Glomerular filtration rate abnormalities in sickle cell disease
 Nowah Afangbedji
Nowah Afangbedji Marina Jerebtsova
Marina Jerebtsova- 1Department of Physiology and Biophysics, Howard University, Washington, DC, United States
- 2Department of Microbiology, Howard University, Washington, DC, United States
Sickle cell disease (SCD) is a group of inherited blood disorders affecting the β-globin gene, resulting in the polymerization of hemoglobin and subsequent sickling of the red blood cell. Renal disease, the most common complication in SCD, begins in childhood with glomerular hyperfiltration and then progresses into albuminuria, a fast decline of glomerular filtration, and renal failure in adults. This mini-review focuses on glomerular filtration abnormalities and the mechanisms of hyperfiltration, explores genetic modifiers and methods of estimating glomerular filtration rates, and examines novel biomarkers of glomerular filtration in SCD.
Introduction
Sickle cell disease (SCD) is caused by a single nucleotide mutation in the β-globin gene resulting in the substitution of valine for glutamic acid leading to the polymerization of hemoglobin and sickling of red blood cells (RBCs) under low oxygen conditions. The pathophysiology of SCD is characterized by chronic hemolysis, vaso-occlusion, and organ damage. SCD may occur as homozygous hemoglobin S (HbSS), compound heterozygous inheritance of HbS with mutant hemoglobin C (HbSC), or mutations that result in decreased or absent β-globin (hemoglobin β+ and β0 thalassemia). HbSS and HbSβ0 genotypes are clinically similar disorders associated with severe anemia and disease complications, whereas HbSC and HbSβ+ thalassemia are relatively less severe. SCD is widespread in the malaria-endemic belt: sub-Saharan Africa; South America, the Caribbean, and Central America; Saudi Arabia; India; and the Mediterranean countries (1). It is also common among individuals whose ancestors came from these regions. Approximately 5% of the world’s population carries HbS mutation (2). The World Health Organization estimates that 300,000 children are born with SCD each year, 75% of whom are in sub-Saharan Africa, and this number could rise to 400,000 by 2050. In the USA, SCD affects approximately 100,000 people, primarily of African descent (3). Large-scale epidemiological studies demonstrated a strong geographical link between the highest HbS allele frequencies and high malaria endemicity on a global scale (1, 4–6).
Renal disease is one of the most common complications of SCD (2, 7). Approximately 30% of SCD patients develop chronic kidney disease (CKD), and 14–18% progress to end-stage kidney disease (8–10). Sickle cell nephropathy (SCN) is a progressive disease that begins in childhood as glomerular hyperfiltration leading to albuminuria, loss of renal function, and renal failure (11–15). Before the introduction of antibiotics prophylaxis in the 1970s, the largest mortality in SCD occurred during the first 5 years of life and was associated with bacterial infections (16). In adult patients, stroke and renal failure are common causes of death (16). Current improvements in patient care allow SCD patients to survive relatively longer, leading to an increased incidence of CKD and renal failure (17). Mortality due to renal failure in adult SCD patients is about 10.5% (10).
Hyperfiltration in children and fast decline of glomerular filtration in adult sickle cell disease patients
Early studies demonstrated that glomerular changes in SCD patients are characterized by high renal blood flow and glomerular hyperfiltration and hypertrophy, starting in infancy and declining to a normal range during the first two decades of life (18–22). Glomerular hyperfiltration is defined as estimated glomerular filtration rate (eGFR) above 130 ml/min/1.73 m2 for women and 140 ml/min/1.73 m2 for men. There is no definition for hyperfiltration based on measured GFR (mGFR). The assessment of mGFR in 176 SCD infants demonstrated significantly higher values than in non-SCD infants (125.2 ± 34.4 in SCD vs. 91.5 ± 17.8 ml/min/1.73 m2 in controls). Schwartz formula for eGFR produced even higher values (184.4 ± 55.5 ml/min/1.73 m2) (22). Both measured and estimated GFR indicates increased prevalence (25–43%) of hyperfiltration in young SCD patients (12, 23–27), with the highest occurrence found in HbSS patients (28, 29).
In young SCD adult patients, hyperfiltration is common, particularly in early adulthood but declines into a normal range. The decline into a normal range is faster in men than women and represents a drop in renal function rather than an improvement. Hyperfiltration assessed by mGFR occurred in 51% of adult SCD patients, and the prevalence of hyperfiltration was higher in men (60%) than in women (42%) (28). We recently reported a high prevalence of hyperfiltration in a cohort of 51 adult SCD patients, with the highest occurrence in HbSS 67% and HbSC 50% patients (30), This result concords with a study of 193 SCD patients where the prevalence of hyperfiltration was 61% (13).
Glomerular enlargement and congestion, mainly in the juxtamedullary glomeruli, are more severe in children (31) than in adults, in whom glomerular scarring and fibrosis are developed (32). Hyperfiltration together with glomerular hypertrophy can induce glomerulosclerosis, leading to the reduction of GFR. GFR decline begins early and progresses more rapidly in individuals with SCD compared with the general population (33, 34).
A retrospective study over 4.01 years median follow-up in 331 patients demonstrated an annual eGFR decline of 2.05 ml/min/1.73 m2 for severe genotypes (HbSS and HbSβ0) and 1.16 ml/min/1.73 m2 for mild genotypes (HbSC and HbSβ+) (14). A higher rate of the annual eGFR decline (2.35 ml/min/1.73 m2) was observed by Xu et al. in 193 out of 288 SCD patients over the course of 5-years follow-up; a rate two-fold higher than in African American adults without SCD (13). The sex-associated renal decline suggests a higher and more common rate in men than women (13, 29). Even more, Asnani et al. found in their cohort an annual rate for mGFR decline in SCD patients to be even higher (3.2 ± 2.83 ml/min) (35).
The proportion of patients with fast eGFR decline was similar between patients with hyperfiltration and normal filtration, and eGFR values at baseline did not correlate with the rate of decline (13). Thus, the decline in eGFR is likely caused by intrinsic factors inducing the loss of renal function and does not reflect the difference in baseline glomerular filtration or improvement of hyperfiltration (13, 29). Because of hyperfiltration and higher GFR in young patients with HbSS and HbSβ0 genotypes, a longer time is needed to reach a GFR level of less than 60 ml/min/1.73 m2 that defines stage 3 CKD, and severe kidney injury may occur at higher GFR levels (14). Therefore, the rate of GFR decline may be a better predictor of progressive renal disease in SCD than the absolute value of eGFR.
Mechanism of hyperfiltration in sickle cell disease
Hyperfiltration and renal injury in SCD result from a cascade of events starting with chronic RBC sickling and hemolysis, leading to increased blood viscosity, microvascular obstruction, anemia, oxidative stress, and inflammation (36). The most common histopathological change in the SCD kidney is the dilation of glomerular and interstitial capillaries filled with sickle RBCs (37). The mechanism leading to hyperfiltration is associated with enhanced renal blood flow (20, 25, 38, 39), reflecting an increased cardiac output due to anemia and indicated by the positive correlation of hyperfiltration with low hemoglobin levels (40, 41). SCD is characterized by a paradoxical coexistence of hypoperfusion in the microvasculature and a hyperperfusion observed systemically. The kidney exemplifies this paradox with enhanced perfusion in the whole kidney and the glomeruli while the vasa recta remain hypoperfused. The hypoxic, acidic, and hyperosmolar environment of the renal medulla promotes the sickling of RBCs and slow blood rheology. In the vasa recta, slow blood rheology aggravates vaso-occlusion and leads to ischemia that can advance to infarction. As a compensatory mechanism, medullary prostaglandins and nitric oxide are secreted, leading to an increased renal cortical flow (42–44) (Figure 1). A screening of 14 HBSS patients using inulin, paraaminohippuric acid, and dextran clearances demonstrated increased renal perfusion and a 65% increase in ultrafiltration coefficient (Kf) in SCD patients compared to non-SCD controls (25). An elevated Kf suggests an increased glomerular filtration area and supports glomerular enlargement in SCD. Administration of prostaglandins synthesis inhibitor indomethacin in SCD patients significantly reduced GFR (45). Indomethacin inhibits cyclo-oxygenase (COX), which is required for the synthesis of prostaglandins. However, non-steroidal anti-inflammatory drugs (NSAIDs) are associated with renal, gastrointestinal, and cardiovascular toxicity (46). In the general population, NSAIDs were associated with CKD and a rapid decline in GFR (47–49). In pediatric SCD cohorts, NSAIDs increased the risk of micro-albuminuria and, in one reported case, irreversible renal failure (50, 51). Thus, the use of prostaglandins inhibitors to reduce GFR is more detrimental than beneficial for SCD patients.
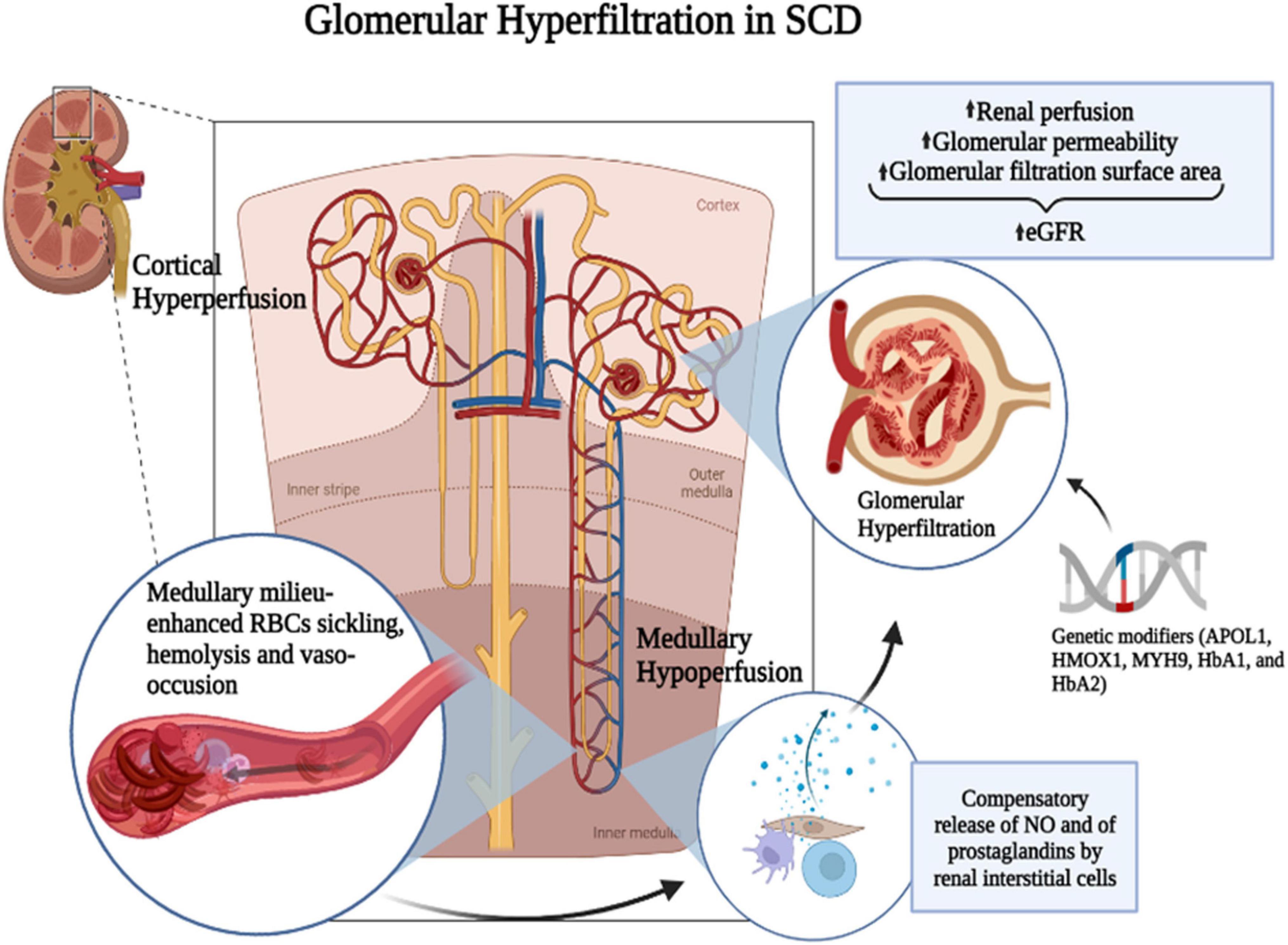
Figure 1. Mechanism of SCD-induced glomerular hyperfiltration. The hypoxic, acidic, and hyperosmolar environment of the renal medulla promotes the polymerization of HbS and the sickling of RBCs. Slow blood rheology in the vasa recta aggravates the vaso-occlusion and leads to ischemia that can advance to infarction. As a compensatory mechanism, medullary prostaglandins and nitric oxide (NO) are secreted leading to an increased renal cortical flow. It was suggested that prostaglandins are released by renal interstitial cells. Genetic modifiers such as APOL1, HMOX1, MYH9, HbA1, and HbA2 and increased renal perfusion coupled with increased glomerular permeability and glomerular filtration surface area may enhance glomerular hyperfiltration in SCD. Figure created with BioRender.com.
Additionally, intravascular hemolysis reduces NO bioavailability through binding to plasma-free hemoglobin and activation of arginase. Several studies demonstrated that markers of chronic hemolysis, such as low total hemoglobin and fetal hemoglobin levels, correlate with hyperfiltration (28, 52, 53). On the contrary, in a transgenic SCD mouse model, nitric oxide synthase 2 (iNOS) induction was shown in the glomeruli and distal nephron. Elevation of iNOS may reflect a feedback mechanism activated by low NO, but it increases NO synthesis, leading to vasodilation that contributes to hyperfiltration (54).
Sickle cell disease increases inflammation and oxidative stress, which induce endothelial injury and glomerulopathy. We and others demonstrated that in the mouse model, SCD nephropathy is associated with glomerular endothelial inflammation induced by activation of the endothelin and RON kinase receptors (37, 55). Treatment with endothelin receptor antagonist or RON kinase inhibitor significantly ameliorated glomerular endothelial injury and hypertrophy. Additionally, statin (atorvastatin) treatment improved urine concentrating ability and glomerular filtration rate, decreased endothelial activation markers, and ameliorated oxidation stress (56). In a pilot study involving 13 SCD patients, treatment with atorvastatin resulted in a small increase in eGFR (57). The protective effects of statins may be due to their anti-inflammatory and antioxidant function.
There is also some evidence that glomerular permeability is increased in SCD, caused by a change in the glomerular porosity: changes in the number of glomerular membrane pores, the size, and the selectivity of the pores. Such findings may suggest a unique, additional, non-hemodynamic-related cause of hyperfiltration in SCD (39).
High plasma glucose and blood pressure (BP) levels are associated with hyperfiltration in diabetic and obese patients (58, 59). In contrast, SCD patients have normal glucose regulation and low BP (25, 26, 38). The incidence of hypertension in patients with HbSS is significantly lower than in non-SCD African Americans in the USA (2–6% in SCD vs. 28% in non-SCD). The slightly increase in BP values considered normal in non-SCD individuals may be a risk factor for cardiovascular complications in SCD patients (60). In the general population, obesity is a risk factor for the progression of renal disease. However, in SCD patients, a higher body mass index is associated with decreased odds of rapid eGFR decline (21, 61).
Genetic modifiers of renal disease in sickle cell disease
Plasma level of fetal hemoglobin (HbF) is a best-characterized modifier of SCD that negatively correlates with the severity of complications. Low HbF levels are associated with more hemolysis (62). Single nucleotide polymorphisms in the promoters of gene encoding HbF, BCL11A, and HMIP-2 are strongly associated with increased HbF levels (63, 64). Hydroxyurea treatment increases HbF levels (65, 66) and is recommended for almost all patients with HbSS and HbSβ0 disease starting in the first year of life. However, high levels of endogenous HbF or hydroxyurea treatment do not significantly improve GFR or reduce hyperfiltration (67). Multiple genetic modifiers, including APOL1, HMOX1, MYH9, HbA1, and HbA2 variants, are implicated in the development and progression of SCN (33, 68–70). The results of these studies are not consistent. Interestingly, data analysis of 326 SCA patients from the Democratic Republic of Congo indicated that APOL1 high-risk genotypes (G1/G1, G2/G2, and G1/G2) were significantly associated with hyperfiltration, but HMOX1 GT-dinucleotide long repeats were associated with lower eGFR (71). In contrast, a cohort of 521 African American SCD patients only demonstrated a weak correlation between MYH9 and APOL1 genotype and eGFR (72).
Estimated glomerular filtration rate calculation methods
Currently, no consensus exists regarding the optimal eGFR equation for SCD patients (73, 74). The evaluation of eGFR with formulas based on the serum creatinine and/or cystatin C concentration is unreliable for SCD patients because of the increased glomerular filtration, lower muscle mass, and increased tubular secretion of low molecular mass organic molecules (25, 36, 75, 76). Plasma creatinine is affected by physiological and analytical factors, and the extreme deviation of eGFR from mGFR is frequent. Non-SCD African Americans also have higher serum creatinine levels and urinary creatinine excretion than the general population (74).
In children with SCD, Schwartz equation based on height and serum creatinine is commonly used for eGFR calculation. A study in 176 HbSS infants found no correlation between mGFR and eGFR estimated by Swartz equation (22). Additionally, Schwartz estimation is highly dependent on the accuracy of the creatinine detection (77). A study in 79 SCD children demonstrated that eGFR calculation based on creatinine detected by mass-spectrometry is better correlated with mGFR (77).
Several equations for eGFR have been used for adult SCD patients, including Cockcroft–Gault (CG), the Modification of Diet in Renal Disease (MDRD), and Chronic Kidney Disease Epidemiology-Collaboration (CKD-EPI) equations with and without adjustment for race (73, 78–80). A study in 59 adult SCD patients demonstrated that CG and MDRD formulas overestimated creatinine clearance compared to mGFR (78). Similar results were obtained in 48 adult HbSS patients, where 66% of the cohort had hyperfiltration assessed by mGFR compared to 72% using MDRD formula (28, 79). The serum creatinine-based CKD-EPI equation performed relatively well but produced a systematic bias of about 45 ml/min/1.73 m2 (79). The use of race for eGFR calculation produces an additional bias because SCD predominantly affects Africans and African Americans and, equations with adjustment for the black race overestimate eGFR (73, 79). New eGFR equations based on creatinine and cystatin C without race have recently been developed (80). The new equations slightly underestimate eGFR in African Americans and overestimate it in the general population. The new creatinine-cystatin C equations without race were more accurate than new creatinine equations, with smaller differences between race groups (21, 80). Also, because the tubular secretion is the predominant mode of creatinine excretion when the GFR is less than 40 ml/min/1.73 m2, serum creatinine levels will produce false values in assessing GFR in older SCD patients with reduced metabolism and tubular dysfunction (36, 76, 81, 82). Thus, the absolute values of eGFR have limited efficacy for routine evaluation of GFR in SCD children and adult patients.
Markers of renal function in sickle cell disease
Glomerular hyperfiltration in SCD plays an essential role in the progression of renal disease; however, to date, no reliable biomarkers of hyperfiltration exist (78). Common clinical markers of renal function, such as serum creatinine and cystatin C are unreliable in estimating GFR in SCD. Reduced production and increased clearance of plasma creatinine may lead to falsely normal plasma creatinine levels and creatine clearance, and delay the detection of kidney disease. Direct measurement of GFR with the injection of substances that do not undergo metabolism, tubular secretion, and absorption, such as inulin, iohexol, iothalamate, and 51Cr-EDTA is recommended; however, the method is difficult and rarely used in the clinical setting. Recently, several potential biomarkers of glomerular filtration were assessed.
Chronic sickling and hemolysis of RBCs induce multiple mechanisms that cause kidney injury. Thus, hemolysis markers such as hemoglobin (Hb), bilirubin, lactate dehydrogenase (LDH) and reticulocyte, and RBCs Hb levels were explored. Studies provided conflicting evidence of the correlation between GFR and hemolysis: Some studies did not find a significant relationship (62, 66, 83), but others showed a strong correlation (13, 28, 83). In a large patient cohort study of 356 patients from the University of Illinois Chicago and 439 patients from the multi-center cohort, Saraf et al. demonstrated an association of hemoglobinuria with progressively lower eGFR values (83). Using a multiple logistic regression analysis in 280 adult SCA patients, Haymann et al. found that hyperfiltration is independently associated with lower HbF and total Hb levels (28). In contrast, in the Kuwaiti SCD patients, Marouf et al. did not find a correlation between eGFR and HbF concentration (78). Higher platelet and reticulocyte counts, higher systolic BP, lower Hb level, and body mass index were associated with a rapid decline of GFR in the study of 288 adult SCD patients (13). And, though BP levels in SCD are lower than in a general population of African Americans, GFR does not correlate with BP (84).
Metabolomics analysis identified two metabolites (asymmetric dimethylarginine – ADMA and quinolinic acid) associated with a rapid decline of GFR (13). ADMA is the endogenous nitric oxide (NO) synthase inhibitor implicated in the pathogenesis of endothelial dysfunction and CKD progression (85). Quinolinic acid was previously associated with incident CKD in the general population (86).
In our study, mass-spectrometry analysis of urine samples collected from HbSS patients identified orosomucoid (ORM) (87) and ceruloplasmin (CP) (88) which positively correlated with hemoglobinuria, and kringle domain-containing protein HGFL, which positively correlated with GFR (89). ORM is a major acute-phase protein and increased ORM expression and serum concentration is associated with tissue injury, inflammation, infection, cancer, and diabetic and systemic lupus erythematosus-associated renal disease (90, 91). CP is a ferroxidase that plays a central role in iron homeostasis, which is highly affected by SCD. Urinary CP was proposed as a biomarker for early diagnosis of membranous nephropathy, focal segmental glomerulosclerosis, lupus nephritis, and IgA nephropathy (92, 93). Therefore, hemoglobinuria, CP, and ORM reflect increased inflammation and iron metabolism in SCD patients and positively correlate with stages of CKD. Plasma and urine ORM inversely correlated with eGFR in HbSS and HbSC patients (30). Despite considerable efforts in searching for SCN-associated biomarkers, there remains a paucity of reliable biomarkers of GFR in SCD.
Conclusion
In summary, SCN is a common cause of morbidity and mortality in SCD patients (36, 94, 95). Despite severe complications of SCD, it remains the most common inherited hemoglobinopathy due to positive environmental selection. The mechanism of renal disease in SCD is unique (39). The higher GFR in patients with albuminuria is consistent with the hypothesis that high glomerular flows cause renal damage (38, 73). But not all hyperfiltrating SCD patients develop albuminuria and renal disease, suggesting that other mechanisms and genetic risk factors are involved in the disease development and progression. More studies are needed to identify novel mechanisms and consistent genetic factors associated with SCN. Common clinical markers of renal function, such as serum creatinine and cystatin C, are unreliable because of increased GFR, low muscular mass, and increased tubular secretion in SCD patients (28, 75). Although an accurate measure of GFR is not necessary for most clinical purposes, the consequences of eGFR overestimation are extremely important in rapid progressors who may benefit from early treatment. Thus, monitoring of eGFR during routine clinical visits may reveal a fast decline of eGFR better than other markers predicting the development of renal disease in SCD patients (96).
Author contributions
NA and MJ wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH Research Grant SC1HL150685 (for MJ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official view of NHLBI, NIMHD, or NIH.
References
1. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. (2010) 1:104. doi: 10.1038/ncomms1104
2. Palit S, Bhuiyan RH, Aklima J, Emran TB, Dash R. A study of the prevalence of thalassemia and its correlation with liver function test in different age and sex group in the Chittagong district of Bangladesh. J Basic Clin Pharm. (2012) 3:352–7. doi: 10.4103/0976-0105.105339
3. Brousseau DC, Panepinto JA, Nimmer M, Hoffmann RG. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. (2010) 85:77–8. doi: 10.1002/ajh.21570
4. Elguero E, Delicat-Loembet LM, Rougeron V, Arnathau C, Roche B, Becquart P, et al. Malaria continues to select for sickle cell trait in Central Africa. Proc Natl Acad Sci U S A. (2015) 112:7051–4. doi: 10.1073/pnas.1505665112
5. Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, et al. Positive natural selection in the human lineage. Science. (2006) 312:1614–20. doi: 10.1126/science.1124309
6. Williams TN. Human red blood cell polymorphisms and malaria. Curr Opin Microbiol. (2006) 9:388–94. doi: 10.1016/j.mib.2006.06.009
7. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68:263–99. doi: 10.3233/CH-189012
8. Airy M, Eknoyan G. The kidney in sickle hemoglobinopathies. Clin Nephrol. (2017) 87:55–68. doi: 10.5414/CN108991
9. Maigne G, Ferlicot S, Galacteros F, Belenfant X, Ulinski T, Niaudet P, et al. Glomerular lesions in patients with sickle cell disease. Medicine. (2010) 89:18–27. doi: 10.1097/MD.0b013e3181ca59b6
10. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
11. Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine. (2005) 84:363–76. doi: 10.1097/01.md.0000189089.45003.52
12. Lebensburger JD, Aban I, Pernell B, Kasztan M, Feig DI, Hilliard LM, et al. Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol. (2019) 94:417–23. doi: 10.1002/ajh.25390
13. Xu JZ, Garrett ME, Soldano KL, Chen ST, Clish CB, Ashley-Koch AE, et al. Clinical and metabolomic risk factors associated with rapid renal function decline in sickle cell disease. Am J Hematol. (2018) 93:1451–60. doi: 10.1002/ajh.25263
14. Derebail VK, Ciccone EJ, Zhou Q, Kilgore RR, Cai J, Ataga KI. Progressive decline in estimated GFR in patients with sickle cell disease: an observational cohort study. Am J Kidney Dis Off J Natl Kidney Foundation. (2019) 74:47–55. doi: 10.1053/j.ajkd.2018.12.027
15. Barros FB, Lima CS, Santos AO, Mazo-Ruiz MF, Lima MC, Etchebehere EC, et al. 51Cr-EDTA measurements of the glomerular filtration rate in patients with sickle cell anaemia and minor renal damage. Nucl Med Commun. (2006) 27:959–62. doi: 10.1097/01.mnm.0000243373.03636.6e
16. Thomas AN, Pattison C, Serjeant GR. Causes of death in sickle-cell disease in Jamaica. Br Med J. (1982) 285:633–5. doi: 10.1136/bmj.285.6342.633
17. Pardo V, Strauss J, Kramer H, Ozawa T, McIntosh RM. Nephropathy associated with sickle cell anemia: an autologous immune complex nephritis. II. Clinicopathologic study of seven patients. Am J Med. (1975) 59:650–9. doi: 10.1016/0002-9343(75)90226-0
18. Etteldorf JN, Tuttle AW, Clayton GW. Renal function studies in pediatrics. 1. Renal hemodynamics in children with sickle cell anemia. AMA Am J Dis Child. (1952) 83:185–91. doi: 10.1001/archpedi.1952.02040060051005
19. Bhathena DB, Sondheimer JH. The glomerulopathy of homozygous sickle hemoglobin (SS) disease: morphology and pathogenesis. J Am Soc Nephrol JASN. (1991) 1:1241–52. doi: 10.1681/ASN.V1111241
20. Etteldorf JN, Smith JD, Tuttle AH, Diggs LW. Renal hemodynamic studies in adults with sickle cell anemia. Am J Med. (1955) 18:243–8. doi: 10.1016/0002-9343(55)90239-4
21. Ataga KI, Zhou Q, Saraf SL, Hankins JS, Ciccone EJ, Loehr LR, et al. Longitudinal study of glomerular hyperfiltration in adults with sickle cell anemia: a multicenter pooled analysis. Blood Adv. (2022) 6:4461–70. doi: 10.1182/bloodadvances.2022007693
22. Ware RE, Rees RC, Sarnaik SA, Iyer RV, Alvarez OA, Casella JF, et al. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. J Pediatrics. (2010) 156:66–70.e1. doi: 10.1016/j.jpeds.2009.06.060
23. Ataga KI, Derebail VK, Archer DR. The glomerulopathy of sickle cell disease. Am J Hematol. (2014) 89:907–14. doi: 10.1002/ajh.23762
24. Herrera J, Avila E, Marin C, Rodriguez-Iturbe B. Impaired creatinine secretion after an intravenous creatinine load is an early characteristic of the nephropathy of sickle cell anaemia. Nephrol Dial Transplant Off Publ Eur Dial Transplant Assoc Eur Renal Assoc. (2002) 17:602–7. doi: 10.1093/ndt/17.4.602
25. Schmitt F, Martinez F, Brillet G, Giatras I, Choukroun G, Girot R, et al. Early glomerular dysfunction in patients with sickle cell anemia. Am J Kidney Dis Off J Natl Kidney Foundation. (1998) 32:208–14. doi: 10.1053/ajkd.1998.v32.pm9708603
26. Aloni MN, Ngiyulu RM, Gini-Ehungu JL, Nsibu CN, Ekila MB, Lepira FB, et al. Renal function in children suffering from sickle cell disease: challenge of early detection in highly resource-scarce settings. PLoS One. (2014) 9:e96561. doi: 10.1371/journal.pone.0096561
27. Belisario AR, de Almeida JA, Mendes FG, da Silva DMM, Planes W, Rezende PV, et al. Prevalence and risk factors for albuminuria and glomerular hyperfiltration in a large cohort of children with sickle cell anemia. Am J Hematol. (2020) 95:E125–8. doi: 10.1002/ajh.25763
28. Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, et al. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol CJASN. (2010) 5:756–61. doi: 10.2215/CJN.08511109
29. Derebail VK, Zhou Q, Ciccone EJ, Cai J, Ataga KI. Longitudinal study of glomerular hyperfiltration and normalization of estimated glomerular filtration in adults with sickle cell disease. Br J Haematol. (2021) 195:123–32. doi: 10.1111/bjh.17723
30. Jerebtsova M, Taye A, Smith N, Afangbedji N, Stokes D, Niu X, et al. Association between plasma and urinary orosomucoid and chronic kidney disease in adults with sickle cell disease. Br J Haematol. (2020) 190:e45–8. doi: 10.1111/bjh.16702
31. Bernstein J, Whitten CF. A histologic appraisal of the kidney in sickle cell anemia. Arch Pathol. (1960) 70:407–18.
32. Walker BR, Alexander F, Birdsall TR, Warren RL. Glomerular lesions in sickle cell nephropathy. JAMA. (1971) 215:437–40. doi: 10.1001/jama.215.3.437
33. Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol. (2022) 18:361–77. doi: 10.1038/s41581-022-00540-9
34. Aygun B, Mortier NA, Smeltzer MP, Hankins JS, Ware RE. Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatric Nephrol. (2011) 26:1285–90. doi: 10.1007/s00467-011-1857-2
35. Asnani M, Serjeant G, Royal-Thomas T, Reid M. Predictors of renal function progression in adults with homozygous sickle cell disease. Br J Haematol. (2016) 173:461–8. doi: 10.1111/bjh.13967
36. Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney Int. (2000) 57:1–8. doi: 10.1046/j.1523-1755.2000.00806.x
37. Khaibullina A, Adjei EA, Afangbedji N, Ivanov A, Kumari N, Almeida LEF, et al. RON kinase inhibition reduces renal endothelial injury in sickle cell disease mice. Haematologica. (2018) 103:787–98. doi: 10.3324/haematol.2017.180992
38. Thompson J, Reid M, Hambleton I, Serjeant GR. Albuminuria and renal function in homozygous sickle cell disease: observations from a cohort study. Arch Internal Med. (2007) 167:701–8. doi: 10.1001/archinte.167.7.701
39. Guasch A, Cua M, You W, Mitch WE. Sickle cell anemia causes a distinct pattern of glomerular dysfunction. Kidney Int. (1997) 51:826–33. doi: 10.1038/ki.1997.116
40. Aygun B, Mortier NA, Smeltzer MP, Shulkin BL, Hankins JS, Ware RE. Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol. (2013) 88:116–9. doi: 10.1002/ajh.23365
41. Haymann JP, Hammoudi N, Livrozet M, Santin A, Mattioni S, Letavernier E, et al. Hemodynamic and biological correlates of glomerular hyperfiltration in sickle cell patients before and under renin-angiotensin system blocker. Sci Rep. (2021) 11:11682. doi: 10.1038/s41598-021-91161-y
42. Nath KA, Katusic ZS, Gladwin MT. The perfusion paradox and vascular instability in sickle cell disease. Microcirculation. (2004) 11:179–93. doi: 10.1080/10739680490278592
43. Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Investigat. (2008) 118:1924–33. doi: 10.1172/JCI33308
44. de Jong PE, Statius van Eps LW. Sickle cell nephropathy: new insights into its pathophysiology. Kidney Int. (1985) 27:711–7. doi: 10.1038/ki.1985.70
45. de Jong PE, de Jong-Van Den Berg TW, Sewrajsingh GS, Schouten H, Donker AJ, Statius van Eps LW. The influence of indomethacin on renal haemodynamics in sickle cell anaemia. Clin Sci. (1980) 59:245–50. doi: 10.1042/cs0590245
46. Brune K, Patrignani P. New insights into the use of currently available nonsteroidal anti-inflammatory drugs. J Pain Res. (2015) 8:105–18. doi: 10.2147/JPR.S75160
47. Gooch K, Culleton BF, Manns BJ, Zhang J, Alfonso H, Tonelli M, et al. NSAID use and progression of chronic kidney disease. Am J Med. (2007) 120:280.e1–7. doi: 10.1016/j.amjmed.2006.02.015
48. Ingrasciotta Y, Sultana J, Giorgianni F, Fontana A, Santangelo A, Tari DU, et al. Association of individual nonsteroidal anti-inflammatory drugs and chronic kidney disease: a population-based case control study. PLoS One. (2015) 10:e0122899. doi: 10.1371/journal.pone.0122899
49. Sandler DP, Burr FR, Weinberg CR. Nonsteroidal anti-inflammatory drugs and the risk for chronic renal disease. Ann Internal Med. (1991) 115:165–72. doi: 10.7326/0003-4819-115-3-165
50. Mawanda M, Ssenkusu JM, Odiit A, Kiguli S, Muyingo A, Ndugwa C. Micro-albuminuria in Ugandan children with sickle cell anaemia: a cross-sectional study. Ann Trop Paediatr. (2011) 31:115–21. doi: 10.1179/1465328111Y.0000000013
51. Simckes AM, Chen SS, Osorio AV, Garola RE, Woods GM. Ketorolac-induced irreversible renal failure in sickle cell disease: a case report. Pediatric Nephrol. (1999) 13:63–7. doi: 10.1007/s004670050565
52. Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. (2005) 129:465–81. doi: 10.1111/j.1365-2141.2005.05411.x
53. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. (2007) 21:37–47. doi: 10.1016/j.blre.2006.07.001
54. Bank N, Aynedjian HS, Qiu JH, Osei SY, Ahima RS, Fabry ME, et al. Renal nitric oxide synthases in transgenic sickle cell mice. Kidney Int. (1996) 50:184–9. doi: 10.1038/ki.1996.301
55. Kasztan M, Pollock DM. Impact of ET-1 and sex in glomerular hyperfiltration in humanized sickle cell mice. Clin Sci. (2019) 133:1475–86. doi: 10.1042/CS20190215
56. Zahr RS, Chappa P, Yin H, Brown LA, Ataga KI, Archer DR. Renal protection by atorvastatin in a murine model of sickle cell nephropathy. Br J Haematol. (2018) 181:111–21. doi: 10.1111/bjh.15157
57. Ataga KI, Wichlan D, Elsherif L, Derebail VK, Wogu AF, Maitra P, et al. A pilot study of the effect of atorvastatin on endothelial function and albuminuria in sickle cell disease. Am J Hematol. (2019) 94:E299–301. doi: 10.1002/ajh.25614
58. Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol JASN. (2006) 17:1703–9. doi: 10.1681/ASN.2005080872
59. Chagnac A, Herman M, Zingerman B, Erman A, Rozen-Zvi B, Hirsh J, et al. Obesity-induced glomerular hyperfiltration: its involvement in the pathogenesis of tubular sodium reabsorption. Nephrol Dial Transplant Off Publ Eur Dial Transplant Assoc Eur Renal Assoc. (2008) 23:3946–52. doi: 10.1093/ndt/gfn379
60. Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. (1997) 102:171–7. doi: 10.1016/S0002-9343(96)00407-X
61. Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Paradoxical association between body mass index and mortality in men with CKD not yet on dialysis. Am J Kidney Dis Off J Natl Kidney Foundation. (2007) 49:581–91. doi: 10.1053/j.ajkd.2007.02.277
62. Saraf SL, Molokie RE, Nouraie M, Sable CA, Luchtman-Jones L, Ensing GJ, et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatric Respiratory Rev. (2014) 15:4–12. doi: 10.1016/j.prrv.2013.11.003
63. Allard P, Alhaj N, Lobitz S, Cario H, Jarisch A, Grosse R, et al. Genetic modifiers of fetal hemoglobin affect the course of sickle cell disease in patients treated with hydroxyurea. Haematologica. (2022) 107:1577–88. doi: 10.3324/haematol.2021.278952
64. Friedrisch JR, Sheehan V, Flanagan JM, Baldan A, Summarell CC, Bittar CM, et al. The role of BCL11A and HMIP-2 polymorphisms on endogenous and hydroxyurea induced levels of fetal hemoglobin in sickle cell anemia patients from southern Brazil. Blood Cells Mol Dis. (2016) 62:32–7. doi: 10.1016/j.bcmd.2016.11.002
65. Di Maggio R, Hsieh MM, Zhao X, Calvaruso G, Rigano P, Renda D, et al. Chronic administration of hydroxyurea (HU) benefits caucasian patients with sickle-beta thalassemia. Int J Mol Sci. (2018) 19:681. doi: 10.3390/ijms19030681
66. Voskaridou E, Christoulas D, Bilalis A, Plata E, Varvagiannis K, Stamatopoulos G, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood. (2010) 115:2354–63. doi: 10.1182/blood-2009-05-221333
67. Roy NB, Fortin PM, Bull KR, Doree C, Trivella M, Hopewell S, et al. Interventions for chronic kidney disease in people with sickle cell disease. Cochrane Database Syst Rev. (2017) 7:CD012380. doi: 10.1002/14651858.CD012380.pub2
68. Saraf SL, Shah BN, Zhang X, Han J, Tayo BO, Abbasi T, et al. APOL1, alpha-thalassemia, and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica. (2017) 102:e1–6. doi: 10.3324/haematol.2016.154153
69. Geard A, Pule GD, Chetcha Chemegni B, Ngo Bitoungui VJ, Kengne AP, Chimusa ER, et al. Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol. (2017) 178:629–39. doi: 10.1111/bjh.14724
70. Kormann R, Jannot AS, Narjoz C, Ribeil JA, Manceau S, Delville M, et al. Roles of APOL1 G1 and G2 variants in sickle cell disease patients: kidney is the main target. Br J Haematol. (2017) 179:323–35. doi: 10.1111/bjh.14842
71. Adebayo OC, Betukumesu DK, Nkoy AB, Adesoji OM, Ekulu PM, Van den Heuvel LP, et al. Clinical and genetic factors are associated with kidney complications in African children with sickle cell anaemia. Br J Haematol. (2022) 196:204–14. doi: 10.1111/bjh.17832
72. Ashley-Koch AE, Okocha EC, Garrett ME, Soldano K, De Castro LM, Jonassaint JC, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol. (2011) 155:386–94. doi: 10.1111/j.1365-2141.2011.08832.x
73. Arlet JB, Ribeil JA, Chatellier G, Eladari D, De Seigneux S, Souberbielle JC, et al. Determination of the best method to estimate glomerular filtration rate from serum creatinine in adult patients with sickle cell disease: a prospective observational cohort study. BMC Nephrol. (2012) 13:83. doi: 10.1186/1471-2369-13-83
74. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF III, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Internal Med. (2009) 150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006
75. Allon M. Renal abnormalities in sickle cell disease. Arch Internal Med. (1990) 150:501–4. doi: 10.1001/archinte.150.3.501
76. Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol. (2000) 63:205–11. doi: 10.1002/(SICI)1096-8652(200004)63:4<205::AID-AJH8¿3.0.CO;2-8
77. Inusa BPD, Liguoro I, Tayo B, Booth C, Turner C, Dalton NR. Reliability of different estimated glomerular filtration rate as measures of renal function in children with sickle cell disease. Blood Cells Mol Dis. (2021) 91:102590. doi: 10.1016/j.bcmd.2021.102590
78. Marouf R, Mojiminiyi O, Abdella N, Kortom M, Al Wazzan H. Comparison of renal function markers in Kuwaiti patients with sickle cell disease. J Clin Pathol. (2006) 59:345–51. doi: 10.1136/jcp.2005.026799
79. Asnani MR, Lynch O, Reid ME. Determining glomerular filtration rate in homozygous sickle cell disease: utility of serum creatinine based estimating equations. PLoS One. (2013) 8:e69922. doi: 10.1371/journal.pone.0069922
80. Inker LA, Eneanya ND, Coresh J, Tighiouart H, Wang D, Sang Y, et al. New Creatinine- and cystatin C-based equations to estimate GFR without race. N Engl J Med. (2021) 385:1737–49. doi: 10.1056/NEJMoa2102953
81. Aparicio SA, Mojiminiyi S, Kay JD, Shepstone BJ, de Ceulaer K, Serjeant GR. Measurement of glomerular filtration rate in homozygous sickle cell disease: a comparison of 51Cr-EDTA clearance, creatinine clearance, serum creatinine and beta 2 microglobulin. J Clin Pathol. (1990) 43:370–2. doi: 10.1136/jcp.43.5.370
82. de Jong PE, de Jong-van den Berg LT, Sewrajsingh GS, Schouten H, Donker AJ, Statius van Eps LW. Beta-2-microglobulin in sickle cell anaemia. Evidence of increased tubular reabsorption. Nephron. (1981) 29:138–41. doi: 10.1159/000182331
83. Saraf SL, Zhang X, Kanias T, Lash JP, Molokie RE, Oza B, et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br J Haematol. (2014) 164:729–39. doi: 10.1111/bjh.12690
84. Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol JASN. (2006) 17:2228–35. doi: 10.1681/ASN.2002010084
85. Mihout F, Shweke N, Bige N, Jouanneau C, Dussaule JC, Ronco P, et al. Asymmetric dimethylarginine (ADMA) induces chronic kidney disease through a mechanism involving collagen and TGF-beta1 synthesis. J Pathol. (2011) 223:37–45. doi: 10.1002/path.2769
86. Rhee EP, Clish CB, Ghorbani A, Larson MG, Elmariah S, McCabe E, et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol JASN. (2013) 24:1330–8. doi: 10.1681/ASN.2012101006
87. Jerebtsova M, Saraf SL, Soni S, Afangbedji N, Lin X, Raslan R, et al. Urinary orosomucoid is associated with progressive chronic kidney disease stage in patients with sickle cell anemia. Am J Hematol. (2018) 93:E107–9. doi: 10.1002/ajh.25036
88. Jerebtsova M, Saraf SL, Lin X, Lee G, Adjei EA, Kumari N, et al. Identification of ceruloplasmin as a biomarker of chronic kidney disease in urine of sickle cell disease patients by proteomic analysis. Am J Hematol. (2018) 93:E45–7. doi: 10.1002/ajh.24965
89. Nekhai S, Lin X, Soni S, Taye A, Smith N, Afangbedji N, et al. Urinary kringle domain-containing protein HGFL: a validated biomarker of early sickle cell anemia-associated kidney disease. Am J Nephrol. (2021) 52:582–7. doi: 10.1159/000517056
90. Jiang H, Guan G, Zhang R, Liu G, Liu H, Hou X, et al. Increased urinary excretion of orosomucoid is a risk predictor of diabetic nephropathy. Nephrology. (2009) 14:332–7. doi: 10.1111/j.1440-1797.2008.01053.x
91. Watson L, Midgley A, Pilkington C, Tullus K, Marks S, Holt R, et al. Urinary monocyte chemoattractant protein 1 and alpha 1 acid glycoprotein as biomarkers of renal disease activity in juvenile-onset systemic lupus erythematosus. Lupus. (2012) 21:496–501. doi: 10.1177/0961203311431249
92. Moon PG, Lee JE, You S, Kim TK, Cho JH, Kim IS, et al. Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics. (2011) 11:2459–75. doi: 10.1002/pmic.201000443
93. Gudehithlu KP, Hart P, Joshi A, Garcia-Gomez I, Cimbaluk DJ, Dunea G, et al. Urine exosomal ceruloplasmin: a potential early biomarker of underlying kidney disease. Clin Exp Nephrol. (2019) 23:1013–21. doi: 10.1007/s10157-019-01734-5
94. Becton LJ, Kalpatthi RV, Rackoff E, Disco D, Orak JK, Jackson SM, et al. Prevalence and clinical correlates of microalbuminuria in children with sickle cell disease. Pediatric Nephrol. (2010) 25:1505–11. doi: 10.1007/s00467-010-1536-8
95. Bolarinwa RA, Akinlade KS, Kuti MA, Olawale OO, Akinola NO. Renal disease in adult Nigerians with sickle cell anemia: a report of prevalence, clinical features and risk factors. Saudi J Kidney Dis Transplant Off Publ Saudi Center Organ Transplant Saudi Arabia. (2012) 23:171–5.
Keywords: sickle cell disease, glomerular filtration, hyperfiltration, chronic kidney disease, sickle cell nephropathy, biomarkers
Citation: Afangbedji N and Jerebtsova M (2022) Glomerular filtration rate abnormalities in sickle cell disease. Front. Med. 9:1029224. doi: 10.3389/fmed.2022.1029224
Received: 26 August 2022; Accepted: 04 October 2022;
Published: 21 October 2022.
Edited by:
Ana Cusumano, Centro de Educación Médica e Investigaciones Clínicas Norberto Quirno (CEMIC), ArgentinaReviewed by:
Isaac Ejembi Ocheke, Jos University Teaching Hospital, NigeriaCopyright © 2022 Afangbedji and Jerebtsova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Jerebtsova, marina.jerebtsova@howard.edu