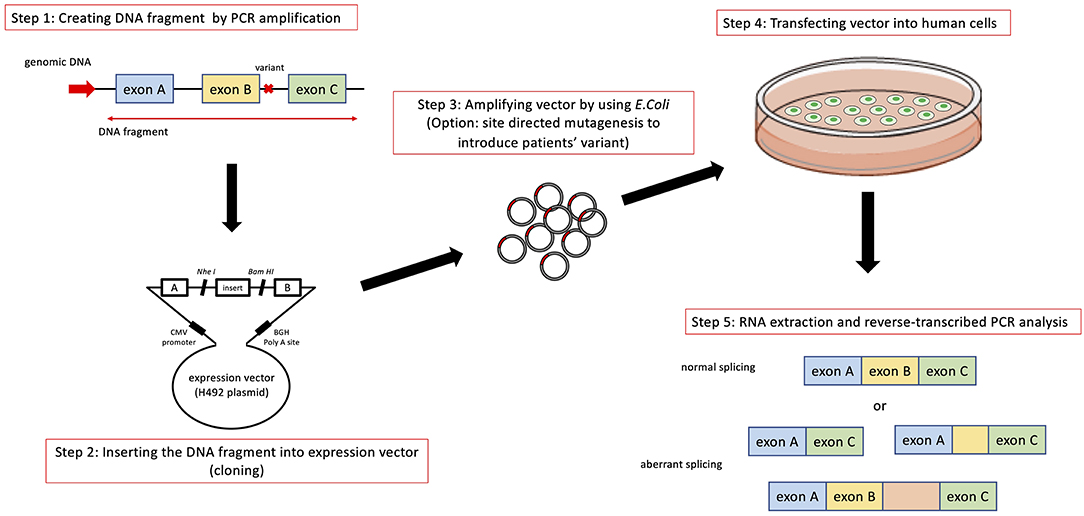
Figure 3. Schematic diagram of minigene analysis to test possible splicing variants. Step 1: Cleaving DNA fragment including the position of mutation by PCR from wild type and patient's gDNA. In addition to the exon close to the mutation being analyzed, we routinely include prier and following exons in DNA fragment. Step 2: Hybrid minigene constructs are created by inserting a test sequence fragment consisting of target exons and its flanking introns into the multicloning site within an intervening intron between two exons (exon A and B) of the minigene construct. Step 3: The hybrid minigene (cloning reaction) is amplified using E. coli. If patient's gDNA is not available, we introduce test mutation by using mutagenesis PCR. Step 4 and 5: These hybrid minigenes (both wild type and mutant) are transfected into human derived cells and culture them to express minigene derived mRNA. Step 5: Total RNA is reverse-transcribed into cDNA and the PCR is performed using specific. PCR products are analyzed by means of electrophoresis and direct sequencing.
The analytical method of this assay is shown in Figure 3. Briefly, hybrid minigene constructs are created by inserting a test sequence fragment consisting of target gDNA region (exons) and the flanking introns into the multiple cloning site within an intervening intron between two exons (exon A and B) of the minigene construct. If a patient sample (or gDNA) is not available, the variant is introduced by site-directed mutagenesis using PCR. Then, hybrid minigenes are transfected into cultured human cells and total RNA was reverse- transcribed into cDNA and the PCR will be performed. Finally, PCR products are analyzed by means of electrophoresis and direct sequencing (Figure 3).
As described earlier, it is often hard to obtain samples for transcript extraction with high expression of target genes for inherited kidney diseases. However, the minigene assay has high flexibility because it does not require any mRNA or even gDNA samples from patients. Variants can be introduced to the minigene by using site directed mutagenesis allowing transcript analysis even if patient sample is not available (33, 34). We have analyzed several novel intronic variants in various inherited kidney diseases using this assay (35–40). As for intronic variants of COL4A5, several studies report using the minigene assay including our studies (11, 15, 41). In addition, most recently, our group applied this in vitro splicing assay for variants located in exons of COL4A5 to clarify the characteristic of exonic splicing variants (22, 24).
Therapeutic Approaches for Targeting Splicing Variants in COL4A5
As described elsewhere (42, 43), there are currently no curative therapies for Alport syndrome including XLAS. Current standard of care is with nephroprotective drugs such as renin-angiotensin-aldosterone system (RAS) blockade. Additional agents including bardoxolone methyl, the mi-RNA 21 inhibitor, endothelin receptor blocker, and sodium-glucose transport protein 2 (SGLT2) inhibitors are being trialed in Alport syndrome. Therapy is important and there is a difference in the treatment effect of RAS blockade for male XLAS patients between genotypes, truncating and non-truncating variants (10). However, non-specific nephroprotective therapy is currently not enough to prevent ESKD in their early age for the patients with truncating variants and the development of disease targeted new therapy is required.
As an inherited disease, the causative gene COL4A5 can be a target of future therapy in addition to non-specific kidney protective therapies. Although one potential gene therapy is direct editing of variants by CRISPR-Cas based system or supplying complete COL4A5 cDNA by using AAV vector, these approaches have not been successfully applied to XLAS so far. In addition, the process of splicing also attracts considerable attention as its potential therapeutic target in various inherited diseases in recent years. Specifically, antisense oligonucleotides (ASOs) therapy has been approved for several inherited diseases such as spinal muscular atrophy (SMA) and Duchene muscular dystrophy (DMD) and its application for Alport syndrome is also expected (44–49).
Our laboratory has already developed the ASO-mediated exon skipping therapy for the male XLAS model mice (with specific Col4a5 nonsense variant) and reported its effects in 2020 (50). As described earlier, male XLAS patients have strong genotype-phenotype correlation that patients with truncating variants show severer kidney phenotype than those with non-truncating variants including in-frame deletion variants even at the transcript level. We aimed to amend the truncating transcript caused by the nonsense variant in COL4A5 to an in-frame transcript by using ASO. Our ASO was designed to combine to the splicing regulatory motif on COL4A5 pre-mRNA and to introduce exon skipping. As a result, we successfully proved its treatment effectiveness in XLAS model mice treated with ASO by showing a significant improvement compared to vehicle treated mice both clinically and pathologically (50).
Exon skipping therapy using ASO is potentially applied for some of splicing variants in COL4A5. About half of these variants result in truncating transcript due to skipping of exon where the nucleotide number is not a multiple of three (out-of-frame deletion). For these variants, additional exon(s) skipping introduced by ASO may be applied as treatment. If the total nucleotide number of skipped exons is a multiple of three, final transcript is in-frame and may lead milder phenotypes. This multiple exon skipping approach has been already considered as a promising treatment for DMD (51–53). In addition, splicing variants which lead to cryptic exon activation are a potential target of this exon skipping approach. A cryptic exon skipped transcript is normal and would result in a much milder phenotype.
In addition to ASO, cell-permeable RNA-targeting small molecules are also attract attention as novel candidates of splicing modifying treatment. A part of the small molecules binds RNA and has an influence on the splicing process. Most recently, risdiplam was approved by FDA as the first small molecule splicing modifier for SMA (54). It was shown that risdiplam analogs directly bind to pre-mRNA of SMN2 to introduce the inclusion of exon 7, which is usually skipped, thereby restoring the production of the SMN2 protein (55). This exon inclusion approach by a small molecule has also been tried for familial dysautonomia caused by intronic variants (IVS20 + 6T>C) in IKBKAP resulting in exon 20 skipping. Two different small molecules have also been reported to increase the normal splicing with inclusion of exon 20 in patients or patient derived cells (56, 57). Small molecules have advantages in terms of the administration route-ASOs are typically administered via intravenous, percutaneous or intrathecal routes, whereas small molecules can be administered orally.
Major problems of developing splicing modifying ASOs or small molecules for XLAS are the specificity of mechanism of action of these drugs and rarity of the treatment-amenable patient population. Each ASO or small molecule can bind a specific region of RNA and shows the action of splicing regulator and one drug may applicable for a few variants. As shown in previously, XLAS has no hotspot region and drugs therefore need to individual exons. However, there has been much interest in the development of individualized treatments for inherited diseases with small numbers of patients, and the FDA has issued a recommendation for the development of individualized ASOs to facilitate progress to individualized therapy.
Conclusion
Splicing variants account for significant proportion of the total variants in XLAS and this proportion might be increasing accompany with advances of gene analysis in future. Thus, it is recommended to conduct RNA sequencing for the patients suspected as having XLAS in whom standard exome sequencing did not detect any variants (6). In addition, even for the cases with obvious splicing variants caused by canonical dinucleotides splice site (GU/AG) variants, transcript analysis by RNA sequencing or in vitro functional splicing assays will clarify the transcript pattern. This is important due to the difference in kidney prognosis of male XLAS patients between transcript types (in-frame vs. out-of-frame). Moreover, it should be noted that even exonic nucleotide substitutions can cause aberrant splicing and we should consider transcript analysis of those who have variants in specific regions (end of each exons) or show an atypical phenotype for their genotype; patients with severe phenotypes accompanied by missense variants or mild phenotype by nonsense mutation. Development of novel disease specific therapy targeting splicing mechanisms for XLAS is expected in the future.
Author Contributions
TH wrote the Section of intronic variants outside the canonical splice site causing abnormal splicing in COL4A5. YA wrote the Section of exonic variants causing abnormal splicing in COL4A5. TY wrote the remaining sections. RL and KN critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grant from the Uehara Memorial Foundation (No. 202040073 to TY).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. (1927) 1:504–6. doi: 10.1136/bmj.1.3454.504
2. Kashtan CE. Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol. (1998) 9:1736–50. doi: 10.1681/ASN.V991736
4. Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. (2019) 23:158–68. doi: 10.1007/s10157-018-1629-4
5. Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine. (1999) 78:338–60. doi: 10.1097/00005792-199909000-00005
6. Yamamura T, Nozu K, Minamikawa S, Horinouchi T, Sakakibara N, Nagano C, et al. Comparison between conventional and comprehensive sequencing approaches for genetic diagnosis of Alport syndrome. Mol Genet Genomic Med. (2019) 7:e883. doi: 10.1002/mgg3.883
7. Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. (2000) 11:649–57. doi: 10.1681/ASN.V114649
8. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. (2010) 21:876–83. doi: 10.1681/ASN.2009070784
9. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. (2002) 17:1218–27. doi: 10.1093/ndt/17.7.1218
10. Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. (2020) 98:1605–14. doi: 10.1016/j.kint.2020.06.038
11. Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Omori T, Nakanishi K, et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. (2018) 29:2244–54. doi: 10.1681/ASN.2018030228
12. Nozu K, Iijima K, Nozu Y, Ikegami E, Imai T, Fu XJ, et al. A deep intronic mutation in the SLC12A3 gene leads to Gitelman syndrome. Pediatr Res. (2009) 66:590–3. doi: 10.1203/PDR.0b013e3181b9b4d3
13. Lo YF, Nozu K, Iijima K, Morishita T, Huang CC, Yang SS, et al. Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome. Clin J Am Soc Nephrol. (2011) 6:630–9. doi: 10.2215/CJN.06730810
14. Nozu K, Vorechovsky I, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al. X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin J Am Soc Nephrol. (2014) 9:1958–64. doi: 10.2215/CJN.04140414
15. Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Nagano C, Sakakibara N, et al. Determination of the pathogenicity of known COL4A5 intronic variants by in vitro splicing assay. Sci Rep. (2019) 9:12696. doi: 10.1038/s41598-019-48990-9
16. Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. (2004) 5:389–96. doi: 10.1038/nrg1327
17. Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. (2003) 12:5–14. doi: 10.1016/S1097-2765(03)00270-3
18. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. (1992) 90:41–54. doi: 10.1007/BF00210743
19. Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. (1999) 64:1617–31. doi: 10.1086/302418
20. Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, et al. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. (2000) 9:237–47. doi: 10.1093/hmg/9.2.237
21. Pagani F, Buratti E, Stuani C, Baralle FE. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J Biol Chem. (2003) 278:26580–8. doi: 10.1074/jbc.M212813200
22. Aoto Y, Horinouchi T, Yamamura T, Kondo A, Nagai S, Ishiko S, et al. Last nucleotide substitutions of COL4A5 exons cause aberrant splicing. Kidney Int Rep. (2021) 7:108–16. doi: 10.1016/j.ekir.2021.10.012
23. Fu XJ, Nozu K, Eguchi A, Nozu Y, Morisada N, Shono A, et al. X-linked Alport syndrome associated with a synonymous p.Gly292Gly mutation alters the splicing donor site of the type IV collagen alpha chain 5 gene. Clin Exp Nephrol. (2016) 20:699–702. doi: 10.1007/s10157-015-1197-9
24. Horinouchi T, Yamamura T, Minamikawa S, Nagano C, Sakakibara N, Nakanishi K, et al. Pathogenic evaluation of synonymous COL4A5 variants in X-linked Alport syndrome using a minigene assay. Mol Genet Genomic Med. (2020) 8:e1342. doi: 10.1002/mgg3.1342
25. King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Hum Genet. (2002) 111:548–54. doi: 10.1007/s00439-002-0830-3
26. Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol Cell Biol. (2000) 20:6414–25. doi: 10.1128/MCB.20.17.6414-6425.2000
27. Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. (2010) 277:841–55. doi: 10.1111/j.1742-4658.2009.07520.x
28. Nagano C, Nozu K, Morisada N, Yazawa M, Ichikawa D, Numasawa K, et al. Detection of copy number variations by pair analysis using next-generation sequencing data in inherited kidney diseases. Clin Exp Nephrol. (2018) 22:881–8. doi: 10.1007/s10157-018-1534-x
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
30. Savige J, Ariani F, Mari F, Bruttini M, Renieri A, Gross O, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. (2019) 34:1175–89. doi: 10.1007/s00467-018-3985-4
31. Lazzeri E, Ronconi E, Angelotti ML, Peired A, Mazzinghi B, Becherucci F, et al. Human urine-derived renal progenitors for personalized modeling of genetic kidney disorders. J Am Soc Nephrol. (2015) 26:1961–74. doi: 10.1681/ASN.2014010057
32. Daga S, Baldassarri M, Lo Rizzo C, Fallerini C, Imperatore V, Longo I, et al. Urine-derived podocytes-lineage cells: a promising tool for precision medicine in Alport Syndrome. Hum Mutat. (2018) 39:302–14. doi: 10.1002/humu.23364
33. Inoue T, Nagano C, Matsuo M, Yamamura T, Sakakibara N, Horinouchi T, et al. Functional analysis of suspected splicing variants in CLCN5 gene in Dent disease 1. Clin Exp Nephrol. (2020) 24:606–12. doi: 10.1007/s10157-020-01876-x
34. Takafuji S, Mori T, Nishimura N, Yamamoto N, Uemura S, Nozu K, et al. Usefulness of functional splicing analysis to confirm precise disease pathogenesis in Diamond-Blackfan anemia caused by intronic variants in RPS19. Pediatr Hematol Oncol. (2021) 2021:1887984. doi: 10.1080/08880018.2021.1887984
35. Nozu K, Iijima K, Kawai K, Nozu Y, Nishida A, Takeshima Y, et al. In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Hum Genet. (2009) 126:533–8. doi: 10.1007/s00439-009-0697-7
36. Nakanishi K, Nozu K, Hiramoto R, Minamikawa S, Yamamura T, Fujimura J, et al. A comparison of splicing assays to detect an intronic variant of the OCRL gene in Lowe syndrome. Eur J Med Genet. (2017) 60:631–4. doi: 10.1016/j.ejmg.2017.08.001
37. Yamamura T, Nozu K, Miyoshi Y, Nakanishi K, Fujimura J, Horinouchi T, et al. An in vitro splicing assay reveals the pathogenicity of a novel intronic variant in ATP6V0A4 for autosomal recessive distal renal tubular acidosis. BMC Nephrol. (2017) 18:353. doi: 10.1186/s12882-017-0774-4
38. Yamamura T, Nozu K, Ueda H, Fujimaru R, Hisatomi R, Yoshida Y, et al. Functional splicing analysis in an infantile case of atypical hemolytic uremic syndrome caused by digenic mutations in C3 and MCP genes. J Hum Genet. (2018) 63:755–9. doi: 10.1038/s10038-018-0436-9
39. Rossanti R, Shono A, Miura K, Hattori M, Yamamura T, Nakanishi K, et al. Molecular assay for an intronic variant in NUP93 that causes steroid resistant nephrotic syndrome. J Hum Genet. (2019) 64:673–9. doi: 10.1038/s10038-019-0606-4
40. Tsuji Y, Nozu K, Sofue T, Hara S, Nakanishi K, Yamamura T, et al. Detection of a splice site variant in a patient with glomerulopathy and fibronectin deposits. Nephron. (2018) 138:166–71. doi: 10.1159/000484209
41. Malone AF, Funk SD, Alhamad T, Miner JH. Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome. Pediatr Nephrol. (2017) 32:997–1003. doi: 10.1007/s00467-016-3565-4
42. Nozu K, Takaoka Y, Kai H, Takasato M, Yabuuchi K, Yamamura T, et al. Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Res Clin Pract. (2020) 39:402–13. doi: 10.23876/j.krcp.20.111
43. Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. (2021) 77:272–9. doi: 10.1053/j.ajkd.2020.03.026
44. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) 377:1723–32. doi: 10.1056/NEJMoa1702752
45. Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. (2018) 378:625–35. doi: 10.1056/NEJMoa1710504
46. Darras BT, Farrar MA, Mercuri E, Finkel RS, Foster R, Hughes SG, et al. An integrated safety analysis of infants and children with symptomatic spinal muscular atrophy (SMA) treated with nusinersen in seven clinical trials. CNS Drugs. (2019) 33:919–32. doi: 10.1007/s40263-019-00656-w
47. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. (2011) 378:595–605. doi: 10.1016/S0140-6736(11)60756-3
48. Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, Smith EC, et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. (2020) 77:982–91. doi: 10.1001/jamaneurol.2020.1264
49. Frank DE, Schnell FJ, Akana C, El-Husayni SH, Desjardins CA, Morgan J, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. (2020) 94:e2270–82. doi: 10.1212/WNL.0000000000009233
50. Yamamura T, Horinouchi T, Adachi T, Terakawa M, Takaoka Y, Omachi K, et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun. (2020) 11:2777. doi: 10.1038/s41467-020-16605-x
51. van Vliet L, de Winter CL, van Deutekom JC, van Ommen GJ, Aartsma-Rus A. Assessment of the feasibility of exon 45-55 multiexon skipping for Duchenne muscular dystrophy. BMC Med Genet. (2008) 9:105. doi: 10.1186/1471-2350-9-105
52. Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. (2016) 18:533–40. doi: 10.1016/j.stem.2016.01.021
53. Lee J, Echigoya Y, Duddy W, Saito T, Aoki Y, Takeda S, et al. Antisense PMO cocktails effectively skip dystrophin exons 45-55 in myotubes transdifferentiated from DMD patient fibroblasts. PLoS ONE. (2018) 13:e0197084. doi: 10.1371/journal.pone.0197084
54. Jaklevic MC. Oral drug approved for spinal muscular atrophy. JAMA. (2020) 324:1026. doi: 10.1001/jama.2020.16783
55. Wang J, Schultz PG, Johnson KA. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc Natl Acad Sci USA. (2018) 115:E4604–12. doi: 10.1073/pnas.1800260115
56. Axelrod FB, Liebes L, Gold-Von Simson G, Mendoza S, Mull J, Leyne M, et al. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr Res. (2011) 70:480–3. doi: 10.1203/PDR.0b013e31822e1825
Keywords: alport syndrome, COL4A5, splicing, genotype phenotype correlation, minigene
Citation: Yamamura T, Horinouchi T, Aoto Y, Lennon R and Nozu K (2022) The Contribution of COL4A5 Splicing Variants to the Pathogenesis of X-Linked Alport Syndrome. Front. Med. 9:841391. doi: 10.3389/fmed.2022.841391
Received: 22 December 2021; Accepted: 10 January 2022;
Published: 08 February 2022.
Edited by:
Oliver Gross, University Medical Center Göttingen, GermanyReviewed by:
Yanqin Zhang, Peking University First Hospital, ChinaDaw-Yang Hwang, National Institute of Cancer Research, National Health Research Institutes, Taiwan
Copyright © 2022 Yamamura, Horinouchi, Aoto, Lennon and Nozu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomohiko Yamamura, tomohiko@med.kobe-u.ac.jp